
Abstract
Progression through the cell cycle must be coordinated with crucial cell fate decisions, including the ability of a cell to exit the cell cycle and differentiate. Not surprisingly, deregulation of the G1/S transition is a well-established hallmark of cancer. While the basic mechanisms involved in this transition have been extensively characterized, it is now evident that components of the core cell cycle machinery, including cyclin D1, are functionally integrated into complex signaling and metabolic pathways not always directly related to cell cycle. In cells at risk of becoming cancerous, this complexity may underlie the cellular variability in the specific tumor suppressive processes that are implemented in response to oncogenic insults. Among these processes, autophagy has generated much debate because it may serve both as a tumor suppressive and as a pro-survival mechanism depending on the stage of tumor formation or the cell type under scrutiny. Nevertheless, a better understanding of the role of autophagy in tumorigenesis, and the functional connection of autophagy with the cell cycle and the metabolic status of the cell, may be necessary for the implementation of more rational regimens to treat cancer. In particular, recent reports have begun to unravel cyclin D1’s involvement in the regulation of the autophagy-senescence balance, as well as the role of cyclin D1 function in metabolic responses. The emerging picture is concordant with the idea that cyclin D1 participates in the integration and transduction of inputs provided by both growth factors and metabolic substrates. The proper integration of these signals, in turn, may be necessary to achieve an appropriate proliferative response. To what extent these functions are exclusively dependent on cyclin D1’s ability to bind and activate CDK4/CDK6, however, remains unclear.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
Alterations in the regulatory circuits that govern the G1-S cell cycle transition are universal features of cancer [39]. At the center of these circuits are cyclin-dependent kinases (CDKs) , a group of serine/threonine kinases that require the binding of short-lived cyclins in order to become catalytically active [19, 68, 69]. In mammalian cells, the G1 CDKs, CDK4 and CDK6, form active complexes with D-type cyclins (cyclin D1, D2, and D3) in early G1, whereas CDK2 becomes activated by E-type cyclins (cyclins E1 and E2 in mammalian cells) in late G1 [69]. Classic substrates for CDK4/CDK6- and CDK2-containing complexes are members of the so-called “pocket protein ” family of repressors, which include the retinoblastoma protein (pRB), p107 and p130. Of these substrates, pRB has become the prototype and, so far, the only one directly involved in human cancer [18]. According to the most accepted model, mitogenic signals dependent on growth factors result in an increase in the expression or the half-life of D-type cyclins, allowing the formation of active cyclin D-CDK4/CDK6 complexes. These complexes, together with cyclin E-CDK2 complexes formed in late G1, help to secure the complete phosphorylation-mediated inactivation of pRB, a step necessary for the derepression or release of E2F factors responsible for driving the G1-S transition [68, 100]. While this simple model still holds true, there is evidence that, depending on the cell type or the experimental conditions used, cyclin D-CDK4/CDK6 complexes may also phosphorylate a collection of substrates not directly involved in cell cycle regulation. These substrates include transcription factors with cell-type specific functions during differentiation (e.g., FoxM1, SMAD3, members of the RUNX family , GATA-4, and MEF-2), chromatin-modifying proteins (e.g., MEP50), and proteins involved in DNA repair processes (e.g., BRCA1) [2, 7, 14, 50, 73, 90].
Given its role in G1-S transition , it is hardly surprising that cyclin D1 can be found overexpressed in a wide spectrum of human cancers, including a large proportion of luminal-type breast tumors [8, 76, 81]. In fact, recent analyses have confirmed CCDN1, the gene encoding cyclin D1, as one of the most frequently amplified loci in human cancer genomes [12]. Most often, however, overexpression of cyclin D1 in cancer cells takes place in the absence of any detectable genomic alteration [76]. As one would anticipate, common mechanisms of overexpression that do not involve genomic alterations include the activation of growth factor-dependent signaling pathways upstream of cyclin D1 or the loss of micro-RNAs that normally target cyclin D1 for degradation [3, 11, 15, 29, 59]. For example, most ERBB2-expressing human breast cancers display moderate to strong cyclin D1 expression [3, 87], and, conversely, mice lacking cyclin D1 are resistant to breast cancer induced by ERBB2 [114]. Moreover, Choi et al. [23] showed that acute deletion of cyclin D1 blocks the progression of ERBB2-driven mammary tumors , an indication that the continued presence of cyclin D1 is necessary to sustain tumor growth in this model [23]. Taken together, these observations are in agreement with a model in which cyclin D1 serves as an integrator of growth-promoting signals, in such a way that its levels in early and mid-G1 dictate the probability of S-phase entrance.
It was long assumed that the oncogenic properties of cyclin D1 (and other D-type cyclins) were mostly dependent on its ability to activate CDK4 and CDK6. However, several observations and experimental findings have challenged this presumption. For example, increased levels of cyclin D1 only moderately correlate with pRB inactivation and proliferation in human tumors [1, 32, 57]. In fact, some of the oncogenic properties of D-type cyclins may depend on its ability to participate in processes other than the cell cycle in a CDK-independent manner [76, 82]. For example, cyclin D1 (like other D-type cyclins) can interact with a variety of proteins in a CDK-independent manner [25, 34], and the resulting cyclin D1-containing complexes seem to participate in cellular functions as diverse as transcriptional regulation, DNA repair, cell migration, and protein folding [14, 57, 61, 74, 79, 119]. Therefore, besides cell cycle control, deregulated expression of cyclin D1 may also affect other cellular processes in ways that could have important oncogenic consequences. It seems therefore likely that the relative contribution of the CDK-associated function of cyclin D1 to tumorigenesis may vary depending on the cell type or the specific constellation of accompanying genetic alterations (Fig. 5.1).

The functions of D-type cyclins . The G1/S cell cycle transition is regulated by the sequential activation of CDK4, CDK6, and CDK2. CDK4 and CDK6 (CDK4/6) are activated by D-type cyclins in early G1, whereas CDK2 is activated by E-type cyclins in late G1. As shown in (a) and (b), both complexes contribute to the phosphorylation-mediated inactivation of pRB, a step necessary for entering S-phase. The activities of CDK4 or CDK6 may be inhibited by members of the INK4 family of CDK inhibitors (p16INK4a, p15INK4b, p18INK4c, and p19INK4d, not shown here), which act by competing with D-type cyclins. Likewise, members of the CIP1/KIP1 family of inhibitors (p21WAF1, p27KIP1, and p57KIP2) form inhibitory complexes with CDK2 and cyclin E. Under some circumstances, p21WAF1 and p27KIP1 also contribute to the stabilization and, therefore, activation of cyclin D-CDK4/6 complexes. In this case, cyclin D-CDK4/6 complexes may titrate p21WAF1 and p27KIP1 away from cyclin E-CDK2 complexes, thus allowing CDK2 activation (b). As shown in (c), D-type cyclins in general, and cyclin D1 in particular, can interact with a variety of proteins in a CDK-independent manner and the resulting cyclin D-containing complexes seem to participate in cellular functions as diverse as transcriptional regulation (shown here), DNA repair, cell migration and protein folding
In spite of these complexities, the in vivo data do suggest that the ability of cyclin D1 to bind and activate CDK4/CDK6 is still relevant for tumor formation, at least in some experimental settings. For example, mammary epithelial cells derived from knockout mice that are deficient in CDK4 (the main G1 CDK that forms complexes with cyclin D1 in the mouse mammary epithelium) are resistant to ERBB2-dependent breast cancer in a manner that is similar to the tumor resistance reported for cyclin D1-deficient mice [115]. Overall, these reports suggested that the formation of cyclin D1-CDK4 complexes, and presumably their associated kinase activities, is required for ERBB2-dependent neoplastic transformation in the mammary epithelium . However, it is important to notice that cyclin D1-CDK4 complexes may also play a non-catalytic function by way of sequestering members of the WAP/CIP family of CDK inhibitors, particularly p21WAP1 and p27KIP1, away from cyclin E-CDK2 complexes [21, 91, 92]. This means that the resistance to ERBB2-induced cancer observed in cyclin D1- or CDK4-deficient mammary tissues could in part be a consequence of the inhibition of cyclin E-CDK2 complexes by p27KIP1 or p21WAP1 in the absence of titrating complexes (see Fig. 5.1b). Therefore, in order to demonstrate that ERBB2-driven tumors are specifically dependent on the ability of cyclin D1 to activate CDK4/CDK6 (i.e., its kinase-associated function), more refined mouse models were needed.
In order to dissect the kinase-dependent functions of cyclin D1 in vivo, Landis et al. [58] generated a knockin mouse carrying a single amino acid substitution at the CDK4 binding region of cyclin D1 [58]. While this mutant protein (cyclin D1-K112E) still binds to CDK4 or CDK6, thus retaining the titrating function of cyclin D1-CDK4/CDK6 complexes, these complexes become enzymatically inactive [10, 42]. Importantly, similar to cyclin D1- and CDK4-deficient mice [95, 115], these kinase dead mice (referred to as cyclin D1 KE/KE mice) are also resistant to breast cancer initiated by ERBB2 [58]. This finding demonstrates that the kinase-dependent function of cyclin D1 is necessary for tumor formation, at least breast tumor formation that is dependent on ERBB2. Interestingly, a further characterization of cyclin D1 KE/KE mice revealed that the ablation of cyclin D1 activity in these animals led to a dramatic reduction in the number, as well as the differentiation capabilities, of a subset of mammary progenitors previously identified as the targets for ERBB2-mediated tumorigenesis [45]. Moreover, at the cellular level, cyclin D1 KE/KE mammary epithelial cells display important alterations in the balance between cellular senescence and autophagy, two processes commonly disrupted in cancer cells [17]. Altogether, these studies have provided new links between the kinase activity dependent on cyclin D1 and cellular mechanisms involved in tumorigenesis, namely, self-renewal of progenitor cells, cellular senescence , and autophagy. Given the catabolic nature of autophagy , it has also become apparent that cyclin D1 activity might be functioning as a regulator of metabolism in some cell types. As explained later in this chapter, autophagy elicited in response to reduced cyclin D1-associated kinase activity could serve as a potential target for cancer treatment, although the long-term metabolic consequences of targeting autophagy are complex and highly context dependent.
5.2 The G1-S Cell Cycle Transition , Cyclin D1, and Autophagy
The ability of cancer cells to adjust their metabolism to the energy and biosynthetic demands imposed by cell proliferation is a well-known hallmark of cancer and an important contributor to anticancer drug resistance [16, 39, 107]. So far, extensive metabolic reprograming has been documented in connection with cellular processes commonly altered in cancer, including cellular senescence, autophagy, and stem cell self-renewal [30, 49, 104, 108]. Nevertheless, the mechanisms linking cell cycle deregulation (as observed in the context of cyclin D1 overexpression) and metabolic reprogramming , as well as the consequences of this metabolic reprogramming for the adaptation of cancer cells to their microenvironment, remain poorly characterized. For example, activation of the pRB pathway is one of the first steps in the implementation of cellular senescence [77], yet the functional links between the metabolic changes associated with cellular senescence and tumor suppression remain unclear. It is envisioned that a better understanding of these functional relationships will provide novel targets that can be used in the development of more efficacious anticancer drugs. In the following sections, we provide evidence that deregulation of the pRB pathway impinges on autophagy and metabolism. In particular, new evidence connecting cyclin D1 function, autophagy, and metabolism will be discussed.
5.2.1 Autophagy
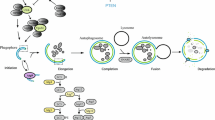
Autophagy is a highly dynamic, evolutionarily conserved, catabolic process that involves the sequestration, and subsequent lysosomal-mediated degradation, of organelles and long-lived proteins and protein complexes [35, 53, 108]. The morphological hallmark of autophagy is the formation of double-membrane vacuoles, also known as autophagosomes, which transport cytoplasmic cargo to the lysosomes for degradation and substrate recycling [102] (Fig. 5.2). The entire process of autophagy involves the orderly assembly of more than 30 autophagy-related (ATG) gene products, each functioning in the implementation of a distinct step [41]. Among these steps, the formation of autophagosomes is probably the best characterized at the molecular level (Fig. 5.2).

The dynamic process of autophagy. Autophagy begins with the sequestration of organelles and long-lived proteins or protein complexes into rudimentary membranous structures known as phagophores, which subsequently mature into LC3B-containing autophagosomes. A cytosolic form of LC3B (LC3-I) is conjugated to phosphatidyl-ethanolamine to form LC3-phosphatidyl-ethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. Autophagosomes fuse with lysosomes to form autolysosomes, and intra-autophagosomal components are degraded by lysosomal hydrolases. At the same time, LC3-II in the autolysosomal lumen is degraded. Thus, lysosomal turnover of the autophagosomal marker LC3-II reflects starvation-induced autophagic activity, and detecting LC3B by immunoblotting or immunofluorescence has become a reliable method for monitoring autophagy and autophagy-related processes. The phagophore-autophagosome transition is partially regulated by a Beclin-1 (ATG-6)/class III PI3K (Vps34) complex (Beclin-1-containing complex) whose activation requires the participation of an ULK kinase-containing complex. Autophagy is regulated by a complex signaling network, which encompasses stimulatory and inhibitory inputs. Autophagy is also negatively regulated by the mTORC1 (mechanistic target of rapamycin) kinase complex, a multi-protein complex that integrates both metabolic and growth-promoting signals conveyed, among others, by AMP (AMPK) and PI3K/AKT kinases, respectively. Activation of growth factor receptors (such as the insulin receptor) stimulates PI3K, leading to the activation of AKT, which inhibits the TSC1/TSC2 complex. Inhibition of this complex leads to the stabilization of the Rheb GTPase, which in turns activates mTORC1. Activation of mTORC1 causes inhibition of autophagy through several mechanisms, including mTORC1-dependent inactivation of proteins involved in autophagosome formation (i.e., ULK1, AMBRA1, and ATG14) and the repression of transcription factors required for lysosomal biogenesis (not shown). Energy depletion causes activation of the AMP-activated protein kinase (AMPK), and this event is necessary to induce autophagy in some cell types. AMPK phosphorylates and activates TSC1/TSC2 complex, resulting in mTORC1 inhibition. AMPK also mediates the phosphorylation-dependent activation of ULK1 and Beclin-1, two positive regulators of autophagy. As shown here, p53 can also regulate autophagy, a function that depends on its ability to transcriptionally control various pathways that converge on mTORC1 and lysosomal regulation (not shown)
Rates of autophagy are tightly coupled to fluctuations in the intracellular concentration of specific metabolic substrates or metabolic by-products, including ATP, glucose, amino acids, fatty acids, and ammonia (one of the main by-products of amino acid catabolism) [35]. These substrates modify the activity of enzymatic complexes that function as “metabolic sensors.” One of these sensors, the serine/threonine kinase mTOR (mechanistic target of rapamycin ), couples nutrients and growth factor availability to cell growth and proliferation [118]. mTOR is found in two complexes, mTORC1 and mTORC2, of which mTORC1 is the most widely studied metabolic sensor [94, 118]. Other proteins or protein complexes, including AMPK (AMP-dependent protein kinase), Rag-GTPases, and Sirtuins, cooperate with, or provide inputs to, the mTOR-dependent pathway [35]. As expected, mTORC1-dependent anabolic responses are accompanied by reduced rates of autophagy. Mechanisms responsible for this reduction include mTORC1-dependent inactivation of proteins involved in autophagosome formation (i.e., ULK1, AMBRA1, and ATG14) [78, 116], as well as transcription factors (i.e., TFEB) required for lysosomal biogenesis [94]. Conversely, mTORC1 inhibition due to a multitude of starvation signals leads to higher rates of autophagy. For example, lack of glucose (which leads to reduced rates of ATP synthesis) results in the accumulation of AMP (and to a lesser extent ADP) and the subsequent activation of AMPK [40]. AMPK in turn activates TSC2, a major suppressor of mTORC1, through phosphorylation. AMPK also mediates the phosphorylation-dependent activation of ULK1 and Beclin-1 , two positive regulators of autophagy [52]. Thus, reduced levels of nutrients (particularly amino acids and glucose), or the pharmacologic inhibition of mTORC1, upregulate autophagy through direct activation of factors involved in the initiation of autophagy and the induction of a lysosomal/autophagic transcriptional program [94].
Under normal nutrient conditions, basal or constitutive levels of autophagy provide a quality control mechanism that prevents the accumulation of protein aggregates or damaged organelles, thus ameliorating the endoplasmic reticulum (ER) stress response and maintaining cellular homeostasis [70,71,72, 108]. On the other hand, in cells subjected to starvation or other forms of stress (e.g., therapeutic stress), above-basal levels of autophagy provide basic biochemical substrates that can be utilized for energy production or to feed biosynthetic reactions, thus ensuring short-term survival [20]. Underscoring the importance of this metabolic function, autophagy-deficient mice die shortly after birth due to a failure to overcome the brief period of postnatal starvation [56]. In addition to these “pro-survival” functions , there is also evidence that under extreme conditions autophagy can serve as a mechanism of cell death [28, 31, 65]. For example, studies carried out in apoptosis-deficient mice have shown that cell lineages that would normally be eliminated through apoptosis still die while displaying an autophagic morphology, suggesting that autophagy-mediated cell death might compensate when apoptosis is compromised [93, 113].
Defects in autophagy are commonly observed in the course of aging and age-related pathologies, such as cancer and neurodegenerative diseases [22, 27]. In line with this association, global inactivation of autophagy in several animal models is accompanied by signs of premature aging, presumably as a result of the accumulation of damaged macromolecules and organelles [75].
With regard to the role of autophagy in cancer, this appears to be highly context dependent. Indeed, the available evidence suggests that autophagy may have opposite functions at different stages of tumor evolution [54, 108]. First, cells with reduced levels of autophagy, due to genetic or pharmacologic manipulations, may have a higher risk of becoming tumorigenic. This outcome is supported by several observations. For example, the hemiallelic loss of the essential autophagy gene Beclin-1/Atg6 has been documented in up to 75% of breast, ovary, and prostate cancers [4]. Similarly, beclin-1+/− mice, which are deficient in autophagy, display an increased frequency of spontaneous malignancies [84, 117]. Moreover, Takamura et al. [97] reported the development of liver adenomas in mice carrying mosaic or liver-specific deletions of the essential autophagy regulators ATG5 or ATG7 [97]. Mechanistically, these effects have been linked to an impairment in the capacity of autophagy-deficient cells to degrade damaged organelles or misfolded proteins, leading to oxidative stress, tissue damage, inflammation, and genomic instability [108]. In addition to these rather indirect mechanisms of tumorigenesis, the inhibition of autophagy in some in vitro models has been shown to impair the orchestration of oncogene-induced senescence, leading directly to the acquisition of a proliferative advantage that may accelerate tumor formation [110, 112]. These observations indicate that autophagy may be involved in the orchestration of at least some of the phenotypic features of senescent cells. However, as mentioned later in this chapter, there are examples in which inhibition of autophagy correlates with the induction or exacerbation, rather than inhibition, of senescence [80], suggesting tissue- or cell-type-based variation in the response of cells to autophagy deficiency.
In contrast to the role of autophagy in suppressing tumorigenesis, other lines of evidence support the idea that autophagy may actually promote tumorigenesis by sustaining metabolism, proliferation, or survival of fully transformed cells, especially if these cells are subjected to starvation or other forms of metabolic stress [37, 109]. However, the exact metabolic consequences of inducing autophagy in these cancer cells are not well defined. Unlike quiescent or terminally differentiated cells, which are dependent on oxidative phosphorylation-mediated ATP synthesis in order to maximize energy production in conditions of limited supply of growth factors [86, 103, 107], actively proliferating cancer cells, in which growth-promoting signals are abundant, show an increase in nutrient uptake and a switch to anabolic metabolism. This metabolic reprogramming is critical to supplying nucleotides, proteins, and lipids for cell division. Moreover, this transition from oxidative phosphorylation to glycolysis, even in the presence of adequate levels of oxygen (a phenomenon known as the Warburg effect), still requires functional mitochondria for the synthesis of metabolic precursors [103, 107]. Taken together, autophagy functions as an adaptive mechanism that sustains cell viability by providing metabolic substrates for biosynthesis. It follows from this idea that inhibition of autophagy would lead to a reduction in the survival and proliferation of cancer cells. Likewise, blocking autophagy would be expected to enhance the therapeutic outcome of drugs (particularly, cancer drugs) that induce autophagy in cancer cells as a pro-survival mechanism of adaptation [6, 44].
Recently, the pro-survival role of autophagy in cancer cells has been corroborated in a variety cancer models. First, it was reported that the overexpression of oncogenic RAS in cancer cell lines is accompanied by high rates of autophagy. In these cells, the constant oncogenic stress associated with RAS activation renders mitochondrial metabolism particularly dependent on autophagy [36, 64]. Accordingly, RAS-expressing cancer cells in which autophagy has been blocked show a reduction in their tumor-forming capacity, which is associated with low levels of tricarboxylic acid (TCA) cycle metabolites and impaired mitochondrial function [36, 111]. Moreover, deletion of the essential autophagy genes Atg5 or Atg7 in a RAS-dependent mouse model of pancreatic cancer retards progression to high-grade intra-epithelial neoplasias and ductal adenocarcinomas in a p53-dependent manner [88]. Underscoring the role of p53 in this phenotype, deletion of p53 accelerates tumor formation in these mice. Similarly, deletion of Atg7 in a K-RAS-driven mouse model of non-small-cell lung cancer (NSCLC) gives rise to more benign tumors characterized by the accumulation of defective mitochondria (oncocytomas), activation of p53, and proliferative arrest [37]. Of note, unlike the RAS-dependent model of pancreatic cancer, the deletion of p53 only partially rescued the tumor suppressive phenotype associated with Atg7 loss in the lungs. From a metabolic standpoint, Atg7- and p53-deficient lung tumors display reduced fatty acid oxidation (FAO) and increased sensitivity to FAO inhibition, indicating that RAS-driven lung tumors require autophagy for mitochondrial function and lipid catabolism [37]. Of note, the involvement of p53 in autophagy and metabolism likely depends, at least in part, on p53’s ability to modulate various pathways that converge on mTOR-containing complexes and lysosomal biogenesis [26, 33, 47, 51].
Overall, its ability to provide metabolic and biosynthetic substrates in situations of nutrient starvation, together with its ability to prevent the accumulation of damaged organelles, particularly mitochondria, renders autophagy necessary for tumorigenesis. As to the predominant cellular response to autophagy inhibition, this may vary depending on cell type and context. Such responses include apoptosis, necrosis (in cells that are deficient in apoptosis), and, most prominently, senescence. As already mentioned, given that autophagy has been considered an effector mechanism of senescence in some models [112], the fact that cellular senescence can be induced or exacerbated following autophagy inhibition is surprising and perhaps points to the existence of different types of senescence.
5.2.2 The G1/S Cell Cycle Transition and Autophagy
In order to grow and proliferate, cells must first sense and interpret a diverse collection of environmental signals. Depending on the availability and proper transduction of these signals, a decision has to be made as to whether a cell enters a reversible (quiescent) or irreversible (senescent, differentiated) form of cell cycle arrest, or simply continues to the next cycle of cell division. Intimately associated with these cell fate decisions, particularly in situations of metabolic stress, autophagy is emerging as a key process that might explain some of the adaptive consequences of cell cycle deregulation. As most of these cell fate decisions take place at the G1/S cell cycle transition, the use of mice in which regulators of this transition, including cyclin D1, were knocked out or functionally modified has been particularly informative.
An important starting point in the assessment of autophagy in mouse models of cancer was the reevaluation of some of the phenotypes displayed by RB-deficient mouse embryos [63]. RB-deficient embryos die at midgestation while exhibiting several developmental defects, including ectopic proliferation and increased apoptosis in the nervous system, lens, and liver [43]. The increased apoptosis observed in RB-deficient tissues was partially dependent on E2F-mediated activation of p53 [38]. Therefore, while RB−/−; E2F1−/− embryos still die in uterus, they do so at a considerable later stage of development than RB−/− embryos, which also correlate with a significant suppression of apoptosis, S-phase entry, and p53 activation [101]. Interestingly, at least some of the defects originally described in RB-deficient tissues have subsequently been attributed to hypoxia, a known inducer of autophagy, in relation to placental dysfunction [67]. In an attempt to tackle the role of pRB in hypoxic tissues, Tracy et al. reported that RB-deficient liver cells display signs of autophagic cell death in response to experimental hypoxia, and this effect was dependent on E2F-mediated derepression of BNIP3, a gene that codes for a hypoxia-inducible factor [99]. The authors of this work extended this observation to RB-deficient mouse embryonic fibroblasts (MEFs) and RB-deficient human cell lines under hypoxic conditions [99]. Similarly, isolated RB-deficient muscle progenitor cells (myoblasts) can still form myotubes and partially differentiate into muscle fibers in vitro but rapidly degenerate afterward, exhibiting signs of autophagy-mediated cell death [24]. Consistent with these findings, it was shown that E2F1 overexpression directly regulates the induction of autophagy genes and enhances the rates of basal autophagy in vitro [83]. Taken together, these observations are in agreement with a model in which derepression of E2F factors secondary to RB loss can, in some lineages and under specific developmental circumstances such as hypoxia, tilt the balance toward the induction of autophagy rather than apoptosis as a mechanism of cell death. It must be emphasized, however, that it is presently unclear whether autophagy in these cases actually represents a failed mechanism of survival or an apoptosis-independent mechanism of cell death.
In contrast to autophagy associated to RB loss, Jiang et al. have shown that reintroducing RB into RB-deficient cancer cell lines also induces autophagy. In this setting, pRB binding to E2F1 (which maintains transcriptional repression) is required for autophagy induction. Accordingly, overexpression of E2F1 overcomes this effect and tilts the balance toward the induction of apoptosis [46]. Mimicking the reintroduction of RB, autophagy induction was also observed following the overexpression of the cyclin-dependent kinase inhibitors (CKIs) p16INK4a or p27KIP1, suggesting that activation of the pRB pathway is sufficient to induce autophagy in these experimental settings [46]. This is in agreement with previous work linking overexpression of CKIs, metabolic stress, and autophagy induction [55, 62]. Thus, under conditions of metabolic stress, the phosphorylation-mediated stabilization of p27KIP1 by AMPK leads to autophagy upregulation. Conversely, downregulation of p27KIP1 under these conditions results in cell death by apoptosis, suggesting that autophagy represents a pro-survival adaptation to metabolic stress. Importantly, these effects were dependent on p27KIP1-mediated modulation of CDK activity [62]. It is worth mentioning that experimental manipulations involving the restoration of RB or the overexpression of CKIs in RB-deficient or RB-expressing cells, respectively, are well-established models of cellular senescence [5]. Therefore, at least in some models, autophagy and senescence may indeed be part of the same tumor suppressor pathway, a possibility that was first suggested by Young et al. [112]. In this scenario, autophagy may be crucial for the implementation of complex cellular traits in senescent cells, including the senescence-associated secretory phenotype (SASP ) [80]. If this is the case, autophagy-mediated catabolism might play a key part in the metabolic reprograming observed in senescent cells. Indeed, recent work has revealed a major shift to a predominantly mitochondrial, oxidative, metabolism in senescent cells [30, 49, 80]. For example, induction of senescence in human diploid fibroblasts (HDFs) following the expression of the oncogene BRAFV600E is associated with activation of pyruvate dehydrogenase (PDH), an enzyme that catalyzes the pyruvate-to-acetyl-CoA conversion that fuels the TCA cycle and oxidative phosphorylation [49]. Increased mitochondrial activity, oxygen consumption, ATP production, and lipid catabolism have also been documented in models of therapy-induced senescence [30]. Similarly, RAS-induced senescence in human fibroblasts is associated with reduced lipid synthesis, increased fatty acid oxidation, and increased oxygen consumption [85]. Taken together, these studies suggest a metabolic shift toward maximal energy production at the expense of biosynthesis in senescent cells. Whether or not autophagy contributes to this metabolic profile in all forms of senescence, however, remains a matter of debate. As discussed elsewhere, in some settings senescence can actually be induced or exacerbated upon autophagy inhibition.
5.2.3 Cyclin D1 and the Autophagy/Senescence Balance
In order to explore the cellular consequences of reducing cyclin D1 activity in the mammary epithelium, Brown et al. took advantage of the kinase dead cyclin D1 KE/KE mouse model. Contrary to the authors’ expectations, cyclin D1 KE/KE mammary tissues displayed high levels of proliferation along with a failure to induce markers of senescence in response to ERBB2 [17]. These findings indicate that aberrant proliferation can still take place in mutant tissues in response to ERBB2 despite the presence of a canonically “active” pRB pathway, perhaps reflecting the ability of these cells to activate compensatory survival processes in order to cope with reduced levels of cyclin D1-associated kinase activity [17]. Indeed, this aberrant proliferative response was also accompanied by an upregulation of markers of autophagy. That the upregulation of autophagy in cyclin D1 KE/KE mammary epithelium represented a survival adaptation to reduced cyclin D1 activity was suggested by experiments carried out in an immortalized cyclin D1 KE/KE cell line that retained high rates of autophagy in vitro. Thus, reducing the rates of autophagy through shRNA-mediated knockdown of ATG5 in these cells led to an impairment of proliferation due to the reactivation of senescence [17]. Therefore, contrary to previous reports suggesting that senescence and autophagy are part of the same pathway [112], these results indicate that senescence can be induced or exacerbated by autophagy inhibition, at least in cells with reduced cyclin D1 function. Of note, induction of senescence upon autophagy inhibition has also been reported in human fibroblasts [48, 106, 112]. From a metabolic standpoint, these fibroblasts display an increased number of mitochondria and lysosomes, produce higher levels of ROS, and display a reduction in the mitochondrial membrane potential and cellular ATP content [48, 106]. In spite of these findings, the specific metabolic profiles that accompany cellular senescence in the context of autophagy inhibition will likely vary depending on the cell type and experimental context.
The link between cyclin D1 activity and the autophagy-senescence balance may also suggest a more general connection between cyclin D1 and metabolism. Thus, contrary to the view that cyclin D1 exclusively acts downstream of growth factor-derived signals to promote proliferation, functional cyclin D1-CDK4/CDK6 complexes may act as a nexus to indicate both growth factor and nutrient proficiencies appropriate for a proliferative response. In the absence of active cyclin D1-CDK4/CDK6 complexes, induction of autophagy may represent an attempt to respond to growth-promoting signals in the perceived absence of metabolic substrates. This inability of dysfunctional cyclin D1-CDK4/CDK6 complexes to properly sense the environment would in turn trigger a metabolic reprograming characterized by an increase in the rates of autophagy. This general model has been supported by recent reports linking cyclin D1 and metabolism in hepatocytes and mammary epithelial cells (see below). This model also suggests a close functional cooperation between cyclin D1 function and bona fide metabolic sensors and effectors, particularly mTOR-containing complexes.
5.2.4 Cyclin D1 and Metabolism
The link between cyclin D1 function and metabolic reprograming has been recently confirmed in several models [60, 82]. In hepatocytes, cyclin D1-CDK4 complexes modulate metabolic responses, independent of cell division, through the phosphorylation-mediated activation of the histone acetyltransferase GCN5 (general control non-repressed protein 5) [60]. Among other substrates, GCN5 acetylates the transcriptional coactivator PGC-1α (peroxisome-proliferator-activated receptor gamma coactivator 1 alpha), suppressing its transcriptional activity. Conversely, Sirtuin-1 deacetylates and therefore activates PGC-1α [96]. As a transcriptional coactivator, PGC-1α promotes the expression of several genes involved in gluconeogenesis and mitochondrial respiration and, at the same time, induces ROS-detoxifying enzymes [96]. Therefore, inhibition of CDK4 or downregulation of cyclin D1 in hepatocytes increases the pool of deacetylated, active PGC-1α and leads to “fasting-like” state characterized by an increase in glucose production and utilization through transcriptional derepression of PGC-1α-dependent gluconeogenic genes [13, 60]. Conversely, insulin-mediated signaling following refeeding facilitates the formation of cyclin D1-CDK4 complexes, leading to suppression of hepatic gluconeogenesis [60] (see Fig. 5.3). Beyond the liver, morphological and functional changes indicative of metabolic reprograming have also been observed in cyclin D1−/− (null) embryonic fibroblasts (MEFs) and mammary epithelial cells. Overall, these cells display an increase in mitochondrial size and activity, with signs of reduced cytosolic glycolysis [89, 105]. Mechanistically, nuclear respiratory factor 1 (NRF-1) , a transcription factor that induces nuclear-encoded mitochondrial genes, might be inactivated by cyclin D1 in a CDK-dependent manner [105]. Thus, reduced expression of cyclin D1 or CDK4, as well as blocking the activity of cyclin D1-CDK4 complexes, has the effect of increasing mitochondrial respiration at the expense of cytosolic glycolysis (Fig. 5.3). Conversely, mammary tumor cells that overexpress cyclin D1 show an inhibition of mitochondrial activity with an enhancement of cytosolic glycolysis [89]. More recently, a direct involvement of cyclin D1 in mitochondrial function has also been suggested. Thus, cyclin D1 can physically interact, in a CDK-independent manner, with the voltage-dependent anion channel (VDAC) localized at the outer mitochondrial membrane, inhibiting the transport of ATP, ADP, and other metabolites and thus impairing mitochondrial function [98].

Cyclin D1 and metabolism . Cyclin D1-CDK4 complexes modulate metabolism at least in part through phosphorylation-mediated activation of the acetyltransferase GCN5. One of the targets of GCN5 is PGC-1α, a transcriptional coactivator of several gluconeogenic genes. PGC-1α also promotes mitochondrial respiration, inducing, at the same time, ROS-detoxifying enzymes. As shown here, upon acetylation, the transcriptional function of PGC-1α is inhibited. Therefore, inhibition of CDK4 or downregulation of cyclin D1 in hepatocytes leads to a derepression of gluconeogenic genes and an increase in glucose production and utilization. On the other hand, mammary epithelial cells (MECs) specifically deficient in cyclin D1-associated kinase activity display high levels of autophagy, implying that cyclin D1-CDK4 complexes inhibit autophagy under normal conditions. There are also reports showing that cyclin D1-deficient (cyclin D1 null) mouse embryonic fibroblasts (MEFs) and MECs display an increase in mitochondrial size and activity, with signs of reduced cytosolic glycolysis. Mechanistically, nuclear respiratory factor 1 (NRF-1), a transcription factor that induces nuclear-encoded mitochondrial genes, can be inactivated by cyclin D1 in a CDK-dependent manner. Thus, reduced expression of cyclin D1 or CDK4, as well as blocking the activity of cyclin D1-CDK4 complexes, has the effect of increasing mitochondrial respiration at the expense of cytosolic glycolysis. Conversely, mammary tumor cells that overexpress cyclin D1 show an inhibition of mitochondrial activity with an enhancement of cytosolic glycolysis. Furthermore, cyclin D1 can physically interact, in a CDK-independent manner, with the voltage-dependent anion channel (VDAC) localized at the outer mitochondrial membrane, leading to an impairing in mitochondrial function. At present, the relationship between, on the one hand, autophagy, glycolysis, and mitochondrial activity and, on the other hand, the functional status of cyclin D1-CDK complexes is poorly understood (depicted here as bidirectional curved arrows)
In summary, there is compelling evidence from different experimental systems that cyclin D1-CDK complexes are involved in the integration and transduction of metabolic signals (Fig. 5.3). However, how these processes are coordinated with autophagy remains unclear.
5.3 Concluding Remarks and Future Directions
In the preceding sections, we have tried to integrate recent lines of evidence connecting cyclin D/CDK function, metabolism, and autophagy. It has become evident that the mechanisms in which autophagy is activated, as well as the specific cellular effects that autophagy activation may have, can vary depending on cell type or the specific stimulus involved. In tumors, this variability likely reflects both the nature of the mutational events that a cancer cell has already experienced and the changes of the coevolving microenvironment. Although the ultimate mechanisms by which cell cycle deregulation may affect autophagy are far from being completely understood, we speculate that part of the answer will come from a careful reevaluation of already existing animal models. This analysis will give us invaluable information about the interplay between autophagy, differentiation, senescence, and apoptosis during development in the absence of cell cycle regulators.
As mentioned in this chapter, cyclin D1-CDK-pRB-E2F deregulation can induce or repress autophagy depending on the cell type and specific stress conditions. In particular, the contrasting outcomes observed between “primary” cells and cancer cells propagated in vitro may well be a reflection of the mutational histories of different cell lineages, a fact that needs to be considered when interpreting findings. Loss or gain of function mutations specifically designed to target members of the CDK-pRB-E2F pathway will be necessary to clarify the role of these proteins in autophagy regulation and tumorigenesis. It is plausible that many phenotypes that have been described in cell cycle mutant mice (including embryonic lethality) might need a reinterpretation in the context of regulation of autophagy and metabolism.
The last decade has witnessed important advances toward the development of specific CDK4/CDK6 inhibitors for cancer treatment [9]. As must be evident from the preceding sections, however, disrupting the cyclin D-CDK4/CDK6 complexes can trigger an extensive metabolic reprogramming, which may result, in some cell types, in an upregulation of autophagy. Taking into account these new findings, and given the dual function of autophagy during cancer initiation and progression, the incorporation of pharmacological modulators of autophagy as anticancer drugs must be cautious. Many of the current anticancer therapies, including drugs that inhibit CDK4/CDK6 kinases, have been shown to induce autophagy in tumor cells. However, there is an ongoing debate as to whether autophagy is required for the efficient killing of tumor cells following chemo- or radiotherapies or whether autophagy represents an adaptive response that enables tumor cells to survive the therapeutic insult [20]. Obviously, inhibition of autophagy will lead to opposite therapeutic outcomes depending on which one of these possibilities applies. Nonetheless, most studies seem to indicate that autophagy inhibition sensitizes tumor cells to a wide spectrum of therapies [20, 66]. Thus, a better understanding of the metabolic and growth suppressive pathways that may be enhanced by autophagy inhibition will be necessary to expand the therapeutic window of current therapeutic regimens and to confront the almost certain development of drug resistance.
References
Agarwal R, Gonzalez-Angulo AM, Myhre S, Carey M, Lee JS, Overgaard J, Alsner J, Stemke-Hale K, Lluch A, Neve RM, et al. Integrative analysis of cyclin protein levels identifies cyclin b1 as a classifier and predictor of outcomes in breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:3654–62.
Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18:329–40.
Ahnstrom M, Nordenskjold B, Rutqvist LE, Skoog L, Stal O. Role of cyclin D1 in ErbB2-positive breast cancer and tamoxifen resistance. Breast Cancer Res Treat. 2005;91:145–51.
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65.
Alexander K, Hinds PW. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol. 2001;21:3616–31.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36.
Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–34.
Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23:4215–24.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46.
Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle. 2005;4:330–8.
Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L, Kappeler A, Brunner T, Vassella E. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009;69:5553–9.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Bhalla K, Liu WJ, Thompson K, Anders L, Devarakonda S, Dewi R, Buckley S, Hwang BJ, Polster B, Dorsey SG, et al. Cyclin D1 represses gluconeogenesis via inhibition of the transcriptional coactivator PGC1alpha. Diabetes. 2014;63:3266–78.
Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463:374–8.
Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D'Urso L, Pagliuca A, Biffoni M, Labbaye C, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–7.
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–9.
Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C, Hinds PW. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012;72:6477–89.
Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82.
Casimiro MC, Crosariol M, Loro E, Li Z, Pestell RG. Cyclins and cell cycle control in cancer and disease. Genes Cancer. 2012;3:649–57.
Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett. 2010;584:1427–35.
Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci U S A. 1998;95:1091–6.
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–51.
Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol. 2010;191:291–301.
Coqueret O. Linking cyclins to transcriptional control. Gene. 2002;299:35–55.
Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34.
Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–13.
Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–88.
Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI, Dickson R, Furth P, Hunter K, Kucherlapati R, et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci U S A. 2002;99:6967–72.
Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–5.
Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35.
Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9:4153–63.
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9.
Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–47.
Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–76.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemmons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–61.
Hakem R, Mak TW. Animal models of tumor-suppressor genes. Annu Rev Genet. 2001;35:209–41.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006.
Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300.
Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–39.
Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, Hinds PW. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010;17:65–76.
Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E, Xu J, McDonnell TJ, Shinojima N, Fueyo J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010;70:7882–93.
Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48.
Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One. 2011;6:e23367.
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–12.
Kehn K, Berro R, Alhaj A, Bottazzi ME, Yeh WI, Klase Z, Van Duyne R, Fu S, Kashanchi F. Functional consequences of cyclin D1/BRCA1 interaction in breast cancer cells. Oncogene. 2007;26:5060–9.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–31.
Kim J, Guan KL. AMPK connects energy stress to PIK3C3/VPS34 regulation. Autophagy. 2013;9:1110–1.
Kim KH, Lee MS. Autophagy – a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322–37.
Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010.
Komata T, Kanzawa T, Takeuchi H, Germano IM, Schreiber M, Kondo Y, Kondo S. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br J Cancer. 2003;88:1277–80.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6.
Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell. 2003;114:323–34.
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22.
Lee RJ, Albanese C, Fu M, D'Amico M, Lin B, Watanabe G, Haines GK 3rd, Siegel PM, Hung MC, Yarden Y, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000;20:672–83.
Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–51.
Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol. 2006;26:4240–56.
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24.
Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18:7873–82.
Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78.
Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48.
Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88.
MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol. 2003;23:1044–53.
Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–31.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007a;7:961–7.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75.
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007b;21:1367–81.
Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–31.
McMahon C, Suthiphongchai T, DiRenzo J, Ewen ME. P/CAF associates with cyclin D1 and potentiates its activation of the estrogen receptor. Proc Natl Acad Sci U S A. 1999;96:5382–7.
Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30.
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72.
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16.
Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–16.
Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997;17:5338–47.
Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14:547–58.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52.
Pestell RG. New roles of cyclin D1. Am J Pathol. 2013;183:3–9.
Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20.
Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, Rovira II, Mohney RP, Karoly ED, Finkel T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle. 2012;11:1383–92.
Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–92.
Reis-Filho JS, Savage K, Lambros MB, James M, Steele D, Jones RL, Dowsett M. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: an immunohistochemical and chromogenic in situ hybridisation analysis. Modern Pathol Off J US Can Acad Pathol. 2006;19:999–1009.
Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300.
Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006;26:5449–69.
Shen R, Wang X, Drissi H, Liu F, O'Keefe RJ, Chen D. Cyclin D1-cdk4 induce runx2 ubiquitination and degradation. J Biol Chem. 2006;281:16347–53.
Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12.
Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–711.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8.
Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–62.
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–30.
Spiegelman BM. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 2007;287:60–63; discussion 63–69.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800.
Tchakarska G, Roussel M, Troussard X, Sola B. Cyclin D1 inhibits mitochondrial activity in B cells. Cancer Res. 2011;71:1690–9.
Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–42.
Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20.
Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;2:293–304.
Tsuchihara K, Fujii S, Esumi H. Autophagy and cancer: dynamism of the metabolism of tumor cells and tissues. Cancer Lett. 2009;278:130–8.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32.
Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 2006;103:11567–72.
Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A. 2012;109:13325–30.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308.
White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10.
White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27:2065–71.
White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784–7.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2.
Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21.
Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32.
Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9:1983–95.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35.
Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–15.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Valenzuela, C., Brown, N.E. (2018). Cyclin D1, Metabolism, and the Autophagy-Senescence Balance. In: Hinds, P., Brown, N. (eds) D-type Cyclins and Cancer. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-319-64451-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-64451-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64449-3
Online ISBN: 978-3-319-64451-6
eBook Packages: MedicineMedicine (R0)