
Abstract
Arrestins are structurally flexible and functionally versatile proteins that regulate the activity of hundreds of different G protein-coupled receptors (GPCRs). A hallmark of arrestin function is that these proteins are only activated for binding the active receptor upon interaction with receptor-attached phosphate groups. Recent years have yielded crystal structures of pre-activated arrestins and arrestin in complex with an active receptor, which provide insight into the arrestin activation mechanism. At the same time, functional studies indicate that arrestin employs different binding modes along the path to tight receptor binding, and the structure of the arrestin-receptor complex is modulated by the activation and phosphorylation state of the active receptor. In this chapter we discuss our current understanding of the receptor-binding mechanism of arrestin, from the initial interaction with the phosphorylated receptor to the structural transformation required for tight binding to the active receptor.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
About 800 different G protein-coupled receptors ( GPCRs ) are expressed in the human body and play a central role in multiple sensory and physiological systems. These receptors bind a wide variety of ligands and share a common structure of seven transmembrane (TM) helices. The receptor exists in a conformational equilibrium comprised of multiple states of varying degrees of activity. In the basal inactive state, the helical bundle is bound together by several hydrogen bond networks and electrostatic interactions. The binding of agonist favours an active receptor conformation, in which the cytoplasmic face of the receptor is open for interaction with cytosolic proteins. The binding partners of the receptor are namely G protein, GPCR kinase (GRK), and arrestin.
G proteins consist of three subunits, termed α, β and γ, and different subunit combinations give rise to about 20 distinct G proteins. Binding of the heterotrimeric G protein to the active receptor induces nucleotide exchange within the G protein and dissociation of the subunits, which then interact with other components of the cell signalling machinery. In essence, the receptor transduces the signal of agonist binding across the cell membrane, and the G protein amplifies this signal within the cytoplasm.
Seven different GRKs exist, each of which phosphorylates a different subset of GPCRs. The kinase activity of GRKs is stimulated by interaction with activated GPCRs . GRKs phosphorylate several serine and threonine residues within the cytoplasmic C-terminal tail of the receptor. Some receptors are also phosphorylated on their cytoplasmic loops. Only three receptor-attached phosphates are required to stimulate arrestin binding to the active GPCR (Vishnivetskiy et al. 2007), yet most GPCRs contain 7 or more phosphorylation sites. The extra sites confer another level of regulation, as different phosphorylation patterns (also called “barcodes”) control the multiple functions of arrestins [reviewed in Reiter et al. (2012)]. Different phosphorylation barcodes arise from different C-terminal primary sequence, tissue-specific or ligand-selected GRK activity, or sequential rounds of receptor activation.
The arrestin family is composed of only four members. Arrestin-1 and arrestin-4, respectively called rod and cone arrestin, are expressed primarily in the retina and interact with light-sensitive GPCRs called opsins. Arrestin-2 and arrestin-3, respectively called β-arrestin 1 and β-arrestin 2, are expressed ubiquitously and interact with hundreds of different GPCRs. This attribute means the β-arrestins must be versatile and relatively promiscuous binding partners. A primary role of arrestin binding is to block G protein coupling and thereby stop GPCR signalling. The β-arrestins are additionally able to induce receptor internalization by recruiting clathrin and other elements of the cellular internalization machinery, as well as mediate their own signalling cascades by scaffolding signalling kinases. In recent years evidence has emerged that the pattern of receptor phosphorylation affects both the conformation of bound arrestin (Shenoy et al. 2009; Nobles et al. 2011) as well as the stability of the arrestin-GPCR complex (Zindel et al. 2014; Oakley et al. 2001; Tohgo et al. 2003). These factors presumably control arrestin-mediated receptor internalization and trafficking (Oakley et al. 2001; Zindel et al. 2014) and arrestin -mediated MAP kinase activity (Tohgo et al. 2003; Ren et al. 2005; Nobles et al. 2011) [reviewed in Tobin et al. (2008)].
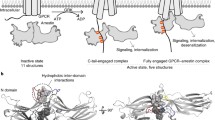
Arrestins are composed of two cup-like domains composed of β-sheets, called the N- and C-domain (Fig. 8.1). The tips of the domains are capped with flexible loops, which compose the central crest (finger loop, middle loop and C-loop) and the C-edge (344-loop and 197-loop). The interdomain interface is stabilized by hydrophobic interactions and hydrogen-bond networks. These include the polar core (Han et al. 2001), the YKS(N)D(A) motif (Kim et al. 2013), the middle loop (a.k.a. 139-loop) (Vishnivetskiy et al. 2013), and the long Loop 17-18 (Kim et al. 2013; Han et al. 2001), which winds between the two domains and makes multiple contacts with both the N- and C-domains. Loop 17-18 also includes the gate loop, which is a functional part of the polar core (Fig. 8.1). A third functionally critical domain of arrestin is the auto-inhibitory C-terminal tail (C-tail). The distal C-tail contains a large number of acidic residues and is highly dynamic, as it is not visualized in any known structure of arrestin. In contrast, the proximal C-tail is stably anchored to the body of the N-domain by the hydrophobic 3-element interaction (Vishnivetskiy et al. 2000) and a salt bridge within the polar core. Together, the interdomain interface and the C-tail restrict flexibility within arrestin , namely of the central crest loops and between the N- and C-domains, and thereby prevent interaction with the active receptor.

Crystal structures of basal arrestin (PDB code 1CF1, molecule D) and pre-activated arrestin p44 (PDB code 4J2Q, molecule B) (Kim et al. 2013; Hirsch et al. 1999). The N-domain is coloured blue and the C-domain is coloured green. The C-tail of arrestin is red, and Loop 17-18 is magenta. Note that only the proximal C-tail is resolved in the arrestin crystal structure, and the unresolved dynamic distal portion is illustrated with a red dashed line. The loop linking the C-domain to the C-tail is also not resolved in the arrestin crystal structure. Notable loops that are discussed in the text are labelled with numbers, which are defined in the legend to the right. Important intramolecular interactions that stabilize the basal state are illustrated by sidechains with Van der Waal surfaces and labelled with capital letters (defined in legend). Note how these networks are broken in the p44 structure, which results in a ~21° rotation of the C-domain compared to its orientation in the basal arrestin
It has long been believed that arrestin activation entails a displacement of the C-tail by the phosphorylated receptor C-terminus (Rpp), based on changes in protease susceptibility (Palczewski et al. 1991a, c), distances measured between spin-labels (Hanson et al. 2006; Vishnivetskiy et al. 2010), and changes in the NMR spectrum of arrestin (Zhuang et al. 2010, 2013). This theory is consistent with the phosphorylation-independent receptor binding behaviour of p44, a naturally occurring splice variant of arrestin-1 that lacks the entire C-tail (Palczewski et al. 1994; Pulvermüller et al. 1997). Supposedly C-tail displacement by the Rpp breaks the polar core and loosens the intramolecular interactions that hold arrestin in an inactive state. Two crystal structures of pre-active arrestin, p44 (Kim et al. 2013) and arrestin-2 bound to a peptide analogue of the Rpp (Shukla et al. 2013), indicate the conformational changes that occur in arrestin upon full C-tail displacement (Fig. 8.1). These changes include a twisting displacement of the gate loop that breaks the polar core, release of the central crest loops from their restricted basal conformations, and a 21° rotation of the two domains relative to one another.
In the recently published crystal structure of arrestin-1 in complex with an active GPCR (Kang et al. 2015), it is clear how these structural changes facilitate arrestin binding of the active receptor (discussed in more detail below). The complex consists of a constitutively active mutant of human rhodopsin fused on its N-terminus to T4 lysozyme and fused on its C-terminus to a constitutively active mutant of mouse arrestin-1 via a 15 amino-acid linker. Although the receptor in this structure lacks ligand (opsin), it is obviously in an active state (Ops*). Likewise, the arrestin in this structure shows all the hallmarks of activation seen in the pre-activated arrestin structures (Kim et al. 2013; Shukla et al. 2013), even though the receptor in the complex is not phosphorylated. Despite the artificial nature of the Ops*/arrestin-1 fusion complex structure, it provides a first glimpse of arrestin bound tightly to an active receptor.
This chapter focuses on the molecular mechanisms by which arrestin couples to an active, phosphorylated GPCR . We discuss the initial interaction of arrestin with the phosphorylated receptor, which we call the pre-complex, and the conformational changes in arrestin involved in formation of the high-affinity complex. We have gained insight into these functionally distinct complexes using the long-studied GPCR rhodopsin and its binding partner arrestin-1, which are expressed in the rod cells of the retina and mediate dim-light vision. Dark-state inactive rhodopsin consists of the aporeceptor opsin and a covalently attached inverse agonist, 11-cis-retinal. Light absorption converts the ligand to the agonist all-trans-retinal, which results in the active receptor form Metarhodopsin II (Meta II). Meta II is phosphorylated by GRK1 on its cytoplasmic C-terminal tail. As an experimental model of the pre-complex, we study the interaction of arrestin-1 with phosphorylated inactive forms of the receptor, specifically dark-state phosphorylated rhodopsin (Rho-P) and the phosphorylated aporeceptor opsin (Ops-P). Using these receptor forms, we can experimentally probe arrestin-receptor interactions that occur before full arrestin activation and formation of the high-affinity complex. Transition to the high-affinity complex is induced by irradiation of the sample with visible light, which converts Rho-P to Meta II-P. Methods we have used to study arrestin-receptor interactions include site-directed fluorescence spectroscopy , Fourier transform infrared spectroscopy (FTIR), and alanine scan mutagenesis. The functional insights we gain from these studies are interpreted within the context of the crystal structures of different arrestins in basal (Hirsch et al. 1999; Granzin et al. 1998; Han et al. 2001; Granzin et al. 2012), pre-activated (Granzin et al. 2015; Kim et al. 2013; Shukla et al. 2013), and fully activated states (Kang et al. 2015). This approach has yielded valuable insights that allow us to propose a mechanistic model of arrestin activation and receptor coupling.
The Pre-complex
The initial interaction of the phosphorylated receptor and arrestin recruits arrestin to the active receptor and activates arrestin for binding the helical bundle of the receptor. Currently very little is known regarding the structure and organization of the pre-complex. Experimental evidence suggests the primary sites of interaction are the phosphorylated receptor C-terminus (Gurevich and Benovic 1993, 1995; Vishnivetskiy et al. 2000; Hanson and Gurevich 2006; Peterhans et al. 2016) and the membrane (Lally et al. 2017). The pre-complex was first proposed by Alexander Pulvermüller, Klaus Peter Hofmann and colleagues (IMPB Charité, Berlin) based on kinetic binding experiments comparing arrestin-1 and p44 (Schröder et al. 2002). This proposal was based on the conclusion that p44 can bypass the initial binding step, which serves normally to displace the C-tail of arrestin . Subsequently two independent studies have presented time-resolved fluorescence data supporting a multi-step binding mechanism, in which arrestin is recruited to the receptor before undergoing a significant conformational change (Kirchberg et al. 2011; Nuber et al. 2016). We interpret these two stages to represent, first formation of the pre-complex, and second, transition to the high-affinity complex.
It has been long known that arrestins intrinsically bind poly-anions, including heparin and inositol phosphate (Palczewski et al. 1991b), as well as receptor-attached phosphate groups [but not simple anions like phosphate (Wilson and Copeland 1997)]. The concave surfaces of all four arrestin subtypes are studded with basic residues that could serve as binding sites for poly-anions. Crystallographic analysis indicated that two molecules of IP6 bind arrestin-2, one each within the concave surfaces of the N- and C-domains (Milano et al. 2006). IP6 binding facilitates homo- and hetero-dimerization of arrestin -2 and arrestin-3. IP6 also binds arrestin-1, although with much lower affinity [KD in the range of 0.2–160 μM (Palczewski et al. 1991b; Wilson and Copeland 1997; Zhuang et al. 2010)]. NMR analysis indicated that IP6 binds within the N-domain of arrestin-1 (Zhuang et al. 2010). This NMR analysis did not detect any binding of IP6 within the C-domain, although site-directed fluorescence experiments suggest a second low-affinity IP6 binding site, most likely in the C-domain (Sommer, unpublished work).
Mutagenesis has long been applied to arrestin to identify potential receptor-interacting sites, and to differentiate which sites are sensitive to the phosphorylation and activation states of the receptor [reviewed in (Gurevich et al. 2011)]. Alanine scan mutagenesis is an unbiased and comprehensive method that has been recently applied to arrestin-1 by the group of Joerg Standfuss (PSI, Switzerland) to identify sites of interaction and functionally important areas that undergo structural changes during arrestin activation and receptor binding (Ostermaier et al. 2014a; Peterhans et al. 2016). For the most part, the alanine-scan studies confirmed the findings from previous mutagenesis studies (Ostermaier et al. 2014a, b). In collaboration with the Standfuss group, we found evidence that the phosphorylated receptor C-terminus binds arrestin differently in the pre-complex and the high-affinity complex (Fig. 8.2). For the interaction of arrestin-1 with Ops-P, which mirrors the pre-complex, mutation of ten different basic residues within the cup of the N-domain significantly decreased arrestin-1 binding to Ops-P. The large number of implicated phospho-sensing residues is congruent with the fact that the affinity of arrestin-1 for Ops-P is directly proportional to the degree of receptor phosphorylation (i.e. the more phosphates per receptor, the higher the affinity) (Vishnivetskiy et al. 2007). This binding mode stands in contrast to that used for Meta II-P, in which fewer residues along the lateral side of the N-domain are implicated (Fig. 8.2). The Rpp binding mode utilized by Meta II-P is discussed in more detail in the section below.

Different “Rpp-binding footprints” for the pre-complex and the high-affinity complex. Alanine scan mutagenesis identified different sets of positively charged residues used in binding the phosphorylated receptor C-terminus (Rpp) in binding inactive Ops-P (pre-complex, left) and light-activated Meta II-P (high-affinity complex, right) (Peterhans et al. 2016). The implicated residues are plotted in blue on the structures of basal arrestin (α-conformer, PDB code 1CF1) for the pre-complex and p44 (PDB code 4J2Q). Note that for basal arrestin , the distal C-tail (rose) does not block access to the implicated phosphate-binding residues, which is consistent with our hypothesis that only the proximal C-tail is displaced in the pre-complex. The putative Rpp-binding crevice that was identified in the crystal structure of activated arrestin-2 bound to a peptide analogue of the Rpp (Shukla et al. 2013) is indicated by a dashed red line. Lateral views of arrestin and p44 are shown in the top panels, and top-down views (showing receptor binding interface) are presented in the bottom panels
If Rpp binds only within the cup of the N-domain, then it is possible that only the distal C-tail of arrestin is displaced in the pre-complex. This assumption is supported by site-directed fluorescence experiments using a mutant designed to monitor the relative position of the gate loop and, by extension, the state of the polar core (Kim et al. 2013; Peterhans et al. 2016). These experiments suggest that the polar core is not broken and thus the proximal C-tail is likely not displaced when arrestin is bound to Ops-P in a pre-complex. Only upon activation of the receptor and transition to the high-affinity complex is the polar core broken. Furthermore, site-directed fluorescence experiments employing mutants that report on other conformational changes associated with activation (e.g. release of central crest loops and interdomain rotation) suggest that arrestin is bound to the receptor in a conformational state resembling the basal state in the pre-complex (Lally and Sommer, unpublished work). This supposition is consistent with differences seen in the functional maps for pre-complex and high-affinity complex derived from alanine-scan mutagenesis (Peterhans et al. 2016). Collectively the fluorescence and mutagenesis data argue against the common assumption that Rpp binding in the pre-complex displaces the entire C-tail and induces activating conformational changes in arrestin (Shukla et al. 2013; Kang et al. 2015; Schröder et al. 2002).
The other site of interaction in the pre-complex is the anchoring of the C-edge of arrestin in the membrane. This interaction has recently been characterized by our group using site-directed fluorescence spectroscopy (Lally et al. 2017). Briefly, the proximity of bimane fluorophores placed at specific sites on loops within the C-edge to the membrane was probed using spin-labelled fatty acids, which were incorporated into native rod outer segment membranes containing phosphorylated rhodopsin. Spin-labels quench bimane fluorescence when in close proximity, and fatty acids with spin label at different positions on the acyl chain allowed the differentiation of deep and shallow membrane anchoring. Our results clearly show that the 344-loop embeds deep within the hydrophobic layer of the membrane when arrestin is bound to dark-state Rho-P in a pre-complex. Furthermore, membrane anchoring is dependent on the presence of phosphorylated receptor, suggesting that Rpp binding either activates the C-edge of arrestin for membrane anchoring or is required to recruit arrestin to the membrane. The relative levels of quenching for neighbouring sites suggest an extended 344-loop conformation (Lally et al. 2017), similar as in the “α-conformer” of the basal arrestin-1 structure (Hirsch et al. 1999).
Membrane anchoring in the pre-complex involves the embedding of leucine residues on the 344-loop within the hydrophobic interior of the membrane. Intriguingly, rhodopsin flips negatively charged acidic phospholipids to the cytoplasmic side upon activation (Hessel et al. 2000, 2001), and acidic phospholipids have been shown by us and others to be necessary for arrestin-1 binding of light-activated phosphorylated rhodopsin (Sommer et al. 2006; Bayburt et al. 2011). Furthermore, we have failed to detect any pre-complex formation of arrestin -1 with dark-state Rho-P in the absence of acidic phospholipids (Sommer, unpublished work). These data indicate that acidic phospholipids are required for arrestin to interact with the phosphorylated receptor. Notably, several basic residues line the C-domain (e.g. K235, K236, K238, K267, K330, and K332 in bovine arrestin-1), which we hypothesize help attract the C-edge of arrestin for membrane anchoring.
To summarize the experimental evidence, in the pre-complex arrestin is bound by the Rpp within the cup of the N-domain and by the membrane at the C-edge (Fig. 8.3). Given the relatively deep insertion of the membrane anchor, the peripheral engagement of the Rpp, and the fact that the central crest loops do not engage the receptor, arrestin is likely loosely associated with the receptor in the pre-complex. A great degree of rotational freedom and a highly dynamic complex is expected. Interestingly, a “hanging” interaction mode of arrestin-2 with a chimeric phosphorylated GPCR was observed by negative-stain electron microscopy (Shukla et al. 2014). The authors of this study hypothesized this complex represents tethering of arrestin solely by the Rpp. Due to the artificial nature of the Fab-stabilized complexes and the absence of membrane (receptors were solubilized in a neutral detergent), we postulate the visualized complexes represent the pre-complex without the benefit of membrane anchoring. Hence future investigations of the overall organization and structure of the pre-complex should include membrane or a membrane mimic in order to engage the C-edge of arrestin.

Model of the pre-complex and high-affinity complex. Binding of arrestin to rhodopsin is illustrated. Basal arrestin (blue N-domain and green C-domain) has an intact polar core and does not interact with dark-state rhodopsin (red). Upon receptor light-activation (Meta II, yellow) and phosphorylation by GRK, arrestin interacts in a pre-complex that displaces the distal C-tail and embeds the C-edge membrane deep in the membrane. However, no significant activating conformational changes (e.g. interdomain rotation) take place. Upon transition to the high-affinity complex, the Rpp moves from the cup of the N-domain to the putative Rpp-binding trench located along the side of the N-domain. This movement displaces the entire C-tail and results in significant conformational changes linked to interdomain rotation
Notably, pre-complex formation as the initial interaction of arrestin and the receptor is fairly nonspecific and likely similar for all GPCRs. The interaction with the Rpp and the membrane is primarily electrostatic and hydrophobic, respectively. The nonspecific nature of these interactions allow arrestins to interact with a wide variety of GPCRs , since active receptors are normally phosphorylated in their active state, and all receptors reside in a membrane. This attribute makes sense for the β-arrestins (arrestin-2 and arrestin-3), which couple to hundreds of different GPCRs.
However, the functional relevance of the pre-complex is not limited to being an obligate step before forming the high-affinity complex. Arrestin also forms complexes with phosphorylated inactive receptors, which occur when the receptor returns to an inactive state after ligand dissociation and before receptor dephosphorylation occurs (Lee et al. 2010; Sommer et al. 2014; Zhuang et al. 2013). Inactive receptors in the vicinity of active receptors can also be phosphorylated by activated GRKs, a phenomenon called high-gain phosphorylation (Shi et al. 2005; Binder et al. 1990, 1996). The interaction of arrestin with inactive phosphorylated receptors could play a significant physiological role. For example, in the visual system binding of arrestin-1 to Ops-P could quench the residual G protein-activating ability of opsin (Sommer et al. 2014; Zhuang et al. 2013). In addition, our group discovered that arrestin-1 facilitates the uptake of all-trans-retinal in Ops-P (Sommer et al. 2012), which could limit the build-up of potentially toxic levels of retinal in rod cells in bright continuous light (Sommer et al. 2014). The fact that most GPCRs have seven or more phosphorylation sites suggests the interaction of arrestins with highly phosphorylated aporeceptors might be prevalent within the larger GPCR family and could play a role in regulating ligand binding affinity (Gurevich et al. 1997).
Transition to the High-Affinity Complex
Pre-complex formation serves to recruit arrestin to the receptor, where arrestin is anchored at the N-domain by the Rpp and at the C-edge by the membrane (Fig. 8.3). Although arrestin is not fully activated in the pre-complex, the displacement of the distal C-tail increases flexibility and mobility of the central crest loops. Importantly, pre-complex formation brings the receptor-binding elements of arrestin close to the helical core of the receptor. Hence the two proteins can interact with one another as they sample their individual conformational spaces. Transition from the pre-complex to the high-affinity complex involves a substantial intramolecular conversion in arrestin, which mutually stabilizes the active form of the receptor. Agonist binding favours an active conformation of the receptor, and engagement by arrestin further stabilizes this active conformation. These conclusions are supported by spectroscopic studies of arrestin-1 binding to light-activated phosphorylated rhodopsin, which are briefly summarized below.
Over twenty five years ago, Klaus Peter Hofmann and co-workers developed an absorbance-based assay called “extra Meta II” to observe the formation of the high-affinity arrestin -1/Meta II-P complex (Schleicher et al. 1989). Light-activation of rhodopsin results in an equilibrium of two species, the active state Meta II and its inactive precursor Meta I. At low temperature and high pH (e.g. 2 °C and pH 8), the Meta I photoproduct is heavily favoured. When the receptor is phosphorylated and arrestin-1 interacts, Meta II is stabilized at the expense of Meta I. In other words, arrestin-1 binding shifts the Meta I ↔ Meta II equilibrium in favour of Meta II. Importantly, the extra Meta II assay can be used to quantitatively describe the formation of the high-affinity arrestin-receptor complex. Arrestin-1 activation and formation of the high-affinity complex entails a high activation energy (140 kJ/mol). For p44, the energy required for Meta II-P binding is halved (70 kJ/mol) (Pulvermüller et al. 1997), suggesting that about half the energy of activation is involved in releasing the intramolecular restrictions of the C-tail. At sufficiently high concentrations, Meta II-P binding by arrestin-1 and p44 occur at the same rate, which suggests that both arrestin-1 and p44 undergo the same intramolecular conversion, and that this step is rate-limiting for formation of the high-affinity complex (Schröder et al. 2002). Our group has recently characterized this interaction using FTIR spectroscopy (Beyriere et al. 2015). The observed spectral signatures confirmed the stabilization of the active Meta II species by arrestin-1 and additionally indicated a loss of beta-sheet, most likely arising from changes in the secondary structure of arrestin-1. Importantly, Meta II stabilization and beta-sheet loss in arrestin occurred at the same rate, indicating that both proteins mutually stabilize one another in their active states during formation of the high-affinity complex. Moreover, p44 and arrestin showed the same rates of Meta II binding and spectral signatures of beta-sheet loss, indicating that they undergo the same structural transition during high-affinity complex formation (Beyriere et al. 2015).
The comparison of the structure of basal arrestin to that in the Ops*/arrestin-1 fusion complex indicates the intramolecular conversions that arrestin undergoes during tight receptor binding. Below we summarize the major conformational changes, and how these changes facilitate coupling to the active phosphorylated receptor:
Displacement of the proximal C-tail—In the pre-complex, the Rpp is bound within the cup of the N-domain, and transition to the high-affinity complex is accompanied by a movement of the Rpp to a positively charged binding trench on the lateral side of the N-domain (Fig. 8.3). This trench is normally obscured by the proximal C-tail, and the Rpp presumably displaces the C-tail. A peptide analogue of the Rpp has been observed by X-ray protein crystallography to bind within this trench (Shukla et al. 2013). This crystal structure shows that peptide-attached phosphate groups interact with many of the residues identified as phospho-sensors for Meta II-P binding (e.g. K14, K15, R29, K110, K300) (Fig. 8.2) (Peterhans et al. 2016). Displacement of the C-tail and binding of the Rpp induces a forward twisting movement of the gate loop, which breaks the polar core and brings a phosphate-binding lysine residue on the gate loop (K300 in arrestin-1, K294 in arrestin-2) into the Rpp-binding trench.
Interdomain rotation—The binding of the Rpp within the putative binding trench is associated with a movement of the gate loop. This conformational change is transmitted along the entire length of Loop 17-18, which winds between the N- and C-domains and forms multiple contacts that stabilize the basal-state interdomain interface (Fig. 8.1). Loop 17-18 is pulled down and away from the central crest region, thereby causing a lateral displacement of the middle loop away from the body of arrestin (Kim et al. 2012; Kang et al. 2015) and a movement of the C-loop (Y247-Y254 in arrestin-1) down and away from the central crest. These movements break the YKS(N)D(A) network, which stabilizes basal arrestin by linking the C-loop (Y247-Y254 in arrestin-1) within the C-domain to the middle and finger loops within the N-domain. Along with the breaking of the polar core, these conformational rearrangements dramatically increase flexibility between the two domains of arrestin. Hence a 21° rotation of the C-domain is observed in the crystal structures of pre-activated and receptor-bound arrestin (Kim et al. 2013; Shukla et al. 2013; Kang et al. 2015). Interdomain rotation has two major effects on the arrestin structure: a crevice opens within the central crest, and the C-edge adopts a different orientation and conformation. These changes facilitate arrestin binding to the receptor and the adjacent membrane.
Opening of receptor-binding crevice—The lateral displacement of the middle loop and the downward movement of the C-loop opens a cleft that accommodates intercellular loop 2 (CL2) of the receptor. In the Ops*/arrestin-1 fusion complex structure, CL2 forms a short helix that makes hydrophobic and van der Waals contacts with the middle loop and C-loops of arrestin . In addition to this interaction, the downward displacement Loop 17-18 opens space for the TM5/6 bundle. In the crystal structure of the complex, a few hydrogen bond contacts are present between TM5/6 and arrestin. In essence, coupling to the helical bundle of the receptor is a direct result of interdomain rotation that allows a deformation of the flexible loops of arrestin. Furthermore, the sparseness of specific hydrogen bonds or salt bridges within the arrestin -receptor interface hints at how the β-arrestins are able to bind so many different GPCRs. Notably, the interdomain hydrogen bond networks of arrestin-2 and arrestin-3 are much weaker than in arrestin-1 (Kim et al. 2013), meaning these arrestins are already “half-way on” (Gurevich et al. 2011) and explains their much lower dependence on receptor phosphorylation for activation and receptor binding.
Engagement of the finger loop—In the pre-complex, the displacement of the distal C-tail by the Rpp likely increases mobility of the finger loop (Hanson et al. 2008). The flexible finger loop is positioned close to the open cytoplasmic face of the receptor, where it can explore the binding cavity. It is possible that binding of the finger loop and stabilization of the active state of the receptor follows a similar stepwise and mutual reduction of conformational space as recently described for G protein (Elgeti et al. 2013). In the Ops*/arrestin-1 fusion complex structure, the finger loop is observed to adopt a helical conformation within the cytoplasmic crevice of the active receptor (Kang et al. 2015). A similar binding mode was observed in a crystal structure of Ops* bound to a peptide analogue of the finger loop (Szczepek et al. 2014). In this structure, specific hydrogen bonding is observed between the finger loop and critical functional motifs on the receptor (e.g. NPxxY(x)5,6F and E(D)RY), which are highly conserved among GPCRs . Consistently, the finger loop sequence is highly conserved among all four arrestins. Engagement of the finger loop is crucial for stabilizing the active form of the receptor (Sommer et al. 2012) and hence formation of the high affinity complex.
Membrane anchoring of the C-edge—Rotation of the C-domain results in a different alignment of the C-edge that changes how this functional domain interacts with the membrane as compared to the pre-complex. In the pre-complex, the 344-loop adopts an extended conformation and is deeply inserted into the hydrophobic layer of the membrane. In the high-affinity complex, this loop adopts a folded conformation as seen in the p44 structure and interacts more shallowly with the membrane. In addition, the rotation of the C-domain allows the 197-loop to engage the membrane in the high-affinity complex. These differences in C-edge membrane engagement in the pre-complex and high-affinity complex were clearly observed in site-directed fluorescence experiments (Lally et al. 2017) and are consistent with differences in functional maps for Meta II-P binding and Ops-P binding derived from alanine scan mutagenesis (Peterhans et al. 2016).
In summary, formation of the arrestin-receptor complex occurs by a multistep mechanism (Fig. 8.3). Initial electrostatic attraction of arrestin to the Rpp and the negatively charged membrane surface recruits arrestin to the membrane, where the N-domain binds the Rpp and the C-edge anchors within the membrane. This interaction brings the central crest of arrestin within close proximity of the receptor helical bundle, so that the two proteins can interact and mutually affect one another. Transition to the high-affinity complex involves multiple conformational rearrangements in arrestin, which allow arrestin to both specifically stabilize the active form of the receptor as well as flexibly accommodate the cytoplasmic face of the receptor. This flexibility is likely critical for making the β-arrestins versatile binding partners for so many different GPCRs.
References
Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV (2011) Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem 286(2):1420–1428
Beyriere F, Sommer ME, Szczepek M, Bartl FJ, Hofmann KP, Heck M, Ritter E (2015) Formation and decay of the arrestin-rhodopsin complex in native disc membranes. J Biol Chem 290(20):12919–12928
Binder BM, Biernbaum MS, Bownds MD (1990) Light activation of one rhodopsin molecule causes the phosphorylation of hundreds of others. A reaction observed in electropermeabilized frog rod outer segments exposed to dim illumination. J Biol Chem 265(25):15333–15340
Binder BM, O’Connor TM, Bownds MD, Arshavsky VY (1996) Phosphorylation of non-bleached rhodopsin in intact retinas and living frogs. J Biol Chem 271(33):19826–19830
Elgeti M, Rose AS, Bartl FJ, Hildebrand PW, Hofmann KP, Heck M (2013) Precision vs flexibility in GPCR signaling. J Am Chem Soc 135(33):12305–12312. doi:10.1021/ja405133k
Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G (1998) X-ray crystal structure of arrestin from bovine rod outer segments. Nature 391(6670):918–921. doi:10.1038/36147
Granzin J, Cousin A, Weirauch M, Schlesinger R, Büldt G, Batra-Safferling R (2012) Crystal structure of p44, a constitutively active splice variant of visual arrestin. J Mol Biol 416:611–618
Granzin J, Stadler A, Cousin A, Schlesinger R, Batra-Safferling R (2015) Structural evidence for the role of polar core residue Arg175 in arrestin activation. Sci Rep 5:15808. doi:10.1038/srep15808
Gurevich VV, Benovic JL (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem 268(16):11628–11638
Gurevich VV, Benovic JL (1995) Visual arrestin binding to rhodopsin. Diverse functional roles of positively charged residues within the phosphorylation-recognition region of arrestin. J Biol Chem 270(11):6010–6016
Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ (1997) Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem 272(46):28849–28852
Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog Retinal Eye Res 30(6):405–430. doi:10.1016/j.preteyeres.2011.07.002
Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C (2001) Crystal structure of beta-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure 9(9):869–880
Hanson SM, Gurevich VV (2006) The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem 281(6):3458–3462. doi:10.1074/jbc.M512148200
Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A 103(13):4900–4905. doi:10.1073/pnas.0600733103
Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV (2008) A model for the solution structure of the rod arrestin tetramer. Structure 16(6):924–934. doi:10.1016/j.str.2008.03.006
Hessel E, Herrmann A, Muller P, Schnetkamp PP, Hofmann KP (2000) The transbilayer distribution of phospholipids in disc membranes is a dynamic equilibrium evidence for rapid flip and flop movement. Eur J Biochem 267(5):1473–1483
Hessel E, Muller P, Herrmann A, Hofmann KP (2001) Light-induced reorganization of phospholipids in rod disc membranes. J Biol Chem 276(4):2538–2543. doi:10.1074/jbc.M009061200
Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 Å crystal structure of visual arrestin: a model for arrestin’s regulation. Cell 97(2):257–269
Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523(7562):561–567. doi:10.1038/nature14656
Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL (2012) Conformation of receptor-bound visual arrestin. Proc Natl Acad Sci U S A 109(45):18407–18412. doi:10.1073/pnas.1216304109
Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME (2013) Crystal structure of pre-activated arrestin p44. Nature 497(7447):142–146. doi:10.1038/nature12133
Kirchberg K, Kim TY, Moller M, Skegro D, Dasara Raju G, Granzin J, Buldt G, Schlesinger R, Alexiev U (2011) Conformational dynamics of helix 8 in the GPCR rhodopsin controls arrestin activation in the desensitization process. Proc Natl Acad Sci U S A 108(46):18690–18695. doi:10.1073/pnas.1015461108
Lally CC, Bauer B, Selent J, Sommer ME (2017) C-edge loops of arrestin function as a membrane anchor. Nat Commun 8:14258. doi:10.1038/ncomms14258
Lee KA, Nawrot M, Garwin GG, Saari JC, Hurley JB (2010) Relationships among visual cycle retinoids, rhodopsin phosphorylation, and phototransduction in mouse eyes during light and dark adaptation. Biochemistry 49(11):2454–2463. doi:10.1021/bi1001085
Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C (2006) Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem 281(14):9812–9823. doi:10.1074/jbc.M512703200
Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ (2011) Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal 4(185):ra51. doi:10.1126/scisignal.2001707
Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C (2016) Beta-arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531(7596):661–664. doi:10.1038/nature17198
Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem 276(22):19452–19460. doi:10.1074/jbc.M101450200
Ostermaier MK, Peterhans C, Jaussi R, Deupi X, Standfuss J (2014a) Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1319402111
Ostermaier MK, Schertler GF, Standfuss J (2014b) Molecular mechanism of phosphorylation-dependent arrestin activation. Curr Opin Struct Biol 29C:143–151. doi:10.1016/j.sbi.2014.07.006
Palczewski K, Buczylko J, Imami NR, McDowell JH, Hargrave PA (1991a) Role of the carboxyl-terminal region of arrestin in binding to phosphorylated rhodopsin. J Biol Chem 266(23):15334–15339
Palczewski K, Pulvermüller A, Buczylko J, Gutmann C, Hofmann KP (1991b) Binding of inositol phosphates to arrestin. FEBS Lett 295(1–3):195–199
Palczewski K, Pulvermüller A, Buczylko J, Hofmann KP (1991c) Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J Biol Chem 266(28):18649–18654
Palczewski K, Buczylko J, Ohguro H, Annan RS, Carr SA, Crabb JW, Kaplan MW, Johnson RS, Walsh KA (1994) Characterization of a truncated form of arrestin isolated from bovine rod outer segments. Protein Sci 3(2):314–324
Peterhans C, Lally CC, Ostermaier MK, Sommer ME, Standfuss J (2016) Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci Rep 6:28686. doi:10.1038/srep28686
Pulvermüller A, Maretzki D, Rudnicka-Nawrot M, Smith WC, Palczewski K, Hofmann KP (1997) Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry 36(30):9253–9260. doi:10.1021/bi970772g
Reiter E, Ahn S, Shukla AK, Lefkowitz RJ (2012) Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Ann Rev Pharmacol Toxicol 52:179–197. doi:10.1146/annurev.pharmtox.010909.105800
Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ (2005) Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc Natl Acad Sci U S A 102(5):1448–1453. doi:10.1073/pnas.0409534102
Schleicher A, Kühn H, Hofmann KP (1989) Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry 28(4):1770–1775
Schröder K, Pulvermüller A, Hofmann KP (2002) Arrestin and its splice variant Arr1-370A(p44). Mechanism and biological role of their interaction with rhodopsin. J Biol Chem 277(46):43987–43996. doi:10.1074/jbc.M206211200
Shenoy SK, Modi AS, Shukla AK, Xiao K, Berthouze M, Ahn S, Wilkinson KD, Miller WE, Lefkowitz RJ (2009) Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci U S A 106(16):6650–6655. doi:10.1073/pnas.0901083106
Shi GW, Chen J, Concepcion F, Motamedchaboki K, Marjoram P, Langen R (2005) Light causes phosphorylation of nonactivated visual pigments in intact mouse rod photoreceptor cells. J Biol Chem 280(50):41184–41191. doi:10.1074/jbc.M506935200 (M506935200)
Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ (2013) Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497(7447):137–141. doi:10.1038/nature12120
Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL Jr, Kobilka BK, Skiniotis G, Lefkowitz RJ (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512(7513):218–222. doi:10.1038/nature13430
Sommer ME, Smith WC, Farrens DL (2006) Dynamics of arrestin-rhodopsin interactions: acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. J Biol Chem 281(14):9407–9417. doi:10.1074/jbc.M510037200
Sommer ME, Hofmann KP, Heck M (2012) Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat Commun 3:995. doi:10.1038/ncomms2000
Sommer ME, Hofmann KP, Heck M (2014) Not just signal shutoff: the protective role of arrestin-1 in rod cells. Handb Exp Pharmacol 219:101–116. doi:10.1007/978-3-642-41199-1_5
Szczepek M, Beyriere F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, von Stetten D, Heck M, Sommer ME, Hildebrand PW, Scheerer P (2014) Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun 5:4801. doi:10.1038/ncomms5801
Tobin AB, Butcher AJ, Kong KC (2008) Location, location, location… site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci 29(8):413–420. doi:10.1016/j.tips.2008.05.006
Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM (2003) The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278(8):6258–6267. doi:10.1074/jbc.M212231200
Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV (2000) An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J Biol Chem 275(52):41049–41057. doi:10.1074/jbc.M007159200
Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem 282(44):32075–32083. doi:10.1074/jbc.M706057200
Vishnivetskiy SA, Francis D, Van Eps N, Kim M, Hanson SM, Klug CS, Hubbell WL, Gurevich VV (2010) The role of arrestin alpha-helix I in receptor binding. J Mol Biol 395(1):42–54. doi:10.1016/j.jmb.2009.10.058
Vishnivetskiy SA, Baameur F, Findley KR, Gurevich VV (2013) Critical role of central 139-loop in stability and binding selectivity of arrestin-1. J Biol Chem. doi:10.1074/jbc.M113.450031
Wilson CJ, Copeland RA (1997) Spectroscopic characterization of arrestin interactions with competitive ligands: study of heparin and phytic acid binding. J Protein Chem 16(8):755–763
Zhuang T, Vishnivetskiy SA, Gurevich VV, Sanders CR (2010) Elucidation of inositol hexaphosphate and heparin interaction sites and conformational changes in arrestin-1 by solution nuclear magnetic resonance. Biochemistry 49(49):10473–10485. doi:10.1021/bi101596g
Zhuang T, Chen Q, Cho MK, Vishnivetskiy SA, Iverson TM, Gurevich VV, Sanders CR (2013) Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc Natl Acad Sci U S A 110(3):942–947. doi:10.1073/pnas.1215176110
Zindel D, Butcher AJ, Al-Sabah S, Lanzerstorfer P, Weghuber J, Tobin AB, Bunemann M, Krasel C (2014) Engineered hyperphosphorylation of the beta2-adrenoceptor prolongs arrestin-3 binding and induces arrestin internalization. Mol Pharmacol. doi:10.1124/mol.114.095422
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Sommer, M.E. (2017). How Arrestin Recognizes and Binds Active GPCRs. In: Gurevich, V. (eds) The Structural Basis of Arrestin Functions. Springer, Cham. https://doi.org/10.1007/978-3-319-57553-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-57553-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57552-0
Online ISBN: 978-3-319-57553-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)