
Abstract
Glycosylation of peptides and proteins has emerged as a promising strategy to improve the pharmacokinetic profile of peptide- and protein-based therapeutics. The synthesis of pure homogeneous N-linked glycopeptides and glycoproteins is a challenging task, and efficient routes to access them are in high demand. Endo-β-N-acetylglucosaminidise catalysed glycosylation of N-acetylglucosamine-tagged peptides, using activated oligosaccharide oxazolines as donors, has recently attracted attention due to the relative simplicity by which the process convergently affords glycoconjugates with complete control of stereo- and regioselectivity. Herein, a brief review of some examples of recent enzyme-mediated N-glycosylation used to synthesise glycopeptides with therapetic potential is provided.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Protein glycosylation is the most complex and diverse form of post-translational modification leading to the formation of N-, O-, S- and C-glycosides, phosphoglycans and glypiated proteins (proteins that are covalently bonded to a glycosylphosphatidylinositol via their C-terminus) [1]. The most common glycan-protein linkages found in nature are formed using either the side chain amide nitrogen of asparagine (Asn) residues or the side chain of serine (Ser) and threonine (Thr) residues to afford N-linked or O-linked glycoproteins, respectively [2]. Examples of C-mannosylation via the indole C-2 carbon atom of tryptophan (Trp) and S-glycosylation using the thiol of cysteine (Cys) are rare and have also been described in the literature (Fig. 1) [3].

Selected examples of N-, O-, C- and S-glycan-peptide linkages found in nature
Glycosylation plays an important role in various biological processes including cell development [4], inflammation [5], cell–cell signalling, adhesion and immune responses [6]. The presence of sugar moieties affects glycoprotein tertiary structures [7, 8], facilitates folding [9] and improves proteolytic stability [9]. Altered glycosylation patterns affect the circulatory lifetime of glycoproteins [10] and are associated with numerous diseases including some cases of congenital disorders [11, 12], leukocyte adhesion deficiency II [1], the aetiology of diabetes [13] and neurodegeneration [14, 15], cancer [16, 17] and Alzheimer’s disease [18, 19].
Glycosylation of peptides and proteins with the intention of improving the pharmacokinetic profile of protein-based drugs has resulted in rapid expansion of the therapeutic peptide and protein market [20,21,22,23,24,25,26]. Increased proteolytic stability has been achieved by glycosylation of glucagon-like peptide-1 (GLP-1) [22], insulin [27], exendin-4 [28] and interferon-β [29]. The composition of protein-bound glycans can modulate the efficacy of protein therapeutics, for example, it has been demonstrated on numerous occasions that multiply sialylated versions of erythropoietin (EPO) [30] possess longer plasma half-lifes as compared to their asialylated counterparts [10].
Robust and general strategies to prepare glycopeptides and glycoproteins in pure forms are highly sought after and have been investigated by many research groups [31,32,33,34,35,36,37,38,39,40,41,42]. N-Linked glycoproteins are prevalent and their glycans encompass diverse structures. They have gained significant scientific attention, not least due to their potential as therapeutic agents and thus are the main focus of this mini-review which describes the use of enzymatic glycosylation to access N-linked glycopeptides [24, 43].
2 Synthesis of Glycopeptides in Nature
The biosynthetic pathways of glycopeptide and glycoprotein synthesis in mammals are complex [2]. Briefly, the initial stage involves scavenging a simple monosaccharide unit, mostly glucose (Glc) but also galactose (Gal), mannose (Man) and glucosamine (GlcN) from the bloodstream by cells throughout the body using protein transporters (sodium-dependent co-transporters, SGLT and sodium independent facilitative transporters, GLUT) located in the plasma membrane of various tissues [2]. This is followed by intracellular de novo synthesis of additional sugar-based building blocks including fucose (Fuc), N-acetyl neuraminic acid (Neu, sialic acid) and N-acetylgalactosamine (GalNAc) by chemical processes including epimerisation, condensation and acetylation [2]. Subsequent phosphorylation of the monosaccharide units and pairing with corresponding nucleotides takes place in the cytosol to afford energy-rich nucleotide sugars required for further glycan synthesis [2]. Organelle-specific transporter proteins traffic nucleotide sugars from the cytosol into the endoplasmic reticulum (ER) and Golgi lumens where assembly of glycans of particular structures takes place. This complex process is mediated via the action of numerous transmembrane glycosyltransferases and glycosidases, the precise mechanisms of which are not yet fully understood [2, 24].
N-Linked glycosylation starts in ER where a dolichol phosphate oligosaccharide [Glc3Man9(GlcNAc)2] is formed and then transferred en block onto an Asn residue on a nascently translated protein via the action of oligosaccharyltransferase (OST) [2]. The glycosylated asparagine residue is invariable within the conserved Asn-X-Thr/Ser peptide sequence (where X is any amino acid residue except proline) of the unfolded protein. The oligosaccharide is then processed by the enzymes glucosidase I and II, affording a truncated GlcMan9(GlcNAc)2 glycoprotein interacting with calnexin and calreticulin and participating in the primary ‘quality control’ system distinguishing native from non-native protein conformations [44]. Subsequent removal of the Glc unit by glucosidase II releases Man9(GlcNAc)2-tagged glycoprotein from the chaperone, which if correctly folded can leave the ER. In case the protein exhibits non-native conformation, association with calnexin and calreticulin is renewed (via GlcMan9(GlcNAc)2 unit containing reattached Glc residue) and ‘quality control’ process is repeated until proper protein conformation is achieved. Glycoproteins permanently misfolded are eliminated from ER for degradation [44].
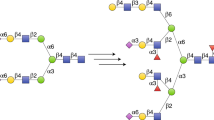
Removal of a terminal α(1-2)-linked mannose unit from either of the two arms of Man9(GlcNAc)2 subsequently takes place and is mediated by mannosidase I or II affording a Man8(GlcNAc)2-bound protein that is then transported to the cis-Golgi apparatus for further processing. Within the Golgi lumen, the common intermediate Man5(GlcNAc)2 is formed by the action of mannosidase IA and IB to remove α(1-2)-linked mannoses. Man5(GlcNAc)2 is then used to assemble, complex and hybrid subclasses of N-glycosides [2]. The partially processed glycans which were not trimmed to Man5(GlcNAc)2, or those which escape remodelling process from Man5(GlcNAc)2 to complex and hybrid N-glycoproteins, fall into high-mannose subclass of N-linked glycoproteins of the type Man(5–9)GlcNAc2 (Fig. 2) [45].

High-mannose, complex and hybrid subclasses of N-glycosides containing the core Man3(GlcNAc)2 pentasaccharide [46]
O-Linked glycosylation occurs in the Golgi apparatus and starts with the attachment of a monosaccharide unit (most often GalNAc) to a Ser or Thr residue present within the sequence of an already folded protein [2]. The subsequent formation of more complex oligosaccharides structures from the Ser/Thr(GalNAc) (Tn-antigen) core is then achieved via the sequential action of a variety of glycosyltransferases [2].
Protein glycosylation, unlike pure protein and oligonucleotide synthesis, is not template mediated and so depends on the activity and concentration of sugar substrates, the structural and conformational properties of glycosylation sites and the differential activities of numerous enzymes [41]. This means that glycoproteins are invariably produced as heterogeneous mixtures, termed glycoforms, where proteins possessing the same peptide chain vary in glycan structure [42, 47] and which may also vary in site occupancy. Access to well-defined homogeneous glycoproteins for subsequent structural and functional studies is, therefore, a challenging task.
Literature methods to prepare homogeneous glycopeptides and glycoproteins include the use of recombinant technology, fully synthetic techniques using chemical methods, enzymatic approaches and combinations of all the above. Each of these methodologies has its own advantages and limitations, and in depth discussion and progress to date has been summarised by a number of recent reviews [32, 33, 35, 36, 41, 42]. Our own endeavours towards the synthesis of glycosylated peptides and proteins with natural and non-natural glycan-peptide linkages began in 2008 and were mainly focused on using fully synthetic techniques [48,49,50,51,52,53,54,55,56]. Our current interest is now directed at combining synthetic techniques with chemoenzymatic methods to achieve the convergent synthesis of complex glycopeptides, [57, 58] and this is the primary focus of the current report. A comprehensive review on the topic, comprising elegant examples of chemoenzymatic approaches by other research groups, has been recently published by Wang and Amin [33].
3 Recombinant Approach to Access Glycopeptides and Glycoproteins
Recombinant methods using either fungal, plant or insect-based systems to produce N-glycoproteins seem promising but suffer from the limitation that heterogeneous products are invariably obtained, as well as the differences in glycosylation patterns between species, and batch-to-batch variability [24]. Expression systems based on bacteria are restricted to the synthesis of non-glycosylated proteins only (insulin, for example) owing to their inability to glycosylate due to the absence of glycosylation machinery [24]. Up to this point in time mammalian cell lines, for example, typically from Chinese hamster ovary (CHO), have been extensively used for the production of therapeutic glycoproteins due to their potential to produce certain human-like glycosylation patterns [24]. Other mammalian systems used for the synthesis of N-glycoproteins include baby hamster kidney (BHK-21) and murine myeloma (NS0 and Sp2/0) cells. Their use, however, is limited; BHK cell lines, similarly to CHO cells are not able to produce α(2,6)–linked terminal sialic acids present in human glycans, and immunogenity concerns are associated with the use of murine myeloma cells [24]. Nevertheless, recombinant methods using mammalian cell lines are routinely used to produce marketed therapeutic monoclonal antibodies [59]. Adalimumab (Humira®), the worlds best selling drug in 2015 [60] used for the treatment of rheumatoid arthritis [61], is expressed in CHO cell lines; Golimumab (Simponi®), used as an immunosuppressant [62], is expressed in Sp2/0 cells [59].
Human cell lines derived from embryonic kidney (HEK293), embryonic retinoblasts (PER.C6) or hybrid HKB11 cell lines composed of embryonic kidney cells (293S) and modified Burkitt’s lymphoma cells (2B8) are attractive but expensive alternatives to CHO cells [63]. Significant scientific focus has therefore been directed into engineering effective and more straightforward yeast-based systems [64, 65]. It was found that ‘humanized’ Pichia pastoris-derived cell lines can be used to express homogeneous human N-linked glycoproteins bearing truncated complex [(GlcNAc)2Man3(GlcNAc)2] units [65] and full-length complex sialylated glycans [64]. These studies have opened up further possibilities to access therapeutic glycoproteins via recombinant techniques using alternative yeast-derived expression systems [64, 65]. However, complete control of glycosylation, i.e. in order to obtain strictly homogeneous glycoproteins via recombinant methods is still challenging [66].
4 The Use of Chemical Synthesis to Access Homogeneous Glycopeptides and Glycoproteins
The use of synthetic techniques to counter challenges faced in the preparation of homogeneous glycopeptides and glycoproteins is under investigation by several research laboratories worldwide [37,38,39,40, 56, 67,68,69,70,71]. Solid phase peptide synthesis technique (SPPS) is the method of choice for the preparation of glycopeptides [32]. Despite the extensive development that SPPS has undergone [72] since its first discovery [73], it is still limited to the preparation of up to 30- to 50-residue long glycopeptides that carry relatively small oligosaccharide units. However, the combination of SPPS and ligation techniques, particularly native chemical ligation (NCL) [74] or expressed protein ligation (EPL) [75], has enabled synthetic access to more complex structures including large glycoproteins [35, 36]. The synthesis of 40- and 80 amino acid MUC1 glycoproteins bearing eight GalNAc units at corresponding Thr11 and Thr19 of tandem repeats was accomplished using a combination of 9-fluorenylmethoxycarbonyl (Fmoc) SPPS and a serine/threonine ligation technique [76,77,78]. The first total synthesis of glycocin F, an antimicrobial 43 amino acid glycopeptide with two βGlcNAc moieties at Ser18 and Cys43 was successfully undertaken by Brimble et al. [56]. The synthetic protocol involved initial Fmoc SPPS of three glycocin F fragments incorporating O- and S-linked GlcNAc unit using either Fmoc-Ser[GlcNAc(OAc)3] or Fmoc-Cys[GlcNAc(OAc)3] building blocks, respectively, accessed via total synthesis. Native chemical ligation [74] was then used to join these fragments which was followed by oxidative folding to effect the desired C-amidated glycocin F [56]. Many other examples of the synthesis of larger N-glycoproteins using ligation techniques have been reported in the literature [35, 36]. Notable examples include the synthesis of 166 amino acid interferon-β-1a [79], the 72 amino acid glycosylated analogue of interleukin-8 which was used in folding studies [80], the hydrophobic glycoprotein saposin C (80 amino acids) [81], and the 124 amino acid bovine ribonuclease (RNAse) C accessed via semisynthetic methods (EPL and NCL) [82] or total synthesis and NCL [83].
In general two approaches may be used to generate a linkage between an oligosaccharide and a peptide chain. The so-called ‘linear approach’ involves initial preparation of a glycosylated amino acid building block, which is then incorporated into a growing resin-bound peptide chain that is then typically extended using SPPS [32, 38, 84]. A major limitation of this technique is the significant effort required to prepare suitably protected carbohydrate-bearing building blocks in sufficient quantities for the subsequent coupling steps. Furthermore, the attachment of complex protected oligosaccharides to amino acid residues generates significant steric hindrance, which may diminish the effectiveness of the peptide coupling steps during SPPS, leading to by-product formation. Therefore, only short- to medium-sized glycopeptides, carrying relatively small (and typically O-linked) oligosaccharide units, can be accessed via the linear approach [33, 38, 42]. Nonetheless, this linear approach has been employed for the synthesis of antitumour vaccine candidates based on mucin glycopeptide antigens [68, 85, 86], and for the synthesis of mannosylated peptides as components for synthetic vaccines [48]. Other literature examples that have adopted the linear strategy include the synthesis of fluorescent glycopeptides as biological probes [87, 88], and the synthesis of analogues of antifreeze glycoproteins [53] in addition to others reviewed elsewhere [84]. Kajihara et al. have employed the linear strategy to synthesise N-linked glycopeptides including a 79–85 fragment of EPO (ALLVNSS) bearing a complex biantennary sialyloligosaccharide [89], an EPO (85–95) fragment with two different glycans (asialo- and sialyloligosaccharides) attached [90], ligation partners for subsequent NCL to construct the full-length EPO mutants bearing one, two or three biantennary sialyloligosaccharides [91, 92] and a 38 amino acid cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) fragment 113–150 with two complex-type undecadisialyloligosaccharides [93]. The glycopeptide thioesters bearing N-linked biantennary complex-type nonasaccharide unit required for subsequent ligation (EPL and NCL) to afford RNAse C were synthesised using the linear strategy by the group of Unverzagt [82, 83].
An alternative and convergent strategy involves the initial synthesis of a peptide chain followed by the direct attachment of a carbohydrate unit either on-resin or in-solution [32, 38, 40]. This technique is widely applicable for the preparation of N-linked glycopeptides where glycosylamines are attached to a pre-assembled peptide via the side chain carboxylic acid of embedded aspartic acid (Asp) residues using the Lansbury aspartylation [94, 95]. A notable example that employed this convergent strategy to access an N-linked glycoprotein was reported by Danishefsky et al. [69, 96, 97]. It involved the total synthesis of the 166 amino acid fully glycosylated homogeneous erythropoietin, containing N-linked branched dodecasaccharides at Asn24, Asn38 and Asn83, and an O-linked glycan at Ser126 [69, 96, 97]. A combination of Fmoc SPPS, NCL [74], O-mercaptoaryl ester rearrangement [96] and metal-free desulfurisation [98] was used to deliver the synthetic target. The highly complex N-linked oligosaccharides were prepared by total synthesis and introduced using a convergent selective amidation of the corresponding aspartic acid residue using a ‘one flask’ aspartylation approach [94, 95, 99, 100]. Further examples of the synthesis of glycopeptides have also been reported [32, 33, 36, 38, 42].
5 Enzymatic Approach to Access Homogeneous Glycopeptides and Glycoproteins
Enzymatic approaches to the glycosylation of peptides and proteins are increasing in popularity due to their simplicity, and excellent stereo- and regiochemical control [101]. This technique may employ a variety of enzymes including glycosidases, glycosyltransferases and glycosynthases to generate desired oligosaccharide structures and sugar-peptide linkages [101]. Glycosidases are responsible for glycosidic bond hydrolysis and catalyse the breakdown of either terminal sugar units, from their non-reducing end, or internal glycosidic bonds (exo- and endo-glycosidases, respectively) [101]. Glycosyltransferases catalyse the formation of specific glycosidic linkages by transferring monosaccharide units from glycosyl donor substrates to corresponding acceptors [101]. Mutant glycosidases (commonly referred to known as glycosynthases) [102,103,104] may be used for glycopeptide and glycan synthesis. Glycosynthases have the ability to transfer activated oligosaccharides onto acceptors, and unlike glycosidases, possess little or no hydrolytic activity [101].
Endoglycosidases acting on glycan chains of glycoproteins can be further divided into two classes, those which hydrolyse the core region of N-linked oligosaccharides embedded within the glycoprotein chain, and those which recognise O-linkages between the sugar and the protein [105]; endo-β-N-acetylglucosaminidase (Endo-β-GlcNAc-ase, ENGase, endohexosaminidase) and endo-β-N-acetylgalactosaminidase are representative examples from each class of endoglycosidase, respectively [105]. To date, sequence analysis of the most synthetically useful ENGase enzymes employed for the synthesis of glycoproteins has lead to their classification in the carbohydrate-active enzymes (CAZy) database [106], as either members of family 18 or family 85 of the glycoside hydrolases (GH18 or GH85) [107]. The family GH18 enzymes are mostly derived from bacteria and fungi, while GH85 enzymes can be found in organisms ranging from bacteria to mammals [107].
In addition to their hydrolytic activity, endoglycosidases can also effectively transfer oligosaccharides onto corresponding hydroxyl-containing substrates by transglycosylation, or glycosylation. This dual capability makes endoglycosidases, especially the ENGases, valuable tools for the convergent synthesis of oligosaccharides and glycoconjugates [105, 108].
6 Wild-Type Enzymes and Peptide and Protein Glycosylation
A number of Endo-β-GlcNAc-ases have attracted scientific attention [107] due to their synthetic potential, and examples from family GH85 include Endo M (from Mucor hiemalis) [109,110,111,112,113,114,115,116,117,118,119,120], Endo A (from Arthrobacter protophormiae) [117, 121,122,123,124,125,126,127,128], Endo D (from Streptococcus pneumoniae) [129, 130], Endo OM (from Ogataea minuta) [107] and Endo-BH (from Bacillus halodurans) [131]. Selected examples of ENGases from family GH18 are Endo H (from Streptomyces griseus) [132], Endo S (from Streptococcus pyogenes) [133, 134] and Endo F1, F2 and F3 (from Flavobacterium meningosepticum) [135,136,137]. All ENGases cleave the N,N′-diacetylchitobiose unit [GlcNAcβ(1-4)GlcNAc], a common motif present within Asn-linked high-mannose, complex and hybrid N-glycans [105]. Enzyme hydrolysis is substrate specific; Endo H, Endo F1 and Endo A only act on high-mannose and hybrid N-glycans, while Endo F3 also recognises bi- and tri-antennary complex glycans [43].
The synthetic ability of ENGases can, therefore, be used for the preparation of homogeneous glycoproteins in an enzyme-mediated glycoprotein remodelling approach (Scheme 1a) [33, 34, 43, 105, 138]. This approach involves initial enzyme-mediated hydrolysis of the β(1–4) bond of [GlcNAcβ(1-4)GlcNAc] unit, affording a GlcNAc-tagged protein. Subsequent reattachment of another glycan to this GlcNAc acceptor using the transglycosylation activity of an ENGase then gives a glycoprotein with a desired glycan structure [43, 105].

a Glycoprotein remodelling approach and b convergent chemoenzymatic synthesis of glycopeptides using ENGases [43]
The protein remodelling approach can be extended to the convergent, chemoenzymatic synthesis of glycopeptides. Herein, the synthesis of a peptide chain incorporating a GlcNAc moiety as a sugar acceptor is performed first, and is then followed by enzymatic attachment of N-glycans using ENGases (Scheme 1b) [43]. However, the relatively low yields of products obtained during the process and hydrolytic activity of endoglycosidases often diminish their synthetic potential [43, 139].
7 Strategies to Improve Enzymatic Glycosylation
Approaches to effect improvements in ENGase catalysed glycosylation processes include the use of N-glycan oxazolines as activated sugar donors in combination with mutant variants of ENGases with altered activity towards product hydrolysis [43, 140,141,142,143,144,145,146,147,148,149,150].
The successful use of an oxazoline-activated disaccharide was first reported in 2001 by Shoda et al. [151]. The Manβ(1-4)GlcNAc-oxazoline (1) was successfully used as a glycosyl donor in an Endo M [109]- and Endo A [121]-mediated glycosylation reaction using GlcNAcOpNP (2) as the acceptor, affording the corresponding trisaccharide 3 (Scheme 2) [151]. The activity of oxazolines as donors for glycosylation processes catalysed by ENGases is related to the structural and functional similarities they share with oxazolinium ions, which are high energy intermediates in the enzymatic hydrolysis [151].

The use of sugar oxazolines as activated sugar donors in ENGase catalysed glycosylation has attracted the attention of many research laboratories worldwide [130, 133, 139,140,141,142,143, 152,153,154,155,156,157,158,159,160,161,162,163,164,165]. The power of ENGase mediated glycosylation using sugar oxazolines was recently demonstrated by Fairbanks et al. [166] who reported the first synthesis of phosphorylated glycoprotein bearing phosphorylated pentasaccharide unit attached via native N-linkage using the protein remodelling approach. The tetrasaccharide oxazoline in which two terminal mannose residues contained phosphate at the 6 position was accessed via chemical synthesis and attached to GlcNAc-tagged RNase B using Endo A [117, 121,122,123,124,125,126,127,128] glycosylation activity [166].
The extensive research on the utility of saccharide oxazolines for ENGase facilitated glycosylation of peptides and proteins [130, 133, 139,140,141,142,143, 152,153,154,155,156,157,158,159,160,161,162,163,164,165] revealed, however, that only smaller natural sugar oxazolines (di- and tri-saccharides) proved to be effective in glycosylation reactions using wild-type enzymes [139]. When larger, natural N-oligosaccharide oxazolines were used, significant reductions in glycosylation yields were observed due to competitive hydrolysis [139].
One potential solution to this problem might be the use of structurally modified sugar oxazolines. In principle, these highly activated glycan donors should be readily processed by ENGases to afford glycosylation products that may not be substrates for hydrolytically active enzymes due to structural changes [140, 142].
Fairbanks et al. [140] showed that chemically synthesised di-, tri-, tetra- and hexasaccharide-oxazolines derived from the core sections of N-linked high-mannose glycans, containing a glucose moiety in place of a central mannose unit [Manβ(1-4)GlcNAc to Glcβ(1-4)GlcNAc] were substrates for Endo M [109] and Endo A [121], but not Endo H [132], and could be used to effect irreversible glycosylation. However, this approach only produces non-natural glycan structures, which may be considered a limiting factor.
Another strategy to curtail undesired product hydrolysis, and hence improve the yield of the glycosylation step, involves using mutant enzymes [43, 139]. Site-directed mutagenesis of wild-type enzymes is used to generate the requisite mutants [43, 139]. The idea originated from the use of glycosynthases [102,103,104] used for oligosaccharide synthesis, and was further developed to afford mutated versions of various ENGases, including Endo A [153, 167], Endo M [161, 168], Endo D [169] and Endo S [170, 171]. These mutated enzymes allowed the synthesis of glycoproteins [160, 161, 164] containing natural N-linked oligosaccharides. An important example is the commercially available N175Q mutant of Endo M, derived from family GH85 developed in the laboratories of Wang and Yamamoto [168]. Exchanging Asn175 for Gln in Endo M proved superior; the mutant exhibited greater glycosylation activity with significantly reduced hydrolytic activity as compared to wild-type Endo M and other mutants investigated [168]. Endo M N175Q became a valuable tool that may be used to access homogeneous N-liked glycopeptides and glycoproteins carrying natural high-mannose- and biantennary complex-type oligostructures [168].
The enzymatic remodelling of immunoglobulin G (IgG) [133, 170] using Endo S [133, 134] further expanded the synthetic potential of ENGases to access homogeneous antibodies (Abs) bearing well-defined sugar structures. Monoclonal antibodies (mAbs) are an important class of N-linked glycoprotein therapeutic which are produced using recombinant techniques as a mixture of multiple glycoforms of variable abundance and complexity depending on the expression system and cell line used [172]. The glycosylation pattern in the Fc region of mAbs is especially important and affects antibodies functions on immune cells via interaction with FcγR receptors [24]. The presence of biantennary N-linked oligosaccharide units with two terminal α2,6-linked sialic acids at the two Asn297 Fc glycosylation sites enhances the activity of immunoglobulin G against cancer and infectious and inflammatory diseases [173]. Straightforward access to pure glycoforms of mAbs is, therefore, the key to modulate their clinical effects and develop improved antibody-based therapeutics. Wong et al. [173] recently reported the synthesis of homogeneous Rituximab IgG1 (used for the treatment of rheumatoid arthritis and cancer) [174] using an enzymatic remodelling approach where the initial formation of a mono-GlcNAc-tagged antibody, achieved using Endo S [133, 134] and a fucosidase from Bacteroides fragilis, was followed by ligation of the well-defined synthetic glycan oxazolines using an Endo S D233Q mutant [170]. The synthetic utility of an enzymatic approach using mutated enzymes has been also demonstrated by Davis et al. [171], who recently reported the synthesis of a homogeneous form of sialylated mAb Herceptin (Trastuzumab) [175], using a the same Endo S D233Q [170] and optimised reaction conditions (enzyme loading and oxazoline concentration). In addition to well-defined natural glycans, modified sugar oxazolines with handles or tags (such as azides or alkynes) incorporated via amidation of non-reducing terminal sialic acids of a decasaccharide unit were also incorporated onto GlcNAc-tagged Herceptin using the optimised protocol [171].
To broaden the scope and potential applications of enzymatic glycosylation, studies on the use of structurally modified GlcNAc or alternative sugar acceptors for ENGase mediated glycosylation have been undertaken [115, 120, 125, 135, 176, 177]. Fairbanks et al. [177] have recently reported the tolerance of various ENGases to transfer N-glycan oxazolines 4 and 5 to a structurally altered Asn(GlcNAc) acceptor in which the hydroxyl group of the glycan unit was protected with a benzyl ether at C-3 (6), C-4 (7) or C-6 (8) (Fig. 3). The OH-3 fucosylated Asn(GlcNAc) acceptor (9) was also tested but none of the enzymes studied (WT Endo M, N175Q Endo M, Endo A, Endo D) were able to effect this glycosylation [177]. The study revealed subtle structural preferences of each enzyme towards sugar acceptors, a factor which needs to be taken into consideration when choosing reaction partners [177].

Tetrasaccharide- and decasaccharide-oxazolines 4 and 5, respectively, and modified glycosyl acceptors 6–9 targeted during the study [177]
8 Access to N-Linked Oligosaccharides
Despite the availability of various synthetic techniques, the synthesis of glycopeptides is still challenging [37, 38, 40, 67,68,69,70,71]. This is mainly due to the limited access to the oligosaccharide components that are generally obtained via multistep syntheses in specialised laboratories [35]. To date, although remarkable progress in carbohydrate synthesis has been made, reliable and general routes to prepare complex oligosaccharides are still needed [178]. Nevertheless, total synthesis gives access to a wide range of sugar constructs, either with natural or non-natural linkages.
A recent report by Shoda et al. [179] revealed a new and very convenient method to access GlcNAc-terminating oligosaccharide oxazolines. These oxazolines can be prepared from the corresponding 2-acetamido-2-deoxy reducing sugars (10) in water by activation using 2-chloro-1,3-dimethylimidazolinium chloride (DMC) as the dehydrating reagent to afford corresponding activated sugar donors (11) (Scheme 3) [179].

Synthesis of the glycosyl oxazoline from unprotected GlcNAc using DMC in water [179]
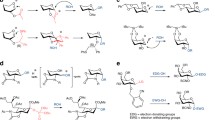
Although labour-intensive, approaches to access full-length N-oligosaccharides have been developed [43]. A general retrosynthetic overview for the synthesis of tetrasaccharide N-oxazolines is shown in Scheme 4 [43]. Due to the susceptibility of glycosyl oxazolines to acid and/or hydrogenation conditions, oxazoline formation must take place before the final base-catalysed removal of protecting groups is effected, or alternatively, the Shoda [179] approach described above can be used to synthesise the oxazoline in the last step. Successive glycan disconnections at C-6, and C-3 of the inner mannose unit allow for the installation of non-symmetrical glycans, which can be achieved using 4,6-benzylidene acetal protection. Subsequent formation of a challenging β-mannosidic linkage takes place by inversion of configuration at C-2 of the selectively synthesised β-glucoside accessed using the neighbouring group participation (NGP) approach. The C-2 hydroxyl of the gluco donor is protected as a levulinate (Lev) ester, which can be removed selectively allowing installation of trifluoromethanesulfonate (triflate) leaving group that is subsequently displaced by acetate affording desired β-mannoside [43, 180].

Generalised retrosynthetic route to N-glycan oxazolines [43]
Selected examples of oligosaccharide oxazolines accessed using these strategies are depicted in Fig. 4 [43].

Selected oligosaccharide oxazolines synthesised [43]
The isolation of large oligosaccharides from natural sources may conveniently bypass laborious total synthesis routes. Some complex Asn-linked N-glycans, such as a sialic acid terminated complex biantennary unit [(NeuAcGalGlcNAcMan)2Man(GlcNAc)2] and a high-mannose glycan [(Man9(GlcNAc)2)] can be obtained in significant quantities from either egg yolks [181] or soy bean flour [182,183,184], respectively. Isolated and purified oligosaccharides can be chemically modified to afford suitably protected or activated building blocks for further incorporation into peptide or protein chains. It has also been shown that the biantennary glycan can be further modified using branch-specific exoglycosidases to access a broad variety of Asn-linked oligosaccharides, thus providing facile access to complex sugar structures [90].
Kajihara et al. [92] reported the chemical synthesis of a mutated EPO variant, with alanine (Ala) residues replacing native glutamic acid (Glu) and glutamine (Gln) residues at position 21 and 78, respectively, using NCL technique [74]. The required asparaginyl sialyloligosaccharide, namely Asn[(NeuAcGalGlcNAcMan)2Man(GlcNAc)2]-(Asn83) was initially isolated from egg yolks. It was then suitably modified with phenacyl (Pac) protecting groups (acid-labile sialic acid residues) and the Nα-amino group of Asn was masked with a tert-butyloxycarbonyl (Boc) protecting group to allow the synthesis of sialylglycopeptide α-thioesters using Boc SPPS. Subsequent ligation of the corresponding peptide fragments using NCL [74] afforded the desired EPO mutant [92]. This synthetic protocol was recently extended by the same research group to the synthesis of full length EPO with one mutation site (Gln78 to Ala) bearing well-defined glycoforms (biantennary sialyloliosaccharide) at one (Asn83), two (Asn38 and Asn83, Asn24 and Asn83, Asn24 and Asn38) and three (Asn24, Asn38, Asn83) native EPO N-glycosylation sites [91].
9 Alternative Access to N-Linked Glycans and N-Linked Glycopeptides Using Glycosidic Bond Mimetics
The synthesis of glycoconjugate mimetics is an alternative approach to construct sugar structures or incorporate glycans onto peptides in a simplified way as compared to a total synthesis approach. The introduction of a glycosidic bond mimetic may improve the stability of the glycoconjugate towards chemical and enzymatic degradation, which is highly beneficial for pharmaceutical applications [185]. The use of the copper(I)-catalysed Huisgen 1,3-dipolar cycloaddition of alkynes and azides to afford a 1,2,3-triazole conjugate (CuAAC ‘click chemistry’) [186, 187] has increased in popularity in recent years, in the peptidomimetic field and as a bioconjugation strategy, due to its simplicity, the mild reaction conditions, its tolerance of various functional groups and its complete regioselectivity to form 1,4-disubstituted products [188].
The syntheses of ‘click’ mimetics of fish antifreeze glycopeptides [49, 189] and a 20 amino acid MUC 1 domain [54] were successfully undertaken in our laboratory [190]. We have also developed a powerful strategy where two ligation techniques, NCL [74] and CuAAC [186, 187], are carried out in a sequential manner to afford ‘click’ neoglycopeptides in a ‘one pot’ fashion [51]. Another highly attractive method which combines the CuAAC strategy [186, 187] and Shoda’s [179] direct synthesis of sugar oxazolines from reducing sugars in water to afford 1,2,3-triazole-linked glycoconjugates in a ‘one pot’ reaction was recently reported [191]. This strategy allows the facile conjugation of reducing sugars with a diverse array of alkynes, including other sugars and peptides. This methodology can potentially be used as a simpler alternative to access homogeneous glycopeptides and possibly glycoproteins in cases where installation of the native N-linkage using an enzymatic approach fails or efficient access to glycosidic bond mimetics is required.
10 Applications of Convergent Enzymatic Glycosylation for the Synthesis of Glycopeptides with Therapeutic Potential
Our on-going interest in the synthesis of peptide-glycoconjugates [48, 52, 53, 56, 87] and glycopeptide mimetics that contain non-natural glycan-peptide linkages (neoglycopeptides) [190] using total synthesis prompted us to investigate the alternative enzymatic approach. Herein, a summary of our recent work on the convergent chemoenzymatic synthesis of N-linked glycopeptides with therapeutic potential is described.
10.1 Synthesis of a Library of Glycosylated Analogues of Pramlintide
Glycosylation of peptides and proteins is an important tool for producing therapeutic peptidomimetics with improved physicochemical and pharmacokinetic profiles [20, 21]. With this idea in mind, we investigated the effect of the N-linked glycosylation of the therapeutic peptide pramlintide (Symlin®), a 37-amino acid synthetic analogue of amylin that is currently used in conjunction with insulin for the treatment of type 1 and type 2 diabetes (Fig. 5) [192,193,194].

Primary sequence of pramlintide and human amylin
Based on the promising previous results obtained from in vitro and in vivo studies on the N-glycosylation of pramlintide at Asn3 and Asn21 with mono-, penta- and undecasaccharides [165], we undertook a systematic investigation into the effect of glycan structure and the position of the attachment of the N-glycan to the pramlintide peptide on the activity of glycosylated peptides to act as agonists of amylin receptors [58]. There are six possible sites for N-glycosylation of pramlintide; Asn3, Asn14, Asn21, Asn22, Asn31 and Asn35. The synthetic strategy was designed to accommodate the presence of GlcNAc units at defined Asn residues within the pramlintide sequence for subsequent enzymatic transfer of more complex sugar structures. In addition, the disulfide bond (Cys2/Cys7) and amidated C-terminus of the peptide had to be installed, as both features are required for biological activity of pramlintide [195].
First, we synthesised a non-glycosylated pramlintide as a control peptide, which was prepared using microwave-enhanced Fmoc SPPS to afford the reduced pramlintide precursor 12. Disulfide bond formation of 12 was subsequently carried out upon the activation using 2,2′-dipyridyl disulfide (DPDS) in dimethyl sulfoxide (DMSO) [196] to afford the cyclic (Cys2/Cys7) and C-amidated pramlintide (13), Scheme 5.

Synthesis of pramlintide 13, and pramlintide analogues 20–25 containing a GlcNAc residue at Asn3, Asn14, Asn21, Asn22, Asn31 or Asn35. Reagents and conditions: a DPDS, DMSO, rt; b DPDS, DMSO; then 13% NH2NH2·1.5 H2O, rt; c 5% NH2NH2·1.5 H2O, 10% DMSO, 85% 6 M Gu·HCl, 17 h, rt; d DPDS, DMSO; then 5% NH2NH2·1.5 H2O, rt [58]
For the synthesis of monoglycosylated pramlintide analogues (20–25), comprising a GlcNAc unit at specific Asn residues, microwave-enhanced Fmoc SPPS was employed. The GlcNAc substitution was introduced using the per-O-acetylated Fmoc-Asn[GlcNAc(OAc)3] building block (38) [197] to give linear, sugar hydroxyl protected pramlintide analogues 14–19. Subsequent use of a ‘one pot approach’ developed by Hojo et al. [198] to form the Cys2/Cys7 disulfide bond with simultaneous removal of the glycan hydroxyl acetate protecting groups required long reaction times (17 h) to obtain the desired product 23. We found that the reaction was significantly accelerated when both reactions were performed sequentially in the same vessel whereby the linear, acetate-protected glycopeptides 14–16, and 18–19 were first treated with DPDS in DMSO to effect disulfide bond formation, then hydrazine hydrate was added to deprotect the sugar hydroxyls [58]. For the synthesis of 24 and 25, the total reaction time was significantly reduced to 5 and 5.5 h, respectively. Faster acetate removal was achieved using a higher concentration of hydrazine hydrate (3 h in total, for analogues 20–22), Scheme 5.
A library of pramlintide analogues 26–31 bearing the core N-glycan pentasaccharide [Man3(GlcNAc)2] was then synthesised using Endo A [121, 128] to transfer tetrasaccharide oxazoline 4 (accessed via total synthesis [140]) to the corresponding GlcNAc-tagged pramlintide 20–25 (Scheme 6).

Synthesis of pramlintide analogues 26–31 containing the core N-glycan pentasaccharide [Man3(GlcNAc)2] at position 3, 14, 21, 22, 31 or 35 [58]
This methodology was then extended to the preparation of pramlintide analogues 32–37 bearing a complex biantennary glycan [(NeuAcGalGlcNAcMan)2Man(GlcNAc)2]. In this case treatment of the decasaccharide-oxazoline 5, synthesised from the corresponding reducing sugar that was isolated from egg yolks [181, 199], with the commercially available Endo M N175Q mutant [168, 177] in the presence of 20–25 enabled the preparation of pramlintide analogues 32–37, (Scheme 7) [58].

Synthesis of pramlintide analogues 32–37 containing a complex biantennary glycan [(NeuAcGalGlcNAcMan)2Man(GlcNAc)2] at position 3, 14, 21, 22, 31 or 35 [58]
A comprehensive series of 18 pramlintide analogues comprising mono-, penta- and undecasaccharides (20–37) were then tested as agonists of amylin receptors and their activity was compared to parent pramlintide (13). The parent pramlintide 13 and analogues 20–25 bearing GlcNAc unit were screened against the three best characterised amylin receptors (CT(a), AMY1(a), and AMY3(a), which contain the CT(a) splice variant of the calcitonin receptor) [200, 201] at which activity of pramlintide is equal to human or rat amylin [202]. Analogues 26–37 containing more complex glycans were only tested against AMY1(a) analogously to the previous study [165].
The study revealed that the presence of N-glycans was well tolerated at Asn21, Asn31 and Asn35 by the AMY1(a) receptor, and that the activity of analogues versus the amylin receptors decreases as the size of the glycan increases (GlcNAc > pentasaccharide > undecasaccharide). It was therefore established that N-glycosylation of pramlintide is a promising tool to afford analogues with improved therapeutic potential. In vivo studies to assess the biological importance of N-glycosylation of pramlintide are under investigation and results will be reported in due course [58].
10.2 Synthesis of Mannosylated Glycopeptides
Our interest in the synthesis of glycopeptide-based vaccine candidates comprising mannose units to target antigen presenting cells (APCs) responsible for initiating an immune response via mannose receptor (MR), led us previously to prepare mono- and di-mannosylated and 5(6)-carboxyfluorescein (5(6)-CF) labelled glycopeptides by chemical synthesis [48, 87]. Subsequent progression of this work involved the synthesis of glycopeptide-based vaccine candidates comprising more complex high-mannose-type N-glycans and testing their ability to bind to APCs [57]. For this purpose, the pp65 protein fragment 491–509 from the cytomegalovirus (CMV) [ILARNLVPMVATVQGQNLK] incorporating peptide epitope pp65495–503 (NLVPMVATV) recognised by human cytotoxic T-lymphocyte (CTL) was chosen as a synthetic target. The target peptide contains two asparagine residues Asn5 and Asn17 conveniently located within the pp65491–509 sequence for potential attachment of sugar residues. To allow detection of the peptides using flow cytometry 5(6)-carboxyfluorescein was attached to the N-terminus of the glycopeptides.
5(6)-CF-labelled control peptide 39, and glycopeptides 40 and 41 comprising either one- or two-GlcNAc units, respectively, were synthesised using microwave-enhanced Fmoc SPPS wherein the Fmoc-Asn[GlcNAc(OAc)3] building block 38 [197] replaced either Asn17 (for 40) or Asn5 and Asn17 (for 41) as required. Subsequent reduction of the methionine sulfoxide of the control peptide, used in place of methionine, was performed following literature procedures [203] and afforded the pp65 protein fragment 491–509 (39). Removal of sugar hydroxyl protecting groups of the per-O-acetylated pp65491–509 precursors of 40 and 41 (sodium methoxide in methanol) was undertaken prior to reduction of the methionine sulfoxide [203] to give GlcNAc-tagged glycopeptides 40 and 41 ready for further enzymatic glycosylation (Fig. 6).

CMV control peptide 39 and glycopeptides 40 and 41 containing a GlcNAc residue either at Asn17, or at both Asn5 and Asn17, respectively [57]
With GlcNAc-tagged glycopeptides 40 and 41 in hand, we next focused on the synthesis of N-glycopeptides bearing a Man3-terminated pentasaccharide. This was successfully undertaken using oxazoline donor 4 [140], glycopeptide acceptors 40 and 41, and the Endo A E173H mutant [153] to afford 42 and 44, bearing either one- or two-pentasaccharide units, respectively, in good yield (68 and 89%, respectively), Scheme 8a.

Synthesis of a glycopeptides 42 and 44 containing a Man3(GlcNAc)2 residue, and b glycopeptides 43 and 45 containing a Man9(GlcNAc)2 residue, either at Asn17, or at both Asn5 and Asn17, respectively. Reagents and conditions: a Endo A E173H, sodium phosphate buffer pH 6.5; b Endo M N175Q, sodium phosphate buffer pH 6.5 [57]
To access full-length high-mannose N-linked glycopeptides 43 and 45 bearing nine mannose units at each glycosylation site, oxazoline donor 46 was used which was conveniently sourced from soy bean flour [161]. Somewhat surprisingly the Endo A E173H mutant [153] proved incapable of transferring the full-length high-mannose oxazoline 46 onto glycopeptide acceptors 40 and 41, possibly due to an altered substrate tolerance of the mutated enzyme as compared to wild-type Endo A [57]. We, therefore, used commercially available Endo M N175Q mutant [168, 177] which produced the desired N-undecasaccharide-glycopeptides 43 and 45 with either one- or two-Man9(GlcNAc)2 units in 48% and 54% yield, respectively (Scheme 8b).
Subsequent analysis to assess glycopeptide binding levels to APCs indicated improved targeting of the peptide cargo to MR-expressing cells due to the presence of the high-mannose N-glycans. This effect was more pronounced for analogues glycosylated at both asparagines (44 and 45) as compared to counterparts bearing a single N-glycan at Asn17 (42 and 43). In addition, stronger binding was observed for glycopeptides bearing the high-mannose unit, Man9(GlcNAc)2 (43 and 45) than those with the truncated glycan, Man3(GlcNAc)2 (42 and 44). Importantly, it was also found that analogues in which the sugars were sited outside the epitope sequence (either Man3(GlcNAc)2 or Man9(GlcNAc)2 at Asn17, 42 and 43, respectively) were readily processed and presented by the APCs to human T cells.
These results provide important evidence that N-glycosylation of peptides using high-mannose glycans may produce superior compounds for vaccine development. Additionally, we have demonstrated the effectiveness of a convergent chemoenzymatic approach to readily obtain complex N-linked glycopeptides with therapeutic potential.
11 Conclusions
The synthesis of homogeneous peptides and proteins is still a complex and onerous task. The synthesis of the sugar component is a limiting factor, especially when laborious total synthesis routes are employed. Fortunately, recent progress in the use of chemoenzymatic techniques using ENGase-mediated glycosylation has demonstrated significant potential. By careful design of reaction conditions and appropriate selection of partners for glycosylation, a wide range of peptide-oligosaccharide structures can be obtained. The use of enzyme-mediated synthesis in combination with chemical synthetic techniques provides a method to access complex, highly desirable glycoconjugates efficiently [101, 204].
References
Spiro RG (2002) Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12:43R–56R
Meledeo MA, Yarema KJ, Begley TP (2007) Glycan biosynthesis in mammals, in wiley encyclopedia of chemical biology. Wiley, New Jersey
Lafite P, Daniellou R (2012) Rare and unusual glycosylation of peptides and proteins. Nat Prod Rep 29:729–738
Haltiwanger RS, Lowe JB (2004) Role of Glycosylation in Development. Annu Rev Biochem 73:491–537
Dube DH, Bertozzi CR (2005) Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat Rev Drug Discov 4:477–488
Dwek RA (1996) Glycobiology: toward understanding the function of sugars. Chem Rev 96:683–720
Imperiali B, O’Connor SE (1999) Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr Opin Chem Biol 3:643–649
Wyss DF, Choi JS (1995) Conformation and function of the N-linked glycan in the adhesion domain of human CD2. Science 269:1270
Opdenakker G, Rudd PM, Ponting CP, Dwek RA (1993) Concepts and principles of glycobiology. FASEB J 7:1330–1337
Erbayraktar S, Grasso G, Sfacteria A, Xie Q-W, Coleman T, Kreilgaard M, Torup L, Sager T, Erbayraktar Z, Gokmen N, Yilmaz O, Ghezzi P, Villa P, Fratelli M, Casagrande S, Leist M, Helboe L, Gerwein J, Christensen S, Geist MA, Pedersen LØ, Cerami-Hand C, Wuerth J-P, Cerami A, Brines M (2003) Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Nat Acad Sci USA 100:6741–6746
Schachter H, Freeze HH (2009) Glycosylation diseases: quo vadis? Biochim Biophys Acta Mol Basis Dis 1792:925–930
Murakami Y, Kinoshita T (2015) Congenital Disorders of Glycosylation: Glycosylphosphatidylinositol (GPI)-Related. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 1229–1236
Akimoto Y, Miura Y, Endo T, Kawakami H, Hart G (2015) Diabetes and O-GlcNAcylation. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 1207–1212
Hart GW, Housley MP, Slawson C (2007) Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446:1017–1022
Zeidan Q, Hart GW (2010) The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci 123:13–22
Stowell SR, Ju T, Cummings RD (2015) Protein Glycosylation in Cancer. Annu Rev Pathol Mech Dis 10:473–510
Korekane H, Taniguchi N (2015) Glycosylation in cancer: enzymatic basis for alterations in N-glycan branching. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 1349–1356
Schedin-Weiss S, Winblad B, Tjernberg LO (2014) The role of protein glycosylation in Alzheimer disease. FEBS J 281:46–62
Gao C, Taniguchi N (2015) Chronic Obstructive Pulmonary Disease (COPD). In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: Biology and Medicine. Springer, Japan, pp 1267–1274
Solá RJ, Griebenow K (2009) Effects of Glycosylation on the Stability of Protein Pharmaceuticals. J Pharm Sci 98:1223–1245
Solá RJ, Griebenow K (2010) Glycosylation of Therapeutic Proteins: an effective strategy to optimize efficacy. BioDrugs 24:9–21
Ueda T, Tomita K, Notsu Y, Ito T, Fumoto M, Takakura T, Nagatome H, Takimoto A, Mihara SI, Togame H, Kawamoto K, Iwasaki T, Asakura K, Oshima T, Hanasaki K, Nishimura SI, Kondo H (2009) Chemoenzymatic Synthesis of Glycosylated Glucagon-like Peptide 1: effect of glycosylation on proteolytic resistance and in vivo blood glucose-lowering activity. J Am Chem Soc 131:6237–6245
Sinclair AM, Elliott S (2005) Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci 94:1626–1635
Costa AR, Rodrigues ME, Henriques M, Oliveira R, Azeredo J (2014) Glycosylation: impact, control and improvement during therapeutic protein production. Crit Rev Biotechnol 34:281–299
Li HJ, d’Anjou M (2009) Pharmacological significance of glycosylation in therapeutic proteins. Curr Opin Biotechnol 20:678–684
Jefferis R (2009) Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov 8:226–234
Sato M, Furuike T, Sadamoto R, Fujitani N, Nakahara T, Niikura K, Monde K, Kondo H, Nishimura SI (2004) Glycoinsulins: Dendritic sialyloligosaccharide-displaying insulins showing a prolonged blood-sugar-lowering activity. J Am Chem Soc 126:14013–14022
Ueda T, Ito T, Tomita K, Togame H, Fumoto M, Asakura K, Oshima T, Nishimura SI, Hanasaki K (2010) Identification of glycosylated exendin-4 analogue with prolonged blood glucose-lowering activity through glycosylation scanning substitution. Bioorg Med Chem Lett 20:4631–4634
Runkel L, Meier W, Pepinsky RB, Karpusas M, Whitty A, Kimball K, Brickelmaier M, Muldowney C, Jones W, Goelz SE (1998) Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-β (IFN-β). Pharm Res 15:641–649
Lappin TRJ, Maxwell AP (1989) Chemistry and assays of erythropoietin. In: Erythropoietin, W. Jelkmann and A. J. Gross, Editors. 1989, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 7–18
Davis BG (2002) Synthesis of glycoproteins. Chem Rev 102:579–602
Gamblin DP, Scanlan EM, Davis BG (2009) Glycoprotein synthesis: an update. Chem Rev 109:131–163
Wang L-X, Amin Mohammed N (2014) Chemical and chemoenzymatic synthesis of glycoproteins for deciphering functions. Chem Biol 21:51–66
Wang L-X, Davis BG (2013) Realizing the promise of chemical glycobiology. Chem Sci 4:3381–3394
Unverzagt C, Kajihara Y (2013) Chemical assembly of N-glycoproteins: a refined toolbox to address a ubiquitous posttranslational modification. Chem Soc Rev 42:4408–4420
Izumi M, Okamoto R, Kajihara Y (2015) Chemical synthesis of homogeneous glycoproteins. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 313–321
Westerlind U (2012) Synthetic glycopeptides and glycoproteins with applications in biological research. Beilstein J Org Chem 8:804–818
Fernández-Tejada A, Brailsford J, Zhang Q, Shieh J-H, Moore MAS, Danishefsky SJ (2015) Total synthesis of glycosylated proteins. In: Liu L (ed) Protein ligation and total synthesis I. Springer International Publishing, Cham, pp 1–26
Kajihara Y, Yamamoto N, Okamoto R, Hirano K, Murase T (2010) Chemical synthesis of homogeneous glycopeptides and glycoproteins. Chem Rec 10:80–100
Payne RJ, Wong C-H (2010) Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins. Chem Commun 46:21–43
Rich JR, Withers SG (2009) Emerging methods for the production of homogeneous human glycoproteins. Nat Chem Biol 5:206–215
Xu C, Li X (2015) Glycopeptide/glycoprotein synthesis. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 323–330
Fairbanks AJ (2013) Endohexosaminidase-catalyzed synthesis of glycopeptides and proteins. Pure Appl Chem 85:1847–1863
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181
Stanley P, Schachter H, Taniguchi N (2009) N-glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (eds) Essentials of glycobiology. Cold Spring Harbor Laboratory Press, New York
Herzner H, Reipen T, Schultz M, Kunz H (2000) Synthesis of glycopeptides containing carbohydrate and peptide recognition motifs. Chem Rev 100:4495–4538
Rudd PM, Dwek RA (1997) Glycosylation: heterogeneity and the 3D structure of proteins. Crit Rev Biochem Mol Biol 32:1–100
Brimble MA, Kowalczyk R, Harris PWR, Dunbar PR, Muir VJ (2008) Synthesis of fluorescein-labelled O-mannosylated peptides as components for synthetic vaccines: comparison of two synthetic strategies. Org Biomol Chem 6:112–121
Miller N, Williams GM, Brimble MA (2009) Synthesis of fish antifreeze neoglycopeptides using microwave-assisted “click chemistry”. Org Lett 11:2409–2412
Kowalczyk R, Harris PWR, Dunbar RP, Brimble MA (2009) Stability of 5(6)-carboxyfluorescein in microwave-assisted synthesis of fluorescein-labelled O-dimannosylated peptides. Synthesis 2009:2210–2222
Lee DJ, Mandal K, Harris PWR, Brimble MA, Kent SBH (2009) A one-pot approach to neoglycopeptides using orthogonal native chemical ligation and click chemistry. Org Lett 11:5270–5273
Lee DJ, Harris PWR, Kowalczyk R, Dunbar PR, Brimble MA (2010) Microwave-assisted synthesis of fluorescein-labelled GalNAcα1-O-Ser/Thr (Tn) glycopeptides as immunological probes. Synthesis 2010:763–769
Peltier R, Evans CW, DeVries AL, Brimble MA, Dingley AJ, Williams DE (2010) Growth habit modification of ice crystals using antifreeze glycoprotein (AFGP) analogues. Cryst Growth Des 10:5066–5077
Lee DJ, Harris PWR, Brimble MA (2011) Synthesis of MUC1 neoglycopeptides using efficient microwave-enhanced chaotrope-assisted click chemistry. Org Biomol Chem 9:1621–1626
Lee DJ, Yang S-H, Williams GM, Brimble MA (2012) Synthesis of multivalent neoglyconjugates of MUC1 by the conjugation of carbohydrate-centered, triazole-linked gllycoclusters to MUC1 peptides using click chemistry. J Org Chem 77:7564–7571
Brimble MA, Edwards PJ, Harris PWR, Norris GE, Patchett ML, Wright TH, Yang S-H, Carley SE (2015) Synthesis of the antimicrobial S-linked glycopeptide, glycocin F. Chem Eur J 21:3556–3561
McIntosh JD, Brimble MA, Brooks AES, Dunbar PR, Kowalczyk R, Tomabechi Y, Fairbanks AJ (2015) Convergent chemo-enzymatic synthesis of mannosylated glycopeptides; targeting of putative vaccine candidates to antigen presenting cells. Chem Sci 6:4636–4642
Kowalczyk R, Brimble MA, Tomabechi Y, Fairbanks AJ, Fletcher M, Hay DL (2014) Convergent chemoenzymatic synthesis of a library of glycosylated analogues of pramlintide: structure-activity relationships foramyl in receptor agonism. Org Biomol Chem 12:8142–8151
Reichert JM (2012) Marketed therapeutic antibodies compendium. mAbs 4:413–415
http://top101news.com/2015-2016-2017-2018/news/health/best-selling-drugs-world/. Top 10 Best selling Drugs in the World
Bang LM, Keating GM (2004) Adalimumab a review of its use in rheumatoid arthritis. BioDrugs 18:121–139
Mazumdar S, Greenwald D (2009) Golimumab. mAbs 1:422–431
Swiech K, de Freitas M, Covas D, Picanço-Castro V (2015) Recombinant glycoprotein production in human cell lines. In: García-Fruitós E (ed) Insoluble proteins. Springer, New York, pp 223–240
Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi B-K, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU (2006) Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313:1441–1443
Hamilton SR, Bobrowicz P, Bobrowicz B, Davidson RC, Li H, Mitchell T, Nett JH, Rausch S, Stadheim TA, Wischnewski H, Wildt S, Gerngross TU (2003) Production of complex human glycoproteins in yeast. Science 301:1244–1246
Wang L-X, Lomino JV (2012) Emerging technologies for making glycan-defined glycoproteins. ACS Chem Biol 7:110–122
Fernandez-Tejada A, Danishefsky SJ (2014) Chapter 25 Development of cancer vaccines from fully synthetic mucin-based glycopeptide antigens. A vision on mucins from the bioorganic chemistry perspective. In: Carbohydrate Chemistry, vol 40. The Royal Society of Chemistry, pp 533–563
Gaidzik N, Westerlind U, Kunz H (2013) The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem Soc Rev 42:4421–4442
Payne RJ (2013) Total synthesis of erythropoietin through the development and exploitation of enabling synthetic technologies. Angew Chem Int Ed 52:505–507
Kajihara Y, Okamoto R, Yamamoto N, Izumi M (2010) Chapter twenty-four—synthesis of glycopeptides. In: Minoru F (ed) Methods in enzymology. Academic Press, pp 503–519
Panda SS, Jones RA, Dennis Hall C, Katritzky AR (2015) Applications of chemical ligation in peptide synthesis via acyl transfer. In: Liu L (ed) Protein ligation and total synthesis I. Springer International Publishing, Cham, pp 229–265
Behrendt R, White P, Offer J (2016) Advances in Fmoc solid-phase peptide synthesis. J Pept Sci 22:4–27
Merrifield RB (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc 85:2149–2154
Dawson P, Muir T, Clark-Lewis I, Kent S (1994) Synthesis of proteins by native chemical ligation. Science 266:776–779
Muir TW (2003) Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem 72:249–289
Xu C, Lam HY, Zhang Y, Li X (2013) Convergent synthesis of MUC1 glycopeptides via serine ligation. Chem Commun 49:6200–6202
Zhang Y, Xu C, Lam HY, Lee CL, Li X (2013) Protein chemical synthesis by serine and threonine ligation. Proc Nat Acad Sci USA 110:6657–6662
Li X, Lam HY, Zhang Y, Chan CK (2010) Salicylaldehyde ester-induced chemoselective peptide ligations: enabling generation of natural peptidic linkages at the serine/threonine sites. Org Lett 12:1724–1727
Sakamoto I, Tezuka K, Fukae K, Ishii K, Taduru K, Maeda M, Ouchi M, Yoshida K, Nambu Y, Igarashi J, Hayashi N, Tsuji T, Kajihara Y (2012) Chemical synthesis of homogeneous human glycosyl-interferon-β that exhibits potent antitumor activity in vivo. J Am Chem Soc 134:5428–5431
Izumi M, Makimura Y, Dedola S, Seko A, Kanamori A, Sakono M, Ito Y, Kajihara Y (2012) Chemical synthesis of intentionally misfolded homogeneous glycoprotein: a unique approach for the study of glycoprotein quality Control. J Am Chem Soc 134:7238–7241
Hojo H, Tanaka H, Hagiwara M, Asahina Y, Ueki A, Katayama H, Nakahara Y, Yoneshige A, Matsuda J, Ito Y, Nakahara Y (2012) Chemoenzymatic Synthesis of hydrophobic glycoprotein: synthesis of saposin c carrying complex-type Carbohydrate. J Org Chem 77:9437–9446
Piontek C, Ring P, Harjes O, Heinlein C, Mezzato S, Lombana N, Pöhner C, Püttner M, Varón Silva D, Martin A, Schmid FX, Unverzagt C (2009) Semisynthesis of a homogeneous glycoprotein enzyme: Ribonuclease C: part 1. Angew Chem Int Ed 48:1936–1940
Piontek C, Varón Silva D, Heinlein C, Pöhner C, Mezzato S, Ring P, Martin A, Schmid FX, Unverzagt C (2009) Semisynthesis of a homogeneous glycoprotein enzyme: Ribonuclease C: part 2. Angew Chem Int Ed 48:1941–1945
Haase C, Seitz O (2007) Chemical synthesis of glycopepticles. In: Wittmann V (ed) Glycopeptides and glycoproteins: synthesis, structure, and application. Springer, Berlin, pp 1–36
Palitzsch B, Gaidzik N, Stergiou N, Stahn S, Hartmann S, Gerlitzki B, Teusch N, Flemming P, Schmitt E, Kunz H (2016) A synthetic glycopeptide vaccine for the induction of a monoclonal antibody that differentiates between normal and tumor mammary cells and enables the diagnosis of human pancreatic cancer. Angew Chem Int Ed 55:2894–2898
Hartmann S, Palitzsch B, Glaffig M, Kunz H (2014) Chapter 24 tumour-associated glycopeptide antigens and their modification in anticancer vaccines. In: Carbohydrate chemistry: volume 40. The Royal Society of Chemistry, pp 506–532
Kowalczyk R, Harris PWR, Dunbar RP, Brimble MA (2009) Stability of 5(6)-carboxyfluorescein in microwave-assisted synthesis of fluorescein-labelled o-dimannosylated peptides. Synthesis, 2210–2222
Lee DJ, Harris PWR, Kowalczyk R, Dunbar PR, Brimble MA (2010) Microwave-assisted synthesis of fluorescein-labelled GalNAc α 1-O-Ser/Thr (Tn) glycopeptides as immunological probes. Synthesis-Stuttgart, 763–769
Yamamoto N, Ohmori Y, Sakakibara T, Sasaki K, Juneja LR, Kajihara Y (2003) Solid-phase synthesis of sialylglycopeptides through selective Esterification of the sialic acid residues of an asn-linked complex-type sialyloligosaccharide. Angew Chem Int Ed 42:2537–2540
Kajihara Y, Suzuki Y, Yamamoto N, Sasaki K, Sakakibara T, Juneja LR (2004) Prompt chemoenzymatic synthesis of diversecomplex-type oligosaccharides and Its application to the solid-phase synthesisof a glycopeptide with asn-linked sialyl-undeca- and asialo-nonasaccharides. Chem Eur J 10:971–985
Murakami M, Kiuchi T, Nishihara M, Tezuka K, Okamoto R, Izumi M, Kajihara Y (2016) Chemical synthesis of erythropoietin glycoforms for insights into the relationship between glycosylation pattern and bioactivity. Sci Adv 2:1–12
Murakami M, Okamoto R, Izumi M, Kajihara Y (2012) Chemical synthesis of an erythropoietin glycoform containing a complex-type Disialyloligosaccharide. Angew Chem Int Ed 51:3567–3572
Yamamoto N, Takayanagi A, Yoshino A, Sakakibara T, Kajihara Y (2007) An approach for a synthesis of asparagine-linked sialylglycopeptides having intact and homogeneous complex-type undecadisialyloligosaccharides. Chem Eur J 13:613–625
Cohen-Anisfeld ST, Lansbury PT (1993) A practical, convergent method for glycopeptide synthesis. J Am Chem Soc 115:10531–10537
Anisfeld ST, Lansbury PT (1990) A convergent approach to the chemical synthesis of asparagine-linked glycopeptides. J Org Chem 55:5560–5562
Wang P, Dong S, Brailsford JA, Iyer K, Townsend SD, Zhang Q, Hendrickson RC, Shieh J, Moore MAS, Danishefsky SJ (2012) At Last: erythropoietin as a single Glycoform. Angew Chem Int Ed 51:11576–11584
Wang P, Dong S, Shieh J-H, Peguero E, Hendrickson R, Moore MAS, Danishefsky SJ (2013) Erythropoietin derived by chemical synthesis. Science 342:1357–1360
Yan LZ, Dawson PE (2001) Synthesis of peptides and proteins without cysteine Residues by native chemical ligation combined with desulfurization. J Am Chem Soc 123:526–533
Wang P, Aussedat B, Vohra Y, Danishefsky SJ (2012) An advance in the chemical synthesis of homogeneous N-linked glycopolypeptides by convergent aspartylation. Angew Chem Int Ed 51:11571–11575
Ullmann V, Rädisch M, Boos I, Freund J, Pöhner C, Schwarzinger S, Unverzagt C (2012) Convergent solid-phase synthesis of NGlycopeptides facilitated by pseudoprolines at consensus-sequence Ser/Thr residues. Angew Chem Int Ed 51:11566–11570
Katoh T, Yamamoto K (2015) Glycoenzymes in glycan analysis and synthesis. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 379–389
Malet C, Planas A (1998) From β-glucanase to β-glucansynthase: glycosyl transfer to α-glycosyl fluorides catalyzed by a mutant endoglucanase lacking its catalytic nucleophile. FEBS Lett 440:208–212
Mackenzie LF, Wang Q, Warren RAJ, Withers SG (1998) Glycosynthases: mutant glycosidases for oligosaccharide synthesis. J Am Chem Soc 120:5583–5584
Hancock SM, Vaughan MD, Withers SG (2006) Engineering of glycosidases and glycosyltransferases. Curr Opin Chem Biol 10:509–519
Yamamoto K (2015) Endo-enzymes. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 391–399
http://www.cazy.org. carbohydrate-active enZYmes database
Murakami S, Takaoka Y, Ashida H, Yamamoto K, Narimatsu H, Chiba Y (2013) Identification and characterization of endo-β-nacetylglucosaminidas from methylotrophic yeast Ogataea minuta. Glycobiology 23:736–744
Yamamoto K (2001) Chemo-Enzymatic synthesis of bioactive glycopeptide using microbial endoglycosidase. J Biosci Bioeng 92:493–501
Kadowaki S, Yamamoto K, Fujisaki M, Kumagai H, Tochikura T (1988) A novel endo-β-N-acetylglucosaminidase acting on complex oligosaccharides of glycoproteins in a fungus. Agric Biol Chem 52:2387–2389
Yamamoto KJ, Kadowaki S, Watanabe J, Kumagai H (1994) Transglycosylation activity of mucor hiemalis endo-β-N-acetylglucosaminidase which transfers complex oligosaccharides to the N-Acetylglucosamine moieties of peptides. Biochem Biophys Res Commun 203:244–252
Haneda K, Inazu T, Yamamoto K, Kumagai H, Nakahara Y, Kobata A (1996) Transglycosylation of intact sialo complex-type oligosaccharides to the N-acetylglucosamine moieties of glycopeptides by Mucor hiemalis endo-β-N-acetylglucosaminidase. Carbohydr Res 292:61–70
Yamamoto K, Fujimori K, Haneda K, Mizuno M, Inazu T, Kumagai H (1997) Chemoenzymatic synthesis of a novel glycopeptide using a microbial endoglycosidase. Carbohydr Res 305:415–422
Mizuno M, Haneda K, Iguchi R, Muramoto I, Kawakami T, Aimoto S, Yamamoto K, Inazu T (1999) Synthesis of a glycopeptide containing oligosaccharides: chemoenzymatic synthesis of eel calcitonin analogues having natural N-linked oligosaccharides. J Am Chem Soc 121:284–290
Haneda K, Inazu T, Mizuno M, Iguchi R, Yamamoto K, Kumagai H, Aimoto S, Suzuki H, Noda T (1998) Chemo-enzymatic synthesis of calcitonin derivatives containing N-linked oligosaccharides. Bioorg Med Chem Lett 8:1303–1306
Yamanoi T, Tsutsumida M, Oda Y, Akaike E, Osumi K, Yamamoto K, Fujita K (2004) Transglycosylation reaction of mucor hiemalis endo-β-N-acetylglucosaminidase using sugar derivatives modified at C-1 or C-2 as oligosaccharide acceptors. Carbohydr Res 339:1403–1406
Osumi K, Makino Y, Akaike E, Yamanoi T, Mizuno M, Noguchi M, Inazu T, Yamamoto K, Fujita K (2004) Mucor hiemalis endo-β-N-acetylglucosaminidase can transglycosylate a bisecting hybrid-type oligosaccharide from an ovalbumin glycopeptide. Carbohydr Res 339:2633–2635
Haneda K, Takeuchi M, Tagashira M, Inazu T, Toma K, Isogai Y, Hori M, Kobayashi K, Takeuchi M, Takegawa K, Yamamoto K (2006) Chemo-enzymatic synthesis of eel calcitonin glycosylated at two sites with the same and different carbohydrate structures. Carbohydr Res 341:181–190
Makimura Y, Watanabe S, Suzuki T, Suzuki Y, Ishida H, Kiso M, Katayama T, Kumagai H, Yamamoto K (2006) Chemoenzymatic synthesis and application of a sialoglycopolymer with a chitosan backbone as a potent inhibitor of human influenza virus hemagglutination. Carbohydr Res 341:1803–1808
Yamanoi T, Yoshida N, Oda Y, Akaike E, Tsutsumida M, Kobayashi N, Osumi K, Yamamoto K, Fujita K, Takahashi K, Hattori K (2005) Synthesis of mono-glucose-branched cyclodextrins with a high inclusion ability for doxorubicin and their efficient glycosylation using Mucor hiemalis endo-β-N-acetylglucosaminidase. Bioorg Med Chem Lett 15:1009–1013
Tomabechi Y, Inazu T (2011) Preparation of pseudo glycoamino acid and its application to glycopeptide synthesis. Tetrahedron Lett 52:6504–6507
Takegawa K, Nakoshi M, Iwahara S, Yamamoto K, Tochikura T (1989) Induction and purification of endo-β-N-acetylglucosaminidase from arthrobacter protophormiae grown in ovalbumin. Appl Environ Microbiol 55:3107–3112
Fan J-Q, Huynh LH, Reinhold BB, Reinhold VN, Takegawa K, Iwahara S, Kondo A, Kato I, Lee YC (1996) Transfer of Man9GlcNAc tol-fucose by endo-β-N-acetylglucosaminidase from arthrobacter protophormiae. Glycoconjugate J 13:643–652
Takegawa K, Tabuchi M, Yamaguchi S, Kondo A, Kato I, Iwahara S (1995) Synthesis of neoglycoproteins using oligosaccharide-transfer activity with endo-β-N-acetylglucosaminidase. J Biol Chem 270:3094–3099
Fan J-Q, Quesenberry MS, Takegawa K, Iwahara S, Kondo A, Kato I, Lee YC (1995) Synthesis of Neoglycoconjugates by Transglycosylation with Arthrobacter protophormiae endo-β-N-acetylglucosaminidase: demonstration of a macro-cluster effect for mannose-binding proteins. J Biol Chem 270:17730–17735
Fan J-Q, Takegawa K, Iwahara S, Kondo A, Kato I, Abeygunawardana C, Lee YC (1995) Enhanced transglycosylation activity of arthrobacter protophormiae endo-β-N-acetylglucosaminidase in media containing organic solvents. J Biol Chem 270:17723–17729
Takegawa K, Fujita K, Fan J-Q, Tabuchi M, Tanaka N, Kondo A, Iwamoto H, Kato I, Lee YC, Iwahara S (1998) Enzymatic synthesis of a neoglycoconjugate by transglycosylation with arthrobacter endo-β-N-acetylglucosaminidase: a substrate for colorimetric detection of endo-β-N-acetylglucosaminidase activity. Anal Biochem 257:218–223
Fujita K, Tanaka N, Sano M, Kato I, Asada Y, Takegawa K (2000) Synthesis of neoglycoenzymes with homogeneous Nlinked oligosaccharides using Immobilized endo-β-N-acetylglucosaminidase A. Biochem Biophys Res Commun 267:134–138
Takegawa K, Yamabe K, Fujita K, Tabuchi M, Mita M, Izu H, Watanabe A, Asada Y, Sano M, Kondo A, Kato I, Iwahara S (1997) Cloning, sequencing, and expression of arthrobacter protophormiae endo-β-N-acetylglucosaminidase in escherichia coli. Arch Biochem Biophys 338:22–28
Muramatsu H, Tachikui H, Ushida H, Song X-J, Qiu Y, Yamamoto S, Muramatsu T (2001) Molecular cloning and expression of endo-β-N-acetylglucosaminidase D, which acts on the core structure of complex type asparagine-linked oligosaccharides. J Biochem 129:923–928
Parsons TB, Patel MK, Boraston AB, Vocadlo DJ, Fairbanks AJ (2010) Streptococcus pneumoniae endohexosaminidase D; feasibility of using N-glycan oxazoline donors for synthetic glycosylation of a GlcNAc-asparagine acceptor. Org Biomol Chem 8:1861–1869
Fujita K, Takami H, Yamamoto K, Takegawa K (2004) Characterization of endo-β-N-acetylglucosaminidase from alkaliphilic bacillus halodurans C-125. Biosci Biotechnol Biochem 68:1059–1066
Tarentino AL, Maley F (1974) Purification and properties of an endo-β-N-acetylglucosaminidase from streptomyces griseus. J Biol Chem 249:811–817
Goodfellow JJ, Baruah K, Yamamoto K, Bonomelli C, Krishna B, Harvey DJ, Crispin M, Scanlan CN, Davis BG (2012) An endoglycosidase with alternative glycan specificity allows broadened glycoprotein remodelling. J Am Chem Soc 134:8030–8033
Collin M, Olsén A (2001) EndoS, a novel secreted protein from streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J 20:3046–3055
Huang W, Li J, Wang L-X (2011) Unusual transglycosylation activity of flavobacterium meningosepticum endoglycosidases enables convergent chemoenzymatic synthesis of core fucosylated complex N-glycopeptides. ChemBioChem 12:932–941
Trimble RB, Tarentino AL (1991) Identification of distinct endoglycosidase (endo) activities in flavobacterium meningosepticum: endo F1, endo F2, and endo F3. Endo F1 and endo H hydrolyze only high mannose and hybrid glycans. J Biol Chem 266:1646–1651
Tarentino AL, Plummer TH Jr (1994) [4] Enzymatic deglycosylation of asparagine-linked glycans: purification, properties, and specificity of oligosaccharide-cleaving enzymes from flavobacterium meningosepticum. In: Methods in enzymology, vol 230. Academic Press, Cambridge, pp 44–57
Zhao G, Liu Y, Wu Z, Zhu H, Yu Z, Fang J, Wang P (2015) Chemoenzymatic synthesis of glycoproteins. In: Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H (eds) Glycoscience: biology and medicine. Springer, Japan, pp 427–435
Wang L-X (2008) Chemoenzymatic synthesis of glycopeptides and glycoproteins through endoglycosidase-catalyzed transglycosylation. Carbohydr Res 343:1509–1522
Rising TWDE, Heidecke CD, Moir JWB, Ling ZL, Fairbanks AJ (2008) Endohexosaminidase-catalysed glycosylation with oxazoline donors: fine tuning of catalytic efficiency and reversibility. Chem Eur J 14:6444–6464
Rising TWDF, Claridge TDW, Davies N, Gamblin DP, Moir JWB, Fairbanks AJ (2006) Synthesis of N-glycan oxazolines: donors for endohexosaminidase catalysed glycosylation. Carbohydr Res 341:1574–1596
Rising TWDF, Claridge TDW, Moir JWB, Fairbanks AJ (2006) Endohexosaminidase M: exploring and exploiting enzyme substrate specificity. ChemBioChem 7:1177–1180
Parsons TB, Moir JWB, Fairbanks AJ (2009) Synthesis of a truncated bi-antennary complextype N-glycan oxazoline; glycosylation catalysed by the endohexosaminidases endo A and endo M. Org Biomol Chem 7:3128–3140
Huang W, Ochiai H, Zhang XY, Wang LX (2008) Introducing N-glycans into natural products through a chemoenzymatic approach. Carbohydr Res 343:2903–2913
Li B, Song HJ, Hauser S, Wang LX (2006) A highly efficient chemoenzymatic approach toward glycoprotein synthesis. Org Lett 8:3081–3084
Li B, Zeng Y, Hauser S, Song HJ, Wang LX (2005) Highly efficient endoglycosidase-catalyzed synthesis of glycopeptides using oligosaccharide oxazolines as donor substrates. J Am Chem Soc 127:9692–9693
Ochiai H, Huang W, Wang LX (2009) Endo-β-N-acetylglucosaminidase-catalyzed polymerization of β-Glcp-(1->4)-GlcpNAc oxazoline: a revisit to enzymatic transglycosylation. Carbohydr Res 344:592–598
Wang LX, Song HJ, Liu SW, Lu H, Jiang SB, Ni JH, Li HG (2005) Chemoenzymatic synthesis of HIV-1 gp41 glycopeptides: effects of glycosylation on the anti-HIV activity and alpha-helix bundle-forming ability of peptide C34. ChemBioChem 6:1068–1074
Wei YD, Li CS, Huang W, Li B, Strome S, Wang LX (2008) Glycoengineering of human IgG1-Fc through combined yeast expression and in vitro chemoenzymatic glycosylation. Biochemistry 47:10294–10304
Zeng Y, Wang JS, Li B, Hauser S, Li HG, Wang LX (2006) Glycopeptide synthesis through endo-glycosidasecatalyzed oligosaccharide transfer of sugar oxazolines: probing substrate structural requirement. Chem Eur J 12:3355–3364
Fujita M, Shoda S-i, Haneda K, Inazu T, Takegawa K, Yamamoto K (2001) A novel disaccharide substrate having 1,2-oxazoline moiety for detection of transglycosylating activity of endoglycosidases. Biochim Biophys Acta Gen Subj 1528:9–14
Fairbanks AJ (2011) Endohexosaminidase catalysed glycosylation with oxazoline donors: the development of robust biocatalytic methods for synthesis of defined homogeneous glycoconjugates. C R Chim 14:44–58
Heidecke CD, Ling Z, Bruce NC, Moir JWB, Parsons TB, Fairbanks AJ (2008) Enhanced glycosylation with mutants of endohexosaminidase A (endo A). ChemBioChem 9:2045–2051
Heidecke CD, Parsons TB, Fairbanks AJ (2009) Endohexosaminidase-catalysed glycosylation with oxazoline donors: effects of organic co-solvent and pH on reactions catalysed by endo A and endo M. Carbohydr Res 344:2433–2438
Li B, Zeng Y, Hauser S, Song H, Wang L-X (2005) Highly efficient endoglycosidase-catalyzed synthesis of glycopeptides using oligosaccharide oxazolines as donor substrates. J Am Chem Soc 127:9692–9693
Li H, Li B, Song H, Breydo L, Baskakov IV, Wang L-X (2005) Chemoenzymatic synthesis of HIV-1 V3 glycopeptides carrying two N-glycans and effects of glycosylation on the peptide domain. J Org Chem 70:9990–9996
Wang L-X, Song H, Liu S, Lu H, Jiang S, Ni J, Li H (2005) Chemoenzymatic synthesis of HIV-1 gp41 glycopeptides: effects of glycosylation on the anti-HIV activity and α-helix bundle-forming ability of peptide C34. ChemBioChem 6:1068–1074
Li B, Song H, Hauser S, Wang L-X (2006) A highly efficient chemoenzymatic approach toward glycoprotein synthesis. Org Lett 8:3081–3084
Zeng Y, Wang J, Li B, Hauser S, Li H, Wang L-X (2006) Glycopeptide synthesis through endo-glycosidase-catalyzed oligosaccharide yransfer of sugar pxazolines: probing substrate structural requirement. Chem Eur J 12:3355–3364
Ochiai H, Huang W, Wang L-X (2008) Expeditious chemoenzymatic synthesis of homogeneous N-glycoproteins carrying defined oligosaccharide ligands. J Am Chem Soc 130:13790–13803
Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang L-X, Yamamoto K (2008) Mutants of mucor hiemalis endo-β-N-acetylglucosaminidase show enhanced transglycosylation and glycosynthaselike activities. J Biol Chem 283:4469–4479
Li H, Singh S, Zeng Y, Song H, Wang L-X (2005) Chemoenzymatic synthesis of CD52 glycoproteins carrying native N-glycans. Bioorg Med Chem Lett 15:895–898
Wei Y, Li C, Huang W, Li B, Strome S, Wang L-X (2008) Glycoengineering of human IgG1-Fc through combined yeast expression and in vitro chemoenzymatic glycosylation. Biochemistry 47:10294–10304
Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang L-X (2009) Glycosynthases enable a highly efficient chemoenzymatic eynthesis of N-glycoproteins carrying intact natural N-glycans. J Am Chem Soc 131:2214–2223
Tomabechi Y, Krippner G, Rendle PM, Squire MA, Fairbanks AJ (2013) Glycosylation of pramlintide: synthetic glycopeptides that display in vitro and in vivo activities as amylin receptor agonists. Chem Eur J 19:15084–15088
Priyanka P, Parsons TB, Miller A, Platt FM, Fairbanks AJ (2016) Chemoenzymatic synthesis of a phosphorylated glycoprotein. Angew Chem Int Ed. doi:10.1002/anie.201600817
Fujita K, Takegawa K (2001) Tryptophan-216 is essential for the transglycosylation activity of endo-β-N-acetylglucosaminidase A. Biochem Biophys Res Commun 283:680–686
Umekawa M, Li CS, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX (2010) Efficient glycosynthase mutant derived from mucor hiemalis endo-b-N-acetylglucosaminidase capable of transferring oligosaccharide from both sugar oxazoline and natural N-glycan. J Biol Chem 285:511–521
Fan S-Q, Huang W, Wang L-X (2012) Remarkable transglycosylation activity of glycosynthase mutants of endo-D, an endo-β-N-acetylglucosaminidase from streptococcus pneumoniae. J Biol Chem 287:11272–11281
Huang W, Giddens J, Fan S-Q, Toonstra C, Wang L-X (2012) Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J Am Chem Soc 134:12308–12318
Parsons TB, Struwe WB, Gault J, Yamamoto K, Taylor TA, Raj R, Wals K, Mohammed S, Robinson CV, Benesch JLP, Davis BG (2016) Optimal synthetic glycosylation of a therapeutic antibody. Angew Chem Int Ed 55:2361–2367
Rosati S, van den Bremer ETJ, Schuurman J, Parren PWHI, Kamerling JP, Heck AJR (2013) In-depth qualitative and quantitative analysis of composite glycosylation profiles and other micro-heterogeneity on intactmonoclonal antibodies by high-resolution native mass spectrometry using a modified orbitrap. mAbs 5:917–924
Lin C-W, Tsai M-H, Li S-T, Tsai T-I, Chu K-C, Liu Y-C, Lai M-Y, Wu C-Y, Tseng Y-C, Shivatare SS, Wang C-H, Chao P, Wang S-Y, Shih H-W, Zeng Y-F, You T-H, Liao J-Y, Tu Y-C, Lin Y-S, Chuang H-Y, Chen C-L, Tsai C-S, Huang C-C, Lin N-H, Ma C, Wu C-Y, Wong C-H (2015) A common glycan structure on immunoglobulin G for enhancement of effector functions. Proc Nat Acad Sci USA 112:10611–10616
Saini KS, Azim HA Jr, Cocorocchio E, Vanazzi A, Saini ML, Raviele PR, Pruneri G, Peccatori FA (2011) Rituximab in Hodgkin lymphoma: is the target always a hit? Cancer Treat Rev 37:385–390
Ellis P (2008) Trastuzumab (Herceptin) a treatment for HER2-positive breast cancer. In: Handbook of therapeutic antibodies. Wiley-VCH Verlag GmbH, pp 1109–1130
Tomabechi Y, Odate Y, Izumi R, Haneda K, Inazu T (2010) Acceptor specificity in the transglycosylation reaction using endo-M. Carbohydr Res 345:2458–2463
Tomabechi Y, Squire MA, Fairbanks AJ (2014) Endo-b-N-acetylglucosaminidase catalysed glycosylation: tolerance of enzymes to structural variation of the glycosyl amino acid acceptor. Org Biomol Chem 12:942–955
Boltje TJ, Buskas T, Boons G-J (2009) Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat Chem 1:611–622
Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda S-I (2009) Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J Org Chem 74:2210–2212
Watt GM, Boons G-J (2004) A convergent strategy for the preparation of N-glycan core di-, tri-, and pentasaccharide thioaldoses for the site-specific glycosylation of peptides and proteins bearing free cysteines. Carbohydr Res 339:181–193
Seko A, Koketsu M, Nishizono M, Enoki Y, Ibrahim HR, Juneja LR, Kim M, Yamamoto T (1997) Occurrence of a sialylglycopeptide and free sialylglycans in hen's egg yolk. Biochim Biophys Acta Gen Subj 1335:23–32
Evers DL, Hung RL, Thomas VH, Rice KG (1998) Preparative purification of a High-mannose typeN-glycan from soy bean agglutinin by hydrazinolysis and tyrosinamide derivatization. Anal Biochem 265:313–316
Liener IE (1955) The photometric determination of the hemagglutinating activity of soyin and crude soybean extracts. Arch Biochem Biophys 54:223–231
Wang L-X, Ni J, Singh S, Li H (2004) Binding of high-mannose-type oligosaccharides and synthetic oligomannose clusters to human antibody 2G12. Cell Chem Biol 11:127–134
Specker D, Wittmann V (2007) Synthesis and application of glycopeptide and glycoprotein mimetics. In: Wittmann V (ed) Glycopeptides and glycoproteins: synthesis, structure, and application. Springer Berlin Heidelberg, Berlin, Heidelberg, pp 65–107
Tornoe CW, Christensen C, Meldal M (2002)Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67:3057–3064
Rostovtsev VV, Green LG, Fokin VV, Sharpless KB (2002) A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed 41:2596–2599
Trabocchi A, Guarna A (2014) Click chemistry: the triazole ring as a privileged peptidomimetic scaffold. In: Peptidomimetics in organic and medicinal chemistry. Wiley, New Jersey, pp 99–121
Miller N, Williams GM, Brimble MA (2010) Synthesis of fish antifreeze neoglycopeptides using microwave-assisted “Click Chemistry”. Org Lett 12:1375–1376
Wojnar JM, Lee DJ, Evans CW, Mandal K, Kent SBH, Brimble MA (2013) Neoglycoprotein synthesis using the copper-catalyzed azide–alkyne click reaction and native chemical ligation. In: Click chemistry in glycoscience. Wiley, New Jersey, pp 251–270
Lim D, Brimble MA, Kowalczyk R, Watson AJA, Fairbanks AJ (2014) Protecting-group-free one-pot synthesis of glycoconjugates directly from reducing sugars. Angew Chem Int Ed 53:11907–11911
Grunberger GJ (2013) Novel therapies for the management of type 2 diabetes mellitus: part 1. Pramlintide and bromocriptine-QR. J Diabetes 5:110–117
Younk LM, Mikeladze M, Davis SN (2011) Pramlintide and the treatment of diabetes: a review of the data since its introduction. Expert Opin Pharmacother 12:1439–1451
Young AA, Vine W, Gedulin BR, Pittner R, Janes S, Gaeta LSL, Percy A, Moore CX, Koda JE, Rink TJ, Beaumont K (1996) Preclinical pharmacology of pramlintide in the rat: Comparisons with human and rat amylin. Drug Dev Res 37:231–248
Roberts AN, Leighton B, Todd JA, Cockburn D, Schofield PN, Sutton R, Holt S, Boyd Y, Day AJ, Foot EA, Willis AC, Reid KBM, Cooper GJS (1989) Molecular and functional-characterization of amylin, a peptide associated with type-2 diabetes-mellitus. Proc Nat Acad Sci USA 86:9662–9666
Maruyama K, Nagasawa H, Suzuki A (1999) 2,2'-bispyridyl disulfide rapidly induces intramolecular disulfide bonds in peptides. Peptides 20:881–884
Inazu T, Kobayashi K (1993) A new simple method for the synthesis of N(α)-Fmoc-N(b)-glycosylated-l-asparagine derivatives. Synlett, 869–870
Katayama H, Asahina Y, Hojo H (2011) Chemical synthesis of the S-linked glycopeptide, sublancin. J Pept Sci 17:818–821
Umekawa M, Higashiyama T, Koga Y, Tanaka T, Noguchi M, Kobayashi A, Shoda S, Huang W, Wang LX, Ashida H, Yamamoto K (2010) Efficient transfer of sialo-oligosaccharide onto proteins by combined use of a glycosynthase-like mutant of mucor hiemalis endoglycosidase and synthetic sialo-complex-type sugar oxazoline. Biophys Acta Gen Subj 1800:1203–1209
Hay DL, Christopoulos G, Christopoulos A, Poyner DR, Sexton PM (2005) Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol 67:1655–1665
Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, Sexton PM (2000) Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J Pharmacol Exp Ther 294:61–72
Gingell JJ, Burns ER, Hay DL (2014) Activity of pramlintide, rat and human amylin but not Ab1-42 at human amylin receptors. Endocrinology 155:21–26
Vilaseca M, Nicolas E, Capdevila F, Giralt E (1998) Reduction of methionine sulfoxide with NH4I/TFA: compatibility with peptides containing cysteine and aromatic amino acids. Tetrahedron 54:15273–15286
Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons G-J (2013) A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 341:379–383
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Kowalczyk, R., Kaur, H., Fairbanks, A.J., Brimble, M.A. (2018). Synthesis of N-Linked Glycopeptides Using Convergent Enzymatic Glycosylation Combined with SPPS. In: Witczak, Z., Bielski, R. (eds) Coupling and Decoupling of Diverse Molecular Units in Glycosciences. Springer, Cham. https://doi.org/10.1007/978-3-319-65587-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-65587-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65586-4
Online ISBN: 978-3-319-65587-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)