
Abstract
The absorption of ionizing radiation by living cells can directly disrupt atomic structures, producing chemical and biological changes. It can also act indirectly through radiolysis of water, thereby generating reactive chemical species that may damage nucleic acids, proteins, and lipids. Together, the direct and indirect effects of radiation initiate a series of biochemical and molecular signaling events that may repair the damage or culminate in permanent physiological changes or cell death. In efforts to gain insight into the mechanisms underlying these effects, we observed a prominent upregulation of the Translationally Controlled Tumor Protein (TCTP) in low dose/low dose rate 137Cs γ-irradiated cells that was associated with adaptive responses that reduced chromosomal damage to a level lower than what occurs spontaneously. Therefore, TCTP may support the survival and genomic integrity of irradiated cells through a role in the DNA damage response. Consistent with this postulate, TCTP was shown to physically interact with ATM, an early sensor of DNA damage, and to exist in a complex with γH2A.X, in agreement with its distinct localization with the foci of the DNA damage marker proteins γH2A.X, 53BP1, and P-ATM. Cells lacking TCTP failed to repair chromosomal damage induced by γ-rays. Further, TCTP was shown to interact with the DNA-binding subunits, Ku70 and Ku80, of DNA-PK, a major participant in nonhomologous end joining of DNA double strand breaks. Moreover, TCTP physically interacted with p53, and its knockdown attenuated the radiation-induced G1 delay, but prolonged the G2 delay. Here, we briefly review the biochemical events leading to DNA damage by ionizing radiation and to its sensing and repair, and highlight TCTP’s critical role in maintaining genomic integrity in response to DNA-damaging agents.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


12.1 Introduction
Cells in the human body are challenged by a variety of harmful compounds generated by normal physiological processes and through exposure to environmental agents. For example, we are constantly exposed to low levels of ionizing radiation from natural sources, and we may also be exposed to radiation released to the environment from nuclear fallouts and man-made sources, including consumer products, and discharges of radioactive waste. In addition, humans may become exposed to radiation during occupational activities related to nuclear technology, mining, high altitude airline travel, and deep space exploration. In particular, with the explosive growth in the use of diagnostic radiology, an increasing numbers of individuals are being repeatedly exposed to low and moderate doses of radiation during conventional as well as computed tomography, and nuclear medicine imaging procedures (NCRP 2009). In addition to low dose exposures, the use of different irradiation modalities (e.g., external beam therapies, brachytherapy with various sources of radiation) remains an effective and widely used means to treat cancer and other pathological conditions such as arteriovenous malformations (Coutard 1937; Hacein-Bey et al. 2014). Currently, 20–60% of all new cancer cases worldwide are treated with external photon beam radiotherapy as a standard option (Tyldesley et al. 2001; Delaney et al. 2005).
Exposures to ionizing radiation are, therefore, an inevitable part of the environment and increasingly of modern life. Hence, elucidation of the mechanisms underlying the cellular responses to low dose radiation is essential for estimating long-term health risks of low level exposures, for understanding the basis of normal tissue toxicities that arise following therapeutic exposures, and for enhancing the efficacy of radiotherapy through combined treatment modalities. Importantly, the study of the biochemical and molecular events underlying cellular and tissue responses to radiation have been major contributors to our knowledge of the stress response to radiation exposure, in particular the DNA damage response.
When cells are exposed to ionizing radiation, nucleic acids, proteins, lipids and other cellular constituents undergo chemical modifications. Of all the cellular constituents, stability of DNA is undoubtedly critical. Damage to the nuclear genome by direct interaction of DNA with radiation, or indirectly by its interaction with reactive compounds (e.g., reactive oxygen and nitrogen species) generated following cellular exposure to radiation, can cause a variety of adverse chemical/structural changes. Depurinations, depyrimidinations, base oxidation and strand breaks are some of the DNA damages that may occur (Dizdaroglu 2012). If unrepaired or misrepaired and propagated to progeny cells, these changes can lead to mutations that promote the development of cancer and degenerative diseases (Georgakilas 2011; Hall and Giaccia 2006; Little 2000).
The early radiation effects on the nuclear genome may be enhanced by excess oxidative stress resulting from persistent perturbations in oxidative metabolism, which can result in excess generation of reactive chemical species that exacerbate harmful conditions (Azzam et al. 2012; Spitz et al. 2004). In response to these stresses, cells have evolved multiple mechanisms to prevent the chemical/structural changes to DNA from occurring and/or repair the damage once it has occurred, thus maintaining genomic integrity. Cells react to genotoxic stress by activating a large network of signaling pathways that sense specific types of damage and trigger a coordinated and complex response to the chromosomal insults by activation of DNA damage sensing and repair pathways that restitute the damage or attenuate its level (Harper and Elledge 2007; Jeggo et al. 2016). In addition, cells activate antioxidant defenses to scavenge reactive chemical species (Petkau 1987) and cell cycle checkpoints to provide additional time for the defense mechanisms to operate (Little 1968; Zhou and Elledge 2000). In exposed tissues, immune responses may be triggered to promote repair and/or eliminate damaged cells (Jackson and Bartek 2009). In addition, apoptosis, necrosis, or mitotic death pathways may also be induced, particularly when the damage is severe (Okada and Mak 2004). These protective pathways have been under intense investigation and have been a fertile field for discovery of new players in the cellular defense against exogenous and endogenous stresses.
The Translationally Controlled Tumor Protein (TCTP) was identified over 25 years ago in Ehrlich’s ascites tumor and in mouse erythroleukemia (Yenofsky et al. 1983; Chitpatima et al. 1988; Thomas et al. 1981). The protein was also named histamine releasing factor (HRF), fortilin, tpt1, Q23, P21, and P23 by independent groups, based on its multiple characteristics and activities (Thomas et al. 1981; Yenofsky et al. 1982; Bohm et al. 1989; MacDonald et al. 1995; Li et al. 2001). TCTP is an evolutionarily conserved molecule expressed in many eukaryotic cells (Bommer and Thiele 2004), which highlights its central biological importance. The complex nature of TCTP regulation during normal physiological functions and in response to stress continues to evolve. The various chapters in this book comprehensively describe the role of TCTP in various physiological functions and in cancer. TCTP has been implicated in transcription (Koziol et al. 2007), protein synthesis (Cans et al. 2003), cell cycle control (Brioudes et al. 2010), cytoskeleton regulation (Gachet et al. 1999; Burgess et al. 2008; Tsarova et al. 2010), immune responses (Macdonald 2012; Kashiwakura et al. 2012), development (Chen et al. 2007a, b; Hsu et al. 2007), viability (Susini et al. 2008), and cancer induction and reversion (Chan et al. 2012; Jung et al. 2011; Tuynder et al. 2002, 2004). Our emerging work has identified a novel role for TCTP in the repair of radiation-induced DNA damage, which is critical to health and disease (Zhang et al. 2012): Unrepaired DNA damage leads to genomic instability, which is a hallmark of cancer (Huang et al. 2003). As a background to this role, we briefly review the cellular effects of ionizing radiation. In particular, we discuss the induction of radiation-induced DNA damage and its repair, and the role of TCTP in the repair of DNA damage. Throughout, we identify areas where research may further illuminate the role of TCTP in this critical field.
12.2 Primary Effects of Ionizing Radiation
12.2.1 Direct and Indirect Effects of Ionizing Radiation
In mammalian cells, significant chemical modifications take place during or shortly after (within 10−15 to 10−6 s) the radiation exposure (Barendsen 1964; O’Neill and Wardman 2009; Meesungnoen et al. 2001). These modifications occur through direct interaction of the radiation with components of the exposed cells (e.g., DNA) or indirectly through generation of oxidizing species from water radiolysis (Fig. 12.1). Water is the major (∼80%) constituent of cells. A thorough knowledge of water radiolysis is therefore critical for understanding radiobiological effects (Zimbrick 2002; LaVerne and Pimblott 1993).

The direct and indirect cellular effects of ionizing radiation on macromolecules. Absorption of ionizing radiation by living cells directly disrupts atomic structures, producing chemical and biological changes and indirectly through radiolysis of cellular water and generation of reactive chemical species by stimulation of oxidases and nitric oxide synthases. Ionizing radiation may also disrupt mitochondrial functions significantly contributing to persistent alterations in lipids, proteins, nuclear DNA (nDNA), and mitochondrial DNA (mtDNA)
The absorption of energetic radiations by water results in both excitations and ionizations leading to production of free radicals that in turn can attack other critical molecules (indirect effect). The schematic in Fig. 12.1 describes the complex events that accompany the absorption of high-energy photons or the passage of fast charged particles through water. These events can be divided into four, more or less clearly demarcated, consecutive, temporal stages (Platzman 1958). During the first or “physical” stage, the energy deposition is caused by the incident radiation and secondary electrons are generated. The resulting species are extremely unstable and undergo fast reorganization in the second or “physicochemical” stage. These processes produce radical and molecular products of radiolysis that are distributed in a nonhomogeneous track structure. The initial (∼10−12 s) spatial distribution of reactants is then directly used as the starting point for the so-called stage of “nonhomogeneous chemistry.” During this third stage, the various chemically reactive species diffuse and react with one another or with the environment (∼10−6 s). Finally, in a physiologic system, the “biological” stage follows, in which the cells respond to the damage resulting from the products formed in the preceding stages. During this stage (∼10−3 s or longer, depending very much upon the medium), the biological responses affecting the long-term consequences of radiation exposure are induced. In an aerobic cellular environment at physiological pH, the major reactive species at homogeneity (∼10−6 s) include O2 •−, •OH, and H2O2 [reviewed in Azzam et al. (2012)]. Whereas ∼1/3 of DNA damage from sparsely ionizing radiation (low linear energy transfer type such as energetic X- or γ-rays) emanates from direct interaction of the DNA with radiation, ∼2/3 results from indirect effects involving water radiolysis products (Fig. 12.1) (Hall and Giaccia 2006). In the case of densely ionizing radiations such as alpha particles emanating from environmental radon gas, or high atomic number (Z) and high energy (E) (HZE) particles used in modern radiotherapy (Newhauser and Durante 2011) or encountered by astronauts during deep space travel (Li et al. 2014), it is commonly accepted that the cellular effects of HZE particles on macromolecules are mainly due to direct rather than indirect effects involving water radiolysis products (Hall and Giaccia 2006). Regardless, in cells exposed to such particulate radiations, the concentration of radiolytic species is very dense in and around the particle track (Goodhead 1988; Muroya et al. 2006; Chatterjee and Schaefer 1976), causing extensive covalent modifications in affected macromolecules (Li et al. 2014). Therefore, it would be of great interest to investigate the protective role of TCTP in the cellular defense against the damages induced by either sparsely or densely ionizing radiations.
In addition to a role of protecting against DNA damage induced through the direct effect of radiation, TCTP may have a role in promoting the scavenging of the reactive/DNA-damaging species generated during the biological stage of water radiolysis, which is consistent with TCTP’s antioxidant role and the general cellular stress response (Gnanasekar and Ramaswamy 2007). While TCTP may act at early stages following irradiation, it could also exert an antioxidant role for longer times to help alleviate oxidative stress induced as a result of activation of oxidases/perturbations in oxidative metabolism (Spitz et al. 2004). Interestingly, although the family of TCTP proteins showed no primary sequence homology to any other protein family, the core domain of TCTP displays remarkable structural similarity with three families of proteins: Mss4/Dss4 proteins, which bind to the GDP/GTP-free form of Rabs proteins (Thaw et al. 2001), methionine sulfoxide reductases, and RNA helicases (Amson et al. 2013). Notably, methionine sulfoxide reductases play an important role in antioxidant defense, protein regulation, and survival (Moskovitz 2005). It is, therefore, attractive to speculate that TCTP may protect cells against oxidative stress by a mechanism that is yet to be discovered due to its structural similarity with methionine-R-sulfoxide reductase B1. Furthermore, it is of interest that one of the TCTP genes deposited in GenBank is designated as a PO2 related protein (accession no. AAM51565) (Oikawa et al. 2002).
12.3 Endogenous and Radiation-Induced DNA Alterations
A strong emphasis thus far has been on the effect of exogenous agents such as ionizing radiation on DNA damage. However, improvements in the sensitivity of analytic methods to measure oxidative damage (Cadet et al. 2011) have revealed altered bases and nucleotides in the DNA of normal cells that have not been exposed to ionizing radiation or other mutagens (Weinberg 2007). The analyses have shown that endogenous biochemical processes greatly contribute to genome mutations. The reactive oxygen species (ROS) produced during normal cellular metabolic processes (mainly O2 •− and H2O2) cause extensive depurinations and, to a lesser extent, depyrimidinations. In addition, ROS can oxidize bases in DNA, such as the oxidation of deoxyguanosine (dG) to 8-hydroxyguanine (8-oxodG), with ∼100–500 of such lesions being formed per day in a human cell (Lindahl 1993). The rate of occurrence of these alterations has been closely linked to the rate of oxidative metabolism: higher oxygen consumption in different species were correlated with an increased rate of base oxidation in DNA (Ames 1989). A failure to repair oxidized bases creates a risk of mutation during DNA replication. For example, 8-oxodG mispairs with deoxyadenosine (dA) rather than deoxycytosine (dC), resulting in a C–A point mutation. Notably, oxidatively induced DNA lesions and DNA repair proteins have been suggested as potential biomarkers for early detection, cancer risk assessment, prognosis, and for monitoring therapy (Dizdaroglu 2012).
Several cellular defenses act to restore DNA integrity. Interestingly, the knockdown of TCTP by RNA interference (RNAi) in normal unirradiated human diploid fibroblasts led to an increase in the spontaneous rate of DNA damage in the form of micronuclei, which was validated by an increase in the average number of γH2A.X foci per cell [(Zhang et al. 2012); see results described in Fig. 12.5]. Micronuclei arise mainly from DNA double strand breaks (DSBs), a serious DNA lesion that leads to cell death (Fenech and Morley 1985; Baumstark-Khan 1993). Therefore, it is attractive to speculate that the increase in micronuclei upon knockdown of TCTP is likely due to abrogation of its antioxidant function, leading to the accumulation of oxidized bases that eventually result in DNA breaks.
12.3.1 DNA Damage Response Pathways and DNA Damage Repair Mechanisms
As highlighted above, DNA is continuously exposed to damaging agents from endogenous and external environmental stresses, along with lifestyle factors. This constant assault on DNA yields tens of thousands of DNA lesions per day in every human cell (Weinberg 2007). These DNA lesions must be repaired to prevent loss or incorrect transmission of genetic material, which can lead to tumorigenesis and other pathologies (Jackson and Bartek 2009; Lin et al. 2012).
As illustrated in Fig. 12.2, the direct interaction of DNA with ionizing radiation and the radiation-induced ROS induces a wide range of DNA damage of varying levels of complexity, such as base damage, single strand breaks (SSBs), abasic sites, DNA–protein cross-links, and DSBs (Nikjoo et al. 2001). Figure 12.3 shows that the choice of the repair system depends on the type of DNA lesion. Single strand breaks or single-base damage (i.e., DNA lesions on a single strand that do not significantly disrupt the helical structure) are generally repaired by base excision repair (BER) (Chou et al. 2015), whereas DNA damage that significantly distorts the DNA helix (e.g., bulky lesions and crosslinks) is repaired by nucleotide excision repair (NER) (Petruseva et al. 2014). Small chemical changes affecting a single base are repaired via direct repair (DR) (Yi and He 2013), and mismatches in base pairing caused by DNA replication errors are repaired by mismatch repair (MMR) (Larrea et al. 2010). Finally, DSBs are repaired via homologous recombination (HR) and/or nonhomologous end joining (NHEJ). The choice of repair system for DSB repair depends on the phase of the cell cycle and the expression, availability, and activation of DNA repair proteins (Lieber 2008; Shah and Mahmoudi 2015). Regardless of the type of lesion and the mechanisms required for its repair, cells initiate a complex signaling cascade that includes activation of DNA repair pathways, cell cycle arrest to allow time for repair of DNA damage, and in certain cases, initiation of senescence or apoptosis. These series of coordinated events are known as the DNA damage response (DDR) pathways (Jackson and Bartek 2009). Upon induction of DSBs, the central components of DDR activation are ATM, ATR, and DNA-dependent protein kinase (DNA-PK), members of the phosphatidyl inositol 3-kinase-like kinase (PIKK) family. ATM and DNA-PK are predominantly activated by DNA DSBs, whereas other types of DNA damage (e.g., replication-induced DSBs, base adducts, and cross-links) activate ATR (Branzei and Foiani 2008; Nam and Cortez 2011; Shiloh and Ziv 2013). Our studies have shown that TCTP interacts with components of both HR and NHEJ to promote repair of DNA damage (Zhang et al. 2012).

DNA lesions induced by ionizing radiation. Cellular exposure to ionizing radiation induces a wide range of damage in DNA including single strand breaks (SSB), base damage, abasic sites, DNA-protein cross-links, and double strand breaks (DSB)

Types of DNA lesions and their corresponding DDR pathways. SSBs or single-base damage (i.e., DNA lesions on a single strand that do not significantly disrupt the helical structure) are generally repaired by base excision repair (BER), whereas DNA damage that significantly distorts the DNA helix (e.g., bulky lesions and crosslinks) is repaired by nucleotide excision repair (NER). Small chemical changes affecting a single base are repaired via direct repair (DR), and mismatches in base pairing caused by DNA replication errors are repaired by mismatch repair (MMR). Finally, DSBs are repaired via homologous recombination (HR) and nonhomologous end joining (NHEJ). AGT = O6-alkylguanine-DNA alkyltransferase; GG-NER = global genome NER; O6MeG = O6-methylguanine; TC-NER = transcription-coupled NER [adapted from Postel-Vinay et al. (2012)]
As shown in Fig. 12.3, DNA-PK and ATM are activated by the recruitment of Ku70/Ku80 and the MRN complex, respectively, to DSBs. Ku70/Ku80 and DNA-PK promote NHEJ repair of DSBs. The DNA-PK catalytic subunit (DNA-PKcs) keeps the broken DNA ends in close proximity during NHEJ repair and recruits end-processing factors (e.g., Artemis, PNKP, APE1, and TDP1), which prepare the DNA ends for re-ligation by the XRCC4–XLF–LIG4 complex (Fig. 12.4) (Postel-Vinay et al. 2012; Panier and Boulton 2014). In recent years, alternative end-joining pathways that repair DSBs independently of one or more core components of this classical-NHEJ machinery have been described (Decottignies 2013; Badie et al. 2015). Our work has indicated that cells lacking TCTP failed to repair chromosomal damage induced by γ-rays (Fig. 12.5), which as will be shown below, perhaps as a result of decreased binding of the Ku proteins to damaged DNA. Significantly, defects in DNA repair genes have long been associated with human disease and in cellular sensitivity to DNA-damaging agents (Jackson and Bartek 2009; McKinnon 2009; Hoeijmakers 2009; Jasin 2015; Weichselbaum et al. 1980).

DNA DSB repair pathways. The two main DNA DSB repair pathways in eukaryotic cells: nonhomologous end joining (NHEJ; part a) and homologous recombination (part b). APE1 = AP endonuclease 1; BLM = Bloom’s syndrome helicase; BRCA1/2 = breast cancer 1/2; CtIP = CtBP-interacting protein; DNA2 = DNA replication ATP-dependent helicase; DNAPKcs = DNA-PK catalytic subunit; EXO1 = exonuclease 1; LIG4 = DNA ligase 4; MRN (MRE11–RAD50–NBS1) complex; PNKP = polynucleotide kinase/phosphatase; RMI = RecQ-mediated genome instability protein 1; RPA = replication protein A; SDSA = synthesis-dependent strand annealing; ssDNA = single-stranded DNA; TDP1 = tyrosyl–DNA phosphodiesterase 1; TOP3α = topoisomerase 3α; XLF = XRCC4-like factor; XRCC4 = X-ray repair cross complementing protein 4 [adapted from Panier and Boulton (2014)]

Involvement of TCTP in protection against DNA damage in γ-irradiated AG1522 cells. (a) Micronucleus formation in confluent cells that were subcultured at 0.25 or 4 h after acute 50 cGy exposure. The cells were transfected with scrambled (Scr) or TCTP siRNA. Scrambled siRNA-transfected cells were also treated with PJ34 or Nu7441 that inhibit DNA repair. (b) Analyses of γH2A.X foci in scrambled or TCTP siRNA transfected cells at different time point after irradiation with 50 cGy. 75–100 cells were counted for each time point. Data represent mean ± SD of three independent experiments. (c) Clonogenic survival of γ-irradiated cells transfected with Scr or TCTP siRNA [adapted from Zhang et al. (2012)]
As a consequence of DSB induction, ATM is activated (Shiloh 2003) and phosphorylates the histone H2A.X (to form γH2A.X), which leads to both structural alterations to the chromatin around the damaged site to allow repair proteins access to the damaged regions of the DNA and the recruitment and retention of key DDR factors (Stucki and Jackson 2006). In addition, accumulating evidence indicates that γH2A.X may also be involved in functions that are not directly related to its function as a DNA DSB marker [reviewed in detail in Turinetto and Giachino (2015)]. γH2A.X foci are formed within minutes after exposure to ionizing radiation in a dose-dependent manner, peak at <1 h post-irradiation, and then rapidly decay to baseline levels within one to several days, depending on the dose received (Rogakou et al. 1998). H2A.X phosphorylation leads to recruitment of many checkpoint and repair factors, such as MDC1, MRN, and the ubiquitin ligases RNF8 and UBC13 (Postel-Vinay et al. 2012; Panier and Boulton 2014). These factors promote the recruitment of 53BP1, BRCA1, and more ATM to facilitate the spreading of the DDR signal through the nucleus. These proteins go on to initiate the phosphorylation and dimerization of checkpoint kinases CHK2/CHK1, which targets effectors including p53, CDC25A, and CDC25C that in turn activates cell cycle checkpoints or induce apoptosis (Raynaud et al. 2008; Thompson 2012). While NHEJ is active in all phases of the cell cycle, HR is restricted to the S and G2 phases when sister chromatids are available in close proximity as repair templates (Branzei and Foiani 2008; Symington and Gautier 2011). Significantly, we have shown that upon exposure of normal human cells that are in different phases of the cell cycle to low dose γ-rays, the TCTP protein level was greatly increased, with a significant enrichment in nuclei. TCTP upregulation occurred in a manner dependent on ATM and DNA-PK (cells deficient in ATM or DNA-PKcs function failed to upregulate TCTP) and was associated with protective effects against DNA damage and cell killing (Fig. 12.5). In chromatin of irradiated cells, TCTP was found to physically interact with ATM and to exist in a complex with γH2A.X, in agreement with its distinct localization with the foci of the DNA damage marker proteins γH2A.X, 53BP1, and p-ATM (Fig. 12.6). Importantly, compared to cells transfected with Scr siRNA, depletion of TCTP by siRNA resulted in opposite abundance patterns of Ku proteins in cytoplasm and nucleus of γ-irradiated cells. The dramatic reduction in TCTP level was associated with radiation dose-dependent decrease in Ku70 and Ku80 in the nucleus. Furthermore, relative to Scr siRNA-treated cells, the decreases in Ku70 and Ku80 abundance in nuclei of irradiated siTCTP-transfected cells were associated with significant attenuation (>50%, p < 0.001) in the DNA-binding activity of Ku70 and Ku80 from extracts of irradiated cells (Fig. 12.7) (Zhang et al. 2012). These findings constitute previously unrecognized roles for TCTP in maintaining genome integrity under stressful conditions (Zhang et al. 2012; Bommer et al. 2010; Nagano-Ito et al. 2009; Gnanasekar et al. 2009). However, the exact function of TCTP in DNA damage sensing and the different modes of DNA repair still remains to be clearly elucidated.

TCTP interacts with components of DNA damage sensing and repair. (a) Benzonase-treated chromatin-enriched fractions that were isolated 30 min after irradiation (IR) with 0 (−) or acute 50 cGy (+) from confluent U2OS cells were immunoprecipitated with anti-ATM or anti-TCTP antibodies, or normal mouse or rabbit serum (PI). Immunoblots were then reacted with antibodies against ATM, TCTP, γH2A.X, or H2AX. (b) Benzonase-treated nuclear extracts isolated 30 min after exposure of U2OS confluent cells to 0 or acute 50 cGy were immunoprecipitated with anti-TCTP, anti-p53 or control anti-TBP antibodies. Mouse or rabbit preimmune serum (PI) was used as a control. Immunoblotting was performed using antibodies against p53, TCTP, or TBP. (c) Immunoblotting of TCTP, Ku70, and Ku80 in benzonase-treated nuclear extracts of control unirradiated U2OS confluent cells after immunoprecipitation with either normal serum (PI) or antibodies against TCTP, Ku70, or Ku80. (d) Untreated or γ-irradiated (acute 100 cGy) AG1522 asynchronous cells were pre-extracted, fixed 1 h later, and immunostained in situ with anti-TCTP, anti-P-ATM (S1981), anti-γH2A.X, or anti 53BP1 antibodies. Bars, 10 μm. (e) Quantitative assessment of co-localization of TCTP foci with those of P-ATM (S1981) (left panel), γH2A (middle panel), and 53BP1 (right panel) in AG1522 asynchronous cells at 1 h after exposure to 50, 100, or 200 cGy [adapted from Zhang et al. (2012)]

TCTP is involved in regulation of DNA-binding activity of Ku70 and Ku80 after γ-irradiation. Ku70 or Ku80 DNA-binding activity in irradiated U2OS confluent cells expressing scrambled or TCTP siRNA at 30 min after irradiation
12.4 TCTP and the Sensing of Genotoxic Stress
There is extensive evidence indicating that TCTP is abundantly expressed in eukaryotes and interacts with several proteins to exert various physiological functions (Amson et al. 2012). Following exposure to environmental insults, the cells in most tissues dramatically increase the production of a small group of proteins that are collectively known as “heat-shock” or stress proteins. Their increased expression in tissues that are subjected to various proteotoxic stressors is an adaptive response that enhances cell survival (Whitesell and Lindquist 2005). In numerous experimental settings and biological systems, it was established that TCTP levels are highly regulated in response to a wide range of extracellular signals and cellular conditions, including heat, heavy metals, hypoxia, and oxidative stress (Bommer and Thiele 2004). Since both human TCTP (HuTCTP) and a TCTP homolog from Schistosoma mansoni (SmTCTP) can bind to a variety of denatured proteins and protect them from the harmful effects of thermal shock, it has been suggested that TCTP may belong to a novel heat shock protein with chaperone-like activity (Gnanasekar et al. 2009). Our immuno-precipitation (IP) and mass spectrometry (MS) analyses indicated that TCTP interacts with heat shock 90 kDa, 70 kDa, and 60 kDa proteins in irradiated cells (Zhang et al. 2012), which may also imply a chaperone role of TCTP.
Low doses of toxic agents often induce protective mechanisms that enhance the ability of the organism to cope with stress from normal metabolism or from exogenous agents (Azzam 2011; Azzam et al. 2016). A study by Lucibello et al. proposed TCTP as a “stress hallmark” in cancer cells (Lucibello et al. 2011): TCTP levels were upregulated in cells surviving mild oxidative stress and were downregulated when cells were treated with severe oxidative stress, which was followed by cell death. In our studies, we showed that doses of γ-rays as low as even 1 cGy, particularly when delivered at very low dose-rate (0.2 cGy/h), upregulate TCTP expression and decrease the frequency of micronuclei formation to below the spontaneous rate in normal human fibroblasts (Zhang et al. 2012). These significant changes in TCTP expression were also detected when normal cells were treated with low levels of t-butyl hydroperoxide and hyperthermia (data unpublished). Like ionizing radiation and t-butyl hydroperoxide, some effects of hyperthermia are mediated by ROS (Katschinski et al. 2000). In contrast, cells exposed to UVC, which induces the formation of DNA photoproducts, did not change TCTP levels (data unpublished). Thus, these results support the association of TCTP upregulation by low levels of environmental stresses with oxidative stress. Cells, especially embryonic mouse stem cells exposed to dioxin, a potent toxic synthetic environmental pollutant that induces production of ROS, experience significant upregulation of the expression and secretion of TCTP (Oikawa et al. 2002), which also support the role of TCTP in sensing and defending cells against oxidative stress.
12.5 TCTP and the Repair of DNA Damage
Presence of abundant amounts of TCTP in the nucleus strongly suggests that this ubiquitous molecule may have a role in protecting the DNA from insults. Our data showed the induction of TCTP occurred in both the soluble nuclear fraction and the chromatin-enriched fraction of irradiated cells. When nucleoplasmic proteins were removed by detergent treatment, nuclear TCTP foci were clearly visible, and their number increased as a function of radiation dose, strongly supporting a role for TCTP in repair and/or sensing of DNA damage induced by radiation (Zhang et al. 2012). Studies by Ramaswamy demonstrated that the entry of TCTP into the nucleus is important for its antioxidant function, and TCTP transport into the nucleus is mediated by sumoylation (Munirathinam and Ramaswamy 2012). Also, Rid et al. showed that H2O2-dependent translocation of TCTP into the nucleus enables its interaction with the vitamin D receptor (VDR) in human keratinocytes (Rid et al. 2010). However, how TCTP translocates into the nucleus soon after cellular exposure of normal or tumor cells to ionizing radiation remains unknown. It is attractive to speculate that this may occur either through its chaperone function or by sumoylation.
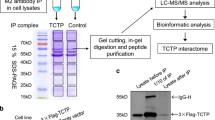
As discussed above, eukaryotes have evolved complex mechanisms to repair DSBs through coordinated actions of protein sensors, transducers, and effectors. Our data established a functional relation between TCTP and several key proteins such as ATM, Ku70, Ku80, and p53 that participate in DSB sensing and repair. Exposure to ionizing radiation induced a significant TCTP protein enrichment in nuclei, which was dependent on early sensors of DNA damage, specifically ATM and DNA-PK (Bakkenist and Kastan 2004). Importantly, this induction was associated with protective effects against DNA damage. Like in the case of cells treated with DNA repair inhibitors, repair of γ-ray-induced chromosomal damage was compromised in TCTP-deficient cells (Fig. 12.5). In chromatin of irradiated cells, TCTP was found to exist in complex with ATM and γH2AX, in agreement with its distinct localization with the foci of the DNA damage marker proteins γH2A.X, 53BP1, and p-ATM (Fig. 12.6) (Zhang et al. 2012). However, the exact nature of the interaction (direct or indirect) between TCTP and ATM kinase remains unknown. Although TCTP contains putative PI3K phosphorylation sites (e.g., T39, S46 and S53), in experiments involving IP of TCTP from AG1522 normal human fibroblasts (wildtype ATM), U2OS human bone osteosarcoma cells (wildtype ATM) or AG4405 human fibroblasts (mutated ATM) exposed to γ-rays, followed by in-gel trypsin digestion, and analysis by liquid chromatography/mass spectrometry (LC-MS/MS) on “Orbitrap velos instrument,” we did not detect any phosphorylation site within the protein. Therefore, the issue of how TCTP might be recruited to the ends of DBSs remains open.
Ku proteins are of central importance to DNA repair in eukaryotes. Ku70/86 heterodimer is the first component of NHEJ as it directly binds DNA and recruits other NHEJ factors to promote the repair of the broken ends (Downs and Jackson 2004). TCTP is required for the DNA-binding activity of Ku70 and Ku80 in response to irradiation. Such important effects of TCTP on Ku proteins also highlight the role of TCTP in NHEJ; furthermore, inactivation of Ku proteins leads to defects in telomere maintenance and chromosomal end fusion (Williams et al. 2009). Further, Ku has a key role in a number of other fundamental cellular processes such as transcription and apoptosis (Downs and Jackson 2004). Therefore, a possible chaperone role of TCTP in Ku translocation may imply additional functions of TCTP (Gnanasekar et al. 2009).
Interestingly, IP/MS experiments also indicate that TCTP interacts with filamin-A (Zhang et al. 2012). Filamin-A interacts with the DNA damage response proteins BRCA1 and BRCA2 and therefore may be required for efficient HRR. Defects in filamin-A impair the repair of DSBs resulting in sensitization of cells to ionizing radiation (Yue et al. 2009). The role of TCTP in such critical mechanisms that maintain genomic integrity may explain in part why homozygous mutation in TCTP is embryonically lethal (Chen et al. 2007a). However, more work will be required to establish how the TCTP/filamin-A complex functions in DNA repair.
12.6 TCTP and Control of Cell Cycle Progression Under Normal and Stress Conditions
TCTP is a conserved mitotic growth integrator in animals and plants (Brioudes et al. 2010). Overexpression of TCTP resulted in growth retardation of cells and affected microtubule stabilization and cell morphology (Gachet et al. 1999). Several nuclear proteins involved in mitotic progression have been proposed to interact with TCTP, either regulating or being regulated by TCTP (Bommer 2012). Association of TCTP with microtubules, the important apparatus of the mitotic spindle, has been demonstrated through binding tubulin (Gachet et al. 1999) and actin (Bazile et al. 2009) in a cell cycle-dependent manner. While TCTP is bound to the mitotic spindle, predominantly to the poles, to stabilize spindle microtubules, it is detached from the spindle during metaphase–anaphase transition (Gachet et al. 1999; Burgess et al. 2008; Yarm 2002). Two phosphorylation sites, for mitotic polo-like kinase (Plk-1) in the flexible loop of the TCTP structure, have been identified (Yarm 2002). Phosphorylation decreases the microtubule-stabilizing activity of TCTP and promotes the increase in microtubule dynamics that occurs after metaphase. Expression of a TCTP protein mutated in these sites led to severe disturbance of mitotic progression and to the formation of multinucleated cells (Yarm 2002; Johnson et al. 2008). Phospho-TCTP-ser46 was even confirmed as a marker for Plk-1 activity in vivo (Cucchi et al. 2010). Notably, Johansson et al. described the interaction of TCTP with the two nuclear proteins, nucleophosmin and nucleolin, in embryonic stem cells (Johansson et al. 2010a). In the case of nucleophosmin, the interaction was shown to be independent of phosphorylation by Plk-1 (Johansson et al. 2010a, b), suggesting the involvement of additional unrecognized mechanisms.
Deregulated microtubule dynamics and chromosome segregation enhances genomic instability (Rao et al. 2009). CHFR (checkpoint protein with FHA and RING domains) is a modulator of the mitotic stress checkpoint that delays entry into metaphase (Scolnick and Halazonetis 2000). Chfr is a tumor suppressor that ensures chromosomal stability by controlling the expression levels of key mitotic proteins such as Aurora A (Yu et al. 2005). Interestingly, the interaction of Chfr with TCTP occurs throughout the cell cycle, but it could be diminished by depolymerization of the microtubules (Burgess et al. 2008). Although Chfr could be the sensor that detects microtubule disruption and then activates the prophase checkpoint, it remains to be examined whether TCTP binding to Chfr protects the latter from being degraded. Recently, a novel pathway CHD1L/TCTP/Cdc25C/Cdk1 involved in hepatocellular carcinoma development has been identified. Overexpression of TCTP transcriptionally induced by CHD1L promoted the ubiquitin–proteasome degradation of Cdc25C during mitotic progression, which caused a failure in dephosphorylation of Cdk1 and decreased Cdk1 activity. Consequently, a faster mitotic exit and chromosome mis-segregation led to chromosomal instability (Chan et al. 2012).
To maintain genome integrity, cells need to adequately respond to various modes of genotoxic stress. DNA damage is known to trigger cell cycle arrest in the G1, S, or G2 phases of the cell cycle through activation of DNA-damage checkpoints (Iliakis et al. 2003). This arrest can be reversed once the damage has been repaired, but irreparable damage can promote apoptosis or senescence. Alternatively, cells can reenter the cell cycle before repair has been completed, giving rise to mutations (Medema and Macurek 2012). Our study (Zhang et al. 2012) showed that TCTP physically associates with p53, a protein with essential function in radiation-induced G1 checkpoint (Sengupta and Harris 2005). Depletion of TCTP greatly attenuated the magnitude of radiation-induced G1 delay in normal human fibroblasts, as well as the induction of p21Waf1, a cyclin-dependent kinase inhibitor (Abbas and Dutta 2009). The loss of normal G1 checkpoint control could disrupt DNA repair and is an early step in carcinogenesis (Syljuasen et al. 1999), which highlights the role for TCTP in p53-dependent mechanisms that maintain genome integrity under stressful conditions.
Further, our studies have shown that knockdown of TCTP modulates the γ-ray-induced G2 checkpoint. While an earlier entry into G2 phase may be a consequence of faster progression through G1 to S phase in irradiated cells with downregulated levels of TCTP, the longer delay in G2 following exposure to ionizing radiation is likely due to a greater level of DNA damage (Zhang et al. 2012). Clearly, additional studies are needed to clarify the role of TCTP in checkpoint control in irradiated cells. As mentioned previously, Phospho-TCTP-ser46 is a marker for Plk-1 activity in vivo (Cucchi et al. 2010). Plk-1 activation can promote mitotic entry in an unperturbed cell cycle, but following a DNA-damaging insult, cells come to completely rely on Plk1 to reenter the mitotic cycle following G2 arrest (van Vugt et al. 2010). Indeed, it has been shown that Plk-1 phosphorylates G2- and S-phase-expressed protein-1, which acts as a negative regulator of p53, thus suggesting that Plk1 activity contributes to suppression of p53 during checkpoint recovery (Liu et al. 2010). Therefore, it will be of interest to see if phosphorylated TCTP would also be a new negative regulator of p53. Interestingly, overexpression of TCTP was shown in lung carcinoma cells to destabilize p53 (Rho et al. 2011). Our results in normal human fibroblasts therefore open an exciting possibility that TCTP effects on p53 may differ in different cell lines/strains subjected to genotoxic stress.
12.7 TCTP and Cell Death
Several models have been proposed to explain how TCTP functions as a mediator of programmed cell death. During early mammalian development, it plays a role in anti-apoptotic activity through functional antagonism of the BMP4 pathway (Koide et al. 2009). As a Ca++-scavenger and molecular chaperone, TCTP protects cells under Ca++-stress (Graidist et al. 2007) or heat shock conditions (Gnanasekar et al. 2009). In addition, it has been shown that TCTP blocks the cleavage of poly(ADP-ribose)-polymerase (PARP) (Tuynder et al. 2002), a key event in apoptosis (Lazebnik et al. 1994). Also, TCTP protects ovarian carcinoma cells against TSC-22-mediated apoptosis (Lee et al. 2008).
TCTP proteins contain a H2–H3 helices structural similarity to channel-forming helices (Petros et al. 1644) of the pro-apoptotic protein Bax (Suzuki et al. 2000). Investigations showed that TCTP exerts its anti-apoptotic function by insertion into the mitochondrial membrane and inhibiting the dimerization of Bax (Susini et al. 2008). Also, it has been shown that TCTP interacts with Bcl-xL (Yang et al. 2005) and Mcl-1 (Liu et al. 2005; Zhang et al. 2002), two other anti-apoptotic proteins of the Bcl-2 family. The N-terminal region of TCTP and the BH3 domain of Bcl-xL are thought to mediate the interaction between these two proteins (Yang et al. 2005), which may inhibit T-cell apoptosis by preventing the phosphorylation/inactivation of Bcl-xL. However, the interaction between TCTP and Mcl-1 is debatable; while some suggested that the two proteins stabilize each other (Liu et al. 2005; Zhang et al. 2002), others showed that they exert their anti-apoptotic function independently of each other (Graidist et al. 2004).
Recently, a novel function of TCTP in intercellular signaling leading to anti-apoptotic effects was proposed by Sirois et al. (2011), which sheds light on a new direction in bystander effects research, which is under intense investigation in radiation studies (Azzam et al. 2003; Mothersill and Seymour 2004; Prise et al. 2005). Interestingly, TCTP was identified on the surface of extracellular vesicles purified from medium conditioned by apoptotic endothelial cells, and caspase-3 activation plays a key role for the release of TCTP when these cells are dying by apoptosis (Sirois et al. 2011). Further, the nanovesicles, which are different from apoptotic blebs, induced an extracellular signal-regulated kinase 1/2 (ERK 1/2)-dependent anti-apoptotic phenotype in vascular smooth muscle cells (VSMC) (Sirois et al. 2011). During cancer radiotherapy, activated caspase-3 in dying tumor cells has been shown to regulate the release of prostaglandin E2 (PGE2), which can potently stimulate growth of surviving tumor cells (Huang et al. 2011). It would be interesting to determine if TCTP also plays an anti-apoptotic function by intercellular signaling in radiation-induced bystander effects.
12.8 Perspective
In humans, DNA can be damaged by various endogenous and environmental agents, leading to various disorders. Mechanisms must, therefore, exist to protect or repair DNA. At high doses, ionizing radiation is known to cause excessive DNA damage, often followed by cancer or degenerative diseases. We studied cellular responses to low doses of ionizing radiation that are typical of certain occupational activities or diagnostic radiography. Surprisingly, we observed significant adaptive responses when normal human cells were exposed to low doses of cesium-137 γ-rays (Azzam et al. 1996; de Toledo et al. 2006) and identified TCTP as a specific protein involved in this response (Zhang et al. 2012).
In our initial irradiation tests, we found that irradiated cells harbored lower levels of chromosomal damage than what occurred spontaneously at the basal level (de Toledo et al. 2006). This unexpected finding prompted us to use a proteomic approach to identify proteins that are differentially expressed in cells after exposure to 10 cGy of cesium-137 γ-rays delivered at a low dose over 50 h. TCTP was found to be upregulated and appeared most sensitive in this context. The precise pro-survival mechanism mediated by TCTP remains poorly understood.
To this end, we tested the hypothesis that TCTP plays a critical role in response to DNA damage and that this function is essential particularly for the survival and genomic integrity of irradiated cells. We found that upon exposure to doses as low as 1 cGy of cesium-137 γ-rays (a dose received in many diagnostic procedures), the TCTP level was greatly increased in normal human cells, with a significant enrichment in the nuclei. The protein level was similarly upregulated in tissues of low-dose-irradiated mice. Moreover, this upregulation was induced by moderate and high doses of different types of ionizing radiation.
Interestingly, TCTP upregulation was dependent on the early sensors of DNA damage, specifically the protein ATM and the enzyme DNA-PK. Importantly, this upregulation was associated with protective effects against DNA damage. As shown in the case of cells treated with DNA repair inhibitors in previous experiments, repair of γ-ray-induced chromosomal damage was compromised in TCTP-deficient cells. In the chromatin of irradiated cells, TCTP was found to exist in complex with ATM and γH2A.X, a protein that marks the sites of DNA damage. This finding is in agreement with TCTP’s distinct localization with the foci of the DNA damage marker proteins γH2A.X, 53BP1, and p-ATM. Furthermore, TCTP was shown to interact with the DNA-binding subunits Ku70 and Ku80 of DNA-PK, a protein with a major role in repair of DNA DSBs, a particularly harmful form of DNA damage (Zhang et al. 2012).
Our findings are consistent with the observation that TCTP knockdown led to decreased levels of Ku70 and Ku80 in the nuclei of irradiated cells and attenuated the DNA-binding activity of DNA-PK. Interestingly, the protective effects of TCTP were not confined to low-dose-irradiated cells, but were observed even against the lethal effects of therapeutic doses of γ-rays. This may explain why knockdown of TCTP increased the failure of normal human cells to divide, or reproduce, when exposed to 200 or 400 cGy (doses received during cancer radiotherapy) (Zhang et al. 2012).
In normal cells, TCTP did not affect such cell cycle progression towards division under normal, homeostatic conditions. However, TCTP had a prominent effect on stress-induced cell cycle checkpoints, which ensure that the cell cycle progresses without any DNA damage. We found that TCTP interacted with p53, a critical protein component of such checkpoints that maintains genomic integrity. Furthermore, TCTP knockdown shortened the radiation-induced delay in the G1 phase of the cell cycle, which is the pre-DNA synthesis phase. The latter effect was associated with attenuated induction of p21Waf1, an inhibitor of master regulators of the cell cycle. The loss of the normal G1 checkpoint control disrupts DNA repair and is an early step in carcinogenesis, thus highlighting the role of TCTP in maintaining healthy survival. In addition, TCTP has a role in the post-DNA synthesis (G2) phase, where it modulates the duration of the radiation-induced G2 checkpoint. Cells with downregulated TCTP entered G2 phase faster than control cells and were arrested longer in G2 phase (Zhang et al. 2012).
Together, our results identify TCTP as a new member of a group of proteins involved in DNA damage response (Fig. 12.8). Our results also point to a chaperone-like role of TCTP, where it interacts with several stress-induced molecular chaperones/heat-shock proteins in irradiated cells. The new role of TCTP in sensing and repairing radiation-induced DNA damage will aid in understanding the system responses to low-dose radiation exposures and in turn help in estimating health risks of such exposures. It may also aid in understanding the molecular events induced by therapeutic doses of radiation. Clearly, future studies need to address the exact role of TCTP in HR, NHEJ, and other modes of DNA repair. To this end, the use of cells that are proficient or deficient in either of the latter DNA repair mechanisms and where TCTP levels are altered should be informative.

The role of TCTP in DNA damage sensing and repair. TCTP is upregulated by ionizing radiation; it interacts with elements of DNA damage sensing and repair and modulates radiation-induced cell cycle checkpoints (IR ionizing radiation, P phosphorylation, MRN MRE11–RAD50–NBS1, NHEJ nonhomologous end joining, HR homologous recombination, HSPs heat shock proteins)
References
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9:400–414
Ames BN (1989) Endogenous oxidative DNA damage, aging, and cancer. Free Radic Res Commun 7:121–128
Amson R, Pece S, Lespagnol A et al (2012) Reciprocal repression between P53 and TCTP. Nat Med 18:91–99
Amson R, Pece S, Marine JC et al (2013) TPT1/TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol 23:37–46
Azzam EI (2011) Exposure to low level environmental agents: the induction of hormesis. Mutat Res 726:89–90
Azzam EI, de Toledo SM, Raaphorst GP et al (1996) Low-dose ionizing radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in C3H 10T1/2 cells. Radiat Res 146:369–373
Azzam EI, de Toledo SM, Little JB (2003) Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene 22:7050–7057
Azzam EI, Jay-Gerin JP, Pain D (2012) Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 327:48–60
Azzam EI, Colangelo NW, Domogauer JD et al (2016) Is ionizing radiation harmful at any exposure? An echo that continues to vibrate. Health Phys 110:249–251
Badie S, Carlos AR, Folio C et al (2015) BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J 34:410–424
Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118:9–17
Barendsen GW (1964) Impairment of the proliferative capacity of human cells in cultures by a particles of differing linear energy transfer. Int J Radiat Biol 8:453–466
Baumstark-Khan C (1993) X-ray-induced DNA double-strand breaks as lethal lesions in diploid human fibroblasts compared to Chinese hamster ovary cells. Int J Radiat Biol 63:305–311
Bazile F, Pascal A, Arnal I et al (2009) Complex relationship between TCTP, microtubules and actin microfilaments regulates cell shape in normal and cancer cells. Carcinogenesis 30:555–565
Bohm H, Benndorf R, Gaestel M et al (1989) The growth-related protein P23 of the Ehrlich ascites tumor: translational control, cloning and primary structure. Biochem Int 19:277–286
Bommer UA (2012) Several nuclear proteins involved in mitotic progression have been proposed to interact with TCTP, either regulating or being regulated by TCTP. Open Allergy J 5:19–32
Bommer UA, Thiele BJ (2004) The translationally controlled tumour protein (TCTP). Int J Biochem Cell Biol 36:379–385
Bommer UA, Heng C, Perrin A et al (2010) Roles of the translationally controlled tumour protein (TCTP) and the double-stranded RNA-dependent protein kinase, PKR, in cellular stress responses. Oncogene 29:763–773
Branzei D, Foiani M (2008) Regulation of DNA repair throughout the cell cycle. Nat Rev 9:297–308
Brioudes F, Thierry AM, Chambrier P et al (2010) Translationally controlled tumor protein is a conserved mitotic growth integrator in animals and plants. Proc Natl Acad Sci USA 107:16384–16389
Burgess A, Labbe JC, Vigneron S et al (2008) Chfr interacts and colocalizes with TCTP to the mitotic spindle. Oncogene 27:5554–5566
Cadet J, Douki T, Ravanat JL (2011) Measurement of oxidatively generated base damage in cellular DNA. Mutat Res 711:3–12
Cans C, Passer BJ, Shalak V et al (2003) Translationally controlled tumor protein acts as a guanine nucleotide dissociation inhibitor on the translation elongation factor eEF1A. Proc Natl Acad Sci USA 100:13892–13897
Chan TH, Chen L, Liu M et al (2012) Translationally controlled tumor protein induces mitotic defects and chromosome missegregation in hepatocellular carcinoma development. Hepatology 55:491–505
Chatterjee A, Schaefer HJ (1976) Microdosimetric structure of heavy ion tracks in tissue. Radiat Environ Biophys 13:215–227
Chen SH, Wu PS, Chou CH et al (2007a) A knockout mouse approach reveals that TCTP functions as an essential factor for cell proliferation and survival in a tissue- or cell type-specific manner. Mol Biol Cell 18:2525–2532
Chen Z, Zhang H, Yang H et al (2007b) The expression of AmphiTCTP, a TCTP orthologous gene in amphioxus related to the development of notochord and somites. Comp Biochem Physiol B Biochem Mol Biol 147:460–465
Chitpatima ST, Makrides S, Bandyopadhyay R et al (1988) Nucleotide sequence of a major messenger RNA for a 21 kilodalton polypeptide that is under translational control in mouse tumor cells. Nucleic Acids Res 16:2350
Chou WC, Hu LY, Hsiung CN et al (2015) Initiation of the ATM-Chk2 DNA damage response through the base excision repair pathway. Carcinogenesis 36:832–840
Coutard H (1937) The results and methods of treatment of cancer by radiation. Ann Surg 106:584–598
Cucchi U, Gianellini LM, De Ponti A et al (2010) Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res 30:4973–4985
de Toledo SM, Asaad N, Venkatachalam P et al (2006) Adaptive responses to low-dose/low-dose-rate gamma rays in normal human fibroblasts: the role of growth architecture and oxidative metabolism. Radiat Res 166:849–857
Decottignies A (2013) Alternative end-joining mechanisms: a historical perspective. Front Genet 4:48
Delaney G, Jacob S, Featherstone C et al (2005) The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 104:1129–1137
Dizdaroglu M (2012) Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett 327:26–47
Downs JA, Jackson SP (2004) A means to a DNA end: the many roles of Ku. Nat Rev 5:367–378
Fenech M, Morley AA (1985) Measurement of micronuclei in lymphocytes. Mutat Res 147:29–36
Gachet Y, Tournier S, Lee M et al (1999) The growth-related, translationally controlled protein P23 has properties of a tubulin binding protein and associates transiently with microtubules during the cell cycle. J Cell Sci 112(Pt 8):1257–1271
Georgakilas AG (2011) From chemistry of DNA damage to repair and biological significance. Comprehending the future. Mutat Res 711:1–2
Gnanasekar M, Ramaswamy K (2007) Translationally controlled tumor protein of Brugia malayi functions as an antioxidant protein. Parasitol Res 101:1533–1540
Gnanasekar M, Dakshinamoorthy G, Ramaswamy K (2009) Translationally controlled tumor protein is a novel heat shock protein with chaperone-like activity. Biochem Biophys Res Commun 386:333–337
Goodhead DT (1988) Spatial and temporal distribution of energy. Health Phys 55:231–240
Graidist P, Phongdara A, Fujise K (2004) Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-fluorouracil-induced cytotoxicity. J Biol Chem 279:40868–40875
Graidist P, Yazawa M, Tonganunt M et al (2007) Fortilin binds Ca2+ and blocks Ca2+-dependent apoptosis in vivo. Biochem J 408:181–191
Hacein-Bey L, Konstas AA, Pile-Spellman J (2014) Natural history, current concepts, classification, factors impacting endovascular therapy, and pathophysiology of cerebral and spinal dural arteriovenous fistulas. Clin Neurol Neurosurg 121:64–75
Hall EJ, Giaccia AJ (2006) Radiobiology for the radiologist, 6th edn. Lippincott Williams & Wilkins, Philadelphia, PA
Harper JW, Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28:739–745
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475–1485
Hsu YC, Chern JJ, Cai Y et al (2007) Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature 445:785–788
Huang L, Snyder AR, Morgan WF (2003) Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene 22:5848–5854
Huang Q, Li F, Liu X et al (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17:860–866
Iliakis G, Wang Y, Guan J et al (2003) DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 22:5834–5847
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Jasin M (2015) Accolades for the DNA damage response. N Engl J Med 373:1492–1495
Jeggo PA, Pearl LH, Carr AM (2016) DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 16:35–42
Johansson H, Svensson F, Runnberg R et al (2010a) Phosphorylated nucleolin interacts with translationally controlled tumor protein during mitosis and with Oct4 during interphase in ES cells. PloS One 5:e13678
Johansson H, Vizlin-Hodzic D, Simonsson T et al (2010b) Translationally controlled tumor protein interacts with nucleophosmin during mitosis in ES cells. Cell Cycle 9:2160–2169
Johnson TM, Antrobus R, Johnson LN (2008) Plk1 activation by Ste20-like kinase (Slk) phosphorylation and polo-box phosphopeptide binding assayed with the substrate translationally controlled tumor protein (TCTP). Biochemistry 47:3688–3696
Jung J, Kim HY, Kim M et al (2011) Translationally controlled tumor protein induces human breast epithelial cell transformation through the activation of Src. Oncogene 30:2264–2274
Kashiwakura JC, Ando T, Matsumoto K et al (2012) Histamine-releasing factor has a proinflammatory role in mouse models of asthma and allergy. J Clin Invest 122:218–228
Katschinski DM, Boos K, Schindler SG et al (2000) Pivotal role of reactive oxygen species as intracellular mediators of hyperthermia-induced apoptosis. J Biol Chem 275:21094–21098
Koide Y, Kiyota T, Tonganunt M et al (2009) Embryonic lethality of fortilin-null mutant mice by BMP-pathway overactivation. Biochim Biophys Acta 2009:326–338
Koziol MJ, Garrett N, Gurdon JB (2007) Tpt1 activates transcription of oct4 and nanog in transplanted somatic nuclei. Curr Biol 17:801–807
Larrea AA, Lujan SA, Kunkel TA (2010) SnapShot: DNA mismatch repair. Cell 141:730–e731
LaVerne JA, Pimblott SM (1993) Yields of hydroxyl radical and hydrated electron scavenging reactions in aqueous solutions of biological interest. Radiat Res 135:16–23
Lazebnik YA, Kaufmann SH, Desnoyers S et al (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347
Lee JH, Rho SB, Park SY et al (2008) Interaction between fortilin and transforming growth factor-beta stimulated clone-22 (TSC-22) prevents apoptosis via the destabilization of TSC-22. FEBS Lett 582:1210–1218
Li F, Zhang D, Fujise K (2001) Characterization of fortilin, a novel antiapoptotic protein. J Biol Chem 276:47542–47549
Li M, Gonon G, Buonanno M et al (2014) Health risks of space exploration: targeted and non-targeted oxidative injury by high charge and high energy particles. Antioxid Redox Signal 20:1501–1523
Lieber MR (2008) The mechanism of human nonhomologous DNA end joining. J Biol Chem 283:1–5
Lin J, Epel E, Blackburn E (2012) Telomeres and lifestyle factors: roles in cellular aging. Mutat Res 730:85–89
Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362:709–715
Little JB (1968) Delayed initiation of DNA synthesis in irradiated human diploid cells. Nature 218:1064–1065
Little JB (2000) Radiation carcinogenesis. Carcinogenesis 21:394–404
Liu H, Peng HW, Cheng YS et al (2005) Stabilization and enhancement of the antiapoptotic activity of mcl-1 by TCTP. Mol Cell Biol 25:3117–3126
Liu XS, Li H, Song B et al (2010) Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep 11:626–632
Lucibello M, Gambacurta A, Zonfrillo M et al (2011) TCTP is a critical survival factor that protects cancer cells from oxidative stress-induced cell-death. Exp Cell Res 317:2479–2489
Macdonald SM (2012) Potential role of histamine releasing factor (HRF) as a therapeutic target for treating asthma and allergy. J Asthma Allergy 5:51–59
MacDonald SM, Rafnar T, Langdon J et al (1995) Molecular identification of an IgE-dependent histamine-releasing factor. Science 269:688–690
McKinnon PJ (2009) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10:100–112
Medema RH, Macurek L (2012) Checkpoint control and cancer. Oncogene 31:2601–2613
Meesungnoen J, Benrahmoune M, Filali-Mouhim A et al (2001) Monte Carlo calculation of the primary radical and molecular yields of liquid water radiolysis in the linear energy transfer range 0.3–6.5 keV/um: application to 137Cs gamma rays. Radiat Res 155:269–278. Erratum Published in Radiat Res 155 (2001) 2873
Moskovitz J (2005) Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Curr Pharm Des 11:1451–1457
Mothersill C, Seymour CB (2004) Radiation-induced bystander effects--implications for cancer. Nat Rev Cancer 4:158–164
Munirathinam G, Ramaswamy K (2012) Sumoylation of human translationally controlled tumor protein is important for its nuclear transport. Biochem Res Int 2012:831940
Muroya Y, Plante I, Azzam EI et al (2006) High-LET ion radiolysis of water: visualization of the formation and evolution of ion tracks and relevance to the radiation-induced bystander effect. Radiat Res 165:485–491
Nagano-Ito M, Banba A, Ichikawa S (2009) Functional cloning of genes that suppress oxidative stress-induced cell death: TCTP prevents hydrogen peroxide-induced cell death. FEBS Lett 583:1363–1367
Nam EA, Cortez D (2011) ATR signalling: more than meeting at the fork. Biochem J 436:527–536
NCRP (2009) Ionizing radiation exposure of the population of the United States. National Council on Radiation Protection and Measurements, Bethesda
Newhauser WD, Durante M (2011) Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer 11:438–448
Nikjoo H, O’Neill P, Wilson WE et al (2001) Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat Res 156:577–583
Oikawa K, Ohbayashi T, Mimura J et al (2002) Dioxin stimulates synthesis and secretion of IgE-dependent histamine-releasing factor. Biochem Biophys Res Commun 290:984–987
Okada H, Mak TW (2004) Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 4:592–603
O’Neill P, Wardman P (2009) Radiation chemistry comes before radiation biology. Int J Radiat Biol 85:9–25
Panier S, Boulton SJ (2014) Double-strand break repair: 53BP1 comes into focus. Nat Rev 15:7–18
Petkau A (1987) Role of superoxide dismutase in modification of radiation injury. Br J Cancer Suppl 8:87–95
Petros AM, Olejniczak ET, Fesik SW (1644) Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta 2004:83–94
Petruseva IO, Evdokimov AN, Lavrik OI (2014) Molecular mechanism of global genome nucleotide excision repair. Acta Naturae 6:23–34
Platzman R (1958) The physical and chemical basis of mechanisms in radiation biology. In: Claus W (ed) Radiation biology and medicine. Selected reviews in the life sciences. Addison-Wesley, Reading, MA, pp 15–72
Postel-Vinay S, Vanhecke E, Olaussen KA et al (2012) The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nature reviews. Clin Oncol 9:144–155
Prise KM, Schettino G, Folkard M et al (2005) New insights on cell death from radiation exposure. Lancet Oncol 6:520–528
Rao CV, Yamada HY, Yao Y et al (2009) Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis 30:1469–1474
Raynaud CM, Sabatier L, Philipot O et al (2008) Telomere length, telomeric proteins and genomic instability during the multistep carcinogenic process. Crit Rev Oncol Hematol 66:99–117
Rho SB, Lee JH, Park MS et al (2011) Anti-apoptotic protein TCTP controls the stability of the tumor suppressor p53. FEBS Lett 585:29–35
Rid R, Onder K, Trost A et al (2010) H2O2-dependent translocation of TCTP into the nucleus enables its interaction with VDR in human keratinocytes: TCTP as a further module in calcitriol signalling. J Steroid Biochem Mol Biol 118:29–40
Rogakou EP, Pilch DR, Orr AH et al (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868
Scolnick DM, Halazonetis TD (2000) Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 406:430–435
Sengupta S, Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev 6:44–55
Shah NR, Mahmoudi M (2015) The role of DNA damage and repair in atherosclerosis: a review. J Mol Cell Cardiol 86:147–157
Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3:155–168
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev 14:197–210
Sirois I, Raymond MA, Brassard N et al (2011) Caspase-3-dependent export of TCTP: a novel pathway for antiapoptotic intercellular communication. Cell Death Differ 18:549–562
Spitz DR, Azzam EI, Li JJ et al (2004) Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev 23:311–322
Stucki M, Jackson SP (2006) GammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5:534–543
Susini L, Besse S, Duflaut D et al (2008) TCTP protects from apoptotic cell death by antagonizing bax function. Cell Death Differ 15:1211–1220
Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103:645–654
Syljuasen RG, Krolewski B, Little JB (1999) Loss of normal G1 checkpoint control is an early step in carcinogenesis, independent of p53 status. Cancer Res 59:1008–1014
Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45:247–271
Thaw P, Baxter NJ, Hounslow AM et al (2001) Structure of TCTP reveals unexpected relationship with guanine nucleotide-free chaperones. Nat Struct Biol 8:701–704
Thomas G, Thomas G, Luther H (1981) Transcriptional and translational control of cytoplasmic proteins after serum stimulation of quiescent Swiss 3T3 cells. Proc Natl Acad Sci USA 78:5712–5716
Thompson LH (2012) Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 751:158–246
Tsarova K, Yarmola EG, Bubb MR (2010) Identification of a cofilin-like actin-binding site on translationally controlled tumor protein (TCTP). FEBS Lett 584:4756–4760
Turinetto V, Giachino C (2015) Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res 43:2489–2498
Tuynder M, Susini L, Prieur S et al (2002) Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci USA 99:14976–14981
Tuynder M, Fiucci G, Prieur S et al (2004) Translationally controlled tumor protein is a target of tumor reversion. Proc Natl Acad Sci USA 101:15364–15369
Tyldesley S, Boyd C, Schulze K et al (2001) Estimating the need for radiotherapy for lung cancer: an evidence-based, epidemiologic approach. Int J Radiat Oncol Biol Phys 49:973–985
van Vugt MA, Gardino AK, Linding R et al (2010) A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 8:e1000287
Weichselbaum RR, Nove J, Little JB (1980) X-ray sensitivity of fifty-three human diploid fibroblast cell strains from patients with characterized genetic disorders. Cancer Res 40:920–925
Weinberg RA (2007) The biology of cancer. Garland Science, Taylor & Francis Group, LLC, New York
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Williams ES, Klingler R, Ponnaiya B et al (2009) Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res 69:2100–2107
Yang Y, Yang F, Xiong Z et al (2005) An N-terminal region of translationally controlled tumor protein is required for its antiapoptotic activity. Oncogene 24:4778–4788
Yarm FR (2002) Plk phosphorylation regulates the microtubule-stabilizing protein TCTP. Mol Cell Biol 22:6209–6221
Yenofsky R, Bergmann I, Brawerman G (1982) Messenger RNA species partially in a repressed state in mouse sarcoma ascites cells. Proc Natl Acad Sci USA 79:5876–5880
Yenofsky R, Cereghini S, Krowczynska A et al (1983) Regulation of mRNA utilization in mouse erythroleukemia cells induced to differentiate by exposure to dimethyl sulfoxide. Mol Cell Biol 3:1197–1203
Yi C, He C (2013) DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol 5:a012575
Yu X, Minter-Dykhouse K, Malureanu L et al (2005) Chfr is required for tumor suppression and Aurora A regulation. Nat Genet 37:401–406
Yue J, Wang Q, Lu H et al (2009) The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res 69:7978–7985
Zhang D, Li F, Weidner D et al (2002) Physical and functional interaction between myeloid cell leukemia 1 protein (MCL1) and Fortilin. The potential role of MCL1 as a fortilin chaperone. J Biol Chem 277:37430–37438
Zhang J, de Toledo SM, Pandey BN et al (2012) Role of the translationally controlled tumor protein in DNA damage sensing and repair. Proc Natl Acad Sci USA 109:E926–E933
Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433–439
Zimbrick JD (2002) Radiation chemistry and the Radiation Research Society: a history from the beginning. Radiat Res 158:127–140
Acknowledgements
This work was supported by grant CA049062 from the National Institutes of Health, grant NNX15AD62G from the National Aeronautics and Space Administration, and the Program for Changjiang Scholars and Innovative Research Team, University of Ministry of Education, China.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Zhang, J., Shim, G., de Toledo, S.M., Azzam, E.I. (2017). The Translationally Controlled Tumor Protein and the Cellular Response to Ionizing Radiation-Induced DNA Damage. In: Telerman, A., Amson, R. (eds) TCTP/tpt1 - Remodeling Signaling from Stem Cell to Disease. Results and Problems in Cell Differentiation, vol 64. Springer, Cham. https://doi.org/10.1007/978-3-319-67591-6_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-67591-6_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67590-9
Online ISBN: 978-3-319-67591-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)