
Abstract
The extracellular matrix (ECM) in tumors is highly dynamic and contributes to tumor evolution. Fibronectin (FN) is a key component of the ECM in tumors that ligates and stimulates integrins on tumor cells, fibroblasts and endothelial cells in the tumor microenvironment. FN induced integrin activity is reduced by fibulin-5 (Fbln5), a matricellular protein that competes with FN for integrin binding but does not stimulate integrin signaling. A consequence of FN-induced integrin activation is the generation of reactive oxygen species (ROS), which can promote cell survival or apoptosis pending the microenvironment. The tumor microenvironment Fbln5 can be viewed as a molecular rheostat that tunes FN stimulated integrin-induced ROS generation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1.1 The Function and Composition of the Extracellular Matrix
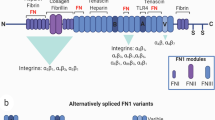
The extracellular matrix (ECM) is a dynamic collection of secreted molecules, which occupies the space between cells and provides the structural framework necessary to maintain tissue integrity. That structural framework includes proteins such as collagens, fibronectin, laminin, and elastin. Collagens are the most abundant protein in the ECM in the human body. There are multiple types of collagen found within the ECM, and they exist as fibrillar proteins, providing stiffness to the tissue (Vogel 2001). Fibronectins (FN) are glycoproteins that bind to other ECM proteins and help connect cells to the ECM and promote cell movement (Pankov 2002). Laminins are glycoproteins found in the basal laminae of all cell types, where they form weblike networks that provide tensile strength to the tissue (Beck et al. 1990). Finally, elastins are proteins that provide flexibility to the tissue, allowing the tissue to retain shape after stretching or contraction (Mithieux and Weiss 2005). Many diverse cell types can secrete ECM molecules; however, tissue fibroblasts are typically the source of the majority of ECM synthesis and secretion.
The most common and well-studied functions of the ECM are cell adhesion and cell-to-cell communication. ECM proteins are anchored to the cell through cell surface receptors, the most intensively studied of which are integrins. Integrins are heterodimeric transmembrane receptors consisting of α and β subunits. In mammals, there are at least 18 α subunits and 8 β subunits, generating 24 unique integrins (Harburger and Calderwood 2009), which have ligand specificity and, as a result, distinct functions. For example, α5β1 (a major FN receptor) is important during angiogenesis (Kim et al. 2000), whereas α1β1 and α2β1 (major collagen receptors) are critical for leukocyte adhesion and inflammation (de Fougerolles et al. 2000; Krieglstein et al. 2002).
In addition to providing a structural framework for tissues, the ECM also regulates a myriad of important signaling pathways. As integrins are ligated, they form clusters at the membrane, referred to as focal adhesions. Focal adhesion complexes are essential to induce downstream signaling, as the cytoplasmic tails of integrins do not harbor any detectable enzymatic activity (Harburger and Calderwood 2009). Integrin clustering results in the reorganization of the actin cytoskeleton and the activation of downstream signaling complexes. Integrins are unique in that they relay signals from inside the cell to the outside (inside-out signaling) and vice versa (outside-in signaling) (Giancotti 1999; Topalovski and Brekken 2015). Integrin contact with the ECM results in outside-in signaling, whereas protein complexes from inside the cell bind to integrin tails, which is thought to prime integrins for ECM interaction (inside-out signaling).
Integrin clustering and focal adhesion assembly activate protein tyrosine kinases such as focal adhesion kinase (FAK) (Lipfert et al. 1992). FAK was one of the first phosphorylation targets discovered downstream of integrin activation (Masur et al. 1995). An immediate downstream target of FAK activity is the major protein kinase Src, which promotes survival, proliferation, and migration (Schlaepfer et al. 1994). FAK is a central signaling scaffold that activates downstream signaling pathways, specifically the Rho-family GTPases, which are critical for actin cytoskeletal dynamics and cell movement. Furthermore, the activation of integrins also promotes growth factor receptor activation. For example, optimum cell stimulation with epidermal growth factor (EGF) requires integrin-mediated cell adhesion (Eliceiri 2001). Therefore, integrins regulate a range of important biological processes from cell migration to cell growth and proliferation.
1.2 The Role of the ECM in Tumor Development with Emphasis on FN
The ECM is also critical in the formation and maintenance of solid tumors. Most solid tumors display increased deposition of ECM proteins and ECM remodeling compared to their normal tissue counterparts. The ECM affects tumorigenic processes and functions, including tumor cell proliferation, survival, apoptosis, migration, adhesion, angiogenesis, and chemoresistance (Topalovski and Brekken 2015; Aguilera et al. 2014; Akiyama et al. 1995; Bachem et al. 2005; Han et al. 2006; Itano et al. 2008; Lu et al. 2012; Miyamoto et al. 2004). Desmoplasia, the robust deposition of ECM, is induced in tumors by growth factors, such as transforming growth factor β (TGF-β), basic fibroblast growth factor, connective tissue growth factor, interleukin-1β (IL-1β), and platelet-derived growth factor (Hocevar et al. 1999; Leask and Abraham 2004; Bonner 2004; Zhao et al. 2012), and environmental conditions including hypoxia (Aguilera et al. 2014). The ECM is a major facet of the tumor microenvironment (TME), a collective term referring to the immediate environment surrounding tumor cells. In addition to ECM proteins, the TME contains multiple types of non-cancer cells such as immune cells, fibroblasts, and endothelial cells that facilitate tumor progression (Weis and Cheresh 2011; Mao et al. 2013; Gajewski et al. 2013).
In normal tissue, cells remain anchored to their surrounding ECM; detachment from this supportive matrix results in a form of cell death termed anoikis (Frisch and Screaton 2001). However, tumor cells have evolved to circumvent this anchorage dependence leading to the presence of circulating tumor cells that can metastasize to distant organs (Guadamillas et al. 2011). When metastatic tumor cells reach their new site, they must create a microenvironment conducive for survival and growth. Recent work in a mouse model of pancreatic ductal adenocarcinoma (PDA) shows that FN is critical in supporting the engraftment of metastatic cancer cells in target organs (e.g., the liver and lung) (Costa-Silva et al. 2015).
The expression of FN is elevated in many solid tumors, especially PDA (Ramakrishnan et al. 2006; Ramaswamy et al. 2003; Stenman and Vaheri 1981). In this context, FN contributes to cancer cell survival, invasion, metastasis, chemoresistance, and angiogenesis (Topalovski and Brekken 2015). For instance, human PDA cell lines Panc-1 and Capan-1 showed increased resistance to cytotoxic agents including gemcitabine, cisplatin, and doxorubicin when grown on FN-coated plates (Miyamoto et al. 2004). The pro-survival effect of FN has been attributed to the activation of the PI3K/AKT/mTOR pathway (Han et al. 2006; Chen and Guan 1994). Moreover, it was reported that FN stimulates reactive oxygen species (ROS) in PDA cells, which led to increased survival that could be reversed by antioxidant treatment (Edderkaoui et al. 2005). ROS, in moderate amounts, serve as signaling molecules that stimulate proliferation through the Ras-Raf-MEK-ERK pathway and promote survival through the NF-κB pathway (Reuter et al. 2010).
FN is also an important contributor to angiogenesis, the formation of new blood vessels from preexisting vessels. In addition to growth factors such as vascular endothelial growth factor (VEGF), ligation of the ECM via integrin receptors is essential for angiogenesis. Genetic deletion of integrins β1, αv, α4, or α5 in mice individually completely hindered angiogenesis (Schaffner et al. 2013). Integrin α5 knockout mice die at embryonic day E10.5 due to severe vascular defects (Yang et al. 1993), and the deletion of FN results in vascular abnormalities and death at day E9.5 (George et al. 1993). Tumors require angiogenesis to persist and thrive. Although still under active investigation, FN has been shown to be important in tumor angiogenesis as well. Early studies revealed that targeting the FN-α5β1 interaction via antibody antagonists blocked angiogenesis in the chick chorioallantoic membrane assay (Kim et al. 2000). Moreover, α5β1-targeting antibodies, such as volociximab, inhibit tumor growth in various animal models of cancer, but these promising preclinical results have yet to be recapitulated in patients (Ramakrishnan et al. 2006; Besse et al. 2012; Bhaskar et al. 2008; Bhaskar et al. 2007; Cranmer et al. 2005; Kuwada 2007; Ricart 2008).
At the molecular level, FN may stimulate angiogenesis by various mechanisms. Integrin adhesion to the ECM, and to FN specifically, triggers endothelial cell migration and microvessel elongation (Nicosia et al. 1993). Furthermore, the alternatively spliced extra domain A of FN can stimulate VEGF-C expression in a PI3K/Akt-dependent manner in colorectal carcinoma (Xiang et al. 2012). Along these lines, FN can stimulate ROS production (Edderkaoui et al. 2005; Wang et al. 2015; Chiarugi et al. 2003), mainly in the form of hydrogen peroxide, which is known to induce VEGF expression when present in moderation (Zhu et al. 2002).
1.3 Matricellular Proteins: Extracellular Modulators of the ECM
Integrin activation by abundant ECM proteins such as FN and collagen is controlled by regulatory proteins found within the ECM referred to as matricellular proteins. Matricellular proteins as a class do not function as structural components of the ECM; instead, they mediate ECM-receptor interactions (Bornstein and Sage 2002; Wong and Rustgi 2013). Some major examples of matricellular proteins are thrombospondin-1 and thrombospondin-2 (TSP-1 and TSP-2), tenascin-C (TN-C), secreted protein acidic and rich in cysteine (SPARC), osteopontin (OPN), and the fibulin family of proteins. Matricellular proteins can regulate ECM function by directly binding to cell surface receptors or structural and soluble proteins within the ECM. For example, SPARC binds directly to collagen and blocks collagen-mediated signaling via the discoidin domain receptors (Aguilera et al. 2014; Arnold et al. 2010). In this context, the absence of SPARC has enhanced collagen-mediated tumor progression. Moreover, TSP-1 blocks FN-induced focal adhesions in endothelial cells (Murphy-Ullrich et al. 1993). Similarly, TN-C interferes with FN-integrin interaction and reduces the FN-mediated adhesion of fibroblasts (Chiquet-Ehrismann et al. 1988; Huang et al. 2001).
Matricellular proteins are typically expressed abundantly during development and reactivated during wound healing and other tissue remodeling events (Bornstein and Sage 2002). Phenotypes of mice lacking a particular matricellular protein are usually mild, reinforcing the fact that these proteins do not contribute to the structural integrity of tissues (Chapman et al. 2009; Nakamura et al. 2002). However, deficiencies in response to wound healing and tissue repair are often seen (Kyriakides and Bornstein 2003). The abnormal expression of matricellular proteins is seen in certain pathologies, such as cancer. The matricellular protein fibulin-5 (Fbln5) is aberrantly expressed in many cancers (Wang et al. 2015; Hwang et al. 2013; Lee et al. 2008; Shi et al. 2014; Tang 2015). Fbln5 has important contributions to normal physiology and development as well as cancer.
1.4 Fibulin-5
Fbln5 is a 66 kDa matricellular glycoprotein. It is a member of the fibulin family of ECM proteins, which are characterized by six calcium-binding EGF-like repeats (cbEGF) at the N-terminus (for protein stability and protein interaction) and a globular C-terminal fibulin module (Yanagisawa et al. 2009). Similar to other ECM proteins, Fbln5 is a TGF-β- and hypoxia-inducible gene (Kuang et al. 2006; Guadall et al. 2011). Fbln5 is distinct among the fibulins in that it contains an RGD-integrin binding domain (Fig. 1.1).

Domain structure of Fbln5. cbEGF (calcium-binding EGF-like domains), RGD (Arg-Gly-Asp; tripeptide binding site of integrins)
Fbln5 was discovered as a gene highly expressed in large vessels during development; however, it is downregulated in most adult tissues except where active tissue remodeling is occurring (Nakamura et al. 1999, 2002; Kowal et al. 1999). Fbln5 is induced by models of vascular injury including balloon withdrawal injury and in atherosclerotic plaques, highlighting its contribution to vascular function and maintenance (Kowal et al. 1999; Nakamura et al. 1999). The generating of Fbln5-deficient mice revealed a major function of Fbln5 in elastic fiber formation (Nakamura et al. 2002; Yanagisawa et al. 2002). Fbln5 -/- mice exhibited general connective tissue defects such as loose skin, tortuous vessels, emphysematous lung, and genital prolapse. This provided the first animal model for congenital elastic fiber disorders (Loeys et al. 2002). These seminal studies laid the foundation for future discoveries regarding Fbln5.
1.4.1 Fbln5 in Cell Adhesion/Migration
As mentioned above, Fbln5 contains an RGD-integrin binding domain and has been reported to ligate RGD-binding integrins including α4β1, α5β1, αvβ3, αvβ5, and α9β1 (Nakamura et al. 2002; Lomas et al. 2007). Fbln5 supports the adhesion of endothelial cells and smooth muscle cells (SMCs) in an RGD-dependent manner (Kowal et al. 1999; Nakamura et al. 1999; Lomas et al. 2007). Furthermore, Fbln5 interacts with multiple extracellular proteins, including extracellular superoxide dismutase (ecSOD) where it tethers ecSOD to endothelial cells where it reduces ROS-induced damage (Nguyen et al. 2004). Fbln5 also binds to tropoelastin and lysyl oxidase-like 1 (LOXL1), which is thought to be critical for normal elastogenesis to occur (Liu et al. 2004).
Fbln5 regulates cell migration in a context-dependent manner. For example, Fbln5 failed to induce migration in SMCs (Lomas et al. 2007). SMCs plated onto Fbln5-coated dishes appeared rounded and less spread compared to cells plated on FN. Immunofluorescence staining showed that focal adhesion and actin stress fiber formation were significantly reduced in the presence of Fbln5 compared to FN. Finally, cells plated on either Fbln5 or FN were analyzed for migratory capacity by imposing a wound in the adherent cultures. Closure of the wound was monitored between the two conditions, which revealed reduced migration in cells plated on Fbln5 (Lomas et al. 2007). Treatment with an activating β1 integrin antibody reversed the negative effects of Fbln5 on migration and stress fiber formation. These results suggest passive ligation of integrins by Fbln5 in SMCs.
Fbln5 also antagonizes endothelial cell migration (Albig and Schiemann 2004). In this context, ectopic expression of Fbln5 in endothelial cells can dampen cell migration as tested by the ability of these cells to move through a Matrigel matrix and also by measuring trans-well migration (in a Boyden chamber assay). Moreover, treating endothelial cells with recombinant Fbln5 blocked angiogenic sprouting in vitro (Albig and Schiemann 2004). In contrast, Fbln5 induced the migration of fibrosarcoma cells (Schiemann 2002). Using a slightly modified version of the Boyden chamber assay where FN was coated on the bottom of the porous membrane, Fbln5-expressing fibrosarcoma cells displayed enhanced migration toward FN, suggesting that Fbln5 promotes a de-adhesive state that facilitates cell migration in certain contexts. These findings highlight cell-type specific effects of Fbln5 signaling that likely reflect the variable integrin expression profile between cell types and whether these specific integrins are subject to inhibition by Fbln5.
1.4.2 Fbln5 in Proliferation/Survival
Similar to the context-specific effects on cell motility, Fbln5 can positively and negatively regulate cell proliferation depending on cell type. For instance, the overexpression of Fbln5 in 3T3 fibroblasts revealed the increased activation of ERK1/2 and p38 mitogen-activated protein kinase (MAPK) (Schiemann 2002). This group also showed that Fbln5 synergized with TGF-β to stimulate proliferation and DNA synthesis in 3T3 cells. Conversely, Fbln5 was shown to induce an antiproliferative response in epithelial cells through the decreased expression of cyclin A, thus abrogating the progression of the cell cycle.
Endothelial cells stimulated with Fbln5 did not show activation of ERK1/2 and p38-MAPK, but Fbln5 did reduce VEGF expression, which resulted in reduced endothelial cell proliferation (Albig and Schiemann 2004). Likewise, Fbln5 failed to induce proliferation in SMCs compared to cells plated on FN due to a reduction in β1 integrin signaling (Lomas et al. 2007). In addition, recent studies performed in ovarian cancer cells also revealed changes in proliferation upon Fbln5 overexpression (Heo et al. 2015). The expression of Fbln5 in SKOV3 ovarian carcinoma cells led to G2/M arrest but did not adversely affect colony-forming ability. Conversely, knockdown of Fbln5 in human gastric cancer cells blocked cell proliferation as tested by BrdU incorporation (Shi et al. 2014). Together, these data support the presence of an antiproliferative effect of Fbln5 in endothelial cells and a pro-proliferative effect in fibroblasts. Yet, it is unclear whether Fbln5 promotes or blocks proliferation in epithelial cells. Since Fbln5 blocks FN-mediated integrin signaling, it is germane to evaluate the effect of integrin-activating or integrin-inhibiting antibodies on proliferation in contexts where Fbln5 has been manipulated.
1.4.3 Fbln5 in Angiogenesis
Fbln5 was identified by two separate groups who were interested in proteins that regulate cardiovascular development and disease (Kowal et al. 1999; Nakamura et al. 1999). Each group identified Fbln5 as being expressed in the embryonic arterial vasculature; however, it was downregulated in most adult vascular beds. They also found that the expression of Fbln5 was reactivated in diseased adult vasculature, namely, atherosclerotic and balloon-injured arteries. Fbln5 -/- mice were created shortly after these original findings, which revealed the important function of Fbln5 in elastogenesis, in that Fbln5-deficient animals had tortuous vessels in addition to loose skin and emphysematous lungs as a result of incomplete elastic fiber formation (Nakamura et al. 2002; Yanagisawa et al. 2002). Elasticity is a major characteristic of blood vessels and critical for proper vessel function. These observations led to the investigation of Fbln5 as a regulator of angiogenesis in several research models.
Sullivan et al. (Sullivan et al. 2007) revealed that polyvinyl alcohol sponges implanted into Fbln5 -/- mice had significantly increased vascular invasion as seen by CD31 (PECAM-1) staining. Interestingly, fibroblast migration into these sponges remained unchanged in the absence of Fbln5. This group then examined a possible mechanism by which Fbln5 was antagonizing vascularization and found that the pro-angiogenic factors, Ang-1, Ang-2, Ang-3, and VEGF, were increased in sponges removed from Fbln5 -/- mice as seen by quantitative PCR. Moreover, vascular smooth muscle cells isolated from WT- and Fbln5-deficient mice also showed an increase of these pro-angiogenic factors in the absence of Fbln5. The authors of this paper did not examine the function of integrins in this phenotype. Therefore, based on these studies alone, it is difficult to determine whether Fbln5 directly antagonizes vascular function or if this is an integrin-dependent phenomenon. Given the evidence that Fbln5 binds to but does not support the activation of α4β1 and α5β1 integrins in SMCs (Lomas et al. 2007), it is probable that enhanced integrin activation in the absence of Fbln5 results in increased endothelial cell migration and proliferation in this context (Lamalice et al. 2007; Clark et al. 1982).
Investigating the function of Fbln5 in tumor angiogenesis has also produced mixed results. The forced expression of Fbln5 by fibrosarcoma cells resulted in tumors that were significantly less vascularized compared to tumors derived from control cells (Albig 2006). These results suggest that Fbln5 antagonizes tumor angiogenesis, which again is likely a consequence of reduced FN-integrin signaling by Fbln5. In contrast, we have reported that subcutaneous and orthotopic pancreatic tumor growth (Pan02) in Fbln5 -/- mice results in reduced tumor blood vessel density (Schluterman et al. 2010). In this study, the loss of Fbln5 in tumors resulted in elevated ROS production that was mediated by FN-integrin signaling. The discrepancy between these two findings is likely attributed to the complete lack of host Fbln5 in the knockout model as well as the lack of Fbln5 expression in Pan02 cells (Wang et al. 2015; Schluterman et al. 2010). In the fibrosarcoma model, it is difficult to distinguish between the function of host- or tumor-derived Fbln5 with regard to the regulation of angiogenesis. Moreover, the loss of Fbln5 function in tumors produces a much different effect on angiogenesis compared to non-tumor tissue (e.g., the polyvinyl alcohol sponge model mentioned above), which may be due to the high amount of FN in tumors versus normal tissue. A major biological outcome of FN-integrin signaling is ROS production (Chiarugi et al. 2003); therefore, Fbln5 functions as a molecular rheostat to control integrin-induced ROS generation.
1.4.4 Fbln5 in Tumor Progression
As described above, the cellular effects of Fbln5 are context dependent, and thus the effect of Fbln5 on tumor progression may also be tumor specific. Initial studies on Fbln5 and tumor growth relied on the forced expression of Fbln5 in cancer cell lines that were then implanted into immunodeficient mice. Lee et al. (2008) revealed increased Fbln5 expression from human breast tumors, and that Fbln5 expression in 4T1 breast cancer cells enhanced tumor growth in mice. Yue et al. (2009) reported that Fbln5-expressing H460 lung cancer cells displayed decreased metastasis to the lungs after IV injection. This study did not examine primary lung tumor growth in the context of Fbln5; however, lung cancer patient samples showed a downregulation of Fbln5 compared to matched normal lung specimens as seen by RT-PCR and immunohistochemistry (IHC) analysis of a tissue microarray.
Fbln5 levels are markedly increased in mouse (Fig. 1.2a) and human PDA (Wang et al. 2015) compared to levels in a normal pancreas. An analysis of various cell types in vitro as well as IHC in PDA samples revealed that stromal cells such as endothelial cells and fibroblasts are the main producers of Fbln5 protein (Wang et al. 2015). Further studies into the factors that control Fbln5 expression in PDA have revealed that the hypoxic TME of PDA induces Fbln5 expression through a TGF-β-PI3K/AKT signaling axis. The inhibition of a TGF-β receptor (TGF-βR), PI3K, or protein kinase B (AKT) was found to block hypoxia-induced Fbln5 expression in vitro. In genetically engineered mouse models (GEMMs) of PDA, therapy-induced hypoxia (via anti-VEGF treatment) elevated Fbln5 expression, while pharmacologic inhibition of TGF-β signaling reduced its expression.

Enhanced ECM and ROS production in mouse PDA. (a) Normal (left column) and tumor (right column) pancreas from WT or Kras LSL-G12D/+; Cdkn2a f/f; p48 Cre/+ (PDA) mice stained for Fbln5 (green) and Meca32 (red), which marks blood vessels. Nuclei were stained with DAPI (blue). (b) Tissues were stained with amylase (green), which stains normal pancreatic acinar tissue, and fibronectin (FN, red). (c) Tissues were stained with dihydroethidium, a ROS-sensitive chemical red dye. Reproduced from Wang et al. (2015)
As mentioned previously, Schluterman et al. (2010) showed that subcutaneous and orthotopic Pan02 tumors implanted into Fbln5 -/- mice had a significant reduction in tumor growth and blood vessel density (Schluterman et al. 2010). Fbln5 regulates ROS production in vascular tissue through binding ecSOD, and the deletion of Fbln5 results in increased ROS production in mouse aortas (Nguyen et al. 2004). Elevated ROS production can be detrimental to vascular function, and consistent with this notion, Schluterman et al. (2010) found elevated levels of ROS production in tumors grown in Fbln5 -/- mice compared to tumors grown in WT mice. Moreover, mice containing a point mutation in the RGD-integrin binding domain of Fbln5 (RGD → RGE) recapitulated the phenotype seen in Fbln5 -/- mice with regard to tumor growth, angiogenesis, and ROS production, highlighting the importance of Fbln5-integrin interaction in this phenotype. Using mouse embryonic fibroblasts in vitro, it was shown that increased ROS production is a consequence of increased integrin signaling by FN in the absence of functional Fbln5. Furthermore, antioxidant treatment restored tumor growth and microvessel density in Fbln5 RGE/RGE mice, confirming that reduced tumor growth and angiogenesis was a direct consequence of elevated ROS production driven by the loss of Fbln5-integrin binding. Expanding on this work, Wang and Topalovski et al. (Wang et al. 2015) found that the mutation of the RGE-integrin binding domain of Fbln5 in the context of a GEMM of PDA also resulted in reduced tumor growth and angiogenesis due to increased ROS production. These data are in line with other evidence that shows Fbln5 competes with FN to negatively regulate β1 integrin function (Lomas et al. 2007).
Elevated oxidative stress is seen in many solid tumors compared to normal tissues, and exploiting this biochemical difference has the potential to enhance the efficacy of anticancer agents (Trachootham et al. 2009). For instance, ROS accumulation after gemcitabine treatment in PDA cells contributes significantly to the cytotoxic activity of this nucleoside analog (Ju et al. 2015). Furthermore, elevated hydrogen peroxide levels were shown to be a mechanism by which paclitaxel killed lung cancer cells (Alexandre et al. 2006). The homeostasis of ROS is important for normal cell function and signaling, but excessive ROS can result in cellular toxicity, and therefore ROS levels must be tightly controlled. ROS can be produced by a number of enzymes, including but not limited to, the electron transport chain (ETC), NADPH oxidase (NOX), 5-lipoxygenase (5-LOX), and nitric oxide synthase (NOS) (Holmstrom and Finkel 2014).
ECM proteins can stimulate the activation of these enzymes and thus indirectly induce ROS production. Chiarugi et al. (Chiarugi et al. 2003) showed that integrin activation by FN induces ROS production in a 5-LOX- and NOX-dependent manner in 3T3 fibroblasts (Chiarugi et al. 2003). Another research group showed that PDA cells are responsive to FN in terms of increased ROS production by 5-LOX and NOX (Edderkaoui et al. 2005). Given that FN can stimulate ROS production via integrin activation and that the loss or mutation of Fbln5 results in higher ROS production, we propose that Fbln5 blocks integrin-induced ROS production. In PDA, where ECM proteins such as FN are abundant (Fig. 1.2b; Bachem et al. 2005; Mahadevan and Von Hoff 2007), oxidative stress levels are much higher compared to those in the normal pancreas (Fig. 1.2c; Blum and Kloog 2014). Thus, Fbln5 is a novel target to investigate as a potential strategy to manipulate ROS levels in the TME.
1.5 Conclusion
As described in this chapter, there are multiple facets of the ECM that contribute to overall tumor progression and response to therapy. The ECM is highly dynamic, and its contribution to disease progression relies on several structural and nonstructural proteins. Nonstructural matricellular proteins, such as Fbln5, can have pro- and anti-tumorigenic activity, which is highly dependent on the nature and composition of the surrounding TME. The large portion of Fbln5 present in the TME is derived from stromal cells; however, it is unclear whether Fbln5 also exerts its effects on nearby tumor cells. It is likely that the expression profile of integrins on the cell will dictate its response to Fbln5 signaling. In PDA, the loss of Fbln5-integrin binding leads to increased FN-integrin signaling, which counterintuitively results in smaller tumors. This is a prime example that highlights the context-specific effects of ECM signaling on tumor growth. Due to the extremely high levels of FN in PDA, the tumor hijacks Fbln5 to protect itself from the harmful by-product of enhanced FN-induced ROS generation. In this situation, too much FN signaling is unfavorable for the tumor, and thus Fbln5 promotes tumor growth by limiting FN-induced ROS production. Given this evidence, multi-faceted approaches to target the ECM while simultaneously augmenting ROS production in the TME may be a viable and effective therapeutic strategy to combat solid tumors such as PDA.
References
Aguilera KY et al (2014) Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res 74(4):1032–1044
Akiyama SK, Olden K, Yamada KM (1995) Fibronectin and integrins in invasion and metastasis. Cancer Metastasis Rev 14(3):173–189
Albig AR (2006) Fibulins 3 and 5 antagonize tumor angiogenesis in vivo. Cancer Res 66:2621–2629
Albig AR, Schiemann WP (2004) Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol 23(6):367–379
Alexandre J et al (2006) Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer 119(1):41–48
Arnold SA et al (2010) Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech 3(1–2):57–72
Bachem MG et al (2005) Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128(4):907–921
Beck K, Hunter I, Engel J (1990) Structure and function of laminin: anatomy of a multidomain glycoprotein. FASEB J 4(2):148–160
Besse B et al (2012) Phase Ib safety and pharmacokinetic study of volociximab, an anti- 5 1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann Oncol 24:90–96
Bhaskar V et al (2007) A function blocking anti-mouse integrin alpha5beta1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J Transl Med 5:61
Bhaskar V et al (2008) Volociximab, a chimeric integrin alpha5beta1 antibody, inhibits the growth of VX2 tumors in rabbits. Investig New Drugs 26(1):7–12
Blum R, Kloog Y (2014) Metabolism addiction in pancreatic cancer. Cell Death Dis 5:e1065
Bonner JC (2004) Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15(4):255–273
Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14(5):608–616
Chapman SL et al (2009) Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler Thromb Vasc Biol 30:68–74
Chen HC, Guan JL (1994) Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 91(21):10148–10152
Chiarugi P et al (2003) Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161(5):933–944
Chiquet-Ehrismann R et al (1988) Tenascin interferes with fibronectin action. Cell 53(3):383–390
Clark RA et al (1982) Blood vessel fibronectin increases in conjunction with endothelial cell proliferation and capillary ingrowth during wound healing. J Invest Dermatol 79(5):269–276
Costa-Silva B et al (2015) Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17:816–826
Cranmer LD, Bedikian A, Ribas A (2005) Phase II study of volociximab (M200), an α5β1 anti-integrin antibody in metastatic melanoma. J Clin Oncol 24 (Abstr 8011)
de Fougerolles AR et al (2000) Regulation of inflammation by collagen-binding integrins alpha1beta1 and alpha2beta1 in models of hypersensitivity and arthritis. J Clin Invest 105(6):721–729
Edderkaoui M et al (2005) Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol 289(6):G1137–G1147
Eliceiri BP (2001) Integrin and growth factor receptor crosstalk. Circ Res 89(12):1104–1110
Frisch SM, Screaton RA (2001) Anoikis mechanisms. Curr Opin Cell Biol 13(5):555–562
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022
George EL et al (1993) Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119(4):1079–1091
Giancotti FG (1999) Integrin signaling. Science 285:1028–1033
Guadall A et al (2011) Fibulin-5 Is Up-regulated by hypoxia in endothelial cells through a hypoxia-inducible factor-1 (HIF-1)-dependent mechanism. J Biol Chem 286:7093–7103
Guadamillas MC, Cerezo A, Del Pozo MA (2011) Overcoming anoikis – pathways to anchorage-independent growth in cancer. J Cell Sci 124(Pt 19):3189–3197
Han S, Khuri FR, Roman J (2006) Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res 66(1):315–323
Harburger DS, Calderwood DA (2009) Integrin signalling at a glance. J Cell Sci 122(Pt 2):159–163
Heo JH et al (2015) Fibulin-5 is a tumour suppressor inhibiting cell migration and invasion in ovarian cancer. J Clin Pathol 69:109–116
Hocevar BA, Brown TL, Howe PH (1999) TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J 18(5):1345–1356
Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15(6):411–421
Huang W et al (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res 61(23):8586–8594
Hwang CF et al (2013) Oncogenic fibulin-5 promotes nasopharyngeal carcinoma cell metastasis through the FLJ10540/AKT pathway and correlates with poor prognosis. PLoS One 8(12):e84218
Itano N, Zhuo L, Kimata K (2008) Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci 99(9):1720–1725
Ju HQ et al (2015) Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther 14(3):788–798
Kim S et al (2000) Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol 156(4):1345–1362
Kowal RC et al (1999) EVEC, a novel epidermal growth factor like repeat-containing protein upregulated in embryonic and diseased adult vasculature. Circ Res 84:1166–1176
Krieglstein CF et al (2002) Collagen-binding integrin alpha1beta1 regulates intestinal inflammation in experimental colitis. J Clin Invest 110(12):1773–1782
Kuang PP et al (2006) Fibulin-5 gene expression in human lung fibroblasts is regulated by TGF-beta and phosphatidylinositol 3-kinase activity. Am J Physiol Cell Physiol 291:C1412–C1421
Kuwada SK (2007) Drug evaluation: Volociximab, an angiogenesis-inhibiting chimeric monoclonal antibody. Curr Opin Mol Ther 9(1):92–98
Kyriakides TR, Bornstein P (2003) Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost 90(6):986–992
Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100(6):782–794
Leask A, Abraham DJ (2004) TGF-beta signaling and the fibrotic response. FASEB J 18(7):816–827
Lee YH et al (2008) Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF- in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 29:2243–2251
Lipfert L et al (1992) Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J Cell Biol 119(4):905–912
Liu X et al (2004) Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet 36(2):178–182
Loeys B et al (2002) Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet 11(18):2113–2118
Lomas AC et al (2007) Fibulin-5 binds human smooth-muscle cells through alpha5beta1 and alpha4beta1 integrins, but does not support receptor activation. Biochem J 405(3):417–428
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196:395–406
Mahadevan D, Von Hoff DD (2007) Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther 6:1186–1197
Mao Y et al (2013) Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 32(1-2):303–315
Masur SK et al (1995) Integrin-dependent tyrosine phosphorylation in corneal fibroblasts. Invest Ophthalmol Vis Sci 36(9):1837–1846
Mithieux SM, Weiss AS (2005) Elastin. Adv Protein Chem 70:437–461
Miyamoto H et al (2004) Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 28(1):38–44
Murphy-Ullrich JE et al (1993) Heparin-binding peptides from thrombospondins 1 and 2 contain focal adhesion-labilizing activity. J Biol Chem 268(35):26784–26789
Nakamura T, Ruiz-Lozano P, Lindner V, Yabe D, Furukawa Y, Taniwaki M, Kobuke K, Tashiro K, Lu Z, Andon NL, Schaub R, Matsumori A, Sasayama S, Chien KR, Honjoa T (1999) DANCE, a novel secreted RGD protein expressed in developing atherosclerotic, and balloon-injured arteries. J Biol Chem 274(32):22467–22483
Nakamura T et al (2002) Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415(6868):171–175
Nguyen AD et al (2004) Fibulin-5 is a novel binding protein for extracellular superoxide dismutase. Circ Res 95(11):1067–1074
Nicosia RF, Bonanno E, Smith M (1993) Fibronectin promotes the elongation of microvessels during angiogenesis in vitro. J Cell Physiol 154(3):654–661
Pankov R (2002) Fibronectin at a glance. J Cell Sci 115:3861–3863
Ramakrishnan V et al (2006) Preclinical evaluation of an anti-alpha5beta1 integrin antibody as a novel anti-angiogenic agent. J Exp Ther Oncol 5(4):273–286
Ramaswamy S et al (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33(1):49–54
Reuter S et al (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–1616
Ricart AD (2008) Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res 14:7924–7929
Schaffner F, Ray A, Dontenwill M (2013) Integrin α5β1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancer 5:27–47
Schiemann WP (2002) Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion. Fibulin-5 is induced by transforming growth factor-beta and affects protein kinase cascades. J Biol Chem 277:27367–27377
Schlaepfer DD et al (1994) Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 372(6508):786–791
Schluterman MK et al (2010) Loss of fibulin-5 binding to 1 integrins inhibits tumor growth by increasing the level of ROS. Dis Models Mech 3:333–342
Shi X-Y et al (2014) Effect of Fibulin-5 on cell proliferation and invasion in human gastric cancer patients. Asian Pac J Trop Med 7:787–791
Stenman S, Vaheri A (1981) Fibronectin in human solid tumors. Int J Cancer 27(4):427–435
Sullivan KM et al (2007) Fibulin-5 functions as an endogenous angiogenesis inhibitor. Lab Invest 87:818–827
Tang J-C (2015) Effect of fibulin-5 on adhesion, migration and invasion of hepatocellular carcinoma cells via an integrin-dependent mechanism. World J Gastroenterol 21:11127
Topalovski M, Brekken RA (2015) Matrix control of pancreatic cancer: new insights into fibronectin signaling. Cancer Lett 381:252–258
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8(7):579–591
Vogel WF (2001) Collagen-receptor signaling in health and disease. Eur J Dermatol 11(6):506–514
Wang M et al (2015) Fibulin-5 blocks microenvironmental ROS in pancreatic cancer. Cancer Res 75(23):5058–5069
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17(11):1359–1370
Wong GS, Rustgi AK (2013) Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer 108(4):755–761
Xiang L et al (2012) The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PLoS One 7(4):e35378
Yanagisawa H et al (2002) Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415(6868):168–171
Yanagisawa H, Schluterman MK, Brekken RA (2009) Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal 3:337–347
Yang JT, Rayburn H, Hynes RO (1993) Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development 119(4):1093–1105
Yue W et al (2009) Fibulin-5 auppresses lung cancer invasion by inhibiting matrix metalloproteinase-7 expression. Cancer Res 69:6339–6346
Zhao JC et al (2012) Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res 22(2):322–331
Zhu JW et al (2002) Upregulation of vascular endothelial growth factor by hydrogen peroxide in human colon cancer. World J Gastroenterol 8(1):153–157
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Topalovski, M., Brekken, R.A. (2017). The Extracellular Matrix of Tumors: A Focus on Fibronectin and Fibulin-5. In: Brekken, R., Stupack, D. (eds) Extracellular Matrix in Tumor Biology. Biology of Extracellular Matrix. Springer, Cham. https://doi.org/10.1007/978-3-319-60907-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-60907-2_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60906-5
Online ISBN: 978-3-319-60907-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)