
Abstract
The importance of chickpea and constraints in chickpea production urged the need of chickpea genome. Varshney and colleagues in 2013 reported the draft genome of chickpea (kabuli). The genome assembly was 532.29 Mb spanning across 7,163 scaffolds and consisted of 28,269 gene models. The estimated size of chickpea genome was 738.09 Mb based on k-mer analysis. The draft genome assembly covered 73.8% of the total estimated genome size for chickpea. Gene annotation was carried for predicted gene models, though the UTRs and promoters have not been yet been predicted. Genome duplication and synteny analysis with other closely related legume crops showed gene conservation and segmental duplications spread across the draft genome assembly. The genome assembly provides resource for targeting genes responsible for disease resistance which are of agronomic importance. The genome assembly has been used for genome-assisted breeding and is further utilized to study the diversity and domestication of chickpea.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
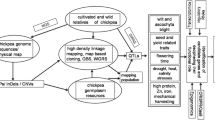

10.1 Introduction
Chickpea (Cicer arietinum L.) is a self-pollinated, diploid (2n = 2x = 16) legume crop primarily grown by resource-poor farmers in the semi-arid regions of the world. The nitrogen fixing ability and high protein content of chickpea make it a crop of high economic importance in developing countries. Based on the grain size and seed coat color, two market classes of chickpea, namely desi and kabuli, are cultivated extensively. Advances in genomics technologies facilitated the adoption of genomics tools in crop improvement referred as genomics-assisted breeding (Goodwin et al. 2016; Varshney et al. 2014; Koboldt et al. 2013; Metzker 2010). The availability of draft genomes of major cereals including rice (Oryza sativa; IRGSP 2002; Goff et al. 2002), sorghum (Sorghum bicolor; Paterson et al. 2008), maize (Zea mays; Schnable et al. 2009), and legumes such as pigeonpea (Cajanus cajan; Varshney et al. 2012) facilitates the deployment of genomic information in crop improvement.
Owing to the economic importance of chickpea and given the usefulness of draft genomes, the International Chickpea Genome Sequencing Consortium (ICGSC) led by ICRISAT decoded the draft genome of kabuli genotype CDC frontier. This chapter mainly summarizes the tools and strategies used for generating the draft genomes and various analyses for understanding the genome architecture of chickpea and synteny with other sequenced legumes. In addition, this chapter also provides a comparative view of both desi and kabuli genomes available.
10.2 Strategies and Tools for Sequencing
The chickpea genome sequencing was carried out using the short reads from Illumina HiSeq 2000 and bacterial artificial chromosome (BAC) end sequencing (Varshney et al. 2013). The Illumina short reads were assembled into contigs which were further used to construct the scaffolds. The BAC end sequencing was used to form the backbone for the scaffolding. Further, the high-density genetic maps were used to anchor the scaffolds on to the pseudomolecules. The unanchored scaffolds and contigs were reported separately along with pseudomolecules, as a part of the final assembly. Paired-end sequencing libraries (11 in total) were formulated with insert sizes of ~170 bp, 500 bp, 800 bp, 2 Kb, 5 Kb, 10 Kb, and 20 Kb. For the development of assembly, scaffolds’ construction and gap closure, SOAPdenovo2 (Luo et al. 2012) was used. The genetic marker sequences along with flanking regions were searched in the assembly using BLASTN (Altschul et al. 1997) and also using e-PCR (Schuler 1997) in case of the presence of only primer sequences, to place these sequences on the scaffolds. The microbial contamination was eliminated from the genome assembly using searches against the bacterial and fungal genomes with the help of Megablast. Further, BLAT (Kent 2002) was used to screen for contamination of organellar DNA, chloroplast genome sequence of chickpea, and Lotus (L. japonicus) mitochondrion in the chickpea genome assembly. The completeness of the genome assembly was verified by mapping the transcriptome assembly contigs to the genome assembly using BLAT. The exome coverage prediction was carried out by mapping the core eukaryotic genes, identified by core eukaryotic gene mapping approach CEGMA v.2.3 (Parra et al. 2007), to the genome assembly.
10.3 Assembly
A total of 153.01 Gb of sequence data was generated for the development of the first draft genome assembly in chickpea. This resulted in coverage of 207.32X from 11 genomic libraries sequenced using Illumina platform with insert sizes ranging from 180 bp to 20 Kb. The high-quality sequence data of 87.65 Gb after filtering was used to assemble into 544.73 Mb of genome sequence scaffolds. The N50 for these scaffolds was 645.3 Kb, and the maximum size of these scaffolds was found to be 6.17 Mb. The chickpea genome is estimated to be of 738.09 in size which shows that the assembled scaffolds were able to cover 73.8% of the genome. The non-assembled genome is believed to be enriched with repetitive sequences as observed by increased read depth in repeat-containing regions in comparison with non-repeat regions and also by having four-fold lower k-mer diversity in non-assembled fraction as compared to non-repetitive assembled fraction. An improved assembly spanning 532.29 Mb with a N50 of 39.99 Mb having 7,163 scaffolds was generated with the help of 46,270 repeat masked paired bacterial artificial chromosome (BAC) end sequences. The anchoring of 65.23% of this assembly to eight genetic linkage groups was carried out with the help of 1,292 genetic markers reported in previous studies. This data was used to obtain eight pseudomolecules namely, Ca1-Ca8. The anchoring of 93.4% of these scaffolds was validated using restriction-site-associated DNA (RAD) single nucleotide polymorphism (SNP) markers that were discovered between two segregating recombinant inbred line populations. This approach resulted in the identification of low-proportion chimeric scaffolds, i.e., 1.7% of the total scaffolds which amounted to 4.6 Mb of mis-assembled genomic sequence. These chimeric scaffolds were processed by excluding the erroneous part of the scaffold sequences and removing them from the pseudomolecule models. Another synteny-based approach was used to anchor the scaffolds onto the pseudomolecules. In this approach, regions lacking genetic support but showing conserved synteny with Medicago (Medicago truncatula) were anchored to pseudomolecules. The regions supported by synteny are hypothetical placements in the pseudomolecules which will be eventually updated upon availability of improved genetic maps supporting these regions or if there are modifications in the assembly of Medicago. The RAD genotyping data was used to anchor 75% of the scaffolds, while the synteny-based approach by comparing scaffolds with Medicago was used to anchor rest of the 25% scaffolds to the pseudomolecules.
10.4 Repetitive Sequences
Repeat regions in the genome were identified using Tandem Repeat Finder (Benson 1999) which resulted in a total of 127,377 such regions. It was observed that 84.9% repeat regions occurred in span of <1 kb, while in gap-spanning clones repeat regions were present in the tracts of 10–103 kb. Out of the total repeat regions identified, 29,018 regions could not be assembled due to low-sequence complexity and the occurrence of such repeats was masked by adding Ns within the pseudomolecules. Nearly half of the chickpea genome consists of transposable elements (TEs) and unclassified repeat elements similar to the percentage observed in other legume crops such as Medicago (30.5%), pigeonpea (Cajanus cajan; 51.6%), and soybean (Glycine max; 59%). The most abundant transposable elements are long terminal repeat (LTR) which covers more than 45% of total nuclear genome. The centromere regions are made up of the microsatellites which are dispersed as tandem repeats. The most found tandem repeats within the genome are 163-bp (18%), 100-bp (30%), and 74-bp (13%) unit repeats and constitute a total of 61% of total tandem repeats identified. The 163-bp and 100-bp units correspond to already identified chickpea microsatellites, CaSat1 and CaSat2, respectively, while 74-bp repeat is similar to dispersed highly repetitive element CaRep2. Tandem repeat finder was used to filter for the genomic regions >3 copies and >60 bp consensus length across the genome assembly. The genome assembly was scanned for the presence of transposable elements combining two approaches of de novo and homology-based searches. LTR_Finder v 1.03 (Xu and Wang 2007), PILER-DF v 1.0 (Edgar and Myers 2005), and RepeatScout v 1.05 (Price et al. 2005), all three de novo software, were used to build a chickpea repeat database. Repeat Masker v 3.2.7 (http://repeatmasker.org/, v 3.2.2) was deployed to identify repeats with the help of the constructed chickpea repeat database and Repbase (Jurka 1995). Along with these approaches, Repbase was also used to identify repeat-related proteins in the genome using RepeatProteinMask (http://repeatmasker.org/, v 3.2.2).
10.5 Gene Annotation
Gene prediction was done using combined approaches of ab initio modeling and homology-based searches with gene sets taken from six closely related legume species and CaTA transcript sequences. These approaches resulted in a non-redundant set of 28,269 gene models where average transcript and coding sequence size were 3,055 bp and 1,166 bp, respectively. Majority of these genes show homology with the gene models present in TrEMBL and Interpro (Zdobnov and Apweiler 2001) databases. The functions were assigned to 89.73% genes, while the rest 2,904 genes remained unannotated. The gene density was observed to be on the rise toward the ends of the pseudomolecules. The nonprotein coding genes resulted in the prediction of 684 tRNA, 478 rRNA, 420 miRNA, and 647 snRNA genes in the genome. The 454/Roche transcriptome data generated for CDC frontier line was mapped to the genome assembly for validation of the gene space capture by the draft genome assembly. The gene coverage is calculated to be ~90.8%. More than 98% homologs for core eukaryotic genes were found to be conserved in the draft genome assembly. BLASTP search using the chickpea proteome as query against the proteomes of Medicago, soybean, pigeonpea, and Lotus (Lotus japonicus) was carried out to estimate the conservation of chickpea gene models present in mentioned species. Proteome of chickpea was found to be most similar to Medicago (89.7% chickpea proteins correspond to Medicago proteins) and least similar to Arabidopsis (Arabidopsis thaliana: 79.2% were found similar to Arabidopsis proteins).
Three approaches homology-based, de novo, and transcript sequence-based were used for the gene prediction. The results of these approaches were fed to GLEAN (Elsik et al. 2007), which after multiple filtration resulted in a gene set of 28,256 genes. Further, CEGMA identified 453 core genes which are highly conserved across all eukaryotes. Out of these 453 core genes, 13 genes did not align to any gene with the set defined by GLEAN and rest were found present in the genome and hence were added to a final set resulting in 28,269 genes. BLASTP against SwissProt and TrEMBL databases (Magrane and Consortium 2011) was used to assign functions to the final predicted gene set. The presence of motifs and domains in genes was detected using InterProScan against protein databases which include Pfam (Punta et al. 2011), PROSITE (Sigrist et al. 2010), SMART (Letunic et al. 2012), PRINTS (Attwood et al. 2003), PANTHER (Thomas et al. 2003), and ProDom (Corpet et al. 2000). Genes were assigned gene ontology IDs, and with the information obtained from KEGG database (Kanehisa and Goto 2000) annotated with their associated pathway. tRNAscan-s.e.m. v1.23 (Lowe and Eddy 1997) was used to scan for tRNA genes, and INFERNAL v0.81 (Nawrocki et al., 2009) was used to predict snRNA and miRNA genes by searches against the Rfam database.
10.6 Genome Duplication
The genome duplication events occur in the genome over the course of evolution for a species. The scanning of the genome sequence for the presence of segmental duplications resulted in 110 syntenic blocks that contained 5 to 62 gene pairs. The divergence time was observed to be 58 million years (Myr) ago based on the rates of synonymous substitution per synonymous site (Ks) for the syntenic blocks. The divergence time is in consistence with genome duplication event that occurred at the base of Papilionoideae. The galegoid (Medicago, Lotus and chickpea) and millettioid (soybean, pigeonpea) clades in this family separated around 54 Myr ago. The chickpea species diverged from Lotus around 20–30 Myr ago and from Medicago around 10–20 Myr ago based on the analysis of four-fold degenerate sites using the calculation of genetic distance–transversion rates.
10.7 Synteny with Allied and Model Genomes
Synteny analysis was carried out for chickpea with 6 other closely related crops, namely Medicago, Lotus, pigeonpea, soybean, Arabidopsis, and grape (Vitis vinifera). The synteny analyses revealed extensive conservation between chickpea, and other species shows that high percentage of chickpea assembly has conserved regions matching with one or more species included in the synteny analysis. The maximum number of conserved syntenic blocks (>10 kb) was seen in Medicago, while it was substantially fragmented with Lotus. When compared with legumes, soybean showed the maximum number of syntenic blocks depicting its recent polyploidy ancestry, while fragmented colinearity with pigeonpea suggests the incompleteness of the pigeonpea genome assembly. The 28,269 gene models of chickpea were compared with 230,161 gene models from four legumes and two non-legumes resulting in 15,441 orthologous groups using reciprocal pairwise approach. Of these, 5,940 orthologous groups were observed having a single chickpea gene indicating simple orthology relationship, while 4,468 chickpea genes were observed in species-specific groups, with no ortholog but having paralogs within the genome. These groups may be attributed to the structural rearrangements that lack simple orthology followed by duplication, as is observed in the case of NBS-LRR disease resistance genes. The percentage of the total predicted gene models which were classified into orthologous groups by OrthoMCL gives insights for the genes which have history of duplication after the divergence of legumes from Arabidopsis and grape. The chickpea genome may be attributed to a series of gene loss and gene duplications as it is the same time interval required for whole-genome duplication event at the base of the Papilionoideae. Several genes from each of the 7 species could not be placed into orthologous groups which may be because of the heterogeneity in gene prediction for each of these species while it may also be due to lineage-specific evolution events. MUMmer (Delcher et al. 2003) and SyMAP (Soderlund et al. 2011) were used in combination for the synteny analysis. Classification of orthologous genes and gene clusters was carried out using OrthoMCL (Li et al. 2003).
10.8 Comparison of Desi and Kabuli Genomes
There was another effort made towards the genome sequencing of chickpea, by whole genome sequencing of the ICC 4958 genotype which is desi type (Jain et al. 2013). There exist various differences in the final assemblies reported by two efforts mentioned above (Table 10.1). The genome size of the kabuli genome was 532.29 Mb, and in case of desi genome, it was 519.84 Mb. The number of gene models reported for the two genomes was similar: 28,269 in kabuli and 27,571 in desi. The number significantly differs for the number of scaffolds assembled and N50 for the two genome assemblies. The number of scaffolds is comparatively too less in case of kabuli genome, and also, the N50 value is comparatively high which states that the kabuli genome is much better in terms of these assembly parameters. As compared to desi, kabuli genome has ~2.8 times sequence anchored at pseudomolecules, and the longest scaffold in kabuli is more than twice the size of longest scaffold observed in desi genome. The number of miRNA, tRNA, and rRNA fragments is significantly higher in the kabuli genome. The GC content is bit higher in the kabuli genome which may be attributed to only Illumina technology used to develop the assembly. The desi genome is more fragmented in comparison with the kabuli genome, and kabuli genome will serve a better resource for genome-based studies in chickpea.
10.9 Subsequent Validation
Both kabuli and desi assemblies were subsequently assessed using a chromosomal genomics approach to determine whether the differences in the genome assemblies represent real differences in genome structure or are artifacts of assembly of one or both genomes. Isolated chromosomes from each of the varieties were sequenced and the data was mapped to the pseudomolecules (Ruperao et al 2014). This analysis demonstrated that the physical genomes of kabuli and desi chickpea types are very similar and the observed differences in the sequence assemblies are due to major errors in the desi genome assembly, including the misplacement of whole chromosomes, portions of chromosomes, and the inclusion of a large portion of sequence assembly which does not appear to be from the genome of chickpea. In contrast, the kabuli assembly is mostly correct. Based on this analysis, updated versions of both kabuli and desi genome assemblies have been produced (http://doi.org/10.7946/P2G596 and http://doi.org/10.7946/P2KW2Q), with GBrowse access at http://www.cicer.info/.
10.10 Conclusion
The chickpea genome sequencing has provided the much needed thrust to genomics based breeding approaches. Further, the re-sequencing of the germplasm will help in better understanding of the diversity present in Cicer species. The resource generated from these sequencing efforts will help in improvement of the genome assembly with enhanced coverage. The improved genome assemblies will help in identification of regions linked to important agronomic traits. These sequencing efforts are expected to enhance the chickpea yield and its resistance to biotic and abiotic stresses.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Attwood TK, Bradley P, Flower DR, Gaulton A, Maudling N, Mitchell AL, Moulton G, Nordle A, Paine K, Taylor P, Uddin A (2003) PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res 31(1):400–402
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573
Corpet F, Servant F, Gouzy J, Kahn D (2000) ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res 28(1):267–269
Delcher AL, Salzberg SL, Phillippy AM (2003) Using MUMmer to identify similar regions in large sequence sets. Current Protocols in Bioinformatics 10–3: doi:10.1002/0471250953.bi1003s00
Edgar RC, Myers EW (2005) PILER: identification and classification of genomic repeats. Bioinformatics 21(1):i152–i158
Elsik CG, Mackey AJ, Reese JT, Milshina NV, Roos DS, Weinstock GM (2007) Creating a honey bee consensus gene set. Genome Biol 8(1):1
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296(5565):92–100
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nature Rev Genet 17(6):333–351
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Yadav M, Kant C, Sharma P, Bhatia S, Tyagi AK, Chattopadhya D (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729
Jurka J (1995) Database of repetitive elements (repbase). NCBI Database Repository
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12(4):656–664
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER (2013) The next-generation sequencing revolution and its impact on genomics. Cell 155(1):27–38
Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40:D302–D305
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13(9):2178–2189
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1(1):1
Magrane M and UniProt Consortium (2011) UniProt Knowledgebase: a hub of integrated protein data. Database p bar009
Metzker ML (2010) Sequencing technologies—the next generation. Nature Rev Genet 11(1):31–46
Nawrocki EP, Kolbe DL, Eddy SR (2009) Infernal 1.0: inference of RNA alignments. Bioinformatics 25(10):1335–1337
Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23(9):1061–1067
Paterson AH, Bowers JE, Bruggmann R, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M (2008) The Sorghum bicolor genome and the diversification of grasses. Nature 457(LBNL-6812E)
Price AL, Jones NC, Pevzner PA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21(suppl 1):i351–i358
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. and Heger A (2011) The Pfam protein families database. Nucleic Acids Res p gkr1065
Ruperao P, Chan KCK, Azam S, Karafiátová M, Hayashi S, Čížková J, Saxena RK, Šimková H, Song C, Vrána J, Chitikineni A, Visendi P, Gaur PM, Millán T, Singh KB, Taran B, Wang J, Batley J, Doležel J, Varshney RK, Edwards D (2014) A chromosomal genomics approach to assess and validate the desi and kabuli draft chickpea genome assemblies. Plant Biotechnol J 12:778–786
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326(5956):1112–1115
Schuler GD (1997) Sequence mapping by electronic PCR. Genome Res 7(5):541–550
Sigrist CJ, Cerutti L, De Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N (2010) PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 38(suppl 1):D161–D166
Soderlund C, Bomhoff M and Nelson WM (2011) SyMAP v3. 4: a turnkey synteny system with application to plant genomes. Nucleic Acids Research p gkr123
Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13(9):2129–2141
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MT, Azam S, Fan G, Whaley AM, Farmer AD (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30(1):83–89
Varshney, RK Song C, Saxena RK, Azam S, Yu S, Sharpe A, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Garcia NC, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CLL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel1 J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31: 240–246
Varshney RK, Terauchi R, McCouch SR (2014) Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biol 12(6):e1001883
Xu Z, Wang H (2007) LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35:W265–W268
Zdobnov EM, Apweiler R (2001) InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17(9):847–848
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Khan, A.W., Thudi, M., Varshney, R.K., Edwards, D. (2017). Sequencing the Chickpea Genome. In: Varshney, R., Thudi, M., Muehlbauer, F. (eds) The Chickpea Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-319-66117-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-66117-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66115-5
Online ISBN: 978-3-319-66117-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)