
Abstract
Only a small fraction (10%) of genetically unselected patients with chemorefractory metastatic colorectal cancer benefits from the anti-EGFR antibodies cetuximab or panitumumab (‘primary’ or ‘de novo’ resistance). Further, almost all patients who initially respond become resistant over the course of treatment (‘secondary’ or ‘acquired’ resistance). Studies in cell lines, patient-derived tumorgrafts, and archival surgical specimens have identified many biomarkers of both primary and acquired resistance to anti-EGFR antibodies, and it is now evident that resistance mechanisms revolve around common genetic lesions and share analogous signaling traits. Here we discuss how resistance to the EGFR blockade is attained in colorectal cancer and elaborate on alternative therapeutic strategies that are now under development to improve response and contrast relapse.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Colorectal cancer
- Epidermal growth factor receptor
- Mitogen-activated protein kinase kinase
- Drug resistance
1.1 Introduction
Colorectal cancer (CRC) is the third commonest cancer worldwide, with approximately 20% of newly-diagnosed patients already presenting with metastatic disease and 50% of patients developing metastasis in subsequent months or years. The median overall survival (OS) is around 20 months [1,2,3,4,5].
The outlook of patients with metastatic colorectal cancer (mCRC ) has been advanced by the introduction in the clinical practice of cetuximab and panitumumab , two monoclonal antibodies (moAbs) that inhibit the epidermal growth factor receptor (EGFR/ErbB1/HER1). These agents are typically administered in combination with chemotherapy in the second- or third-line treatment of individuals who have become resistant to previous rounds of cytotoxic chemotherapy [6,7,8]; in this chemorefractory setting, patients achieve an objective response and disease stabilization rates of approximately 10% and 20%, respectively [7,8,9]. Different from other tumor types, such as non-small cell lung cancers (NSCLCs) or melanomas , in which actionable targets such as EGFR or BRAF are constitutively hyperactive as a consequence of underlying genetic alterations [10, 11], mutational abnormalities in the EGFR gene are extremely infrequent in colorectal tumors (see below).
The 70% of CRC tumors that are intrinsically refractory to EGFR blockade display primary (also known as innate) resistance. Acquired (or secondary) resistance refers to disease progression in the face of an ongoing treatment that was initially effective. In both primary and secondary resistance, lack of response can be explained by compensatory signaling activities driven by mutational events or adaptive mechanisms such as biochemical feedbacks or gene expression changes [12, 13]. In the case of colorectal cancer, acquired resistance typically occurs within 3–18 months after treatment initiation [7, 8]. Starting with seminal observations in 2006–2007 [14, 15], several biomarkers of primary resistance to anti-EGFR moAbs in mCRC patients have been progressively identified and biologically validated, and some of them are now routinely used to exclude a number of molecularly defined nonresponders from unnecessary treatment [16, 17]. The topic of acquired resistance has received preclinical and clinical focus more recently, with the emergence of new critical information only in the last 5 years.
Here, we will survey the current state of the art on primary and acquired resistance to anti-EGFR moAbs in mCRC, from early mechanistic investigations to clinical applications, and will discuss fresh knowledge on how to improve the response and delay the relapse in mCRC patients. This chapter is inspired, with relevant updates, to a review article that we have recently authored [18].
1.2 The Genomic Landscape of Resistance to Anti-EGFR Antibodies in Patients with Metastatic Colorectal Cancer
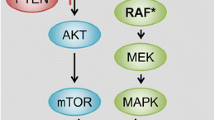
EGFR is a member of the ErbB family of receptor tyrosine kinases (RTKs ), which also includes HER2/neu (ERBB2), HER3 (ErbB3) and HER4 (ErbB4) [19]. Following homo- and hetero-dimerization of EGFR with itself or other ErbB members, induced by EGF or other EGF-like ligands, several downstream signal transduction pathways are activated, including the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR axes, but also SRC-like family kinases, PLCγ-PKC, and STATs [19, 20]. Such activation stimulates key processes involved in tumor growth and progression, including proliferation, survival, angiogenesis, invasion, and metastasis [21] (Fig. 1.1).

EGFR signaling pathways . (a) Following ligand binding and the ensuing receptor homo- and hetero-dimerization, ErbB family members trigger several signaling pathways, including the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR axes, the SRC family kinases, PLCγ-PKC, and STATs. All these signals stimulate cell proliferation and/or survival
Of note, the EGFR gene is very rarely mutated or amplified in CRC . Because ‘addiction’ to the EGFR pathway does not have genetic underpinnings, this dependency may represent an aberrant declination of para-physiological traits typical of normal colonic tissues. In the adult intestine, mucosal renewal after tissue damage is prompted by increased EGFR signaling (through transcriptional induction of the receptor and autocrine production of the cognate ligands) [22,23,24], and is impaired by EGFR inhibition [22]. Importantly, EGFR neutralization curbs the propensity of epithelial cells to undergo neoplastic transformation promoted by inflammatory stimuli [25]. Altogether, these observations suggest that persistent upregulation of EGFR activity during chronic intestinal inflammation —a condition that typically predisposes to colorectal cancer—may act as a pro-tumorigenic cue. This stimulation would positively select for cancer cells relying on EGFR-driven signals for their growth, explaining why a fraction of CRCs are strictly dependent on EGFR activity even in the absence of underlying genetic alterations. On this ground, it comes with no surprise that the increased expression of EGFR and EGFR ligands not only encourages intestinal regeneration during inflammation, but also characterizes ‘EGFR-addicted’ tumors with marked sensitivity to EGFR inhibition [26,27,28].
In the absence of genetic alterations correlating with sensitivity to anti-EGFR antibodies, patient stratification is only applied by subtraction: in general terms, the commonest mechanisms of innate resistance involve genomic alterations affecting EGFR downstream effectors , such as KRAS/NRAS and PIK3CA mutations, with consequent constitutive pathway hyperactivation. The RAS and PI3K signaling cascades can also be triggered by upstream RTKs other than EGFR [29], leading to an oncogenic shift [30]. In this situation, the primary drug target remains unaltered and continues to be inhibited while an alternative signal transducer becomes activated, circumventing the effects of EGFR inhibition [13, 31] (Fig. 1.2a–c).

Mechanisms of resistance to anti-EGFR moAbs in mCRC . (a) By binding the extracellular domain of EGFR, both cetuximab and panitumumab prevent ligand-induced activation of downstream signaling. (b) Activating mutations of genes encoding EGFR transducers such as KRAS (by either point mutations or gene amplification), BRAF, PIK3CA and MAP2K1 (MEK1), or PTEN loss of function, cause relentless activation of downstream signaling that circumvent EGFR inhibition. (c) Excessive activation (by either receptor gene amplification or high ligand expression) of alternative receptors, such as HER2 or MET (not shown), can substitute for EGFR inhibition and activate downstream pathways. (d) Additional genetic alterations within the target receptor may abolish antibody binding (EGFR extra-cellular domain mutations) or mediate EGFR activation even in the presence of the drug (kinase domain mutations)
It is becoming increasingly clear that tumors can contain a high degree of mutational heterogeneity within the same lesion [32]. Thus, secondary resistance can arise not only through stochastic acquisition of de novo genetic lesions along treatment, but also through therapy-induced selection of intrinsically resistant minor subclones already present in the original tumor [33]. If secondary resistance can be re-interpreted as the emergence, under drug pressure, of rare tumor subpopulations featuring primary resistance, then the molecular mechanisms of primary and acquired resistance are expected to be the same. Accordingly, hereinafter we will delineate resistance predictors as absolute traits, specifying, for each determinant, how it contributes to primary or secondary resistance. We will also concentrate on current research efforts that have put forward alternative strategies to bypass such resistances in patients with no other therapeutic options. Table 1.1 summarizes the main predictors of primary and acquired resistance observed in mCRC patients and describes potential approaches for tackling them therapeutically.
1.2.1 RAS
The RAS family includes three small G proteins (KRAS, NRAS, and HRAS) that couple EGFR to downstream activation of the RAF-MEK-ERK pathway [30]. Several retrospective trials have linked KRAS mutations in exon 2 (codons 12 and 13), which are found in approximately 40–45% of CRCs [17, 34], to primary resistance to cetuximab or panitumumab [14, 35,36,37]. The robust predictive significance of such correlations was sufficient for the regulatory approval of companion diagnostics for routine assessment of KRAS exon 2 mutations, and now the clinical use of anti-EGFR moAbs is limited to the subset of patients with KRAS-wild-type colorectal cancers [34, 38,39,40,41,42].
Although the exclusion of patients with KRAS (exon 2)-mutant tumors from anti-EGFR therapy has increased the percentage of responders from 10% to 13–17%, most KRAS (exon 2) wild-type tumors remain insensitive to anti-EGFR moAbs [34, 40]. Rare mutations of KRAS in codons other than 12 and 13, as well as mutations of NRAS , have been found to correlate with therapeutic refractoriness. The relatively high cumulative frequency of such additional mutations, and their successful validation as resistance biomarkers in prospective trials, strongly call for systematic evaluation of these genotypes in clinical practice to enlarge the fraction of patients to be spared anti-EGFR therapy [43]. A very low frequency of KRAS amplification (0.7%) has also been reported and demonstrated to correlate with primary resistance [44].
RAS activating mutations and gene copy number gains are responsible not only for primary resistance but also for acquired resistance in 40–60% of patients who progress on cetuximab or panitumumab [45,46,47]. As mentioned above, such mutations are either pre-existing in minor tumor subclones before treatment initiation [45, 46] or arise as de novo alterations under drug pressure [46, 47]. KRAS mutations could be detected non-invasively 5–10 months before radiographic evidence of disease progression by analyzing cell-free circulating tumor DNA (ctDNA) [45, 46]. Using this methodology, two recent studies have documented the emergence of several independent clones displaying heterogeneous patterns of KRAS and NRAS mutations in concomitance with progressive desensitization to EGFR blockade [48, 49].
At present, patients with KRAS-mutant mCRC are treated with chemotherapy (with or without anti-angiogenic therapy) and, in the chemorefractory setting, with the multi-target inhibitor regorafenib [50, 51]. To date, direct pharmacologic blockade of the mutant KRAS protein has been unsuccessful; therefore, preclinical studies have concentrated on approaches as different as targeting downstream effectors such as MEK and PI3K [52], leveraging synthetic lethal interactions [53,54,55,56,57,58], or deploying high-throughput drug screens [59]. Most of these attempts showed that the combinatorial inhibition of two different pathways induces some anti-cancer effects in KRAS mutant CRC mouse models, albeit seldom with manifest tumor shrinkages [60] (see Table 1.1). Some of these preclinical strategies have been translated in recently completed phase I/II clinical trials (NCT01085331; NCT01390818; NCT02039336), for which the results are eagerly awaited. In the case of acquired resistance due to RAS mutations, preclinical evidence suggests that combination therapies ab initio with EGFR and MEK inhibitors could delay or reverse the emergence of resistance [48, 61].
1.2.2 BRAF
Point mutations of BRAF, which encodes a serine/threonine kinase directly activated by RAS and impinging on the downstream effector MEK , are found in 4–13% of advanced CRCs and are typically mutually exclusive with KRAS mutations [17, 62].
The BRAF V600E mutation has been described as a determinant of poor response to cetuximab and panitumumab [15, 17, 62, 63]. However, the negative predictive power of BRAF mutations is undermined by their low frequency and is further biased by the pervasive role of mutant BRAF as a negative prognostic biomarker [41, 62,63,64]. Overall, the predictive impact of this alteration remains to be established and requires further prospective evaluation before clinical applicability [17, 41, 62, 65].
Unlike RAS, BRAF can be efficiently blocked by clinically approved compounds; BRAF small-molecule inhibitors are extensively and successfully used in BRAF-mutant melanoma, for example, with response rates (RRs) ranging between 48% and 67% [10, 66]. However, selective BRAF inhibitors such as vemurafenib have failed in BRAF-mutant CRCs (RR of 5%) [67]; this lack of efficacy has been ascribed to rapid feedback activation of EGFR following BRAF inactivation , resulting in constitutive signaling through the MAPK–ERK pathway and continued tumor cell proliferation [68, 69]. Accordingly, preclinical studies have demonstrated that BRAF blockade can resensitize to anti-EGFR antibodies [62, 68,69,70]. At the clinical level, interim reports from an ongoing clinical trial have shown 22% RRs in patients with BRAF-mutant mCRC treated with a combination of cetuximab and encorafenib, an investigational BRAF inhibitor [71]. The trial has now entered a phase II expansion cohort (NCT01719380). Investigators are also collecting tumor and blood samples from patients before and after treatment to analyze the drugs’ pharmacodynamic consequences, while a broad genomic survey is planned to identify predictive biomarkers [71]. Other combinatorial approaches under preclinical or clinical evaluation [59, 72,73,74] are listed in Table 1.1.
Intriguingly, some BRAF wild-type CRCs display a gene expression signature and a clinical behavior (poor prognosis) that are very similar to those typifying BRAF-mutant tumors [75]. By applying a loss-of-function genetic screen, cell lines from this specific tumor subtype were shown to have defects in microtubule formation, unveiling a potential vulnerability to microtubule-disrupting agents [76].
BRAF mutations could be also captured non-invasively by ctDNA analysis, together with concomitant KRAS and NRAS mutations [48, 49], in patients who had responded to anti-EGFR antibodies and then progressed. Hence, the emergence of BRAF mutant subclones may also sustain acquired resistance.
1.2.3 PI3K-AKT-PTEN Pathway
PI3Ks include different classes of lipid kinases; in particular, activation of class IA PI3Ks can be triggered by upstream stimulation from RTKs [77], but also through RAS intermediation [78] or signaling from G protein-coupled receptors [19].
Class IA PI3Ks are heterodimeric proteins composed of a regulatory (p85) and a catalytic (p110) subunit [79]. Activating mutations of PIK3CA (encoding p110α) have been detected in 10–20% of CRCs [17, 80,81,82]; most of them occur in exons 9 and 20, respectively, in the helical and kinase domain [80, 83]. In a retrospective analysis of 110 mCRC patients treated with cetuximab or panitumumab, a statistically significant association between primary resistance to EGFR inhibition and PIK3CA mutations (11 in exon 20 and 4 in exon 9, all in KRAS wild-type tumors) was reported [84]. Another study, conducted in a patient cohort with a higher prevalence of exon 9 mutations, did not confirm such a correlation [82]. These discrepant data were then reconciled by a retrospective consortium analysis on a larger collection of 1022 tumor samples; the consensus is now that, in the KRAS wild-type subpopulation, only the PIK3CA exon 20 mutations may be predictive of lack of response to anti-EGFR moAbs [17]. This study also highlighted a strong association between PIK3CA exon 9 (but not exon 20) mutations and KRAS mutations, reinforcing the notion that PIK3CA exon 9 mutations do not have an independent predictive value for anti-EGFR antibody efficacy.
Loss of function of PTEN , a phosphatase that contrasts PI3K activity, occurs in 30% of CRCs through various mechanisms including gene deletion, frameshift or nonsense mutations, and promoter methylation [85, 86]. PTEN inactivation (usually evaluated as lack of protein expression) has been associated with poor sensitivity to anti-EGFR moAbs in mCRC patients in several studies [16, 85, 87, 88], whereas others have only put forward a prognostic role [63]. All in all, both PIK3CA exon 20 mutations and PTEN inactivation are promising predictors of reduced responsiveness to anti-EGFR therapies. However, due to the low incidence of exon 20 mutations (2–5%) [89] and lack of an established method for assessment of PTEN inactivation [17, 85, 88, 90, 91], further prospective trials and methodological efforts are necessary to validate the clinical utility of PI3K pathway activation as a negative response determinant.
In principle, patients with tumors exhibiting PIK3CA mutations or PTEN loss of function, without concomitant KRAS/BRAF mutations, may respond to therapies targeting PI3K or PI3K-downstream transducers, such as mTOR or AKT [92]; however, clinical data have demonstrated only minimal single-agent activity of such therapies at tolerated doses [93,94,95]. Since the PI3K/AKT inhibition is commonly counteracted by feedback activation of tyrosine kinase receptors [96], it is expected that blockade of the PI3K pathway will provide greater benefit when combined with RTK inhibitors [97]. Phase I/II studies testing mTOR inhibitors, such as everolimus or temsirolimus, in combination with RTK inhibitors or anti-EGF moAbs (in some cases, in the presence of a chemotherapy backbone) are presently being conducted or have been recently completed in mCRC patients (NCT01154335; NCT01139138; NCT01387880; NCT00827684).
Finally, prevention studies have shown improved survival by low-dose aspirin in patients with PIK3CA-mutant CRC [98,99,100]; this observation, which demands further prospective evaluation, could be at least partially related to the fact that the PI3K-AKT axis induces NF-ĸB-dependent transcriptional upregulation of COX2, which has been demonstrated to exert pro-survival signals in CRC cells [100,101,102]. Therefore, a PIK3CA-mutant makeup may render CRC cells vulnerable to apoptosis by aspirin-mediated COX2 inhibition.
Recently, the presence of PIK3CA mutations has been also detected in tissue samples from mCRC patients treated with cetuximab who relapsed while on treatment. Of note, such mutations coexisted with other acquired mutations (in KRAS, NRAS or BRAF genes) within the same sample [103].
1.2.4 HER2
When considering the cumulative frequency of KRAS, NRAS, BRAF, and PIK3CA alterations, approximately 60–65% of anti-EGFR resistant cases can be ascribed to the presence of such mutations [16]; in the remaining 30% of ‘quadruple negative’ cases, still-unidentified features sustain lack of response.
HER2 is the only member of the ErbB family that is not bound by growth factor ligands; it is activated through hetero-dimerization with other ligand-stimulated receptors [20], with the most powerful growth-promoting cues generated by HER2-HER3 heterodimers; HER2 overexpression, usually caused by gene amplification, enables HER2 constitutive signaling regardless of the activation state of the other partners [104].
Several preclinical and clinical studies have shown that HER2 amplification is a predictor of poor sensitivity to anti-EGFR antibodies [105, 106]. Based on genotype-response correlations in a platform of patient-derived mCRC tumorgrafts, HER2 amplification was found to be significantly associated with resistance to cetuximab and specifically enriched in the quadruple negative population [91]. Aberrant HER2 signaling (by either HER2 amplification or overproduction of the HER3 ligand heregulin) was confirmed as a mediator of lack of response in an independent report [106]. In retrospective clinical studies, patients with colorectal tumors displaying HER2 amplification or heregulin overexpression and treated with cetuximab or panitumumab had shorter progression-free and overall survival compared with patients with HER2 wild-type tumors [105,106,107]. Notably, in patients with acquired resistance, HER2 amplification was detected in a small fraction (14%) of pretreatment tumor cells and in a much larger proportion of cells (71%) in samples biopsied after anti-EGFR therapy . Similarly, heregulin levels, as assessed in both plasma and tumor specimens, were found to be significantly higher in patients who had relapsed on anti-EGFR therapy with respect to responders [106]. Hence, increased HER2 signaling drives both primary and acquired resistance.
Besides HER2 amplification, also HER2 activating point mutations can confer resistance to EGFR blockade in CRC cell lines and patient-derived tumorgrafts [108]. In both instances (amplification and mutations), monotherapy with either anti-HER2 antibodies or HER2 small-molecule inhibitors was not sufficient to induce regression of patient-derived tumorgrafts in mice, and only a combination of antibodies and chemical inhibitors led to massive tumor shrinkage [108, 109]. At least for HER2 amplification in CRC, trastuzumab (the prototypical anti-HER2 antibody) alone was found to be mainly active against HER3, with minor inhibitory effects on HER2 and EGFR. In contrast, the reversible HER2 small-molecule inhibitor lapatinib prompted rapid and drastic dephosphorylation of all ErbB receptors, but also led to delayed reactivation of HER3 as a compensatory mechanism. Indeed, the stronger effect of the antibody-small molecule combination was attributed to the ability of trastuzumab, through preferential targeting of HER3, to prevent lapatinib-induced HER3 rephosphorylation [109].
These preclinical findings encouraged the design and execution of HERACLES, a clinical trial that assessed the efficacy of the trastuzumab-lapatinib combination in mCRC patients with KRAS wild-type, HER2-amplified, cetuximab-resistant tumors. Eight (30%) patients achieved objective responses, and 12 (44%) had stable disease [110]. Because this patient subpopulation was heavily pretreated and resistant to both conventional chemotherapy and anti-EGFR antibodies, the outcome data are particularly compelling and testify to the potential of HER2 as a viable target in the treatment of colorectal cancer.
Active HER2 also exacerbates the oncogenic properties of HER3 mutations, which have been recently described in about 11% of colon cancers [111]. One could envision a ‘dosage effect’ whereby low-grade HER2 amplification or low levels of heregulin, which alone would not be enough to foster therapeutic resistance, might in fact attenuate sensitivity to EGFR inhibition by cooperating with co-existing HER3 mutations. Investigational anti-HER3 antibodies and small molecules have been shown to productively contrast HER3-mediated signals and tumor progression in preclinical studies in vivo [111] and are now being tested clinically. Therefore, HER3 mutations in CRC merit investigation as new potential biomarkers of resistance to anti-EGFR treatment as well as new predictors of response to other therapeutic options.
1.2.5 MET
Similar to EGFR family members, the MET tyrosine kinase receptor for hepatocyte growth factor (HGF) can activate growth, survival and motility pathways through the RAS- ERK cascade, the PI3K-AKT axis, and stimulation of SRC and STAT [112,113,114]. Excessive MET signaling may occur by several mechanisms, including genetic abnormalities such as MET amplification and exon 14 skipping mutations (splicing variants that result in the deletion of a negative regulatory domain of the MET kinase), but also as a consequence of increased HGF expression/activity [96]. When genetically altered, MET can act both as a primary oncogenic driver and as a determinant of resistance to EGFR tyrosine kinase inhibitors , in particular in NSCLCs harboring EGFR mutations [115,116,117]. MET amplification also sustains tumorigenesis and correlates with response to MET small-molecule inhibitors in gastroesophageal cancer [118].
In CRC, MET amplification has been documented as a mechanism of primary and acquired resistance to cetuximab and panitumumab [119]. In retrospective analyses, MET amplification was detected in around 1% of mCRC samples, in line with previous findings [120]. However, this frequency increased to 12.5% in a subgroup of cetuximab-resistant patient-derived tumorgrafts with wild-type forms of KRAS, NRAS, BRAF, PIK3CA and HER2. Notably, MET-mediated resistance appears to be driven by a dosage effect: only focal, high-grade amplification of the MET locus correlated with overt therapeutic refractoriness, whilst tumors with modest gene copy number gains or polysomy of chromosome 7, where the MET gene is located, were still susceptible to cetuximab [120]. Preclinical trials in MET-positive xenografts from CRC cell lines and patient-derived materials revealed that MET inhibition, with or without concurrent interception of EGFR, led to long-lasting abolition of tumor growth [119, 121]. In this vein, a phase II clinical trial aimed to assess the efficacy and safety of the dual MET-ALK inhibitor crizotinib in patients with solid tumors (including CRCs) harboring MET genetic alterations has been designed and is currently recruiting participants (NCT02034981).
MET amplification was also found in the tumors of three out of seven patients who had developed a form of acquired resistance to the anti-EGFR antibodies that could not be ascribed to the emergence of secondary KRAS mutations. Importantly, the MET amplicon was detected in circulating, cell-free DNA as early as 3 months after treatment initiation, well before relapse was observed radiologically. Similar to HER2 amplification and KRAS mutations, rare MET-amplified cells could be identified in pre-treatment tumor material from one out of three patients with MET-dependent acquired resistance, suggesting that pre-existing subclones were positively selected under the pressure of anti-EGFR therapy [119].
A recent case report suggests that MET amplification in CRC not only precludes sensitivity to upstream EGFR blockade, but also prevents responsiveness to agents targeting the downstream RAS pathway. A patient with a BRAF-mutant mCRC who had initially responded to combined EGFR and BRAF inhibition progressively developed resistance. Genetic analysis of matched biopsies before and after therapy revealed a higher representation of MET-amplified cancer cells in the post-treatment tissue, and dual blockade of both BRAF and MET proved to be clinically effective [122]. Again, these results point to MET hyperactive signaling as a pervasive resistance trait in mCRC, and highlight the value of MET therapeutic targeting to oppose disease progression.
MET activation can attenuate sensitivity to cetuximab also as a consequence of paracrine HGF stimulation, as observed in CRC cell lines [119, 123] or, more recently, in CRC spheroids enriched in cancer stem cells [124]. In these studies, only concomitant inhibition of both MET and EGFR substantially regressed tumors in vivo. This experimental evidence might have clinical relevance, as HGF overexpression correlates with reduced sensitivity to cetuximab in patients [124]. However, the definition of cut-offs to dichotomize HGF-positive versus HGF-negative tumors in the clinic is not trivial, which undermines the portability of assessing HGF levels for patient stratification.
1.2.6 EGFR
Additional genetic alterations within the target oncoprotein, which affect drug binding thus preventing kinase inhibition , are frequently responsible for both primary and acquired resistance in cancer; an emblematic example is represented by the T790M ‘gatekeeper’ secondary mutation in the EGFR gene, which drives resistance to first-generation EGFR small-molecule inhibitors in EGFR-mutant NSCLC [125]. In colorectal cancer, different mutations in the extracellular domain of EGFR have been recently described as a typical mechanism of acquired resistance, namely, S492R, G465E and G465R mutations [126,127,128] (Fig. 1.2d). Structural analyses indicate that while S492 selectively lies in the cetuximab binding site, G465 is located in the center of the region in which the epitopes of both cetuximab and panitumumab overlap. Accordingly, S492R abrogates cetuximab binding but retains panitumumab interaction, whereas G465E and G465R prevent binding of both antibodies. Studies in patient-derived tumorgrafts [128] and cell cultures [129] harboring mutations in the G465 residue have shown that new-generation anti-EGFR antibodies that bind EGFR epitopes different from those recognized by cetuximab and panitumumab are very effective in opposing the growth of these tumors.
Resistance may be also driven by mutations in the EGFR kinase domain: two alterations have been identified as circulating mutations by cell-free DNA analysis [49], and one has been detected in cetuximab-resistant patient-derived tumorgrafts [128]. Treatment of such tumorgrafts with an EGFR small-molecule inhibitor or cetuximab alone was not effective, but the combination resulted in substantial and durable inhibition of tumor growth [128].
1.3 Newly Emerging Biomarkers of Drug Resistance and Sensitivity
A recent systematic survey of molecularly annotated patient-derived tumorgrafts has functionally linked therapeutic responses to EGFR inhibitors with complete exome sequence and copy number analyses as a way to identify new resistance traits and, potentially, new druggable targets. By doing so, in addition to the genetic abnormalities described above, new alterations have been found, including mutations/amplification in FGFR1, PDGFRA and MAP2K1 [128] and outlier overexpression of IGF2 [28]. All these tumorgrafts proved to be susceptible to therapies targeting the resistance-conferring genetic alterations. Another actionable lesion in CRC that has recently received clinical attention is the NTRK1 chromosomal rearrangement, which leads to the synthesis of a highly expressed fusion protein with constitutive NTRK kinase activity. A case report has described a patient with metastatic colorectal cancer harboring an LMNA–NTRK1 rearrangement who achieved a remarkable clinical and radiographic response to entrectinib (RXDX-101), a multikinase inhibitor targeting TRK, ALK, and ROS1, which was followed by the emergence of resistance [130]. Longitudinal monitoring of the LMNA–NTRK1 status by ctDNA analysis revealed the acquisition of two novel NTRK1 kinase domain mutations (G595R and G667C) that were absent from ctDNA collected at the time of treatment initiation. According to structural studies, such mutations are expected to abrogate or reduce entrectinib binding to the catalytic pocket, rendering tumors less vulnerable to this specific inhibitor [131].
While the quest for resistance biomarkers has yielded considerable results in the past years, data remain immature as far as the identification of positive determinants of responsiveness to EGFR blockade is concerned. As noted above, EGFR is very rarely mutated or amplified in CRC, and the only known means to achieve EGFR hyperactivation seems to be increased paracrine/autocrine expression of some EGFR ligands , in particular amphiregulin and epiregulin. Accordingly, high levels of amphiregulin and epiregulin correlate with a better response to anti-EGFR moAbs [26, 27, 29, 132, 133]. However, as already discussed for HGF, the clinical application of this information is hindered by the difficulty in setting thresholds to distinguish ligand-positive versus ligand-negative tumors. Intriguingly, responsive cases appear to be enriched for genetic lesions (mutations or amplification) of IRS2, a cytoplasmic adaptor protein that relays signals from tyrosine kinase receptors to downstream effectors [128]. In functional assays, RNA interference-mediated silencing of IRS2 was accompanied by attenuated sensitivity to cetuximab and reduced activation of EGFR-dependent pathways, in line with the role of IRS2 as an amplifier of tyrosine kinase signals . The clinical applicability of this information for optimized selection of responsive patients remains to be determined.
1.4 Outlook
Although many genetic determinants of resistance to anti-EGFR antibodies have been recently documented, and some of them have been validated as alternative pharmacologic targets, there is still space for the identification of additional druggable alterations and the deployment of further therapeutic strategies. Genome-scale analyses of CRC tumor collections are expected to provide a fresh catalog of new mutations, rearrangements, and copy-number alterations with therapeutically actionable potential [134, 135] and will receive further momentum by proteogenomics data [136]. Moreover, promising results are being offered by treatments that disrupt immune evasion strategies. To stimulate immune suppression, tumor cells often engage immune checkpoint molecules, such as CTLA-4 and PD1, which quench cytotoxic T-cell activation. Antibodies against CTLA-4 (e.g., ipilimumab) or PD1 (e.g., nivolumab, pembrolizumab) have been shown to induce durable tumor regressions [137, 138] in mismatch repair–deficient colorectal cancer, likely because the large number of somatic mutations present in these hypermutated tumors increase the presentation of non-self immunogenic neo-antigens and, hence, sensitize to immune checkpoint blockade [139].
Although several resistance mechanisms have been documented so far, mutant RAS is the only clinically validated biomarker for selection of mCRC patients eligible to treatment with anti-EGFR antibodies. This attrition between experimental discovery and clinical implementation advocates the introduction of new clinical trial designs that capitalize on reliable preclinical findings. In this regard, a successful story is our experience with mCRC cases harboring HER2 amplification: from retrospective identification of this alteration in archival patient material, and after establishing a statistically robust correlation between the occurrence of HER2 amplification and primary resistance to EGFR inhibition, we moved to testing different therapeutic options in HER2-positive patient-derived tumorgrafts and found one treatment that resulted in overt and long-lasting tumor regression [105, 109]. The very same regimen was then applied to patients with HER2-amplified tumors with positive results [110]. In this case, reliable tumor models, stringent endpoint criteria for animal studies, and accurate genetic selection were the ingredients that made this translational effort a winning opportunity.
Future clinical trials will be informed by real-time monitoring of tumor evolution along treatment so as to adjust therapies (likely, combination therapies) to the continuing mutability of cancer. While multi-dimensional analysis of serial biopsies is, in principle, the most informative approach, it should also be considered that an individual tumor biopsy may not be representative of overall intratumor heterogeneity, and post-treatment tumor tissue is difficult to obtain. Such limitations can be overcome by less invasive analyses on ctDNA , which can offer a high degree of sensitivity and specificity to detect the surfacing of resistance-conferring mutations over the course of therapy [49, 140]. The mechanism by which ctDNA is released into the bloodstream and whether multiple metastases, or different regions within the same tumor, shed ctDNA homogeneously are still unclear; however, the proof-of-concept that such an approach is viable and its merit in raising an early warning of acquired resistance are now consolidated [46, 49, 141, 142]. Inevitably, to gather a more comprehensive picture of tumor adaptation to targeted treatment and to more effectively tackle the ever-evolving resistant phenotype at the therapeutic level, mutational analysis needs to be integrated by other molecular approaches that detect changes in gene expression, proteins, and protein activities. While this is feasible, at present, only in bioptic material—with all the hurdles and challenges related to repeated biopsies discussed above—hints are emerging whereby non-invasive techniques may prove useful also to measure RNA and protein/phosphoprotein levels in blood, for example by isolating circulating exosomes [143].
If appropriately dosed in quantity and scheduled in time, new investigational therapies could also leverage tumor heterogeneity to their own advantage: creating a “balance” between drug activity and graded responsiveness of different clones to drug pressure might be useful to retard the onset of resistance and, ideally, to turn cancer into a chronic disease. Intriguingly, the prevalence of KRAS mutant subclones that become detectable in the blood of mCRC patients on anti-EGFR therapy has been demonstrated to decline after treatment withdrawal, leaving space to KRAS wild-type populations that regain drug sensitivity [142]. This could explain why some mCRC patients benefit from multiple challenges with anti-EGFR antibodies.
More than a decade after the introduction of cetuximab in the treatment of metastatic colorectal cancer, much is known about the genetic determinants of primary and acquired resistance to anti-EGFR moAbs in CRC. What is now becoming increasingly clear is that therapeutic resistance is not a fixed, irreversible state, but rather the expression of a resilient phenotype that reacts to drug pressure through manifold sophisticated elusion strategies. The time is ripe to move from a static vision of the disease to a more flexible appraisal of tumor evolution, adaptation and dynamic instability.
Abbreviations
- BRAF:
-
v-Raf murine sarcoma viral oncogene homolog B1
- CRC:
-
Colorectal cancer
- ctDNA:
-
Circulating tumor DNA
- EGFR/ErbB1/HER1:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal regulated kinase
- HER2/neu/ERBB2:
-
V-ERB-B2 avian erythroblastic leukemia viral oncogene homolog 2
- KRAS:
-
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- mCRC:
-
Metastatic colorectal cancer
- MEK:
-
Mitogen-activated protein kinase kinase
- moAbs:
-
Monoclonal Antibodies
- NRAS:
-
Neuroblastoma RAS viral oncogene homolog
- PIK3CA:
-
Phosphatidylinositol 3-kinase, catalytic, alpha
- PTEN:
-
Phosphatase and tensin homolog
- RR:
-
Response rate
- RTKs:
-
Receptor Tyrosine kinases
References
Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18(3):581–92.
Poston GJ, Figueras J, Giuliante F, Nuzzo G, Sobrero AF, Gigot JF, Nordlinger B, Adam R, Gruenberger T, Choti MA, Bilchik AJ, Van Cutsem EJ, Chiang JM, D’Angelica MI. Urgent need for a new staging system in advanced colorectal cancer. J Clin Oncol. 2008;26(29):4828–33.
Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med. 2009;60:207–19.
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–403.
Garden OJ, Rees M, Poston GJ, Mirza D, Saunders M, Ledermann J, Primrose JN, Parks RW. Guidelines for resection of colorectal cancer liver metastases. Gut. 2006;55:iii1–8.
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, HJ A, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–8.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–45.
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658–64.
Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101(19):1308–24.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, Kuhlmann GL, Han M, Goldberg JS, Settleman J, Iafrate AJ, Engelman JA, Haber DA, Johnson BE, Lynch TJ. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–9.
Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214–26.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26.
Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–5.
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67(6):2643–8.
Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani T, Marrapese G, Mazzucchelli L, Lamba S, Veronese S, Frattini M, Bardelli A, Siena S. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One. 2009;4(10):e7287.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–62.
Leto SM, Trusolino L. Primary and acquired resistance to EGFR-targeted therapies in colorectal cancer: impact on future treatment strategies. J Mol Med (Berl). 2014;92(7):709–22.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37.
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–54.
Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7(10):2958–70.
Egger B, Procaccino F, Lakshmanan J, Reinshagen M, Hoffmann P, Patel A, Reuben W, Gnanakkan S, Liu L, Barajas L, Eysselein VE. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology. 1997;113(3):825–32.
Egger B, Büchler MW, Lakshmanan J, Moore P, Eysselein VE. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand J Gastroenterol. 2000;35(11):1181–7.
Brandl K, Sun L, Neppl C, Siggs OM, Le Gall SM, Tomisato W, Li X, Du X, Maennel DN, Blobel CP, Beutler B. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc Natl Acad Sci U S A. 2010;107(46):19967–72.
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–81.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA 3rd, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–7.
Tabernero J, Cervantes A, Rivera F, Martinelli E, Rojo F, von Heydebreck A, Macarulla T, Rodriguez-Braun E, Eugenia Vega-Villegas M, Senger S, Ramos FJ, Roselló S, Celik I, Stroh C, Baselga J, Ciardiello F. Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: biomarker analysis of a phase I dose-escalation study. J Clin Oncol. 2010;28(7):1181–9.
Zanella ER, Galimi F, Sassi F, Migliardi G, Cottino F, Leto SM, Lupo B, Erriquez J, Isella C, Comoglio PM, Medico E, Tejpar S, Budinská E, Trusolino L, Bertotti A. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci Transl Med. 2015;7(272):272ra12.
Tian S, Simon I, Moreno V, Roepman P, Tabernero J, Snel M, van’t Veer L, Salazar R, Bernards R, Capella G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut. 2013;62(4):540–9.
Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11(9):777–92.
Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–400.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13(4):e178–85.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65.
Lièvre A, Blons H, Laurent-Puig P. Oncogenic mutations as predictive factors in colorectal cancer. Oncogene. 2010;29(21):3033–43.
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96(8):1166–9.
Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28(7):1254–61.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–705.
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28(31):4706–13.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–34.
Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–9.
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27(12):2091–6.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34.
Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, Dimartino V, Hobor S, Jacobs B, Ercolani C, Lamba S, Scala E, Veronese S, Laurent-Puig P, Siena S, Tejpar S, Mottolese M, Punt CJ, Gambacorta M, Bardelli A, Di Nicolantonio F. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013;133(5):1259–65.
Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–40.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6.
Osumi H, Matsusaka S, Shinozaki E, Suenaga M, Mingyon M, Saiura A, Ueno M, Mizunuma N, Yamaguchi T. Acquired drug resistance conferred by a KRAS gene mutation following the administration of cetuximab: a case report. BMC Res Notes. 2013;6:508.
Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, Russo M, Buscarino M, Lazzari L, Sartore-Bianchi A, Bencardino K, Amatu A, Lauricella C, Valtorta E, Siena S, Di Nicolantonio F, Bardelli A. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6(224):224ra26.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih M, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson A, Bodoky G, Ciardiello F, D’Hoore A, Diaz-Rubio E, Douillard JY, Ducreux M, Falcone A, Grothey A, Gruenberger T, Haustermans K, Heinemann V, Hoff P, Köhne CH, Labianca R, Laurent-Puig P, Ma B, Maughan T, Muro K, Normanno N, Österlund P, Oyen WJ, Papamichael D, Pentheroudakis G, Pfeiffer P, Price TJ, Punt C, Ricke J, Roth A, Salazar R, Scheithauer W, Schmoll HJ, Tabernero J, Taïeb J, Tejpar S, Wasan H, Yoshino T, Zaanan A, Arnold D. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–422.
Tabernero J, Lenz HJ, Siena S, Sobrero A, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Yoshino T, Goldberg RM, Sargent DJ, Wagner A, Laurent D, Teufel M, Jeffers M, Grothey A, Van Cutsem E. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16(8):937–48.
Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L, Bertotti A. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18(9):2515–25.
Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J, Hancock DC. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22(8):1227–45.
Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, Döhner K, Bullinger L, Sandy P, Boehm JS, Root DE, Jacks T, Hahn WC, Gilliland DG. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34.
Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48.
Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, Song Y, Lifshits E, Hung KE, Shioda T, Dias-Santagata D, Singh A, Settleman J, Benes CH, Mino-Kenudson M, Wong KK, Engelman JA. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–8.
Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148(4):639–50.
Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, Ryan DP, Meyerhardt JA, Benes C, Settleman J, Wong KK, Cantley LC, Engelman JA. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121(11):4311–21.
Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, Kodack DP, Jain RK, Corcoran RB, Rivera MN, Ramaswamy S, Hung KE, Benes CH, Engelman JA. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4(1):42–52. https://doi.org/10.1158/2159-8290.CD-13-0315.
Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, Li C, Wheeler DL. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene. 2011;30(5):561–74.
Misale S, Bozic I, Tong J, Peraza-Penton A, Lallo A, Baldi F, Lin KH, Truini M, Trusolino L, Bertotti A, Di Nicolantonio F, Nowak MA, Zhang L, Wood KC, Bardelli A. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun. 2015;6:8305.
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–12.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouché O, Reid J, Stone S, Penault-Llorca F. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–30.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Köhne CH. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–75.
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, Zalcberg JR, Simes J, Moore MJ, Tebbutt NC, Price TJ, Shapiro JD, Pavlakis N, Gibbs P, Van Hazel GA, Lee U, Haq R, Virk S, Tu D, Lorimer IA, NCIC Clinical Trials Group and the Australasian Gastro-Intestinal Trials Group. PIK3CA, BRAF, and PTEN Status and Benefit from Cetuximab in the Treatment of Advanced Colorectal Cancer—Results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20(3):744–53.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19.
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, Nolop KB, Saltz L. PLX4032 in metastatic colorectal cancer patients with mutant BRAFtumors. J Clin Oncol. 2010;28:15s, (Abstract nr 3534).
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3.
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M, Engelman JA. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–35.
Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, Schostack K, Simcox ME, Kopetz S, Heimbrook D, Lestini B, Bollag G, Su F. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72(3):779–89.
Tabernero J, Van Geel R, Guren TK, Yaeger RD, Spreafico A, Faris JE, Yoshino T, Yamada Y, Kim TW, Bendell JC, Schuler MH, Lenz H-J, Eskens F, Desai J, Hochester HS, Avsar E, Demuth T, Sandor V, Elez E, Schellens JHM. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol. 2016;34:3544, (suppl; abstr 3544).
Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Dayyani F, Gopal YN, Jiang ZQ, Wistuba II, Tang XM, Bornman WG, Bollag G, Mills GB, Powis G, Desai J, Gallick GE, Davies MA, Kopetz S. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–67.
Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ, Bronson RT, Tejpar S, Xavier RJ, Engelman JA, Martin ES, Hung KE. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res. 2013;19(10):2688–98.
Zecchin D, Boscaro V, Medico E, Barault L, Martini M, Arena S, Cancelliere C, Bartolini A, Crowley EH, Bardelli A, Gallicchio M, Di Nicolantonio F. BRAF V600E is a determinant of sensitivity to proteasome inhibitors. Mol Cancer Ther. 2013;12(12):2950–61.
Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, Van Cutsem E, Xie T, Bosman FT, Roth AD, Delorenzi M. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012;30(12):1288–95.
Vecchione L, Gambino V, Raaijmakers J, Schlicker A, Fumagalli A, Russo M, Villanueva A, Beerling E, Bartolini A, Mollevi DG, El-Murr N, Chiron M, Calvet L, Nicolazzi C, Combeau C, Henry C, Simon IM, Tian S, In’t Veld S, D’ario G, Mainardi S, Beijersbergen RL, Lieftink C, Linn S, Rumpf-Kienzl C, Delorenzi M, Wessels L, Salazar R, Di Nicolantonio F, Bardelli A, van Rheenen J, Medema RH, Tejpar S, Bernards R. A vulnerability of a subset of colon cancers with potential clinical utility. Cell. 2016;165(2):317–30.
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–60.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–32.
Kraus S, Nabiochtchikov I, Shapira S, Arber N. Recent advances in personalized colorectal cancer research. Cancer Lett. 2014. https://doi.org/10.1016/j.canlet.2014.01.025.
Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt JA, Fuchs CS, Ogino S. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res. 2012;18(8):2257–68.
Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94(4):455–9.
Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15(9):3184–8.
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–8.
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–7.
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E, Bertario L, Leo E, Pierotti MA, Pilotti S. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20(1):84–90.
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004;64(9):3014–21.
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97(8):1139–45.
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G, Campani D, Floriani I, Cascinu S, Falcone A. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–9.
Okada Y, Miyamoto H, Goji T, Takayama T. Biomarkers for predicting the efficacy of anti-epidermal growth factor receptor antibody in the treatment of colorectal cancer. Digestion. 2014;89(1):18–23.
Negri FV, Bozzetti C, Lagrasta CA, Crafa P, Bonasoni MP, Camisa R, Pedrazzi G, Ardizzoni A. PTEN status in advanced colorectal cancer treated with cetuximab. Br J Cancer. 2010;102(1):162–4.
Inno A, Di Salvatore M, Cenci T, Martini M, Orlandi A, Strippoli A, Ferrara AM, Bagalà C, Cassano A, Larocca LM, Barone C. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer. 2011;10(4):325–32.
Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L, Sasazuki T, Shirasawa S, Geuna M, Frattini M, Baselga J, Gallicchio M, Biffo S, Bardelli A. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120(8):2858–66.
Ganesan P, Janku F, Naing A, Hong DS, Tsimberidou AM, Falchook GS, Wheler JJ, Piha-Paul SA, Fu S, Stepanek VM, Lee JJ, Luthra R, Overman MJ, Kopetz ES, Wolff RA, Kurzrock R. Target-based therapeutic matching in early-phase clinical trials in patients with advanced colorectal cancer and PIK3CA mutations. Mol Cancer Ther. 2013;12(12):2857–63.
Altomare I, Hurwitz H. Everolimus in colorectal cancer. Expert Opin Pharmacother. 2013;14(4):505–13.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–56.
Trusolino L, Bertotti A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: six degrees of separation. Cancer Discov. 2012;2(10):876–80.
Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–83.
Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, Giovannucci E, Fuchs CS, Chan AT, Ogino S. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367(17):1596–606.
Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, Davidson B, Kerr DJ, Tomlinson IP, Midgley R. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol. 2013;31(34):4297–305.
Fuchs CS, Ogino S. Aspirin therapy for colorectal cancer with PIK3CA mutation: simply complex. J Clin Oncol. 2013;31(34):4358–61.
Hutti JE, Pfefferle AD, Russell SC, Sircar M, Perou CM, Baldwin AS. Oncogenic PI3K mutations lead to NF-κB-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012;72(13):3260–9.
Kaur J, Sanyal SN. PI3-kinase/Wnt association mediates COX-2/PGE(2) pathway to inhibit apoptosis in early stages of colon carcinogenesis: chemoprevention by diclofenac. Tumour Biol. 2010;31(6):623–31.
Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I, Iglesias M, Gavilan E, Corti G, Hobor S, Crisafulli G, Salido M, Sánchez J, Dalmases A, Bellmunt J, De Fabritiis G, Rovira A, Di Nicolantonio F, Albanell J, Bardelli A. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21(9):2157–66.
Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16(10):5276–87.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–23.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M, Nakagawa K, Jänne PA. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3(99):99ra86.
Martin V, Landi L, Molinari F, Fountzilas G, Geva R, Riva A, Saletti P, De Dosso S, Spitale A, Tejpar S, Kalogeras KT, Mazzucchelli L, Frattini M, Cappuzzo F. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer. 2013;108(3):668–75.
Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L, Jacobs SA, Bertotti A, Bose R. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–41.
Leto SM, Sassi F, Catalano I, Torri V, Migliardi G, Zanella ER, Throsby M, Bertotti A, Trusolino L. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2-amplified gastrointestinal carcinomas. Clin Cancer Res. 2015;21(24):5519–31.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, Bertotti A, Siravegna G, Torri V, Amatu A, Ghezzi S, Marrapese G, Palmeri L, Valtorta E, Cassingena A, Lauricella C, Vanzulli A, Regge D, Veronese S, Comoglio PM, Bardelli A, Marsoni S, Siena S. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–46.
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ, Seshagiri S. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23(5):603–17.
Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–16.
Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007;213(2):316–25.
Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119(3):629–41.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, CJ Y, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–7.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43.
Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, Santoro A, Varella-Garcia M. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20(2):298–304.
Kwak EL, Ahronian LG, Siravegna G, Mussolin B, Godfrey JT, Clark JW, Blaszkowsky LS, Ryan DP, Lennerz JK, Iafrate AJ, Bardelli A, Hong TS, Corcoran RB. Molecular heterogeneity and receptor coamplification drive resistance to targeted therapy in MET-amplified esophagogastric cancer. Cancer Discov. 2015;5(12):1271–81.
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, Lauricella C, Zanon C, Perera T, Veronese S, Corti G, Amatu A, Gambacorta M, Diaz LA Jr, Sausen M, Velculescu VE, Comoglio P, Trusolino L, Di Nicolantonio F, Giordano S, Siena S. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–73.
Cappuzzo F, Varella-Garcia M, Finocchiaro G, Skokan M, Gajapathy S, Carnaghi C, Rimassa L, Rossi E, Ligorio C, Di Tommaso L, Holmes AJ, Toschi L, Tallini G, Destro A, Roncalli M, Santoro A, Jänne PA. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer. 2008;99(1):83–9.
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, De Palma R, D’Aiuto E, Berrino L, Bianco R, Ciardiello F. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19(24):6751–65.
Pietrantonio F, Oddo D, Gloghini A, Valtorta E, Berenato R, Barault L, Caporale M, Busico A, Morano F, Gualeni AV, Alessi A, Siravegna G, Perrone F, Di Bartolomeo M, Bardelli A, de Braud F, Di Nicolantonio F. MET-driven resistance to dual EGFR and BRAF blockade may be overcome by switching from EGFR to MET inhibition in BRAF-mutated colorectal cancer. Cancer Discov. 2016;6(9):963–71.
Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17(3):472–82.
Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM 3rd, Perera T, Bertotti A, Trusolino L, Comoglio PM, Boccaccio C. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014;74(6):1857–69.
Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–74.
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Serrano S, Himmelbauer H, Bellmunt J, Rovira A, Settleman J, Bosch F, Albanell J. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18(2):221–3.
Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, Iannaccone A, Tatangelo F, Forgione L, Pasquale R, Barbaro A, Botti G, Ciardiello F, Normanno N. The S492R EGFR ectodomain mutation is never detected in KRAS wild type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther. 2013;14(12)
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, Sassi F, Cottino F, Migliardi G, Zanella ER, Ribero D, Russolillo N, Mellano A, Muratore A, Paraluppi G, Salizzoni M, Marsoni S, Kragh M, Lantto J, Cassingena A, Li QK, Karchin R, Scharpf R, Sartore-Bianchi A, Siena S, Diaz LA Jr, Trusolino L, Velculescu VE. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–7.
Arena S, Siravegna G, Mussolin B, Kearns JD, Wolf BB, Misale S, Lazzari L, Bertotti A, Trusolino L, Adjei AA, Montagut C, Di Nicolantonio F, Nering R, Bardelli A. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci Transl Med. 2016;8(324):324ra14.
Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, Raddrizzani L, Palmeri L, Banfi P, Bonazzina E, Misale S, Marrapese G, Leone A, Alzani R, Luo D, Hornby Z, Lim J, Veronese S, Vanzulli A, Bardelli A, Martignoni M, Davite C, Galvani A, Isacchi A, Siena S. Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J Natl Cancer Inst. 2016;108(1)
Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, Corti G, Rospo G, Novara L, Mussolin B, Bartolini A, Cam N, Patel R, Yan S, Shoemaker R, Wild R, Di Nicolantonio F, Bianchi AS, Li G, Siena S, Bardelli A. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6(1):36–44.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, Humblet Y, Pénault-Llorca F, De Hertogh G, Laurent-Puig P, Van Cutsem E, Tejpar S. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–74.
Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, Papamichael D, Xanthakis I, Sgouros J, Televantou D, Kafiri G, Tsamandas AC, Razis E, Galani E, Bafaloukos D, Efstratiou I, Bompolaki I, Pectasides D, Pavlidis N, Tejpar S, Fountzilas G. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer. 2013;13:49.
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJ, Stinson J, Gnad F, Huntley MA, Degenhardt JD, Haverty PM, Bourgon R, Wang W, Koeppen H, Gentleman R, Starr TK, Zhang Z, Largaespada DA, TD W, de Sauvage FJ. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–4.
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD, Rodland KK, Townsend RR, Kinsinger C, Mesri M, Rodriguez H, Liebler DC. Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013;3(10):1108–12.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20.
Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–86.
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, Rosenfeld N. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12.
Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, Valtorta E, Rospo G, Medico E, Motta V, Antoniotti C, Tatangelo F, Bellosillo B, Veronese S, Budillon A, Montagut C, Racca P, Marsoni S, Falcone A, Corcoran RB, Di Nicolantonio F, Loupakis F, Siena S, Sartore-Bianchi A, Bardelli A. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):827.
Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6.
Acknowledgments
Work in the author’s laboratory is supported by AIRC, Associazione Italiana per la Ricerca sul Cancro—2010 Special Program Molecular Clinical Oncology 5 × 1000, project 9970— and AIRC Investigator Grant, project 18532; FPRC, Fondazione Piemontese per la Ricerca sul Cancro, Ministero della Salute 2011; ERA-NET TRANSCAN, project TACTIC.Clinical trials available at:NCT01085331: http://clinicaltrials.gov/ct2/show/NCT01085331?term=NCT01085331&rank=1NCT01390818: http://clinicaltrials.gov/ct2/results?term=NCT01390818&Search=SearchNCT02039336: http://clinicaltrials.gov/ct2/show/NCT02039336?term=NCT02039336&rank=1NCT01154335: http://clinicaltrials.gov/ct2/show/NCT01154335?term=colorectal+cancer&rank=33NCT01139138: http://clinicaltrials.gov/ct2/show/NCT01139138?term=colorectal+cancer&rank=67NCT01387880: http://clinicaltrials.gov/ct2/show/NCT01387880?term=everolimus+AND+colorectal+cancer&rank=2NCT00827684: http://clinicaltrials.gov/ct2/show/NCT00827684?term=everolimus+AND+colorectal+cancer&rank=9NCT02034981: http://clinicaltrials.gov/ct2/results?term=NCT02034981&Search=Search
Conflict of Interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Trusolino, L., Leto, S.M. (2018). Resistance of Colorectal Tumors to Anti-EGFR Antibodies. In: Yarden, Y., Elkabets, M. (eds) Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Resistance to Targeted Anti-Cancer Therapeutics, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-67932-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-67932-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67930-3
Online ISBN: 978-3-319-67932-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)