
Abstract
Proteins are fundamental to most biological processes, which accomplish a vast amount of functions by interacting with other proteins. The research of PPI (protein-protein interaction) and its network has developed into a great importance part in bioinformatics. In the protein-protein interaction networks, most proteins interact with only a few partners, and small number of proteins interact with many partners, these proteins are called hub proteins. The hub proteins can be divided into party hub and date hub. Therefore, in this paper, we do some works about hub proteins. In addition, this paper uses the connectivity and betweenness to classify the hub protein in protein-protein interaction network. On the other hand, the paper studies hub proteins from another perspective (interfaces conformation), which reflects the organization of hot spot residues in hub protein interface.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Proteins interact with other proteins to complete life activities. The study of protein-protein interaction (PPI) network is helpful to understand the evolutionary process of life. Developing computational methods to predict and analyze protein-protein interaction networks is not only superior to traditional experiment, but also crucial for understanding biological functions. Identification is not complete about the physical interactions of proteins and the functions of many protein are not found.
Protein-protein interaction (PPI) have been researched from multiple perspectives [1]. Each protein is defined as a node in the network, and the interaction between proteins is defined as the linkage. Some proteins have distinctive characteristics and special regions for interacting with other protein.
A large number of research results show that protein-protein interaction (PPI) network is power-law connectivity distribution [2]. It indicates that some proteins are highly connected to other proteins which can be called hub protein, while most proteins interact with only a few proteins [1]. Each hub protein has original conformation for interacting in protein-protein interaction (PPI). One of the study perspective in the protein-protein network is how a hub protein can interact with other so many non-hub protein. In some environment, external changes can bring about the new transformation of the space conformation [3], such as PH, temperature, partner concentration and ionic strength. However, since the coverage of the natural protein-protein interaction (PPI) is low, it is still questioned whether the topology structure of the protein-protein interaction (PPI) network can be expressed correctly [4].
In protein-protein interaction (PPI) network, hub protein is a kind of protein with higher number of connections, which plays a key role in driving the evolution of genomes and genetic systems. However, the number of connections does not accurately reflect the role of proteins in protein-protein interaction (PPI) network, because hub proteins with same or similar connect degree are not always equal important role in biological network.
Although the distribution characteristics of protein-protein interaction (PPI) network have not been completely determined, it is obvious that highly connected proteins have certain properties to play an important role [3]. For example, the hub proteins might be crucial in drug design. An understanding of hub protein is necessary for the development of new drugs and drug discovery in modern era. Han [4] indicated that date hub proteins may be responsible for organizing biological modules in the protein-protein interaction (PPI) network.
One of the important characteristic of protein interaction interfaces is the contribution degree of the amino acid to the binding free energy. For many years, a large number of experiments show the binding free energy is not uniformly distributed in the binding of protein-protein interaction. Only a sub-fraction of amino acid residues (these residues are called hot spots) contributes a disproportionately generous to the interaction binding free energy [5]. In addition, some researchers showed that hot spots can be obtained from the alanine mutation energy database [6] to create the hot region models for analyzing the evolutionary mechanism. For the research of hot spot residues in protein-protein interfaces, a lot of computational methods have been proposed [7,8,9]. Hus [10] and Cukuroglu et al. [11] predicted hot regions in protein-protein interactions from the different perspective. Carles Pons [12] identified and analyzed the protein-protein binding regions conformation.
The PPI depends on distinct space conformation (3D structure) of proteins. Thus, the protein spatial 3D structure can help to analyze the characteristics and the rules of the evolution and functions of life. Our research group made contributions to the prediction of protein spatial 3D structure and hot regions in protein-protein interactions. Lin [13] has proposed the local adjust tabu search algorithm to predict protein spatial folding structure with the 2D off-lattice model and 3D off-lattice model. Zhang [14, 15] used the different heuristic algorithms to predict the protein spatial structure in 3D. Nan [16] and Hu [17] predicted the hot regions in protein-protein interaction by complex network and clustering method.
Tuncbag et al. [18] expressed that hot spot residues are the interface residues with △△G >= 2.0 kcal/mol in the protein-protein interaction interface, while nonhot spot residues are the interface residues with △△G < 0.4 kcal/mol. In addition, Ozlem Keskin [5], Reichman [19] and Ahmad et al. [20] defined hot regions by different method. This paper defines hot region which have at least three hot spot residues adjacent to each other in the protein-protein interaction interface. Cukuroglu [11] addressed hub proteins yet from interfaces structural, which investigated how hot spots and hot regions can be organized in hub proteins.
This paper consists of the follows sections. Section 2 describes the method of classification of date hub proteins and party hub proteins. Section 3 analyzes the hot region features. Section 4 gives the experiment results and discusses. Section 5 gives conclusion and future directions.
2 Classification of Date Hub and Party Hub
2.1 Classification Based on Average PCC
Han [4] proposed two types of hub proteins by selecting the average PCC: date hubs and party hubs. The former interacts with different proteins at different times or locations, and they are the global connection point between different groups of biological functions. The latter interacts with most of proteins at the same. Therefore, identification and analysis of date hub proteins are critical to the discovery of hidden biological information in protein-protein interaction (PPI) networks. The mRNA expressions are independent and are uncorrelated [21, 22] if hub proteins connected with other proteins by false-positive interactions [23], these proteins could be identified as date hubs. In order to reduce false-positive, Han [4] obtained a high-quality intersecting dataset. For each hub protein, it is necessary to calculated the average PCC of each mRNA expression between the hub proteins and other proteins.
Where \( E_{x} \), \( E_{y} \) are mRNA expression in different conditions or samples. N is the number of samples. n indicates the number of interaction objects in hub proteins. Party hubs are those with an average PCC higher than the threshold. All other hubs are defined as date hubs.
2.2 Classification Based on Betweenness
According to many researches, the betweenness is also an important topological property beside the connectivity in graph theory. Nodes with higher betweenness values may be the key nodes of the control module in the whole network. The degree of betweenness is the number of the shortest path in a network through a node. Proteins with high betweenness value are the key node to connect multiple important biological pathways in PPI networks. The improved algorithms were proposed to count the betweenness by Girvan and Newman [24, 25]. In this paper, we also use the same definition as Yu [26] that proteins with high betweenness are defined as bottlenecks. To facilitate the comparison and analysis, the proteins with the top 20% betweenness values are selected as bottlenecks, which is consistent with Yu [27]. Then, all proteins in a certain network can be divided into four categories: Hub-Bottleneck Node (HBN, High betweenness and high connectivity), Non-hub-bottleneck node (NHBN, High betweenness and low connectivity), Hub-non-bottleneck node (HNBN, Low betweenness and high connectivity) and Non-hub-non-bottleneck node (NHNBN, Low betweenness and low connectivity).
The connection degree and betweenness of all nodes in the network are calculated, and the betweenness is defined as
Where,\( \upsigma({\text{x}},{\text{y}}) \). represents the shortest path between x and y, \( \upsigma({\text{x}},{\text{y}}|{\text{v}}) \).represents the shortest path between x and y through the node v.
3 Hot Region Features and Classification
Cukuroglu [11] described hub proteins from an interfaces structural point and defined the different types of interfaces. In the study, the interfaces were defined as DD (interfaces between two date hubs), PP (between two party hubs), and NN (between two non-hub proteins) where D, P, N, and X are for date hub, party hub, non-hub, and any protein, respectively [11].
3.1 Feature Selection
Feature selection technique is a crucial step of classification, which has been widely used in protein-protein interactions [28]. It can contribute to avoid overfitting and enhance the accuracy of classification model, because feature selection can preserve the most primitive and optimal features of amino acid residues. The results of feature selection have a certain effect on the reliability of classification results. Many amino acid residues need to be removed because their biological characteristics are useless. The best feature subset can be obtained by feature selection procedure for improving the performance of the classifier. In this paper, properties of protein are estimated by mRMR (minimum Redundancy Maximum Relevance) algorithm [29].
The basic idea of mRMR algorithm is to meet the minimum redundancy and maximum correlation criterion. That is to say, the redundancy between the residues is analyzed based on the correlation measure, and the correlation function is combined into an objective function. When the objective function is optimal, the correlation between the residues is maximum, and the redundancy of the residues is minimum. Given two random variables X and Y, the mutual information is defined as
The maximum correlation criterion is defined as
The minimum redundancy criterion is defined as
Where S is the characteristic set, and C is the category of targets. \( I(f_{i, } {\text{C}}) \) is the mutual information between feature i and category C. \( I(f_{i, } f_{j } ) \) is the mutual information between feature i and feature j. Combining the above two formulas, the criterion about mRMR can be defined the following.
This criterion indicates that a feature subset should be selected with highly correlated and less redundant from the alternative features. Assume that the n feature has been selected as feature subset \( S_{n} \), then the j feature can be selected from alternative feature set \( {\text{S}} - S_{t} \) and satisfies the following formula.
3.2 Classification
Machine learning methods have been widely used in bioinformatics [30,31,32,33]. This paper used the support vector machines (SVM) for classification. Support vector machines, developed by Vapnik, are a set of related supervised learning methods which are used for classification and regression. For many years, SVM classifier are more and more widely used in the field of computational biology for predicting protein-protein interaction sites [34,35,36,37]. The protein-protein interaction interfaces can be classified as hub and non-hub interfaces according to the different conformation of hot spot residues.
The prediction process of hot regions in protein-protein interaction interfaces adopts 10-fold cross validation. The data set can be separate into ten portion. The training set data has nine portion, and test data is the remained one. The average of the correct rates of the 10 results is used as an estimate of the algorithm’s accuracy. The main parameters of the kernel function can be given by the cross validation.
4 Experimental Results and Evaluation
In the experiments, the proportion of various proteins was detected by adjusting the threshold value to classify the date proteins and the party hub protein. According to Fig. 1, under different thresholds, Hub-Bottleneck Node (HBN, High betweenness and high connectivity) class proteins have the highest gene encoding ratio. So, these proteins are most likely to become date hub protein. When the threshold equals 0.1, the proportion of essential gene with Hub-Bottleneck Node (HBN, High betweenness and high connectivity) is the highest. Figure 2 gives the proportion of essential gene with the threshold equals 0.1.

The proportion of four kinds of proteins with different thresholds

The proportion with the threshold equals 0.1
It is generally known that polar and hydrophobic interactions play important role in protein-protein interfaces. Therefore, there are two categories amino acids: polar amino acids and non-polar ones. The former includes R, N, D, E, Q, H, K, S, T and Y, while the latter includes A, C, G, I, L, M, F, P, W and V. For the classification, it is necessary to assess the features used for classification. So, Table 1 lists the description of the alternative features, and Table 2 lists their assessment between different types of PPI. These features can be selected to classify if their values are smaller than 0.05.
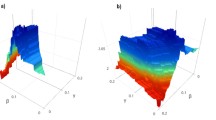
Figure 3 shows that date hub proteins are more inclined to cluster one hot region or more hot regions, which is consistent with Cukuroglu’s conclusion. Results also reveal that there are obvious distinguish between different types of interfaces.

The distribution of the average fraction of hot spots in the hot regions
5 Conclusion
Hub proteins have high connectivity in protein-protein interaction (PPI) network, which are one of the most significant factors in biological system. Nevertheless, the connectivity cannot entirely illuminate the hub protein’s role in protein-protein interaction (PPI) network. The reason is that hub proteins with similar connectivity maybe not equal contribution to the protein-protein interaction (PPI) network. Therefore, the betweenness is considered as the stronger determinant of analyzing and understanding the protein network system. On the other hand, the strong link can be found between hot spot residues or hot regions and hub proteins in the protein-protein interface. There are obviously differences between date hub protein and party hub protein interfaces. One of the future studies is to consider structural properties and energy contribution of different categories of hub proteins.
References
Jeong, H., Mason, S.P., Barabasi, A.L., Oltvai, Z.N.: Lethality and centrality in protein networks. Nature 411, 41–42 (2001)
Han, J.D., Dupuy, D., Bertin, N., Cusick, M.E., Vidal, M.: Effect of sampling on topology predictions of protein-protein interaction networks. Nat. Biotechnol. 23, 839–844 (2005)
Apic, G., Ignjatovic, T., Boyer, S., Russell, R.B.: Illuminating drug discovery with biological pathways. FEBS Lett. 579, 1872–1877 (2005)
Han, J.D., Bertin, N., Hao, T., Goldberg, D.S., Berriz, F., Zhang, L.V., Dupuy, D., Walhout, A.J., Cusick, M.E., Roth, F.P., Vidal, M.: Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 430(6995), 88–93 (2004)
Keskin, O., Ma, B.Y., Mol, R.J.: Hot regions in protein-protein interactions: the organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 345(5), 1281–1294 (2005)
Thorn, K.S., Bogan, A.A.: ASEdb: a data base of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 17(3), 284–285 (2001)
Lise, S., Buchan, D., Pontil, M., Jones, D.T.: Predictions of hot spot residues at protein-protein interfaces using support vector machines. PloS One 6(2), e16774 (2011)
Lise, S., Archambeau, C., Pontil, M., Jones, D.T.: Prediction of hot spot residues at protein-protein interfaces by combining machine learning and energy-based methods. BMC Bioinform. 10(1), 365 (2009)
Tuncbag, N., Keskin, O., Gursoy, A.: HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res. 38, 402–406 (2010)
Hsu, C.M., Chen, C.Y., Liu, B.J., Huang, C.C.: Identification of hot regions in protein-protein interactions by sequential pattern mining. BMC Bioinfor. 8(Suppl 5), S8 (2007)
Cukuroglu, E., Gursoy, A., Keskin, O.: Analysis of hot region organization in hub proteins. Ann. Biomed. Eng. 38(6), 2068–2078 (2010)
Carles, P., Fabian, G., Juan, F.: Prediction of protein-binding areas by small world residue networks and application to docking. BMC Bioinform. 12, 378–388 (2011)
Lin, X.L., Zhang, X.L., Zhou, F.L.: Protein structure prediction with local adjust tabu search algorithm. BMC Bioinform. 15(S15), S1 (2014)
Zhang, X.L., Wang, T., Luo, H.P., Yang, J.Y., Deng, Y.P., Tang, J.S., Yang, M.Q.: 3D protein structure prediction with genetic tabu search algorithm. BMC Syst. Biol. 4(Suppl 1), S6 (2010). doi:10.1186/1752-0509-4-S1-S6
Zhang, X.L., Lin, X.L.: Effective 3D protein structure prediction with local adjustment genetic-annealing. Interdisc. Sci. Comput. Life Sci. 2(3), 256–262 (2010)
Nan, D.F., Zhang, X.L.: Prediction of hot regions in protein-protein interactions based on complex network and community detection. In: IEEE International Conference on Bioinformatics and Biomedicine, pp. 17–23 (2013)
Hu, J., Zhang, X.L., Liu, X.M., Tang, J.S.: Prediction of hot regions in protein-protein interaction by combining density-based incremental clustering with feature-based classification. Comput. Biol. Med. 61, 127–137 (2015)
Tuncbag, N., Gursoy, A., Keskin, O.: Identification of computational hot spots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics 25(12), 1513–1520 (2009)
Reichmann, D., Rahat, O., Albeck, S., Meged, R., Dym, O., Schreiber, G.: The modular architecture of protein-protein binding interfaces. Proc. Natl. Acad. Sci. 102(1), 57–62 (2005)
Ahmad, S., Keskin, O., Sarai, A., Nussinov, R.: Protein-DNA interactions: structural, thermodynamic and clustering patterns of conserved residues in dna-binding proteins. Nucleic Acids Res. 36(18), 5922–5932 (2008)
Kemmeren, P., et al.: Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol. Cell 9, 1133–1143 (2002)
Ge, H., Liu, Z., Church, G.M., Vidal, M.: Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nature Genet. 29, 482–486 (2001)
Von Mering, C., et al.: Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 299–403 (2002)
Girvan, M., Newman, M.E.: Community structure in social and biological networks. Proc. Natl. Acad. Sci. U.S.A. 99, 7821–7826 (2002)
Newman, M.E.: Scientific collaboration networks. II. Shortest paths, weighted networks, and centrality. Phys. Rev. E 64, 016132 (2001)
Yu, H.Y., Kim, P.M., Sprecher, E., et al.: The importance of bottlenecks in protein net-works: correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 3(4), 59 (2007)
Yu, H.Y., Greenbaum, D., Xin Lu, H., Zhu, X., Gerstein, M.: Genomic analysis of essentiality within protein networks. Trends Genet. 20, 227–231 (2004)
Yugandhar, K., Gromiha, M.M.: Feature selection and classification of protein-protein complexes based on their binding affinities using machine learning approaches. Proteins: Struct., Funct., Bioinf. 82(9), 2088–2096 (2014)
Li, B.Q., Feng, K.Y., Li, C., Huang, T.: Prediction of protein-protein interaction sites by random forest algorithm with mRMR and IFS. PloS One 7(8), e43927 (2012)
Abnousi, A., Broschat, S.L., Kalyanaraman, A.: An alignment-free approach to cluster proteins using frequency of conserved K-Mers. In: BCB 2015 Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics, pp. 597–606 (2015)
Broin, P.Ó., Smith, T.J., Golden, A.A.: Alignment-free clustering of transcription factor binding motifs using a genetic-k-medoids approach. BMC Bioinform. 16(1), 1–12 (2015)
Zheng, C.H., Huang, D.S., Zhang, L., Kong, X.Z.: Tumor clustering using non-negative matrix factorization with gene selection. IEEE Trans. Inf. Technol. Biomed. 13(4), 599–607 (2009)
Zhu, L., Guo, W.L., Deng, S.P., Huang, D.S.: ChIP-PIT: enhancing the analysis of ChIP-Seq data using convex-relaxed pair-wise interaction tensor decomposition. IEEE/ACM Trans. Comput. Biol. Bioinform. 13(1), 55–63 (2016)
Cho, K.I., Kim, D., Lee, D.: A feature-based approach to modeling protein-protein interaction hot spots. Nucleic Acids Res. 37(8), 2672–2678 (2009)
Wong, G.Y., Leung, F.H.F., Ling, S.H.: Predicting protein-ligand binding site using support vector machine with protein properties. IEEE/ACM Trans. Comput. Biol. Bioinform. 10(6), 1517–1529 (2013)
Sriwastava, B.K., Basu, S., Maulik, U.: Predicting protein-protein interaction sites with a novel membership based fuzzy SVM classifier. IEEE/ACM Trans. Comput. Biol. Bioinform. 12(6), 1394–1404 (2015)
Wei, Z.S., Han, K., Yang, J.Y., Shen, H.B., Yu, D.J.: Protein-protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 193, 201–212 (2016)
Acknowledgment
The authors thank the members of Machine Learning and Artificial Intelligence Laboratory, School of Computer Science and Technology, Wuhan University of Science and Technology, for their helpful discussion within seminars. This work was supported in part by National Natural Science Foundation of China (No. 61502356, 61273225, 61273303).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Lin, X., Zhang, X., Hu, J. (2017). Classification of Hub Protein and Analysis of Hot Regions in Protein-Protein Interactions. In: Huang, DS., Jo, KH., Figueroa-García, J. (eds) Intelligent Computing Theories and Application. ICIC 2017. Lecture Notes in Computer Science(), vol 10362. Springer, Cham. https://doi.org/10.1007/978-3-319-63312-1_32
Download citation
DOI: https://doi.org/10.1007/978-3-319-63312-1_32
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-63311-4
Online ISBN: 978-3-319-63312-1
eBook Packages: Computer ScienceComputer Science (R0)