
Abstract
Chronic myelomonocytic leukemia is an “overlap” disease with clinical and pathologic features of both a myelodysplastic and a myeloproliferative neoplasm. By definition, there must be a persistent peripheral blood monocytosis; however, the other features may be quite variable from case to case. Over 90% of cases have identifiable genetic mutations and in the absence of morphologic dysplasia, the presence of one of these mutations can be used to satisfy diagnostic criteria. The most commonly implicated mutations involve the TET2, ASXL1, and SRSF2 genes. However, none of the mutations found in CMML are pathognomonic. These mutations can be seen in other myeloid neoplasms as well as in some phenotypically normal older persons. Some patterns are beginning to emerge with regards to the accumulation of mutations during the course of CMML progression as well as in different subtypes of CMML, namely the “dysplastic” type (WBC < 13 × 109) and the “proliferative” type (WBC ≥ 13 × 109). Mutations in spliceosome machinery and/or epigenetic regulation tend to occur in the “dysplastic” subtype or early in the evolution of the disease whereas mutations involving RUNX1, ASXL1, or signal pathways proteins tend to occur in the proliferative phase or later in the disease chronology. Prognostic and therapeutic implications of the molecular basis of CMML are still evolving and the clinical picture largely drives the treatment decisions and prognostic models. The notable exception is frameshift and nonsense mutations of ASXL1, which have shown independent prognostic significance on multivariate analysis in multiple studies.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Chronic myelomonocytic leukemia (CMML)
- ASXL1
- TET2
- SRSF2
- Molecular
- Prognosis
- Therapy
- Epigenetic
- Dysplastic CMML
- Proliferative CMML
- Diagnosis
- Morphology
- Cytogenetics
Introduction
Chronic myelomonocytic leukemia (CMML) is a myeloid neoplasm characterized by a persistent absolute monocytosis, often with a background of dysplastic morphologic features. The diagnosis encompasses a heterogeneous group of cases with considerable variability in morphologic dysplasia, cytopenias, leukocytosis, and the presence or absence of organomegaly. As such, the diagnostic category has features of a myelodysplastic syndrome as well as a myeloproliferative neoplasm . It was originally categorized as a form of myelodysplastic syndrome (MDS) in the early French-American-British (FAB) classification [1]. But in later editions of the World Health Organization (WHO) classification of tumours of haematopoietic and lymphoid tissues , it was placed within the newly created category of “Myelodysplastic/Myeloproliferative Neoplasms” [2].
Clinical
The diagnosis of CMML requires a persistent peripheral blood (PB) monocytosis (≥1 × 109 cells /L and ≥10% of the white blood cell differential count as defined in the 2016 WHO classification of hematopoietic neoplasms) with <20% blasts in the peripheral blood and bone marrow (BM) and the absence of a BCR-ABL1 rearrangement [3]. There should be morphologic dysplasia ; however, if dysplasia is minimal or absent, there should be an acquired clonal cytogenetic or molecular genetic abnormality or monocytosis lasting ≥3 months with exclusion of other causes of monocytosis [2]. Additionally, in cases with eosinophilia , rearrangements involving PDGFRA, PDGFRB, and FGFR1, as well as the PCM1-JAK2 fusion, should be excluded [3]. If eosinophils are >1.5 × 109 cells/L and there is no rearrangement with the aforementioned genes, the diagnosis is CMML with eosinophilia (see Table 12.1).
There is some evidence to support dividing CMML into “dysplastic” (<13 × 109 WBC/L) and “proliferative ” (≥13 × 109 WBC/L) categories based on distinctive molecular and clinical features of each subset [4,5,6,7,8,9,10]. In addition, CMML is categorized by the percentage of blastsFootnote 1 present in the peripheral blood and bone marrow as CMML-0: <2% blasts in PB and <5% blasts in BM; CMML-1: 2–4% blasts in PB and/or 5–9% blasts in the BM; and CMML-2: 5–19% blasts in PB, 10–19% blasts in the BM, and/or presence of any Auer rods [3].
In two large epidemiological studies in Europe and the United States, the incidence of CMML was 3–4.1/1,000,000 person-years [11, 12]. The median age at diagnosis was 76 years with a male predominance [12]. The overall survival at 5 years was 18% [11]. Patients have a spectrum of MDS to MPN-like presentations. The majority have elevated WBC counts , although some have normal or decreased counts. Symptoms include fatigue, weight loss, fever, night sweats, infections, and bleeding. Splenomegaly or hepatomegaly may occur, especially with the myeloproliferative subgroup. Typically, patients present with <5% circulating blasts and <10% BM blasts, corresponding to CMML-0/1 [2].
Morphology and Immunophenotype
In the blood (see Fig. 12.1), a monocytosis of ≥1 × 109cells/L and ≥10% of total WBCs is a requirement, in distinction from chronic myeloid leukemia, BCR-ABL1 positive, which may have an absolute monocytosis, but it is typically <10% of all WBCs [2, 3]. The monocytes of CMML can have abnormal morphology with atypical granulation and nuclear lobation, or immature chromatin that is somewhat denser than that of promonocytes or monoblasts; overall however, the monocytes are usually mature and morphologically unremarkable [2]. Monoblasts are large with abundant gray-to-blue cytoplasm, possible pseudopod formation, and round nuclei with delicate chromatin and prominent nucleoli. Promonocytes also have abundant gray or blue cytoplasm and nuclei with finely reticulated chromatin, but the nuclei have delicate folds with or without a small nucleolus (see Fig. 12.2) [2]. Cytopenias are often present and there may be neutrophilia [2]. There is usually dysgranulopoiesis, which may manifest as nuclear hypolobation, abnormal nuclear lobation, or hypogranular granulocytes [2].

(a) Bone marrow aspirate of CMML-2 (Wright stain, 500× original magnification). (b) Peripheral blood of CMML (Wright stain, 500× original magnification). (c) Bone marrow core biopsy (hematoxylin and eosin, 200× original magnification) with (d) numerous dysplastic megakaryocytes (hematoxylin and eosin, 400× original magnification) (Courtesy of Dr. H. Joyce Rogers, Pathology and Laboratory Medicine Institute, Cleveland Clinic, Cleveland, OH)

Peripheral blood of CMML highlighting a monoblast (arrow) and promonocytes (arrowhead) (Wright stain, 1000× original magnification)
Of note, cases of MPN can be associated with monocytosis or can develop monocytosis during the course of the disease. In these rare situations, a previously documented history of MPN excludes CMML. Additionally, the presence of MPN features in the bone marrow and/or of MPN associated mutations (JAK2, CALR or MPL) tend to support MPN with monocytosis rather than CMML.
The bone marrow (see Fig. 12.1) is usually hypercellular, but it can be normocellular or hypocellular [2]. A granulocytic proliferation is often the most prominent finding and may obscure the monocytic proliferation [2]. Most cases have dysgranulopoiesis and dysmegakaryopoiesis, and many have dyserythropoiesis [2]. Reticulin fibrosis occurs in up to 30% of cases and 20% of cases have nodules of clonally related, neoplastic plasmacytoid dendritic cells [2]. In cases with splenomegaly, the red pulp is typically infiltrated by leukemic cells [2].
Phenotypically, the leukemic cells typically express the myeloid associated antigens CD33 and CD13 [2]. Monocytic antigens such as CD14, CD68, and CD64 are variably expressed [2]. Oftentimes, there is an aberrant immunophenotype on monocytes with ≥2 immunophenotypic aberrancies such as expression of CD56 or CD2, decreased expression of CD14 (possibly indicative of immaturity), and/or decreased expression of HLA-DR, CD13, CD15, CD64, and CD36 [2]. Monoblasts and promonocytes are typically negative for CD34. The monocytes are positive for lysozyme, nonspecific esterase, and are negative for naphthol-ASD-chloroacetate esterase [2].
Cytogenetics and Molecular
Approximately 30% of CMML cases have cytogenetic abnormalities [13, 14]. Among the most common findings are +8, −Y, del(20q), +21, der(3q), and chromosome 7 abnormalities including −7 and del(7q) [14, 15]. Karyotypes having multiple abnormalities are also common [13,14,15]. Isolated abnormalities of chromosome 5 (−5/del(5q)) are relatively rare [13,14,15]. Single nucleotide polymorphism (SNP) arrays have shown increased numbers of chromosomal alterations than appreciated by karyotype analysis alone including frequent copy neutral loss of heterozygosity (LOH) [16, 17].
Over 90% of CMML cases have identifiable gene mutations [18, 19] (see Table 12.2). Molecular genetic or cytogenetic abnormalities can be used as evidence of clonality in cases without significant morphologic dysplasia, especially when involving gene mutations commonly associated with CMML. Many of these genetic mutations can be broadly divided into 3 pathways : epigenetic regulation/histone modification, spliceosome machinery, and cell signaling/transcription factors.
It is important to bear in mind that many of the mutations found in CMML (such as TET2, ASXL1, SRSF2, CBL) can also be found in isolation in hematologically normal appearing patients or patients with cytopenia(s) who do not otherwise meet criteria for a myelodysplastic syndrome (MDS) or CMML [20, 21]. Thus, the presence of one of these mutations should be carefully considered in the context of the duration of the monocytosis, exclusion of other reactive causes of monocytosis, and of the presence of other clinical data such as cytopenias or splenomegaly that might support CMML.
In CMML patients, the most commonly identified mutations in genes encoding proteins involved in epigenetic regulation/histone modification involve TET2, DNMT3A, IDH2, ASXL1, EZH2, and UTX (see Table 12.2) [19, 22, 23]. Of these, TET2 and ASXL1 are the most frequent and most important. TET2 catalyzes the hydroxylation of methylated DNA and as a result, somatic mutations in TET2 are believed to lead to epigenetic dysregulation [24]. TET2 mutations are found in 46–58% of CMML cases [19, 22] and TET2 deletions have been detected in 7% CMML cases, with a higher incidence of cryptic deletions noted than in acute myeloid leukemia (AML) or MDS [25]. Mutations of IDH1, IDH2 and TET2 tend to be mutually exclusive since they are functionally redundant [22, 26]. Mutated IDH1/IDH2 results in abnormal production of the metabolite 2-hydroxyglutarate(2-HG) [24], which inhibits multiple enzymes including TET2, leading to hypermethylation [26]. ASXL1 is thought to affect histone modification through effects on the polycomb group repressive complex proteins (PRC1/2) [27]. The most common mutations associated with ASXL1 are c.1934dupG;p.Gly646TrpfsX12 and 1900_1922_ del [28]. ASXL1 mutations are associated with a higher WBC count, lower hemoglobin, extramedullary disease and an abnormal karyotype [15, 22]. EZH2 is a component of the PRC2 complex [29] and UTX is a lysine specific demethylase with effects on histone H3K27 [23].
The most commonly identified spliceosome mutations involve the following genes: SF3B1, SRSF2, U2AF1, and ZRSR2 [19, 22]. Mutations within this category affect the machinery involved in pre-mRNA splicing [30, 31] and tend to be exclusive of each other in cases of CMML [22, 30, 32]. Of these genes, mutations of SRSF2 are by and far the most common. Mutations of SRSF2 tend to result in alterations at the 95th amino acid residue, normally occupied by proline and are associated with a normal karyotype [15, 30]. Mutations of SF3B1 have an association with der(3q) and, as with cases of MDS, tend to be associated with ring sideroblasts [15, 30]. Recurrent SF3B1 mutations include K700E, H662Q, and K666N [30, 32]. U2AF1 encodes a small nuclear RNA auxiliary factor. Mutations in this gene (most commonly S34F, Q157, [30] are associated with a normal karyotype, but can also be seen with a monosomal karyotype (defined as having loss of two chromosomes or loss of one chromosome plus another structural alteration) [15].
There are several recurring mutations in genes involved in signaling/tyrosine kinase pathways including JAK2, RAS (KRAS + NRAS), CBL, PTPN11, and BRAF [22, 33,34,35]. The RAS gene family is composed of multiple isoforms, including KRAS and NRAS, which have GTPase activity and are involved in cell signaling pathways [36]. BRAF is a kinase intimately involved with RAS signaling and is an important activator of the MEK/ERK pathway [37]. JAK2 is a tyrosine kinase involved with cell signaling and proliferation via the STAT pathway [38]. JAK2 mutated CMML tends to share some morphologic features with JAK2+ myeloproliferative neoplasms such as mild/moderate reticulin fibrosis, erythroid and megakaryocytic hyperplasia, occasional megakaryocytic clustering and atypia, and dilated sinusoids [39]. Overall, these morphologic features appear to be less developed than in a pure MPN. Additional factors that would lend support to a diagnosis of JAK2+ CMML would include a lack of a history of MPN or lack of cell counts consistent with an MPN, and finding morphologic features of dysplasia. CBL regulates receptor tyrosine kinase activity by ubiquitination [34]. It is associated with TET2 mutations and monosomy 7 and tends to associate with wild type JAK2 and KRAS/NRAS [34].
Other reported common mutations found in CMML are of RUNX1, SETBP1, STAG2, and BCOR [19, 22, 33, 40,41,42,43]. The cohesin complex is a multimer composed of four subunits, including STAG2, thought to be involved in cohesion of sister chromatids during cell division, postreplicative DNA repair, and regulation of gene expression [40]. Among myeloid neoplasms, STAG2 mutations are often found with other mutations such as TET2, ASXL1, and EZH2 [40]. RUNX1 encodes the alpha subunit of the core-binding factor and is essential for hematopoiesis/differentiation and helps regulate expression of GCSF and MPO [44]. SETBP1 is a binding partner for SET protein, a protein which has downstream effects on transcription and nucleosome assembly [45, 46]. SETBP1 mutations have an association with mutations of ASXL1 or spliceosome machinery and often occur with a normal karyotype [47].
From a molecular perspective, the myelodysplastic type of CMML(MD-CMML) is associated with mutations of spliceosome proteins such as SRSF2, SF3B1, ZRSR2 and U2AF35 and epigenetic regulators of DNA methylation such as TET2 and IDH1/2 [10]. The myeloproliferative type of CMML (MP-CMML) is associated with mutations of ASXL1 and signal pathway mutations such as CBL, FLT3, JAK2 and KRAS/NRAS, in addition to the mutations involving spliceosome machinery and regulators of DNA methylation [8, 10, 39]. Even in cases originally diagnosed as MD-CMML, the identification of signal pathway mutations (RAS) at the time of diagnosis or during the disease course has been associated with progression to MP-CMML [8]. Investigation of the mutational hierarchy of CMML [18] indicates that mutations affecting epigenetic regulators (such as TET2 and ASXL1) and spliceosome mutations are often associated with early neoplastic clones whereas signal pathway mutations tend to be later mutational hits.
Prognosis and Therapy
Although there have been several large studies that have examined various clinical and pathologic markers for prognostic utility regarding CMML, there is no universally accepted prognostic model for CMML. Factors that have been found to have some prognostic significance at one time or another in multivariate analysis include increased age, high WBC count, increased bone marrow blasts, cytogenetic risk stratification, circulating immature myeloid cells, thrombocytopenia, anemia, and the presence of frameshift or nonsense ASXL1 mutations [6, 22, 28, 47,48,49,50].
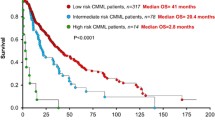
Three prognostic models with external validation include the CPSS (CMML-specific prognostic scoring system) , the GFM (Group Francophone des Myélodysplasies) , and the Mayo Model (see Table 12.3) [22, 28, 49]. The CPSS model identified 4 factors for stratifying overall survival (OS) and (acute) leukemia free survival (LFS) risk: French-American-British (FAB) classification, WHO classification, the CMML-specific cytogenetic risk stratification, and blood transfusion dependency. The CMML-specific cytogenetic risk stratification considered a normal karyotype and –Y as low risk; complex cytogenetics (≥3 chromosomal abnormalities), chromosome 7 abnormalities, and trisomy 8 as poor risk; and other chromosomal abnormalities as intermediate risk [14]. All positive risk factors were assigned a value of 1, except for high risk cytogenetics, which is assigned a value of 2. A total score was obtained from the sum of the individual scores and is placed into 1 of 4 categories: Low risk, Intermediate-1, Intermediate-2, and high risk. The GFM model used five risk factors for OS and LFS: age > 65 years, WBC >15 × 109/L, anemia, platelets <100 × 109/L, and presence of a nonsense or frameshift ASXL1 mutation. Positive risk factors were assigned a value of 1, 2, or 3 and summed for a total score that was placed in one of three categories: low, intermediate, and high risk . The Mayo Model identified four significant prognostic variables for OS and LFS: absolute monocyte count (>10 × 109/L), circulating immature mononuclear cells (defined as any of myeloblasts, promyelocytes, myelocytes, metamyelocytes), anemia (<10 g/dL), and thrombocytopenia (<100 × 109/L). Three prognostic categories were created from this: low risk (0 risk factors), intermediate risk (1 risk factor), and high risk (≥2 risk factors).
Although mutations of various genes (RUNX1, TET2, NRAS, CBL, SETBP1, SRSF2) have been associated with differences in overall survival (OS) or leukemia-free survival (LFS) , the data are inconsistent and further validations are necessary before drawing conclusions [19, 22, 30, 33, 34, 41, 43, 47, 51, 52].
Cytogenetic risk stratifications have also yielded variable results with regard to prognosis, but some common themes have emerged. These common themes include complex karyotypes are associated with a worse prognosis and a normal karyotype or –Y is associated with a better OS [14, 15]. When applied to multiple prognostic models in multivariate analysis, the Mayo-French cytogenetic model retained independent prognostic significance [15]. This model effectively predicted leukemic transformation and stratifies cases into one of three risk groups—high risk: complex karyotype or monosomal (defined as having at least one autosomal monosomy and one more structural abnormality or having at least two autosomal monosomies) karyotype; low risk: normal karyotype, −Y, order(3q); or intermediate risk: all others. Another study [53] examining the prognostic impact of cytogenetic abnormalities acquired during the course of CMML disease showed they were associated with an overall decrease in LFS by multivariate analysis. Acquisition of a complex karyotype was associated with leukemia progression, but del(20q) was associated with stable disease [53].
Therapy for CMML has largely been drawn from treatments for myelodysplastic syndrome and myeloproliferative neoplasms. Supportive care including erythropoietic stimulating agents and transfusions are typically utilized for significant anemia [54]. Hypomethylating agents , including 5-azacitidine and decitabine, have been approved by the United States Food and Drug Administration for treatment of CMML. In several studies, these two drugs collectively have shown overall response rates ranging from 25–69% and median OS from 12–37 months [55,56,57,58,59,60,61]. Proliferative phase CMML is typically treated with hydroxyurea . In a randomized control trial comparing hydroxyurea with etoposide, hydroxyurea was associated with better treatment response (60% versus 36%) and OS (20 months versus 9 months) than etoposide [62]. Even so, treatment outcomes with hypomethylating agents and hydroxyurea are still relatively poor and there is a strong need for more effective therapies. Allogeneic hematopoietic stem cell transplant (HSCT) is the only known cure; however, due to the advanced age and/or comorbidities often associated with CMML patients, this option is often not available [63].
Although TET2 mutations have been associated with response to hypomethylating agents in MDS patients, there are no consistent molecular mutation predictors of response to hypomethylating agents in CMML patients [58, 64,65,66], and analysis of predictive models of methylation patterns have so far yielded mixed results [66]. Numerous investigational therapies such as the JAK2 inhibitor ruxolitinib and RAS pathway inhibitors (farnesyltransferase inhibitors) have been evaluated in patients with CMML with variable but limited responses [67,68,69].
Conclusion
CMML is a myeloid neoplasm with overlapping features of a myelodysplastic and myeloproliferative neoplasm. The diagnosis requires a combination of a persistent absolute monocytosis (>1 × 109 cells/L) and either a background of morphologic dysplasia, clonal cytogenetic/molecular genetic abnormalities or persistence of monocytosis for ≥3 months with exclusion of other causes of monocytosis. There are no disease-defining cytogenetic or molecular genetic abnormalities. However, the presence of a cytogenetic or molecular abnormality may help to make a diagnosis of CMML in the appropriate clinical context. The independent prognostic and therapeutic value of molecular mutations is currently limited. Yet, as our knowledge of the mutational landscape is expanded and refined, and newer therapies become available, our understanding of the molecular basis of CMML may yield additional insight into the treatment potential of CMML.
Notes
- 1.
The blast count in either the PB or BM that results in the highest CMML category should be used.
References
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissue. Lyon: IARC Press; 2008. p. 439.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Schuler E, Schroeder M, Neukirchen J, Strupp C, Xicoy B, Kündgen A, et al. Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemias. Leuk Res. 2014;38:1413–9.
Nösslinger T, Reisner R, Grüner H, Tüchler H, Nowotny H, Pittermann E, et al. Dysplastic versus proliferative CMML--a retrospective analysis of 91 patients from a single institution. Leuk Res. 2001;25:741–7.
Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia : a retrospective analysis of 213 patients. Blood. 2010;99:840–9.
Voglová J, Chrobák L, Neuwirtová R, Malasková V, Straka L. Myelodysplastic and myeloproliferative type of chronic myelomonocytic leukemia — distinct subgroups or two stages of the same disease? Leuk Res. 2001;25:493–9.
Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clinical Cancer Res Off J Am Assoc Cancer Res. 2010;16:2246–56.
Gelsi-Boyer V, Cervera N, Bertucci F, Brecqueville M, Finetti P, Murati A, et al. Molecular similarity between myelodysplastic form of chronic myelomonocytic leukemia and refractory anemia with ring sideroblasts. Haematologica. 2013;98:576–83.
Cervera N, Itzykson R, Coppin E, Prebet T, Murati A, Legall S, et al. Gene mutations differently impact the prognosis of the myelodysplastic and myeloproliferative classes of chronic myelomonocytic leukemia. Am J Hematol. 2014;89:604–9.
Visser O, Trama A, Maynadié M, Stiller C, Marcos-Gragera R, De Angelis R, et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J cancer Oxford England 1990. 2012;48:3257–66.
Srour SA, Devesa SS, Morton LM, Check DP, Curtis RE, Linet MS, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol. 2016;174:382.
Tang G, Zhang L, Fu B, Hu J, Lu X, Hu S, et al. Cytogenetic risk stratification of 417 patients with chronic myelomonocytic leukemia from a single institution. Am J Hematol. 2014;89:813–8.
Such E, Cervera J, Costa D, Solé F, Vallespí T, Luño E, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96:375–83.
Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French consortium study. Am J Hematol. 2014;89:1111–5.
Dunbar AJ, Gondek LP, O'Keefe CL, Makishima H, Rataul MS, Szpurka H, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–57.
Gondek LP, Tiu R, O ‘keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008;111:1534–42.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–98.
Patnaik MM, Lasho TL, Vijayvargiya P, Finke CM, Hanson CA, Ketterling RP, et al. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e385.
Mason CC, Khorashad JS, Tantravahi SK, Kelley TW, Zabriskie MS, Yan D, et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia. 2016;30:906–13.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:1546–58.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:2428–36.
Jankowska AAM, Makishima H, Tiu RRV, Szpurka H, Huang Y, Traina F, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–41.
Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121:3563–72.
Bacher U, Weissmann S, Kohlmann A, Schindela S, Alpermann T, Schnittger S, et al. TET2 deletions are a recurrent but rare phenomenon in myeloid malignancies and are frequently accompanied by TET2 mutations on the remaining allele. Br J Haematol. 2012;156:67–75.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–93.
Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27:1504–10.
Chung YR, Schatoff E, Abdel-Wahab O. Epigenetic alterations in hematopoietic malignancies. Int J Hematol. 2012;96:413–27.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201–6.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9.
Kar SA, Jankowska A, Makishima H, Visconte V, Jerez A, Sugimoto Y, et al. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica. 2013;98:107–13.
Kohlmann A, Grossmann V, Klein H-U, Schindela S, Weiss T, Kazak B, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–65.
Schnittger S, Bacher U, Alpermann T, Reiter A, Ulke M, Dicker F, Eder C, Kohlmann A, Grossmann V, Kowarsch A, Kern W, Haferlach C, Haferlach T. Use of CBL exon 8 and 9 mutations in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative disorders: an analysis of 636 cases. Haematologica. 2012;97:1890.
Zhang L, Singh RR, Patel KP, Stingo F, Routbort M, You MJ, et al. BRAF kinase domain mutations are present in a subset of chronic myelomonocytic leukemia with wild-type RAS. Am J Hematol. 2014;89:499–504.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–9.
Roring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31(11):2629–47.
Aaronson DS, Horvath CMA. Road map for those who don't know JAK-STAT. Science New York NY. 2002;296(5573):1653–5.
Pich A, Riera L, Sismondi F, Godio L, Bonino LD, Marmont F, et al. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol. 2009;62:798–801.
Kon A, Shih L-Y, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45:1232–7.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27:2100–2.
Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122:3169–77.
Kuo M-C, Liang D-C, Huang C-F, Shih Y-S, J-H W, Lin T-L, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–31.
Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–13.
Kutney SN, Hong R, Macfarlan T, Chakravarti DA. Signaling role of histone-binding proteins and INHAT subunits pp32 and set/TAF-Ibeta in integrating chromatin hypoacetylation and transcriptional repression. J Biol Chem. 2004;279:30850–5.
Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119–30.
Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28:2206–12.
Kantarjian H, O'Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original international prognostic scoring system. Cancer. 2008;113:1351–61.
Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Padron E, Garcia-Manero G, Patnaik MM, Itzykson R, Lasho T, Nazha A, et al. An international data set for CMML validates prognostic scoring systems and demonstrates a need for novel prognostication strategies. Blood cancer journal. 2015;5:e333.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120:3080–8.
Tang G, Fu B, Hu S, Lu X, Tang Z, Li S, et al. Prognostic impact of acquisition of cytogenetic abnormalities during the course of chronic myelomonocytic leukemia. Am J Hematol. 2015;90:882–7.
Patnaik MM, Tefferi A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management. Am J Hematol. 2016;91:631–42.
Ades L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–13.
Fianchi L, Criscuolo M, Breccia M, Maurillo L, Salvi F, Musto P, et al. High rate of remissions in chronic myelomonocytic leukemia treated with 5-azacytidine: results of an Italian retrospective study. Leuk Lymphoma. 2013;54(3):658–61.
Costa R, Abdulhaq H, Haq B, Shadduck RK, Latsko J, Zenati M, et al. Activity of azacitidine in chronic myelomonocytic leukemia. Cancer. 2011;117(12):2690–6.
Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–31.
Aribi A, Borthakur G, Ravandi F, Shan J, Davisson J, Cortes J, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer. 2007;109(4):713–7.
Thorpe M, Montalvao A, Pierdomenico F, Moita F, Almeida A. Treatment of chronic myelomonocytic leukemia with 5-Azacitidine: a case series and literature review. Leuk Res. 2012;36(8):1071–3.
Wijermans PW, Rüter B, Baer MR, Slack JL, Saba HI, Lübbert M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008;32:587–91.
Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahé B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Français des Myélodysplasies and European CMML group. Blood. 1996;88:2480–7.
Cheng H, Kirtani VG, Gergis U. Current status of allogeneic HST for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2012;47:535–41.
Patnaik MM, Wassie EA, Padron E, Onida F, Itzykson R, Lasho TL, et al. Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J. 2015;5:e280.
Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–12.
Meldi K, Qin T, Buchi F, Droin N, Sotzen J, Micol J-B, et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J Clin Invest. 2015;125:1857–72.
Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, et al. A multi-institution phase 1 trial of ruxolitinib in patients with chronic myelomonocytic leukemia (CMML). Clin Cancer Res. 2016;22:3746–54.
Feldman EJ, Cortes J, DeAngelo DJ, Holyoake T, Simonsson B, O'Brien SG, et al. On the use of lonafarnib in myelodysplastic syndrome and chronic myelomonocytic leukemia. Leukemia. 2008;22:1707–11.
Fenaux P, Raza A, Mufti GJ, Aul C, Germing U, Kantarjian H, et al. A multicenter phase 2 study of the farnesyltransferase inhibitor tipifarnib in intermediate- to high-risk myelodysplastic syndrome. Blood. 2007;109:4158–63.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Gentry, M., Hsi, E.D. (2018). Chronic Myelomonocytic Leukemia: Clinical and Pathologic Features. In: Chang, CC., Ohgami, R. (eds) Precision Molecular Pathology of Myeloid Neoplasms. Molecular Pathology Library, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-62146-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-62146-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62144-9
Online ISBN: 978-3-319-62146-3
eBook Packages: MedicineMedicine (R0)