
Abstract
The most recently discovered 3′,5′-cyclic nucleotide phosphodiesterase family is the Phosphodiesterase 11 (PDE11) family, which is encoded by a single gene PDE11A. PDE11A is a dual-specific PDE, breaking down both cAMP and cGMP. There are four PDE11A splice variants (PDE11A1-4) with distinct tissue expression profiles and unique N-terminal regulatory regions, suggesting that each isoform could be individually targeted with a small molecule or biologic. PDE11A4 is the PDE11A isoform expressed in brain and is found in the hippocampal formation of humans and rodents. Studies in rodents show that PDE11A4 mRNA expression in brain is, in fact, restricted to the hippocampal formation (CA1, possibly CA2, subiculum, and the adjacently connected amygdalohippocampal area). Within the hippocampal formation of rodents, PDE11A4 protein is expressed in neurons but not astrocytes, with a distribution across nuclear, cytoplasmic, and membrane compartments. This subcellular localization of PDE11A4 is altered in response to social experience in mouse, and in vitro studies show the compartmentalization of PDE11A4 is controlled, at least in part, by homodimerization and N-terminal phosphorylation. PDE11A4 expression dramatically increases in the hippocampus with age in the rodent hippocampus, from early postnatal life to late aging, suggesting PDE11A4 function may evolve across the lifespan. Interestingly, PDE11A4 protein shows a three to tenfold enrichment in the rodent ventral hippocampal formation (VHIPP; a.k.a. anterior in primates) versus dorsal hippocampal formation (DHIPP). Consistent with this enrichment in VHIPP, studies in knockout mice show that PDE11A regulates the formation of social memories and the stabilization of mood and is a critical mechanism by which social experience feeds back to modify the brain and subsequent social behaviors. PDE11A4 likely controls behavior by regulating hippocampal glutamatergic, oxytocin, and cytokine signaling, as well as protein translation. Given its unique tissue distribution and relatively selective effects on behavior, PDE11A may represent a novel therapeutic target for neuropsychiatric, neurodevelopmental, or age-related disorders. Therapeutically targeting PDE11A4 may be a way to selectively restore aberrant cyclic nucleotide signaling in the hippocampal formation while leaving the rest of the brain and periphery untouched, thus, relieving deficits while avoiding unwanted side effects.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- PDE11A
- PDE11
- Phosphodiesterase 11
- cAMP
- cGMP
- Psychiatric illness
- Brain
- Endocrine
- Immune
- Hippocampus
- Memory
- Inflammation
- Lithium
- Tissue expression
- Social Behavior
- Oxytocin
1 Introduction
The most recently discovered phosphodiesterase (PDE) family is PDE11, which hydrolyzes cAMP and cGMP equally well (Hetman et al. 2000; Fawcett et al. 2000; Yuasa et al. 2000; Yuasa et al. 2001a; Weeks et al. 2007). The PDE11 family is comprised of a single gene, PDE11A (Hetman et al. 2000; Fawcett et al. 2000; Yuasa et al. 2000; Yuasa et al. 2001a; Yuasa et al. 2001b). As with most PDE families, the N-terminal domain of PDE11A serves a regulatory function, whereas the C-terminal domain encompasses the catalytic domain (Weeks et al. 2007). The longest isoform of mouse, rat and human PDE11A (a.k.a. PDE11A4) demonstrates ~95% protein sequence homology, suggesting results obtained in preclinical rodent models will be applicable to the human condition. As extensively reviewed elsewhere (Kelly 2015), literature findings surrounding the tissue distribution pattern for the various PDE11A isoforms are highly disparate, in part due to a number of poor quality commercially-available antibodies, but well-controlled studies consistently show select PDE11A isoforms being expressed in brain (particularly the hippocampal formation), the adrenal gland, and the prostate. Studies examining tissues from PDE11A wild-type (WT) and knockout (KO) mice find abundant PDE11A4 protein in the hippocampal formation of brain but not in any of 23 peripheral tissues assessed; whereas, PDE11A1 was found in prostate, PDE11A3 in the seminal vesicles, and PDE11A1 and PDE11A3 in spleen (Kelly 2015). Consistent with this restricted tissue distribution profile, PDE11A appears to regulate brain function, tumor physiology, and, possibly, inflammation (Kelly 2015; Fatemi et al. 2010a; Couzin 2008; Wong et al. 2006; Luo et al. 2009a; Kelly et al. 2010; Hegde et al. 2015; Pathak et al. 2016; Hegde et al. 2016b; Mertens et al. 2015 but see Laje et al. 2009; Perlis et al. 2010; Kelly 2015; Alevizaki and Stratakis 2009; Faucz et al. 2011; Greene et al. 2010; Carney et al. 2010; Almeida and Stratakis 2011; Vezzosi et al. 2012; Horvath et al. 2009; Horvath et al. 2006a; Horvath et al. 2006b; Libe et al. 2008; Libe et al. 2011 but see Bimpaki et al. 2009; Kelly 2015; Pathak et al. 2016; DeWan et al. 2010; Oki et al. 2011; Witwicka et al. 2007; Bazhin et al. 2010). Here, we review how PDE11A may regulate brain function to determine whether or not PDE11A4 holds promise as a future therapeutic target, particularly in the context of neuropsychiatric, neurodevelopmental and/or age-related disease.
2 Molecular Features of PDE11A
The PDE11A gene on chromosome 2q31.2 contains 23 exons and yields four splice variants PDE11A1-4 (Hetman et al. 2000; Fawcett et al. 2000; Yuasa et al. 2000; Yuasa et al. 2001a; Yuasa et al. 2001b), which have been recently schematically reviewed in depth (Kelly 2015). Isoform-specific promoters, coupled with alternative splicing, yield unique N-terminal domains for each PDE11A isoform (Yuasa et al. 2001b) and likely account for the differences in tissue expression that are seen with each PDE11A isoform (Kelly 2015). While the C-terminal region encompasses the catalytic domain of PDE11A, the N-terminal region encompasses various regulatory domains and sites of post-translational modification. Within the N-terminal region are 2 cGMP binding PDE, Anabaena adenylyl cyclase and E. coli FhlA (GAF) domains (Makhlouf et al. 2006). Perhaps not surprisingly, phylogenetic analyses suggests PDE11A is most closely related to other GAF-domain containing PDEs, particularly PDE5A and PDE6A-C as well as PDE2A and PDE10A (Yuasa et al. 2001b; Kelly 2015). As we will see below, the four PDE11A isoforms hydrolyze cAMP and cGMP, and each has a unique N-terminal region with differential representation of the GAF-A domain, which binds cGMP (Gross-Langenhoff et al. 2008; Gross-Langenhoff et al. 2006; Jager et al. 2012; Matthiesen and Nielsen 2009), and the GAF-B domain, which is required for homodimerization.
Although each of the four variants exhibits a unique N-terminal regulatory domain (c.f., Kelly 2015), the C-terminal domain is consistent across the 4 PDE11A isoforms. Exons 8–23 are included in each isoform and encode not only the C-terminal catalytic domain but also a portion of the N-terminal regulatory GAF-B domain (Makhlouf et al. 2006). In addition to exons 8–23, PDE11A1 also uniquely includes exon 7; thus, PDE11A1 includes only a truncated GAF-B domain within its N-terminus (Yuasa et al. 2001b). PDE11A2 includes exons 5 and 6, but not 7, in addition to exons 8–23; thus, PDE11A2 includes a truncated GAF-A domain and a full GAF-B domain. (Hetman et al. 2000; Makhlouf et al. 2006). The PDE11A3 transcript is encoded by exons 1, 2, 4, 5, and 6 in addition to exons 8–23; however, translation of PDE11A3 does not begin until exon 2 (Yuasa et al. 2001b). Thus, similar to PDE11A2, PDE11A3 contains a truncated GAF-A domain and a full GAF-B domain upstream of the C-terminal catalytic domain. PDE11A4 is the only isoform to include exon 3 along with the shared exons 4–6 and 8–23 (Yuasa et al. 2001b). PDE11A4, the isoform expressed in brain, is the longest isoform. Therefore, it is at times erroneously referred to as “full-length” PDE11A, despite the fact that it lacks exons 2 and 7. Importantly, exon 3 contains two validated phosphorylation sites (S117 and S162) and the beginning of the GAF-A domain (Yuasa et al. 2000); thus, PDE11A4 is the only isoform to include the GAF-A domain in its entirety. As we will review in greater detail below, these phosphorylation sites and GAF domains may be important for intramolecular signaling (Weeks et al. 2007; Gross-Langenhoff et al. 2008) and may provide a mechanism by which it will be possible to therapeutically target a single PDE11A isozyme with high selectivity (Kelly 2015). Indeed, isozyme-specific targeting of PDE11A activity has already been demonstrated as vinpocetine can inhibit PDE11A3 but not PDE11A4 (Yuasa et al. 2000).
Under physiological conditions, all PDE11A isoforms hydrolyze both cAMP and cGMP (cf. Makhlouf et al. 2006). As extensively reviewed elsewhere (Kelly 2015), PDE11A1 and PDE11A2 have higher substrate affinities relative to PDE11A3 and PDE11A4 (Yuasa et al. 2001a; Weeks et al. 2007); however, PDE11A3 and PDE11A4 have significantly higher catalytic rates relative to PDE11A1 and PDE11A2 (Yuasa et al. 2001a; Weeks et al. 2007). A comparison of rat PDE11A found similar results to those obtained using Homo sapiens PDE11A (Yuasa et al. 2001a). The net effect of the relative differences in substrate affinities versus turnover rates is that the four PDE11A isoforms hydrolyze cAMP and cGMP comparably well (Yuasa et al. 2001a; Weeks et al. 2007). Site-directed mutagenesis shows that Q869 is required for substrate binding of the PDE11A catalytic domain (Weeks et al. 2009); however, in absence of a resolved crystal structure the full implications of these results remain to be determined. Such knowledge would benefit designing compounds capable of selectively targeting the PDE11A catalytic domain over its nearest neighbor PDE5A.
The N-terminal region of PDE11A clearly regulates catalytic function of the enzyme. For example, as the length of the N-terminus increases, the affinity of the catalytic domain for cAMP and cGMP decreases (Weeks et al. 2007). That is, PDE11A4 has the lowest substrate affinity for cAMP and cGMP and PDE11A1 and PDE11A2 have the highest substrate affinities for cAMP and cGMP (Weeks et al. 2007). Similarly, in a chimeric protein consisting of the PDE11A4 N-terminus linked to the catalytic domain of a bacterial adenylyl cyclase, deletion of the first 196 amino acids (i.e., all amino acids upstream of the GAF-A domain) increases basal catalytic activity (Gross-Langenhoff et al. 2008). Together, these studies support a regulatory role for the PDE11A N-terminal region.
As mentioned above, PDE11A4 is the only PDE11A variant that includes S117 and S162, 2 serines phosphorylated by protein kinase A, protein kinase G (PKA/PKG) (Yuasa et al. 2000) and possibly other enzymes (Kelly 2015). In vitro, PKA and PKG fail to phosphorylate PDE11A3, which lacks S117 and S162, but do phosphorylate PDE11A4 (Yuasa et al. 2000). Further, most of the PKA and PKG-induced phosphorylation of PDE11A4 is ablated by mutating S117 and S162 to phosphomutant alanines (Yuasa et al. 2000). The remaining PKA and PKG-induced phosphorylation observed in the S117A/S162A PDE11A4 mutant likely reflects phosphorylation of S124, which is predicted to be phosphorylated by PKA and PKG (Kelly 2015) and is phosphorylated along with S117 and S162 in COS-1 cells (Pathak et al. 2015).
Emerging evidence suggests these N-terminal phosphorylation sites may play an important role in regulating PDE11A4 function. Gross-Langenhoff et al. used a chimeric protein consisting of the PDE11A4 N-terminus linked to the catalytic domain of a bacterial adenylyl cyclase to explore the functional role S117/S162 phosphorylation (Gross-Langenhoff et al. 2008). In that chimeric protein, the phosphorylation state of the N-terminal regulatory region affected intramolecular regulation of the GAF-A domain. Specifically, when both S117 and S162 were phosphorylated, or were replaced with amino acids that mimicked the phosphorylated state, the downstream GAF-A regulatory domain had an increase in affinity for cGMP (Gross-Langenhoff et al. 2008). Interestingly, deleting the first 176 amino acids similarly increased cGMP affinity for the GAF-A domain (Gross-Langenhoff et al. 2008), suggesting that phosphorylation of S117/S162 may produce a conformational change that exposes the cyclic nucleotide binding pocket of the GAF-A domain. In COS-1 and HEK293T cells, we have shown that introducing a phosphomimic mutation (aspartate, D) to S117 and S124 (S117D, S124D) traffics PDE11A4 into punctate structures and shifts PDE11A4 from the cytosol to the membrane (Pathak et al. 2015). In contrast, a S162D mutation has the completely opposite effect, distributing PDE11A4 throughout the cytosol and shifting PDE11A4 from the membrane to the cytosol (Pathak et al. 2015). Interestingly, S162D completely blocks the effects of S117D/S124D (Pathak et al. 2015). The fact that S162D blocks the effect of S117D in the context of a full-length recombinant protein, yet potentiates the effect of S117D in the context of the chimeric protein, may suggest a complex interplay between these N-terminal phosphorylation events and the C-terminus. Consistent with the fact that S162D shifts PDE11A4 to the cytosol and blocks the ability of S117D/S124D to shift PDE11A4 toward the membrane, we find that wild-type PDE11A found in the cytosol can be phosphorylated at any of the three serines; whereas, PDE11A4 in the membrane shows phosphorylation at serines 117 and 124 but not 162 (Pathak et al. 2015). It will be important to determine whether the ability of these phosphorylation events to control the subcellular compartmentalization of PDE11A4 is related to conformational changes in the enzyme, changes in cGMP binding of the GAF-A domain, and/or the prevention or promotion of other posttranstional modifications (e.g., palmitoylation, ubiquitination, sumoylation).
The functional consequence of cyclic nucleotide binding of a PDE GAF domain differs by PDE family. cGMP binding the GAF-A domain can directly increase catalytic activity (PDE2A, Martinez et al. 2002; Beavo et al. 1971), increases substrate affinity (PDE5A, Francis et al. 2011), indirectly inhibit catalytic activity by strengthening specific inhibitory protein-protein interactions (PDE6, D’Amours and Cote 1999), or have no effect at all (PDE10A, Jager et al. 2012; Matthiesen and Nielsen 2009). Several laboratories have reported that cGMP binds the PDE11A4 GAF-A, but not GAF-B, domain (Gross-Langenhoff et al. 2008; Gross-Langenhoff et al. 2006; Jager et al. 2012; Matthiesen and Nielsen 2009), likely with a lower Ki versus the catalytic domain (Matthiesen and Nielsen 2009). Although cGMP binding of the PDE11A4 GAF-A domain did stimulate catalytic activity of the PDE11A4-cyclase chimeric protein described above (Gross-Langenhoff et al. 2008; Gross-Langenhoff et al. 2006), it did not impact catalytic activity of recombinant full-length PDE11A4 (Jager et al. 2012; Matthiesen and Nielsen 2009). That said, it is interesting to note that the ability of cGMP to allosterically stimulate catalytic activity of the PDE11A4-cyclase chimera was dependent on the presence of an aspartate within a conserved NKFDE motif that is present in all mammlian PDE GAF domains (Gross-Langenhoff et al. 2006). Mutating D355 to an alanine abolished the intramolecular signaling events triggered by cGMP. It remains to be determined whether mutating D355 simply prevents cGMP from binding the GAF-A domain or prevents some downstream consequence of cGMP binding (e.g., a conformational change; Gross-Langenhoff et al. 2006). Although cGMP does not appear to allosterically regulate catalytic activity of PDE11A4, a cGMP analog Rp-8-pCPT-PET-cGMPs does appear to allosterically increase the catalytic activity of both recombinant and native PDE11A4 (Jager et al. 2012). Unfortunately, Rp-8-pCPT-PET-cGMPs inhibits PKG and, likely, many other cGMP-binding molecules; thus, its use as a tool compound is limited. That said, the fact that Rp-8-pCPT-PET-cGMPs is capable of allosterically stimulating PDE11A function provides a key proof-of-concept for developing PDE11A4 activators.
GAF domains not only bind cyclic nucleotides, they also bind proteins, including other molecules of PDEs themselves. All PDE11A isoforms, indeed all PDEs (Francis et al. 2011; Heikaus et al. 2009), exist as oligomers (Weeks et al. 2007). The C-terminal portion of the GAF-B domain is required for dimerization of all PDE11A isoforms (Weeks et al. 2007). PDE11A1, with the shortest N-terminal region, tetramerizes; whereas, PDE11A2-4 homodimerize. It is not completely understood how oligomerization affects PDE11A function. Weeks et al. found that the dimeric PDE11A2 and the tetrameric PDE11A1 have nearly identical substrate affinities for cAMP and cGMP, suggesting that quaternary structure does not greatly impact affinity of the cyclic nucleotides for the catalytic site (Weeks et al. 2007). We have shown that homodimerization controls the subcellular compartmentalization of PDE11A4 (Pathak et al. 2016). A mutation within the GAF-B domain found in select mouse strains that strengthens homodimerization shifts PDE11A4 from the cytosol towards the membrane; whereas, disrupting homodimerization shifts PDE11A4 from the membrane to the cytosol (Pathak et al. 2016). Disrupting homodimerization also increases proteolytic degradation of PDE11A4 (Pathak et al. 2016). Although the mechanism responsible remains to be determined, it is interesting to note that PDE11A4 is predicted to be ubiquitinated and sumoylated (Kelly 2015). Thus, it is possible that disruption of homodimerization may make PDE11A4 more vulnerable to these particular posttranslational modifications that tag a protein for degradation. It remains to be determined whether or not homodimerization directly affects PDE11A4 catalytic activity as well. In vitro studies examining the PDE2, PDE4, and PDE5 families argue that dimerization is not required for catalytic activity; however, dimerization may influence post-translational modifications of the enzymes, thereby altering subcellular localization and/or catalytic activity (Francis et al. 2011; Keravis and Lugnier 2010). Clearly, much work remains to understand the functional role of PDE11A oligomerization. That said, these studies suggest that homodimerization may be a novel mechanism by which we could therapeutically target PDE11A function, particularly in disease states where the enzyme may become mislocalized.
3 Tissue Expression Patterns for PDE11A
Understanding the tissue expression profile of an enzyme is key when considering it as a therapeutic target. As noted above, tissue expression profiling studies for PDE11A have proven highly inconsistent. Poor tools are likely to blame for many of these discrepant reports. We have shown that Western blots of peripheral tissues probed with various commercially-available PDE11A antibodies demonstrate an abundance of non-specific bands at molecular weights that are consistent with the various PDE11A isoforms (Kelly 2015). The fact that the bands do not disappear in tissue from a PDE11A KO mouse shows these signals are non-specific. Some discrepancies may also be related to the fact that studies have not examined the correct sub-compartment of certain organs. For example, initial studies failed to detect PDE11A expression in brain (Fawcett et al. 2000; Yuasa et al. 2000). Subsequent studies, however, have convincingly shown that PDE11A4 is, in fact, expressed within a small, restricted region of the brain (particularly the ventral hippocampal formation, Kelly et al. 2010; Jager et al. 2012; Kelly et al. 2014). Finally, discrepancies could be related to species differences or a differential expression profile under normal versus disease conditions.
As we have extensively reviewed elsewhere (Kelly 2015), tissue expression profiling studies to date suggest PDE11A expression is relatively restricted throughout the body. PDE11A1 and/or PDE11A2 may be expressed at a moderate level in prostate, pancreas and kidney. PDE11A3 is likely expressed within specific compartments of the testes and seminal vesicles at moderate levels. PDE11A4 is certainly expressed at high levels in the ventral hippocampal formation of brain, and may be expressed at moderate levels in the human—but not mouse—adrenal gland. Limited evidence points to low levels of PDE11A4 being expressed in liver, heart, and pituitary, and moderate levels being expressed in human prostate, but our own studies employing 23 peripheral tissues from PDE11A KO mice as negative controls fail to detect PDE11A4 outside of brain (Kelly 2015). With such a limited tissue expression profile, a PDE11A-targeted therapeutic would stand to impact very selective systems of interest without eliciting wide-ranging toxicological side effects.
PDE11A4 mRNA in brain is almost exclusively expressed in neurons of CA1, possibly CA2, the subiculum and the adjacently connected amygdalohippocampal area (Kelly 2015; Kelly et al. 2010; Hegde et al. 2016a; Kelly et al. 2014; Kelly 2014) (Fig. 8.1). Within these neuronal populations, PDE11A4 protein is found in the cell bodies, axons, and select dendrites (Hegde et al. 2016a) (Fig. 8.2, 8.3, 8.4 and 8.5). Trafficking of PDE11A4 protein to the axons explains why PDE11A4 protein can be detected at miniscule levels in brain regions that receive VHIPP projection but, themselves, lack PDE11A4 mRNA (e.g., prefrontal cortex and striatum) (Kelly 2015; Hegde et al. 2016a). PDE11A4 is the only PDE whose expression in brain originates solely from the hippocampal formation (c.f., Kelly et al. 2014; Kelly 2014; Kelly and Brandon 2009; Xu et al. 2011). In the rodent hippocampus, PDE11A4 expression is minimal on postnatal day 7 but dramatically increases with each postnatal week, reaching young adulthood levels by postnatal day 28 (Hegde et al. 2016a). PDE11A expression stabilizes for a short period of time during young adulthood, but then again significantly increases between young, middle, and late adulthood (Kelly et al. 2014). The fact that PDE11A expression is restricted to the hippocampal formation, at least during young adulthood, suggests a PDE11A-targeted therapeutic may be capable of selectively targeting hippocampal function whilst leaving the rest of the brain undisturbed.

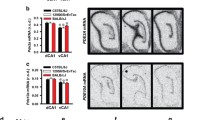
PDE11A4 mRNA expression is restricted to the hippocampal formation, with enrichment in ventral versus dorsal hippocampus. Autoradiographic in situ hybridization was conducted using two 35S-labeled antisense oligonucleotide probes with identical patterns of signal (shown: 5′-gccacctgtctggagatctcccacggtttggtcacggc-3′ recognizing nucleotides 2538-2501 of NM_001081033.1; not shown: 5′-cgcatcaagtaatcttcaaacaactctgggtgcct-3′ recognizing nucleotides 129-95 of NM_001081033.1). Note that no signal is observed in sections from PDE11A knockout (KO) mice, suggesting specificity of the signal detected in sections from PDE11A wild-type (WT) mice. When comparing the autoradiograph to the thionin tissue stain, it is clear that PDE11A mRNA expression is restricted to the hippocampal formation (CA1@, possibly CA2#, subiculum& and the adjacently connected amygdalohippocampal area*—regions indicated on thionin stain with corresponding symbols), with a two to tenfold enrichment in ventral versus dorsal hippocampus. Top panels show sections from ~2.76–2.88 mm lateral from Bregma, and bottom panels show sections from ~2.28–2.40 mm lateral from Bregma. Contrast and brightness were adjusted to enhance the graphical clarity of the images

PDE11A4 protein expression is enriched in the ventral hippocampal formation, with expression throughout the stratum pyramidale, stratum radiatum, and fimbria. Identical staining for PDE11A was obtained in sections from PDE11A wild-type (WT) mice when sections were processed for immunofluorescence using an antibody that recognized all PDE11A isoforms (Fabgennix PD11-112 1:100, shown) or an antibody recognizing only PDE11A4 (AVES PDE11#1 1:5000; Hegde et al. 2016a). Note that minimal staining is observed in sections from PDE11A knockout (KO) mice, suggesting specificity of the signal detected in WT tissue. PDE11A protein is distinctly expressed in CA1@, possibly CA2#, subiculum&, the adjacently connected amygdalohippocampal area*, and the fimbria^), with a two to threefold enrichment in ventral versus dorsal hippocampus. Images collected using a 4× objective. Histogram stretch and gamma were corrected to enhance the graphical clarity of the images. Green—PDE11A, Blue—DAPI nuclear stain

PDE11A4 protein expression within CA1 is found within a subset of neurons, not astrocytes. Immunofluorescence was conducted on sections from PDE11A wild-type (WT) and knockout (KO) mice collected ~2.76–2.88 mm lateral from Bregma. PDE11A protein expression (Fabgennix PD11-112 1:100) clearly colocalizes with a marker of neuronal cytoplasm (AVES neuronal specific enolase (NSE) 1:500) within a subset of neurons in CA1, and possibly CA2, stratum pyramidale (SP; i.e., cell body layer). Note: colocalization of green + red = yellow. PDE11A protein does not appear to colocalize with a marker of astrocytes (AVES glial acidic fibrillary protein (GFAP) 1:500) in SP, stratum radiatum (SR; i.e., the dendritic layer), stratum oriens (SO; i.e., axonal projection layer), or the fimbria (Fi; i.e., projecting axonal bundle). Images collected using a 20× objective. Histogram stretch and gamma were corrected to enhance the graphical clarity of the images. Green—PDE11A, Blue—DAPI nuclear stain, Red—either NSE or GFAP as indicated

PDE11A4 protein expression is seen in the fimbria, a structure mostly composed of hippocampal efferents. Immunofluorescence was conducted on sections from PDE11A wild-type (WT) and knockout (KO) mice collected ~2.76 mm lateral from Bregma. PDE11A protein expression (Fabgennix PD11-112 1:100) is clearly found in the fimbria, as indicated by colocalization with a marker of myelin that labels axon bundles (AVES myelin basic protein (MBP) 1:100). Note: colocalization of green + red = yellow. Two images per section were collected using a 10× objective and then stitched together using Adobe Photoshop. Histogram stretch and gamma were corrected to enhance the graphical clarity of the images. Green—PDE11A, Blue—DAPI nuclear stain; Red—MBP

PDE11A4 protein expression is detected within a subset of dendrites in stratum radiatum. Immunofluorescence was conducted on sections from PDE11A wild-type (WT) and knockout (KO) mice collected ~3.00 mm lateral from Bregma. Punctate PDE11A protein expression (Fabgennix PD11-112 1:100) is clearly found in stratum pyramidale (cell body layer), as indicated by colocalization with a subset of DAPI staining. Fibril-like PDE11A labeling can be found in the stratum radiatum, a structure mostly composed of CA dendrites. Consistent with the fibril-like pattern of PDE11A staining in the stratum radiatum, PDE11A protein expression colocalizes with a marker of dendrites (Neuromics microtubule associated protein 2 (MAP2) 1:2000). Note: colocalization of green + red = yellow. Images were collected using a 20× or 40× objective, as indicated. Histogram stretch and gamma were corrected to enhance the graphical clarity of the images. Green—PDE11A, Blue—DAPI nuclear stain; Red—MAP2
Upon biochemical fractionation of the hippocampus, PDE11A4 can be found in nuclear, cytosolic and membrane compartments (both soluble and insoluble). This compartmentalization of PDE11A4 is modifiable in response to behavioral, genetic, and biochemical manipulations. Relative to single-housed mice, group-housed mice exhibit increased PDE11A4 protein expression in the soluble membrane compartment but not the cytosolic compartment of the VHIPP (Hegde et al. 2016b). Similarly, BALB/cJ mice express significantly more PDE11A4 protein than C57BL/6J mice in the soluble membrane but not cytosolic compartment of VHIPP (Pathak et al. 2016). Although it remains to be determined how social experience elicits such compartment-specific effects on PDE11A4 protein expression, the compartment-specific difference in PDE11A4 protein expression that is observed between C57BL/6J and BALB/cJ mice appears to be largely accounted for by a single point mutation at amino acid 499 within the GAF-B homodimerization domain (Pathak et al. 2016). In the C57BL/6J, 499 is a non-phosphorylatable alanine; however, in the BALB/cJ 499 is a phosphorylatable threonine (Pathak et al. 2016). We showed that the BALB/cJ 499T and corresponding phosphomimic mutation (T499D) shifts PDE11A4 from the cytosol to the membrane, relative to the C57BL/6J 499A, perfectly replicating the PDE11A4 protein expression differences noted between these mouse strains in VHIPP (Pathak et al. 2016). The ability of the 499T mutation to drive PDE11A4 into the membrane appears to be driven by an ability to increase homodimerization of PDE11A4, as 499T increases homodimerization of PDE11A4 relative to 499A and disrupting homodimerization has the opposite effect of the 499T mutation, shifting PDE11A4 from the membrane to the cytosol (Pathak et al. 2016). PDE11A4 can also be shifted between the cytosol and membrane in response to N-terminal phosphorylation events. We have shown that phosphorylation of serines 117 and 124 shifts PDE11A4 from the cytosol to the membrane, while phosphorylation of serine 162 keeps PDE11A4 in the cytosol (Pathak et al. 2016). The ability to therapeutically control the subcellular localization of PDE11A4 may become highly relevant as bipolar disorder, lithium responsiveness, and Alzheimer’s disease have all been associated with subcellular compartment-specific differences in cyclic nucleotide signaling (Rahman et al. 1997; Fields et al. 1999; Chang et al. 2003; Mori et al. 1998; Jensen and Mork 1997; Casebolt and Jope 1991; Bonkale et al. 1999).
Not only is PDE11A4 expression restricted to the hippocampal formation, it is uniquely enriched three to tenfold in ventral versus dorsal hippocampus (Kelly 2015; Kelly et al. 2010; Hegde et al. 2016a; Kelly et al. 2014; Kelly 2014). The extent of the VHIPP versus DHIPP enrichment—that is, whether it is closer to three versus tenfold—depends on the mouse strain examined (Pathak et al. 2016). The ventral hippocampal formation in rodents is analogous to the anterior hippocampal formation in primates (Moser and Moser 1998). As such, we will use the term “ventral-anterior” hippocampal formation (VHIPP) and “dorsal-posterior” hippocampal formation (DHIPP) throughout the remainder of this chapter for consistent referencing of these anatomically and functionally distinct subregions across species.
The VHIPP and DHIPP are functionally, anatomically, and biochemically segregated (e.g., (Roman and Soumireu-Mourat 1988; Papatheodoropoulos and Kostopoulos 2000; Gusev et al. 2005; Bast and Feldon 2003; Fanselow and Dong 2010). While the VHIPP modulates sociality, emotion/affect, motivation, stress, behavioral flexibility, and sensorimotor gating, the DHIPP regulates learning, memory, contextual representation, and spatial navigation (c.f., Moser and Moser 1998; Bast and Feldon 2003; Fanselow and Dong 2010; Tseng et al. 2008; Behrendt 2011) also see (Roman and Soumireu-Mourat 1988; Marquis et al. 2008; Gruber et al. 2010). These separable functions are reflected in, if not driven by, differences in the extrahippocampal connectivity of VHIPP versus DHIPP. Whereas the VHIPP is highly interconnected with the prefrontal cortex, amygdala, olfactory bulb, hypothalamus, and the shell of the nucleus accumbens, the DHIPP is highly interconnected with thalamic subregions, sensory-related cortices, and the core of the nucleus accumbens (c.f., Bast and Feldon 2003; Fanselow and Dong 2010). Biochemical segregation of VHIPP versus DHIPP can easily be visualized when mapping expression of numerous gene products (Fanselow and Dong 2010), including PDE11A4 (Fig. 8.1). Consistent with its enrichment in the VHIPP, emerging evidence from human and rodent studies suggests PDE11A4 is a key regulator of social behaviors and mood stabilization.
4 A Role for PDE11A in Brain Function
Human studies to date largely implicate PDE11A dysfunction in mood disorders (Kelly 2015). Several studies associate PDE11A single nucleotide polymorphisms (SNPs) with major depressive disorder (Wong et al. 2006; Luo et al. 2009b; Cabanero et al. 2009) and 1 study associates an inactivating mutation in PDE11A with suicide risk (Coon et al. 2013). PDE11A has also been nominally associated with fluoxetine and desipramine response (Wong et al. 2006; Luo et al. 2009b), but not with citalopram or duloxetine response (Perlis et al. 2010; Cabanero et al. 2009). Whether the inconsistencies in the latter findings are related to differences in patient populations, differences in the specific antidepressants studied, or some unknown factor remains to be determined. PDE11A SNPs have also been associated with lithium responsiveness in both a retrospective and prospective cohort of patients with bipolar disorder (Couzin 2008; Kelsoe 2010). Further, lithium decreases PDE11A mRNA expression in IPSC-derived hippocampal neurons originating from lithium-responsive patients but not lithium-unresponsive patients (Mertens et al. 2015). As reviewed below, studies in rodents suggest PDE11A may be highly relevant to the social deficits that are associated with mood disorders, and may be particularly important for regulating lithium responsivity.
A link between PDE11A and lithium responsivity is consistent with studies that associate changes in the cAMP cascade with bipolar disorder (Rahman et al. 1997; Fields et al. 1999; Chang et al. 2003; Avissar et al. 1997; Avissar and Schreiber 2006; Schreiber and Avissar 1991; Schreiber et al. 1991; Alda et al. 2013; Dowlatshahi et al. 1999; Fatemi et al. 2010b; Fatemi et al. 2008), including cyclic nucleotide alterations restricted to specific subcellular compartments (Rahman et al. 1997; Fields et al. 1999; Chang et al. 2003) and changes associated with lithium responsivity (Alda et al. 2013; Sun et al. 2004). The link between PDE11A and lithium responsivity is also particularly intriguing in light of the study associating PDE11A SNPs and suicide risk (Coon et al. 2013) because lithium uniquely reduces suicide risk in patients with bipolar disorder (Malhi et al. 2013).
Deletion of PDE11A in mice elicits very selective behavioral deficits. PDE11A KO mice show no differences in basal anxiety- or depression-related behaviors (but see below for drug-induced changes in these behaviors), including elevated plus maze, stress-induced hyperthermia, four-plate, tail suspension test, and forced swim test (Kelly et al. 2010), as well as sucrose preference (WT, 80.3 ± 3.5%; KO, 78.2 ± 3.3%; n = 16-17/genotype; effect of genotype: F(1,28) = 0.022, P = 0.883). They also show no differences in contextual or cued fear conditioning or the motoric response to an acute dopaminergic challenge (Kelly et al. 2010). While single-housed PDE11A KO mice show increased locomotor activity in an open field and normal prepulse inhibition (PPI) of acoustic startle relative to WT littermates (Kelly et al. 2010), group-housed PDE11A KO mice show normal locomotor activity in an open field (WT,11,187 ± 455 cm; KO, 10,978 ± 486 cm; n = 21–25/genotype; effect of genotype (F(1,41) = 0.098, P = 0.756) and modestly lower PPI at the lowest prepulse intensity relative to PDE11A WT mice (PPI at 4 db above background: WT, 16.49 ± 2.76%; KO, 3.85 ± 2.92%; n = 50–55/genotype; genotype × prepulse intensity: F(2207) = 3.59, P = 0.029; Post hoc WT vs. KO within 4 db above background, P = 0.02). The most notable and reliable phenotypes exhibited by PDE11A KO mice, however, are impairments in social behaviors (Kelly et al. 2010; Hegde et al. 2016a; Hegde et al. 2016b).
PDE11A is required for intact social interactions and is a key mechanism by which social experience, particularly social isolation, shapes the brain. Using Brodkin’s version of the 3-chamber social approach assay, we found that deletion of PDE11A reduces social approach behaviors towards stranger mice but not towards cagemates (Kelly et al. 2010; Hegde et al. 2016b). The reduced social approach observed in PDE11A heterozygous (HT) and knockout (KO) mice appears somewhat sensitive to the effects of social buffering and depends on the social context (i.e., the strain of the stimulus mouse). Presence of a cagemate improves social approach behavior of PDE11A KO mice towards a novel mouse from the colony but not towards a novel C57BL/6J; whereas, presence of a cagemate improves social approach behavior of PDE11A HT mice towards a C57BL/6J but not a novel mouse from the colony (Hegde et al. 2016b). Other mice appear to be capable of detecting abnormalities in the social behavior of PDE11A KO mice, because male and female C57BL/6J spend significantly more time with a sex-matched PDE11A WT mouse vs its KO littermate (Hegde et al. 2016b), suggesting the PDE11A KO mice may be socially isolated. We believe this finding is particularly intriguing given the PDE11A SNP that has been associated with increased suicide risk in a Utah pedigree is an inactivating mutation (Coon et al. 2013). As noted above, social isolation decreases PDE11A4 protein expression within the membrane fraction of VHIPP samples relative to group-housed mice, and this isolation-induced decrease in PDE11A4 expression is sufficient to impair subsequent social behavior—both social approach and social memory formation (Hegde et al. 2016b). Consistent with these behavioral phenotypes, RNA sequencing of VHIPP from PDE11A KO and WT littermates shows that PDE11A regulates gene expression in the oxytocin signaling pathway (Hegde et al. 2016b), a key signaling cascade underlying social behaviors (c.f. Ebstein et al. 2012; Viero et al. 2010; Lukas and Neumann 2013; Stoesz et al. 2013).
We have also shown that PDE11A is required for the formation of social memories (Kelly et al. 2010; Hegde et al. 2016a). Although PDE11A KO mice show intact short-term memory for social odor recognition (SOR) or social transmission of food preference (STFP), PDE11A KO mice fail to show any long-term memory for SOR or STFP 24 h following training. Such a pattern suggests PDE11A KO mice retain the ability to learn and retrieve each memory but fail in their ability consolidate these memories to a long-lasting form (Hegde et al. 2016a). In contrast, PDE11A KO mice show perfectly intact contextual fear conditioning (Kelly et al. 2010) and non-social odor recognition memory 24 h after training (Hegde et al. 2016a), suggesting the SOR and STFP memory consolidation impairments are selective and due to the social nature of the stimuli employed. There are several possible explanations for why PDE11A KO mice exhibit such a selective memory deficit. As noted above, the social deficits present in the PDE11A mutant mice are consistent with the fact that PDE11A4 is enriched in the VHIPP. The social deficits are also consistent with our RNA sequencing study showing that PDE11A KO mice exhibit significantly altered gene expression in the oxytocin pathway (Hegde et al. 2016b). Indeed, like our PDE11A KO mice, oxytocin KO mice similarly show an inability to form social memories while their ability to form non-social memories remains intact (Ferguson et al. 2000). The memory impairments observed in PDE11A KO mice may also be related to altered glutamatergic signaling. Previously, we showed that PDE11A KO mice are more sensitive to the hyperactivating effects of the NMDA receptor antagonist MK-801 (Kelly et al. 2010). We have also shown that deleting PDE11A impairs de novo protein synthesis, which is required to transition memory from a short-term to a long-term state. PDE11A KO mice show significantly reduced expression of the ribosomal S6 kinase 2 (RSK2) and phosphorylation of the ribosomal protein S6 at serines 235/236 relative to WT littermates (Hegde et al. 2016a). It will be of interest to future studies to determine which of these mechanisms accounts for social memory deficits in PDE11A KO mice and if those deficits can be reversed.
Interestingly, the manifestation of social memory deficits precedes the manifestation of social approach deficits in PDE11A mutant mice. That is, adolescent PDE11A KO mice already have an inability to form social long-term social memories (Hegde et al. 2016a) but show no differences in their social approach behavior, relative to WT littermates (Hegde et al. 2016b). This raises the interesting possibility that adolescent PDE11A mutant mice have a fundamental inability to form long-term memories for what is and what is not a socially-acceptable behavior, which ultimately leads to alterations in social approach behavior at a later age. Such a relationship has been observed in humans where impaired social skills early in development predict lower quality friendships in adolescents with intellectual disabilities (Tipton et al. 2013). It will be of interest to future studies to determine if social interaction deficits observed in adult PDE11A mutant mice are, in fact, directly related to their inability to form social memories.
As noted above, PDE11A has been genetically and functionally associated with lithium responsivity in patients with bipolar disorder (Couzin 2008; Mertens et al. 2015; Kelsoe 2010). In mice, we have shown that PDE11A4 negatively regulates lithium responsivity (Pathak et al. 2016). PDE11A4 mRNA and protein expression negatively correlate with lithium responsivity such that C57BL/6J mice that respond well to lithium exhibit lower levels of PDE11A4 expression in hippocampus than do BALB/cJ mice that respond poorly to lithium (Pathak et al. 2016). Further, using PDE11A mutant mice, we have shown that decreasing PDE11A expression is, in fact, sufficient to increase lithium response (Pathak et al. 2016). At face value, this may suggest that a PDE11A inhibitor might augment the therapeutic effects of lithium. Indeed, AKT—an enzyme whose activation is required for lithium’s behavioral effects (Pan et al. 2011)—is predicted to phosphorylate PDE11A at serines 117 and 124 (Kelly 2015). As noted above, phosphorylation of S117/S124 would shift PDE11A4 towards the membrane compartment (Pathak et al. 2016), thereby removing it from the cytosol where AKT is selectively located. Shifting PDE11A from the cytosol to the membrane could serve to extend the activation of AKT (i.e., its phosphorylation), because PDE11A4 would no longer be in a position to negatively regulate the cytosolic pools of cAMP that lead to phosphorylation of AKT in neurons via Epac (Nijholt et al. 2008). Of course, deleting PDE11A4 would artificially augment such feedforward signaling. That said, it is equally possible that lowered PDE11A expression may simply trigger a specific pathophysiology that happens to be particularly well-treated by lithium (Pathak et al. 2016). Indeed, both PDE11A KO mice and IPSC-derived hippocampal neurons from bipolar patients exhibit altered signaling in the neuroactive-ligand receptor pathway and increased neural activity (Kelly et al. 2010; Hegde et al. 2016b; Mertens et al. 2015). Further, decreasing PDE11A4 expression is sufficient to upregulate the proinflammatory cytokine IL-6 (Pathak et al. 2016), increased signaling of which has been repeatedly measured in patients with mania, depression, and suicidal ideation (Dowlati et al. 2010; Maes et al. 1997; Maes et al. 1995; Brietzke et al. 2009; Simon et al. 2008; Niculescu et al. 2015; Khandaker et al. 2014) and is reversed by lithium (Watanabe et al. 2014). Thus, studies in mice are most consistent with the notion that lowered levels of PDE11A trigger a specific pathophysiology that is readily treated by lithium.
PDE11A inactivating mutations have been identified in patients with adrenocortical tumors (Vezzosi et al. 2012; Horvath et al. 2006a; Horvath et al. 2006b; Libe et al. 2008; Libe et al. 2011) but see (Bimpaki et al. 2009; Louiset et al. 2010), so it is also worth noting that one study has examined the anterior hippocampus in patients with Cushing Syndrome, albeit not patients genotyped with regard to PDE11A mutations. Maheu and colleagues (Maheu et al. 2008) found heightened functional activation of the VHIPP during encoding of an emotional-faces recognition test in patients with Cushing Syndrome. As noted above, PDE11A KO mice show deficits in social odor recognition memory and social transmission of food preference (Kelly et al. 2010; Hegde et al. 2016a). PDE11A KO mice also show heightened activation of ventral CA1 relative to WT littermates, as indicated by increased expression of the activity-regulated immediate-early gene Arc (Kelly et al. 2010). In this context, it is important to note that our PDE11A KO mice do not appear to develop any adrenal tumor pathology (Kelly et al. 2010), consistent with the fact that the mouse adrenal gland does not express PDE11A (Kelly 2015), suggesting it is the loss of PDE11A4 in the brain, as opposed to adrenal dysfunction, that is driving the neurocognitive deficits of PDE11A KO mice. Studies in patients with Cushing Syndrome have correlated overall hippocampal function with dysregulated cortisol levels (Starkman et al. 1999; Starkman et al. 1992; Forget et al. 2000; Starkman et al. 2001); however, a potential role for aberrant PDE11A signaling within the VHIPP has not yet been examined. Unfortunately, to our knowledge, hippocampal function has not been assessed in patients genotyped with PDE11A inactivating mutations. Thus, it remains to be determined whether hippocampal deficits observed in patients with Cushing Syndrome are simply related to the indirect effects of rising peripheral cortisol levels or the direct effects of PDE11A inactivation within the hippocampus.
Ectopic PDE11A expression in brain regions outside of the hippocampus may also be an important facet of disease pathophysiology. Fatemi and colleagues reported an increase in PDE11A mRNA expression in the cerebellum of patients with bipolar disorder relative to controls (Fatemi et al. 2010a). We are unable to detect PDE11A expression in cerebellum taken from healthy humans or rodents, suggesting that a disease state may drive ectopic expression of PDE11A outside of the hippocampal formation (Kelly 2015). Our recent findings and ongoing studies in the lab suggest that aging drives ectopic expression of PDE11A4 outside of the hippocampal formation (Kelly et al. 2014), with negative consequences. If this holds true, it may be important to not simply modulate catalytic activity of PDE11A4 but rather to restore proper physiological control over the mechanisms controlling PDE11A4’s unique tissue-specific expression profile.
5 PDE11A Pharmacological Tools
As extensively reviewed elsewhere (Kelly 2015), progress has been made in identifying pharmacological activators and inhibitors of PDE11A catalytic activity. Jager and colleagues used an in vitro FRET-based assay to screen compounds for their ability to bind to and initiate a conformational change in a fragment of PDE11A4 that included both GAF domains (Jager et al. 2012). While cAMP failed to bind the PDE11A4 GAF construct, both cGMP and the cGMP analog Rp-8-pCPT-PET-cGMPS did (Jager et al. 2012). In a catalytic activity assay, cGMP failed to stimulate PDE11A4 catalytic activity. In contrast, Rp-8-pCPT-PET-cGMPS stimulated PDE11A catalytic activity of both recombinant human PDE11A4 as well as native mouse PDE11A4 enriched from hippocampus approximately four to fivefold (Jager et al. 2012). Interestingly, Rp-8-pCPT-PET-cGMPS failed to activate PDE11A when the 196 amino acids upstream of the PDE11A4 GAF-A domain were deleted (Jager et al. 2012). This suggests that it will be important for PDE11A compound screens to employ full-length PDE11A4, as opposed to some truncated version of the protein. Although Rp-8-pCPT-PET-cGMPS does not appear to bind the GAF domains of PDE5A or PDE2A (Jager et al. 2010), suggesting specificity in its ability to target the GAF domains of PDE11A4, it is well known to have significant off-target activities, including inhibition of PKG and cGMP-gated ion channels (see product insert for Rp-8-pCPT-PET-cGMPS on www.biolog.de). Although this off-target activity severely limits the use of Rp-8-pCPT-PET-cGMPS as a tool PDE11A activator, this study provides critical proof-of-concept that it is possible to stimulate PDE11A4 catalytic activity.
Ceyhan et al. identified four PDE11A4 inhibitors using a yeast-based high-throughput assay (Ceyhan et al. 2012). All four compounds were fairly potent (IC50s = 0.11–0.33 μM) and BC11-28 and BC11-38, in particular, were highly selective for PDE11A4 versus other PDE families (>350-fold selective for PDE11A) (Ceyhan et al. 2012). One strength of this screen was the fact that it employed full-length PDE11A4, as opposed to an isolated catalytic domain; therefore, these compounds are likely to inhibit the native protein. That said, because the screen was conducted with the full-length PDE11A4, it is not possible to determine whether the inhibitors compete for the substrate binding pocket or modulate catalytic activity via binding to an allosteric site (e.g., the GAF-A domain) (Ceyhan et al. 2012). PDE11A cGMP hydrolytic activity was inhibited by all four compounds in both the yeast-based assay and an enzyme assay. In an H295R cell-based assay, however, only BC11-38 was able to inhibit PDE11A cAMP hydrolytic activity (Ceyhan et al. 2012). The fact that all four compounds were able to inhibit cGMP-PDE11A activity in the yeast-based assay but only one was able to inhibit cAMP-PDE11A activity in the H295R-based assay may simply be related to differential availability of the four PDE11A inhibitors in the two different media employed in the respective assays (i.e, differences in solubility or freely available compound) (Ceyhan et al. 2012). An alternative possibility, reviewed elsewhere in great detail (Kelly 2015), is that certain compounds may be functionally selective in their ability to inhibit cAMP-PDE11A versus cGMP-PDE11A catalytic activity. While many compounds, such as IBMX, zaprinast, dipyrimadole, and a few PDE5A-preferring inhibitors, inhibit cAMP-PDE11A and cGMP-PDE11A activity with equal potency, several other PDE5A-preferring inhibitors require much higher concentrations to inhibit the cAMP-hydrolyzing activity of PDE11A versus its cGMP-hydrolyzing activity (Yuasa et al. 2000; Ahmed et al. 2011; Ahmed et al. 2012; Mohamed et al. 2011). Clearly the BC compounds are membrane-penetrant; however, their ability to cross the blood-brain-barrier has not been determined. Indeed no studies have yet reported in vivo activities of the compounds or their pharmacokinetic profiles. Although it remains to be determined whether these PDE11A inhibitors will prove useful for in vivo experiments, their identification certainly proves it is possible to inhibit PDE11A with a high degree of selectivity versus other PDEs.
6 Future Considerations for PDE11A Research
In recent years we have learned much about PDE11A4 function in the brain, but many questions remain. Considering the number of conflicting findings in the field regarding the tissue expression profile of PDE11A, it will be critical to continue carefully assessing which tissues and cell types express the various PDE11A isoforms, and how those expression patterns may change with age or disease state. Given how dramatically PDE11A4 expression increases in the hippocampus across the lifespan (Kelly et al. 2010; Kelly et al. 2014), the “when and where” of PDE11A4 expression will be critically important when evaluating the efficacy and side effect potential of a PDE11A4-targeted therapeutic. It will also be key in understanding whether PDE11A4 may be considered a therapeutic target in the context of neurodevelopmental, adulthood, or age-related disease. Although emerging evidence shows that phosphorylation events and homodimerization can alter the subcellular compartmentalization of PDE11A4 (Pathak et al. 2016; Pathak et al. 2015), it is not yet clear if these mechanisms also affect catalytic activity and/or whether they can be therapeutically exploited to restore aberrant subcellular localization of the enzyme (e.g., in the case of social isolation; Hegde et al. 2016b). That said, the GAF domains are highly intriguing from a drug target perspective because they are found in no other mammalian proteins other than PDEs (Francis et al. 2011), and the PDE11A4 GAF domains are less than 50% homologous to those in other PDE families (Viero et al. 2010). This, together with the fact that PDE11A4 is the only PDE11A isoform with a full GAF-A domain, suggests it might not only be possible to selectively target PDE11A4 over other PDE families, but also selectively target PDE11A4 relative PDE11A3, A2 or A1. With such specific targeting, it might be possible to reduce, if not eliminate, side effect liability associated with targeting other PDEs or PDE11A signaling outside the brain. Finally, we need to gain a far better understanding of signaling events that lie upstream and downstream of PDE11A, and how coupling to those signaling events may change with age, disease, and acute versus chronic manipulations.
7 Summary
Evidence is mounting that PDE11A critically regulates select aspects of brain function. Our studies in PDE11A KO mice show that PDE11A plays a crucial role in the formation of social memories and lithium responsiveness and is a key mechanism by which social experience feeds back to shape the brain (Kelly et al. 2010; Hegde et al. 2016a; Pathak et al. 2016; Hegde et al. 2016b). These finding are consistent with the fact that human studies associate PDE11A with lithium responsiveness, MDD, and suicide risk (Couzin 2008; Wong et al. 2006; Mertens et al. 2015; Luo et al. 2009b; Cabanero et al. 2009; Coon et al. 2013; Kelsoe 2010; Aitchison et al. 2009). As extensively reviewed elsewhere (Kelly 2015), there are a large number of studies reporting VHIPP dysfunction as well as cyclic nucleotide dysregulation in patients with neuropsychiatric disease, particularly schizophrenia and Alzheimer’s disease (Lee et al. 2004; Pegues et al. 2003; Suddath et al. 1990; Rametti et al. 2007; Schobel et al. 2009a; Shenton et al. 1992; Nesvaderani et al. 2009; Ghose et al. 2009; Zhou et al. 2008; Goldman et al. 2011; Schobel et al. 2009b; Jessen et al. 2003; Hall et al. 2010; Rajarethinam et al. 2001; Rusch et al. 2008; Laakso et al. 2000; Leube et al. 2008; Jansen et al. 1993; Quiroz et al. 2010; Yakushev et al. 2011a; Yakushev et al. 2011b; Yakushev et al. 2010; Rahman et al. 1997; Avissar et al. 1997; Garver et al. 1982; Kafka et al. 1979; Kafka et al. 1986; Kanof et al. 1986; Kafka and van Kammen 1983; Ofuji et al. 1989; Kaiya 1992; Kaiya et al. 1990; Kanof et al. 1989; Kang 1990; Kanof et al. 1987; Bowers and Study 1979; Belmaker et al. 1978; Gattaz et al. 1983; Ebstein et al. 1976; Turetsky and Moberg 2009; Memo et al. 1983; Avissar et al. 2001a; Avissar et al. 2001b; Edmunds et al. 2008; Young et al. 1991; Young et al. 1993; Young et al. 1994; Cowburn et al. 1994; Gurguis et al. 1999a; Gurguis et al. 1999b; Gurguis et al. 1997; Bonkale et al. 1996; Shanahan et al. 1997; Yamamoto et al. 2000; Fowler et al. 1995; Ohm et al. 1989; Baltrons et al. 2004; Ohm et al. 1991; Martinez et al. 1999). Certainly, a PDE11A-targeted therapeutic would be positioned to address both. Clearly, future studies should determine which, if any, neuropsychiatric disorders are associated with alterations in PDE11A expression, localization, and/or catalytic activity or if a PDE11A-targeted therapeutic could compensate for upstream or downstream insults (e.g., at the AMPA receptor). Clearly more studies examining PDE11A in patient tissue are needed to drive a therapeutic indication for a PDE11A modulator, but we believe the data to date suggest PDE11A holds potential as a future therapeutic target (Kelly 2015).
References
Ahmed NS, Gary BD, Tinsley HN, Piazza GA, Laufer S, Abadi AH. Design, synthesis and structure-activity relationship of functionalized tetrahydro-beta-carboline derivatives as novel PDE5 inhibitors. Arch Pharm (Weinheim). 2011;344:149–57.
Ahmed NS, Ali AH, El-Nashar SM, Gary BD, Fajardo AM, Tinsley HN, Piazza GA, Negri M, Abadi AH. Exploring the PDE5 H-pocket by ensemble docking and structure-based design and synthesis of novel beta-carboline derivatives. Eur J Med Chem. 2012;57:329–43.
Aitchison K, Serretti A, Goldman D, Curran S, Drago A, Malhotra AK. The 8th annual pharmacogenetics in psychiatry meeting report. Pharmacogenomics J. 2009;9:358–61.
Alda M, Shao L, Wang JF, Lopez de Lara C, Jaitovich-Groisman I, Lebel V, Sun X, Duffy A, Grof P, Rouleau GA, Turecki G, Young LT. Alterations in phosphorylated cAMP response element-binding protein (pCREB) signaling: an endophenotype of lithium-responsive bipolar disorder? Bipolar Disord. 2013;15:824–31.
Alevizaki M, Stratakis CA. Multiple endocrine neoplasias: advances and challenges for the future. J Intern Med. 2009;266:1–4.
Almeida MQ, Stratakis CA. How does cAMP/protein kinase A signaling lead to tumors in the adrenal cortex and other tissues? Mol Cell Endocrinol. 2011;336:162–8.
Avissar S, Schreiber G. The involvement of G proteins and regulators of receptor-G protein coupling in the pathophysiology, diagnosis and treatment of mood disorders. Clin Chim Acta. 2006;366:37–47.
Avissar S, Nechamkin Y, Barki-Harrington L, Roitman G, Schreiber G. Differential G protein measures in mononuclear leukocytes of patients with bipolar mood disorder are state dependent. J Affect Disord. 1997;43:85–93.
Avissar S, Barki-Harrington L, Nechamkin Y, Roitman G, Schreiber G. Elevated dopamine receptor-coupled G(s) protein measures in mononuclear leukocytes of patients with schizophrenia. Schizophr Res. 2001a;47:37–47.
Avissar S, Roitman G, Schreiber G. Differential effects of the antipsychotics haloperidol and clozapine on G protein measures in mononuclear leukocytes of patients with schizophrenia. Cell Mol Neurobiol. 2001b;21:799–811.
Baltrons MA, Pifarre P, Ferrer I, Carot JM, Garcia A. Reduced expression of NO-sensitive guanylyl cyclase in reactive astrocytes of Alzheimer disease, Creutzfeldt-Jakob disease, and multiple sclerosis brains. Neurobiol Dis. 2004;17:462–72.
Bast T, Feldon J. Hippocampal modulation of sensorimotor processes. Prog Neurobiol. 2003;70:319–45.
Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol. 2010;47:678–84.
Beavo JA, Hardman JG, Sutherland EW. Stimulation of adenosine 3′,5′-monophosphate hydrolysis by guanosine 3′,5′-monophosphate. J Biol Chem. 1971;246:3841–6.
Behrendt R-P. Neuroanatomy of social behavior: an evolutionary and psychoanalytic perspective. London: Karnac Books; 2011.
Belmaker RH, Ebstein RP, Biederman J, Stern R, Berman M, van Praag HM. The effect of L-dopa and propranolol on human CSF cyclic nucleotides. Psychopharmacology. 1978;58:307–10.
Bimpaki EI, Nesterova M, Stratakis CA. Abnormalities of cAMP signaling are present in adrenocortical lesions associated with ACTH-independent Cushing syndrome despite the absence of mutations in known genes. Eur J Endocrinol. 2009;161:153–61.
Bonkale WL, Fastbom J, Wiehager B, Ravid R, Winblad B, Cowburn RF. Impaired G-protein-stimulated adenylyl cyclase activity in Alzheimer’s disease brain is not accompanied by reduced cyclic-AMP-dependent protein kinase A activity. Brain Res. 1996;737:155–61.
Bonkale WL, Cowburn RF, Ohm TG, Bogdanovic N, Fastbom J. A quantitative autoradiographic study of [3H]cAMP binding to cytosolic and particulate protein kinase A in post-mortem brain staged for Alzheimer’s disease neurofibrillary changes and amyloid deposits. Brain Res. 1999;818:383–96.
Bowers MB Jr, Study RE. Cerebrospinal fluid cyclic AMP and acid monoamine metabolites following probenecid: studies in psychiatric patients. Psychopharmacology. 1979;62:17–22.
Brietzke E, Stertz L, Fernandes BS, Kauer-Sant'anna M, Mascarenhas M, Escosteguy Vargas A, Chies JA, Kapczinski F. Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord. 2009;116:214–7.
Cabanero M, Laje G, Detera-Wadleigh S, McMahon FJ. Association study of phosphodiesterase genes in the Sequenced Treatment Alternatives to Relieve Depression sample. Pharmacogenet Genomics. 2009;19:235–8.
Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). 2010; 34: 547-555.
Casebolt TL, Jope RS. Effects of chronic lithium treatment on protein kinase C and cyclic AMP-dependent protein phosphorylation. Biol Psychiatry. 1991;29:233–43.
Ceyhan O, Birsoy K, Hoffman CS. Identification of biologically active PDE11-selective inhibitors using a yeast-based high-throughput screen. Chem Biol. 2012;19:155–63.
Chang A, Li PP, Warsh JJ. Altered cAMP-dependent protein kinase subunit immunolabeling in post-mortem brain from patients with bipolar affective disorder.[erratum appears in J Neurochem 2003 Apr;85(1):286]. J Neurochem. 2003;84:781–91.
Coon H, Darlington T, Pimentel R, Smith KR, Huff CD, Hu H, Jerominski L, Hansen J, Klein M, Callor WB, Byrd J, Bakian A, Crowell SE, McMahon WM, Rajamanickam V, Camp NJ, McGlade E, Yurgelun-Todd D, Grey T, Gray D. Genetic risk factors in two Utah pedigrees at high risk for suicide. Transl Psychiatry. 2013;3:e325.
Couzin J. Science and commerce. Gene tests for psychiatric risk polarize researchers. Science. 2008;319:274–7.
Cowburn RF, Marcusson JO, Eriksson A, Wiehager B, O’Neill C. Adenylyl cyclase activity and G-protein subunit levels in postmortem frontal cortex of suicide victims. Brain Res. 1994;633:297–304.
D’Amours MR, Cote RH. Regulation of photoreceptor phosphodiesterase catalysis by its non-catalytic cGMP-binding sites. Biochem J. 1999;340(Pt 3):863–9.
DeWan AT, Triche EW, Xu X, Hsu LI, Zhao C, Belanger K, Hellenbrand K, Willis-Owen SA, Moffatt M, Cookson WO, Himes BE, Weiss ST, Gauderman WJ, Baurley JW, Gilliland F, Wilk JB, O'Connor GT, Strachan DP, Hoh J, Bracken MB. PDE11A associations with asthma: results of a genome-wide association scan. J Allergy Clin Immunol, 2010; 126: 871-73.e9.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Dowlatshahi D, MacQueen GM, Wang JF, Reiach JS, Young LT. G Protein-coupled cyclic AMP signaling in postmortem brain of subjects with mood disorders: effects of diagnosis, suicide, and treatment at the time of death. J Neurochem. 1999;73:1121–6.
Ebstein RP, Biederman J, Rimon R, Zohar J, Belmaker RH. Cyclic GMP in the CSF of patients with schizophrenia before and after neuroleptic treatment. Psychopharmacology. 1976;51:71–4.
Ebstein RP, Knafo A, Mankuta D, Chew SH, Lai PS. The contributions of oxytocin and vasopressin pathway genes to human behavior. Horm Behav. 2012;61:359–79.
Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30:519–29.
Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19.
Fatemi SH, Reutiman TJ, Folsom TD, Lee S. Phosphodiesterase-4A expression is reduced in cerebella of patients with bipolar disorder. Psychiatr Genet. 2008;18:282–8.
Fatemi SH, Folsom TD, Reutiman TJ, Vazquez G. Phosphodiesterase signaling system is disrupted in the cerebella of subjects with schizophrenia, bipolar disorder, and major depression. 2010a; 119: 266-267.
Fatemi SH, Folsom TD, Reutiman TJ, Vazquez G. Phosphodiesterase signaling system is disrupted in the cerebella of subjects with schizophrenia, bipolar disorder, and major depression. Schizophr Res. 2010b;119:266–7.
Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libe R, Raffin-Sanson ML, Bertherat J, Carraro DM, Soares FA, Molina GD, Campos AH, Alexandre RB, Bendhack ML, Nesterova M, Stratakis CA. Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J Clin Endocrinol Metabol. 2011;96:E135–40.
Fawcett L, Baxendale R, Stacey P, McGrouther C, Harrow I, Soderling S, Hetman J, Beavo JA, Phillips SC. Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc Natl Acad Sci U S A. 2000;97(7):3702.
Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet. 2000;25:284–8.
Fields A, Li PP, Kish SJ, Warsh JJ. Increased cyclic AMP-dependent protein kinase activity in postmortem brain from patients with bipolar affective disorder. J Neurochem. 1999;73:1704–10.
Forget H, Lacroix A, Somma M, Cohen H. Cognitive decline in patients with Cushing's syndrome. J Int Neuropsychol Soc. 2000;6:20–9.
Fowler CJ, Cowburn RF, Garlind A, Winblad B, O’Neill C. Disturbances in signal transduction mechanisms in Alzheimer’s disease. Mol Cell Biochem. 1995;149–150:287–92.
Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91:651–90.
Garver DL, Johnson C, Kanter DR. Schizophrenia and reduced cyclic AMP production: evidence for the role of receptor-linked events. Life Sci. 1982;31:1987–92.
Gattaz WF, Cramer H, Beckmann H. Low CSF concentrations of cyclic GMP in schizophrenia. Br J Psychiatry. 1983;142:288–91.
Ghose S, Chin R, Gallegos A, Roberts R, Coyle J, Tamminga C. Localization of NAAG-related gene expression deficits to the anterior hippocampus in schizophrenia. Schizophr Res. 2009;111:131–7.
Goldman MB, Wang L, Wachi C, Daudi S, Csernansky J, Marlow-O'Connor M, Keedy S, Torres I. Structural pathology underlying neuroendocrine dysfunction in schizophrenia. Behav Brain Res. 2011;218:106–13.
Greene MH, Kratz CP, Mai PL, Mueller C, Peters JA, Bratslavsky G, Ling A, Choyke PM, Premkumar A, Bracci J, Watkins RJ, McMaster ML, Korde LA. Familial testicular germ cell tumors in adults: 2010 summary of genetic risk factors and clinical phenotype. 2010; 17: R109-R121.
Gross-Langenhoff M, Hofbauer K, Weber J, Schultz A, Schultz JE. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J Biol Chem. 2006;281:2841–6.
Gross-Langenhoff M, Stenzl A, Altenberend F, Schultz A, Schultz JE. The properties of phosphodiesterase 11A4 GAF domains are regulated by modifications in its N-terminal domain. FEBS J. 2008;275:1643–50.
Gruber AJ, Calhoon GG, Shusterman I, Schoenbaum G, Roesch MR, O'Donnell P. More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J Neurosci. 2010;30:17102–10.
Gurguis GN, Turkka J, George DT, Linnoila M. Beta-adrenoreceptor coupling to GS protein in alcohol dependence, panic disorder, and patients with both conditions. Neuropsychopharmacology. 1997;16:69–76.
Gurguis GN, Blakeley JE, Antai-Otong D, Vo SP, Orsulak PJ, Petty F, Rush AJ. Adrenergic receptor function in panic disorder. II. Neutrophil beta 2 receptors: Gs protein coupling, effects of imipramine treatment and relationship to treatment outcome. J Psychiatr Res. 1999a;33:309–22.
Gurguis GN, Vo SP, Blakeley J, Orsulak PJ, Rush AJ. Characteristics of norepinephrine and clonidine displacement of [3H]yohimbine binding to platelet alpha2-adrenoreceptors in healthy volunteers. Psychiatry Res. 1999b;85:305–14.
Gusev PA, Cui C, Alkon DL, Gubin AN. Topography of Arc/Arg3.1 mRNA expression in the dorsal and ventral hippocampus induced by recent and remote spatial memory recall: dissociation of CA3 and CA1 activation. J Neurosci. 2005;25:9384–97.
Hall J, Whalley HC, Marwick K, McKirdy J, Sussmann J, Romaniuk L, Johnstone EC, Wan HI, McIntosh AM, Lawrie SM. Hippocampal function in schizophrenia and bipolar disorder. Psychol Med. 2010;40:761–70.
Hegde S, Capell WR, Ibrahim BA, Klett J, Patel NS, Sougiannis AT, et al. Phosphodiesterase 11A4 (PDE11A4) in hippocampus is required for the consolidation of social but not non-social memories. Neuropsychopharmacology. 2016a; 41:2920–31.
Hegde SO, Ji H, Oliver D, Patel NS, Poupore N, Shtutman M, Kelly MP. PDE11A is required for intact social behaviors and is a key mechanism by which social experience sculpts the brain. Neuroscience. 2016b;335:151–69.
Heikaus CC, Pandit J, Klevit RE. Cyclic nucleotide binding GAF domains from phosphodiesterases: structural and mechanistic insights. Structure. 2009;17:1551–7.
Hetman JM, Robas N, Baxendale R, Fidock M, Phillips SC, Soderling SH, Beavo JA. Cloning and characterization of two splice variants of human phosphodiesterase 11A. Proc Natl Acad Sci U S A. 2000;97:12891–5.
Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res. 2006a;66:11571–5.
Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libe R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006b;38:794–800.
Horvath A, Korde L, Greene MH, Libe R, Osorio P, Faucz FR, Raffin-Sanson ML, Tsang KM, Drori-Herishanu L, Patronas Y, Remmers EF, Nikita ME, Moran J, Greene J, Nesterova M, Merino M, Bertherat J, Stratakis CA. Functional phosphodiesterase 11A mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res. 2009;69:5301–6.
Jager R, Schwede F, Genieser HG, Koesling D, Russwurm M. Activation of PDE2 and PDE5 by specific GAF ligands: delayed activation of PDE5. Br J Pharmacol. 2010;161:1645–60.
Jager R, Russwurm C, Schwede F, Genieser HG, Koesling D, Russwurm M. Activation of PDE10 and PDE11 phosphodiesterases. J Biol Chem. 2012;287:1210–9.
Jansen KL, Faull RL, Storey P, Leslie RA. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer’s disease correlates with CA1 pyramidal cell loss. Brain Res. 1993;623:299–302.
Jensen JB, Mork A. Altered protein phosphorylation in the rat brain following chronic lithium and carbamazepine treatments. Eur Neuropsychopharmacol. 1997;7:173–9.
Jessen F, Scheef L, Germeshausen L, Tawo Y, Kockler M, Kuhn KU, Maier W, Schild HH, Heun R. Reduced hippocampal activation during encoding and recognition of words in schizophrenia patients. Am J Psychiatry. 2003;160:1305–12.
Kafka MS, van Kammen DP. alpha-Adrenergic receptor function in schizophrenia. Receptor number, cyclic adenosine monophosphate production, adenylate cyclase activity, and effect of drugs. Arch Gen Psychiatry. 1983;40:264–70.
Kafka MS, van Kammen DP, Bunney WE Jr. Reduced cyclic AMP production in the blood platelets from schizophrenic patients. Am J Psychiatry. 1979;136:685–7.
Kafka MS, Kleinman JE, Karson CN, Wyatt RJ. Alpha-adrenergic receptors and cyclic AMP production in a group of schizophrenic patients. Hillside J Clin Psychiatry. 1986;8:15–24.
Kaiya H. Second messenger imbalance hypothesis of schizophrenia. Prostaglandins Leukot Essent Fatty Acids. 1992;46:33–8.
Kaiya H, Ofuji M, Nozaki M, Tsurumi K. Platelet prostaglandin E1 hyposensitivity in schizophrenia: decrease in cyclic AMP formation and in inhibitory effects on aggregation. Psychopharmacol Bull. 1990;26:381–4.
Kang WH. Assaying the SCF platelets cyclic nucleotides of schizophrenics and analyzing its correlation with pathopschological factors. Zhonghua Shen Jing Jing Shen Ke Za Zhi. 1990;23:266–8. 318
Kanof PD, Johns C, Davidson M, Siever LJ, Coccaro EF, Davis KL. Prostaglandin receptor sensitivity in psychiatric disorders. Arch Gen Psychiatry. 1986;43:987–93.
Kanof PD, Davidson M, Johns CA, Mohs RC, Davis KL. Clinical correlates of platelet prostaglandin receptor subsensitivity in schizophrenia. Am J Psychiatry. 1987;144:1556–60.
Kanof PD, Coccaro EF, Johns CA, Davidson M, Siever LJ, Davis KL. Cyclic-AMP production by polymorphonuclear leukocytes in psychiatric disorders. Biol Psychiatry. 1989;25:413–20.
Kelly MP. Putting together the pieces of phosphodiesterase distribution patterns in the brain: a jigsaw puzzle of cyclic nucleotide regulation. In: Brandon NJ, West AR, editors. Cyclic nucleotide phosphodiesterases in the central nervous system: from biology to disease. Wiley, Hoboken; 2014.
Kelly MP. Does phosphodiesterase 11A (PDE11A) hold promise as a future therapeutic target? Curr Pharm Des. 2015;21:389–416.
Kelly MP, Brandon NJ. Differential function of phosphodiesterase families in the brain: gaining insights through the use of genetically modified animals. Prog Brain Res. 2009;179:67–73.
Kelly MP, Logue SF, Brennan J, Day JP, Lakkaraju S, Jiang L, Zhong X, Tam M, Sukoff Rizzo SJ, Platt BJ, Dwyer JM, Neal S, Pulito VL, Agostino MJ, Grauer SM, Navarra RL, Kelley C, Comery TA, Murrills RJ, Houslay MD, Brandon NJ. Phosphodiesterase 11A in brain is enriched in ventral hippocampus and deletion causes psychiatric disease-related phenotypes. Proc Natl Acad Sci U S A. 2010;107:8457–62.
Kelly MP, Adamowicz W, Bove S, Hartman AJ, Mariga A, Pathak G, Reinhart V, Romegialli A, Kleiman RJ. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell Signal. 2014;26:383–97.
Kelsoe J. Method to predict response to treatment for psychiatric illnesses. In: Office UPT, editors. The regents of the University of California. Oakland, USA; 2010. p. 1.
Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr Pharm Des. 2010;16:1114–25.
Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiat. 2014;71:1121–8.
Laakso MP, Frisoni GB, Kononen M, Mikkonen M, Beltramello A, Geroldi C, Bianchetti A, Trabucchi M, Soininen H, Aronen HJ. Hippocampus and entorhinal cortex in frontotemporal dementia and Alzheimer’s disease: a morphometric MRI study. Biol Psychiatry. 2000;47:1056–63.
Laje G, Perlis RH, Rush AJ, McMahon FJ. Pharmacogenetics studies in STAR*D: strengths, limitations, and results. Psychiatr Serv. 2009;60:1446–57.
Lee JM, Kim SH, Jang DP, Ha TH, Kim JJ, Kim IY, Kwon JS, Kim SI. Deformable model with surface registration for hippocampal shape deformity analysis in schizophrenia. NeuroImage. 2004;22:831–40.
Leube DT, Weis S, Freymann K, Erb M, Jessen F, Heun R, Grodd W, Kircher TT. Neural correlates of verbal episodic memory in patients with MCI and Alzheimer's disease--a VBM study. Int J Geriatr Psychiatry. 2008;23:1114–8.
Libe R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, Rene-Corail F, Groussin L, Bertagna X, Raffin-Sanson ML, Stratakis CA, Bertherat J. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res. 2008;14:4016–24.
Libe R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, Ragazzon B, Guillaud-Bataille M, Groussin L, Clauser E, Raffin-Sanson ML, Siegel J, Moran J, Drori-Herishanu L, Faucz FR, Lodish M, Nesterova M, Bertagna X, Bertherat J, Stratakis CA. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab. 2011;96:E208–14.
Louiset E, Gobet F, Libe R, Horvath A, Renouf S, Cariou J, Rothenbuhler A, Bertherat J, Clauser E, Grise P, Stratakis CA, Kuhn JM, Lefebvre H. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab. 2010;95:18–24.
Lukas M, Neumann ID. Oxytocin and vasopressin in rodent behaviors related to social dysfunctions in autism spectrum disorders. Behav Brain Res. 2013;251:85–94.
Luo HR, Wu GS, Dong C, Arcos-Burgos M, Ribeiro L, Licinio J, Wong ML. Association of PDE11A global haplotype with major depression and antidepressant drug response. 2009a; 5: 163-170.
Luo HR, GS W, Dong C, Arcos-Burgos M, Ribeiro L, Licinio J, Wong ML. Association of PDE11A global haplotype with major depression and antidepressant drug response. Neuropsychiatr Dis Treat. 2009b;5:163–70.
Maes M, Bosmans E, Calabrese J, Smith R, Meltzer HY. Interleukin-2 and interleukin-6 in schizophrenia and mania: effects of neuroleptics and mood stabilizers. J Psychiatr Res. 1995;29:141–52.
Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–8.
Maheu FS, Mazzone L, Merke DP, Keil MF, Stratakis CA, Pine DS, Ernst M. Altered amygdala and hippocampus function in adolescents with hypercortisolemia: a functional magnetic resonance imaging study of Cushing syndrome. Dev Psychopathol. 2008;20:1177–89.
Makhlouf A, Kshirsagar A, Niederberger C. Phosphodiesterase 11: a brief review of structure, expression and function. Int J Impot Res. 2006;18:501–9.
Malhi GS, Tanious M, Das P, Coulston CM, Berk M. Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs. 2013;27:135–53.
Marquis JP, Goulet S, Dore FY. Neonatal ventral hippocampus lesions disrupt extra-dimensional shift and alter dendritic spine density in the medial prefrontal cortex of juvenile rats. Neurobiol Learn Mem. 2008;90:339–46.
Martinez M, Fernandez E, Frank A, Guaza C, de la Fuente M, Hernanz A. Increased cerebrospinal fluid cAMP levels in Alzheimer’s disease. Brain Res. 1999;846:265–7.
Martinez SE, AY W, Glavas NA, Tang XB, Turley S, Hol WG, Beavo JA. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci U S A. 2002;99:13260–5.
Matthiesen K, Nielsen J. Binding of cyclic nucleotides to phosphodiesterase 10A and 11A GAF domains does not stimulate catalytic activity. Biochem J. 2009;423:401–9.
Memo M, Kleinman JE, Hanbauer I. Coupling of dopamine D1 recognition sites with adenylate cyclase in nuclei accumbens and caudatus of schizophrenics. Science. 1983;221:1304–7.
Mertens J, Wang QW, Kim Y, DX Y, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, Eames T, Schafer ST, Boyer L, Marchetto MC, Nurnberger JI, Calabrese JR, Odegaard KJ, McCarthy MJ, Zandi PP, Alba M, Nievergelt CM, Pharmacogenomics of Bipolar Disorder Study, Mi S, Brennand KJ, Kelsoe JR, Gage FH, Yao J. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–9.
Mohamed HA, Girgis NM, Wilcken R, Bauer MR, Tinsley HN, Gary BD, Piazza GA, Boeckler FM, Abadi AH. Synthesis and molecular modeling of novel tetrahydro-beta-carboline derivatives with phosphodiesterase 5 inhibitory and anticancer properties. J Med Chem. 2011;54:495–509.
Mori S, Tardito D, Dorigo A, Zanardi R, Smeraldi E, Racagni G, Perez J. Effects of lithium on cAMP-dependent protein kinase in rat brain. Neuropsychopharmacology. 1998;19:233–40.
Moser MB, Moser EI. Functional differentiation in the hippocampus. Hippocampus. 1998;8:608–19.
Nesvaderani M, Matsumoto I, Sivagnanasundaram S. Anterior hippocampus in schizophrenia pathogenesis: molecular evidence from a proteome study. Aust N Z J Psychiatry. 2009;43:310–22.
Niculescu AB, Levey DF, Phalen PL, Le-Niculescu H, Dainton HD, Jain N, Belanger E, James A, George S, Weber H, Graham DL, Schweitzer R, Ladd TB, Learman R, Niculescu EM, Vanipenta NP, Khan FN, Mullen J, Shankar G, Cook S, Humbert C, Ballew A, Yard M, Gelbart T, Shekhar A, Schork NJ, Kurian SM, Sandusky GE, Salomon DR. Understanding and predicting suicidality using a combined genomic and clinical risk assessment approach. Mol Psychiatry. 2015;20:1266–85.
Nijholt IM, Dolga AM, Ostroveanu A, Luiten PG, Schmidt M, Eisel UL. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. 2008; 20: 1715-1724.
Ofuji M, Kaiya H, Nozaki M, Tsurumi K. Platelet prostaglandin E1 hyposensitivity in schizophrenia: reduction of prostaglandin E1- or forskolin-stimulated cyclic AMP response in platelets. Life Sci. 1989;45:2135–40.
Ohm TG, Bohl J, Lemmer B. Reduced cAMP-signal transduction in postmortem hippocampus of demented old people. Prog Clin Biol Res. 1989;317:501–9.
Ohm TG, Bohl J, Lemmer B. Reduced basal and stimulated (isoprenaline, Gpp(NH)p, forskolin) adenylate cyclase activity in Alzheimer’s disease correlated with histopathological changes. Brain Res. 1991;540:229–36.
Oki NO, Motsinger-Reif AA, Antas PR, Levy S, Holland SM, Sterling TR. Novel human genetic variants associated with extrapulmonary tuberculosis: a pilot genome wide association study. BMC Res Notes. 2011;4:28.
Pan JQ, Lewis MC, Ketterman JK, Clore EL, Riley M, Richards KR, Berry-Scott E, Liu X, Wagner FF, Holson EB, Neve RL, Biechele TL, Moon RT, Scolnick EM, Petryshen TL, Haggarty SJ. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36:1397–411.
Papatheodoropoulos C, Kostopoulos G. Dorsal-ventral differentiation of short-term synaptic plasticity in rat CA1 hippocampal region. Neurosci Lett. 2000;286:57–60.
Pathak GH, Fisher JL, Wilson S, Kelly MP, et al. Homodimerization and N-terminal phosphorylation control the subcellular localization of PDE11A4. Soc Neurosci. 2015;524:17.
Pathak G, Agostino MJ, Bishara K, Capell WR, Fisher JL, Hegde S, Ibrahim BA, Pilarzyk K, Sabin C, Tuczkewycz T, Wilson S, Kelly MP. PDE11A negatively regulates lithium responsivity. Mol Psychiatry. 2016. Epub ahead of print DOI: MP.2016.155.
Pegues MP, Rogers LJ, Amend D, Vinogradov S, Deicken RF. Anterior hippocampal volume reduction in male patients with schizophrenia. Schizophr Res. 2003;60:105–15.
Perlis RH, Fijal B, Dharia S, Heinloth AN, Houston JP. Failure to replicate genetic associations with antidepressant treatment response in duloxetine-treated patients. Biol Psychiatry. 2010;67:1110–3.
Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillon G, Lopera F, Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann Neurol. 2010;68:865–75.
Rahman S, Li PP, Young LT, Kofman O, Kish SJ, Warsh JJ. Reduced [3H]cyclic AMP binding in postmortem brain from subjects with bipolar affective disorder. J Neurochem. 1997;68:297–304.
Rajarethinam R, DeQuardo JR, Miedler J, Arndt S, Kirbat R, Brunberg JA, Tandon R. Hippocampus and amygdala in schizophrenia: assessment of the relationship of neuroanatomy to psychopathology. Psychiatry Res. 2001;108:79–87.
Rametti G, Segarra N, Junque C, Bargallo N, Caldu X, Ibarretxe N, Bernardo M. Left posterior hippocampal density reduction using VBM and stereological MRI procedures in schizophrenia. Schizophr Res. 2007;96:62–71.
Roman F, Soumireu-Mourat B. Behavioral dissociation of anterodorsal and posteroventral hippocampus by subseizure stimulation in mice. Brain Res. 1988;443:149–58.
Rusch N, Tebartz van Elst L, Valerius G, Buchert M, Thiel T, Ebert D, Hennig J, Olbrich HM. Neurochemical and structural correlates of executive dysfunction in schizophrenia. Schizophr Res. 2008;99:155–63.
Schobel SA, Kelly MA, Corcoran CM, Van Heertum K, Seckinger R, Goetz R, Harkavy-Friedman J, Malaspina D. Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophr Res. 2009a;114:110–8.
Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009b;66:938–46.
Schreiber G, Avissar S. Lithium sensitive G protein hyperfunction: a dynamic model for the pathogenesis of bipolar affective disorder. Med Hypotheses. 1991;35:237–43.
Schreiber G, Avissar S, Danon A, Belmaker RH. Hyperfunctional G proteins in mononuclear leukocytes of patients with mania. Biol Psychiatry. 1991;29:273–80.
Shanahan C, Gibson GE, Cowburn RF, Johnston JA, Wiehager B, Lannfelt L, O'Neill C. G protein subunit levels in fibroblasts from familial Alzheimer’s disease patients: lower levels of high molecular weight Gs alpha isoform in patients with decreased beta-adrenergic receptor stimulated cAMP formation. Neurosci Lett. 1997;232:33–6.
Shenton ME, Kikinis R, Jolesz FA, Pollak SD, LeMay M, Wible CG, Hokama H, Martin J, Metcalf D, Coleman M, et al. Abnormalities of the left temporal lobe and thought disorder in schizophrenia. A quantitative magnetic resonance imaging study. N Engl J Med. 1992;327:604–12.
Simon NM, McNamara K, Chow CW, Maser RS, Papakostas GI, Pollack MH, Nierenberg AA, Fava M, Wong KK. A detailed examination of cytokine abnormalities in Major Depressive Disorder. Eur Neuropsychopharmacol. 2008;18(3):230.
Starkman MN, Gebarski SS, Berent S, Schteingart DE. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biol Psychiatry. 1992;32:756–65.
Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE. Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing’s disease. Biol Psychiatry. 1999;46:1595–602.
Starkman MN, Giordani B, Berent S, Schork MA, Schteingart DE. Elevated cortisol levels in Cushing’s disease are associated with cognitive decrements. Psychosom Med. 2001;63:985–93.
Stoesz BM, Hare JF, Snow WM. Neurophysiological mechanisms underlying affiliative social behavior: insights from comparative research. Neurosci Biobehav Rev. 2013;37:123–32.
Suddath RL, Christison GW, Torrey EF, Casanova MF, Weinberger DR. Anatomical abnormalities in the brains of monozygotic twins discordant for schizophrenia. N Engl J Med. 1990;322:789–94.
Sun X, Young LT, Wang JF, Grof P, Turecki G, Rouleau GA, Alda M. Identification of lithium-regulated genes in cultured lymphoblasts of lithium responsive subjects with bipolar disorder. Neuropsychopharmacology. 2004;29:799–804.
Tipton LA, Christensen L, Blacher J. Friendship quality in adolescents with and without an intellectual disability. J Appl Res Intellect Disabil. 2013;26:522–32.
Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DA, O'Donnell P. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci. 2008;28:12691–9.
Turetsky BI, Moberg PJ. An odor-specific threshold deficit implicates abnormal intracellular cyclic AMP signaling in schizophrenia. Am J Psychiatry. 2009;166:226–33.
Vezzosi D, Libe R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, Rene-Corail F, Ragazzon B, Stratakis CA, Vandecasteele G, Bertherat J. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab. 2012;97:E2063–9.
Viero C, Shibuya I, Kitamura N, Verkhratsky A, Fujihara H, Katoh A, Ueta Y, Zingg HH, Chvatal A, Sykova E, Dayanithi G. REVIEW: Oxytocin: Crossing the bridge between basic science and pharmacotherapy. CNS Neurosci Ther. 2010;16:e138–56.
Watanabe S, Iga J, Nishi A, Numata S, Kinoshita M, Kikuchi K, Nakataki M, Ohmori T. Microarray analysis of global gene expression in leukocytes following lithium treatment. Hum Psychopharmacol. 2014;29:190–8.
Weeks JL 2nd, Zoraghi R, Francis SH, Corbin JD. N-Terminal domain of phosphodiesterase-11A4 (PDE11A4) decreases affinity of the catalytic site for substrates and tadalafil, and is involved in oligomerization. Biochemistry. 2007;46:10353–64.
Weeks JL 2nd, Corbin JD, Francis SH. Interactions between cyclic nucleotide phosphodiesterase 11 catalytic site and substrates or tadalafil and role of a critical Gln-869 hydrogen bond. J Pharmacol Exp Ther. 2009;331:133–41.
Witwicka H, Kobialka M, Siednienko J, Mitkiewicz M, Gorczyca WA. Expression and activity of cGMP-dependent phosphodiesterases is up-regulated by lipopolysaccharide (LPS) in rat peritoneal macrophages. Biochim Biophys Acta. 2007;1773:209–18.
Wong ML, Whelan F, Deloukas P, Whittaker P, Delgado M, Cantor RM, McCann SM, Licinio J, Wong ML, Whelan F, Deloukas P, Whittaker P, Delgado M, Cantor RM, McCann SM, Licinio J. Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proc Natl Acad Sci U S A. 2006;103:15124–9.
Xu Y, Zhang HT, O'Donnell JM. Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders. Handb Exp Pharmacol. 2011:447–85.
Yakushev I, Muller MJ, Lorscheider M, Schermuly I, Weibrich C, Dellani PR, Hammers A, Stoeter P, Fellgiebel A. Increased hippocampal head diffusivity predicts impaired episodic memory performance in early Alzheimer’s disease. Neuropsychologia. 2010;48:1447–53.
Yakushev I, Schreckenberger M, Muller MJ, Schermuly I, Cumming P, Stoeter P, Gerhard A, Fellgiebel A. Functional implications of hippocampal degeneration in early Alzheimer’s disease: a combined DTI and PET study. Eur J Nucl Med Mol Imaging. 2011a;38:2219–27.
Yakushev I, Gerhard A, Muller MJ, Lorscheider M, Buchholz HG, Schermuly I, Weibrich C, Hammers A, Stoeter P, Schreckenberger M, Fellgiebel A. Relationships between hippocampal microstructure, metabolism, and function in early Alzheimer’ disease. Brain Struct Funct. 2011b;216:219–26.
Yamamoto M, Gotz ME, Ozawa H, Luckhaus C, Saito T, Rosler M, Riederer P. Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer's disease. Biochim Biophys Acta. 2000;1535:60–8.
Young LT, Li PP, Kish SJ, Siu KP, Warsh JJ. Postmortem cerebral cortex Gs alpha-subunit levels are elevated in bipolar affective disorder. Brain Res. 1991;553:323–6.
Young LT, Li PP, Kish SJ, Siu KP, Kamble A, Hornykiewicz O, Warsh JJ. Cerebral cortex Gs alpha protein levels and forskolin-stimulated cyclic AMP formation are increased in bipolar affective disorder. J Neurochem. 1993;61:890–8.
Young LT, Li PP, Kamble A, Siu KP, Warsh JJ. Mononuclear leukocyte levels of G proteins in depressed patients with bipolar disorder or major depressive disorder. Am J Psychiatry. 1994;151:594–6.
Yuasa K, Kotera J, Fujishige K, Michibata H, Sasaki T, Omori K. Isolation and characterization of two novel phosphodiesterase PDE11A variants showing unique structure and tissue-specific expression. J Biol Chem. 2000;275:31469–79.
Yuasa K, Ohgaru T, Asahina M, Omori K. Identification of rat cyclic nucleotide phosphodiesterase 11A (PDE11A): comparison of rat and human PDE11A splicing variants. Eur J Biochem. 2001a;268:4440–8.
Yuasa K, Kanoh Y, Okumura K, Omori K. Genomic organization of the human phosphodiesterase PDE11A gene. Evolutionary relatedness with other PDEs containing GAF domains. Eur J Biochem. 2001b;268:168–78.
Zhou Y, Shu N, Liu Y, Song M, Hao Y, Liu H, Yu C, Liu Z, Jiang T. Altered resting-state functional connectivity and anatomical connectivity of hippocampus in schizophrenia. Schizophr Res. 2008;100:120–32.
Acknowledgements
The author would like to thank Baher Ibrahim for conducting the immunofluorescence shown herein. Support received from a Research Starter Grant in Pharmacology & Toxicology from the PhRMA Foundation, an ASPIRE award from the Office of the Vice President for Research from the University of South Carolina, a Research Development Fund Award from the University of South Carolina School of Medicine, a NARSAD Young Investigator Award from the Brain & Behavior Research Foundation, and 1R01MH101130 from NIMH (all awards to MPK).
Conflict of Interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Kelly, M.P. (2017). A Role for Phosphodiesterase 11A (PDE11A) in the Formation of Social Memories and the Stabilization of Mood. In: Zhang, HT., Xu, Y., O'Donnell, J. (eds) Phosphodiesterases: CNS Functions and Diseases. Advances in Neurobiology, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-58811-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-58811-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58809-4
Online ISBN: 978-3-319-58811-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)