
Abstract
Development requires cell proliferation, differentiation and spatial organization of daughter cells to occur in a highly controlled manner. The mode of cell division, the extent of proliferation and the spatial distribution of mitosis allow the formation of tissues of the right size and with the correct structural organization. All these aspects depend on cell cycle duration, correct chromosome segregation and spindle orientation. The centrosome, which is the main microtubule-organizing centre (MTOC) of animal cells, contributes to all these processes. As one of the most structurally complex organs in our body, the brain is particularly susceptible to centrosome dysfunction. Autosomal recessive primary microcephaly (MCPH), primordial dwarfism disease Seckel syndrome (SCKS) and microcephalic osteodysplastic primordial dwarfism type II (MOPD-II) are often connected to mutations in centrosomal genes. In this chapter, we discuss the consequences of centrosome dysfunction during development and how they can contribute to the etiology of human diseases.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Centrosome
- Microcephaly
- Animal models of microcephaly
- Autosomal recessive primary microcephaly (MCPH)
- Seckel syndrome (SCKS)
- Microcephalic osteodysplastic primordial dwarfism type II (MOPD-II)
2.1 The Centrosome Duplication Cycle
The centrosome is a non-membranous organelle composed of a pair of orthogonally organized centrioles, which during mitosis organize the pericentriolar material (PCM) [1]. The two centrioles are composed of nine sets of microtubules (MTs), polarized filaments of tubulin. The PCM surrounds the centrioles to support mitotic spindle assembly and consists of a highly organized matrix of more than one hundred proteins [2,3,4,5].
The centrosome is the main MT-organizing centre of animal cells : two centrosomes organize the mitotic spindle during mitosis and—most of the time—it is required to build the MT-cytoskeleton in interphase [6, 7]. In addition, in interphase , the centriole functions as basal body to template the assembly of cilia and flagella [8]. MT nucleation from the centrosome depends on the presence of γ-tubulin containing complexes [9]. The minus-ends of MTs are embedded at the centrosome, while the plus-ends extend in the cytoplasm, forming a polarized network that sustains chromosome (or molecular cargoes) movement. MTs possess an intrinsic dynamic instability and are in general built by 13 polar protofilaments, each being composed by heterodimers of α and β-tubulin [10, 11].
At the end of cell division, each daughter cell contains a single centrosome composed of a pair of orthogonally organized centrioles [12]. This allows the assembly an organized MT network in interphase [13]. To form a bipolar spindle at the next mitosis, the centrosome is duplicated only once per cell cycle in a tightly regulated process (Fig. Fig. 2.1) [15]. Centrosome duplication is licensed by centriole disengagement, which occurs during G1 when centrioles lose their orthogonal configuration [15]. Centriole disengagement allows the assembly—during S-phase—of one daughter procentriole next to each mother. In preparation for mitosis, the centrosomes starts recruiting PCM and the two centrosomes separate and nucleate MTs in order to assemble the mitotic spindle. The very last step of cell division, cytokinesis, will then separate the two centrosomes in two distinct cells.

The centrosome duplication cycle . The centrosome is composed of a pair of centrioles (green cylinders) [12] surrounded by pericentriolar material (PCM) (orange) [14]. It normally duplicates once, in coordination with the cell cycle (composed by sequential G1-, S-, G2- and M-phase) [15]. (1) Centriole disengagement or ciliogenesis . At the end of cell division, each daughter cell contains a single centrosome composed by a pair of orthogonally organized centrioles. This configuration is lost in G1 in a process called “disengagement”, which license the centrosome duplication cycle [15]. Alternatively, the mother centriole can dock at the membrane and form the basal body that templates the assembly of cilia and flagella [8]. (2) Centriole duplication . During S-phase a number of proteins are timely recruited on the disengaged centrioles and trigger the assembly of a new centriole next to each parental one (see Fig. 2.2 for a more detailed description of the process) [17,18,20]. (3) Centriole elongation . The newly formed centrioles continue to elongate during S-phase and the rest of the cell cycle [21]. From G2 onwards, the centrosomes reinforce the recruitment of PCM material, which will serve to organize the mitotic spindle in M-phase [22]. (4) Centrosome separation . At the beginning of M-Phase, centrosomes separate and migrate at opposite side of the cell [23]. Each centrosome will be composed of two centrioles that organize the PCM: an older, parental centriole and a daughter one. (5) Spindle assembly . In mitosis, the centrosomes nucleate MTs and organize the mitotic spindle, a network that contacts the chromosomes and allows their segregation at opposite pole of the cells [24]
2.1.1 Proteins Required for Centrosome Duplication
Proteins involved in the centrosome duplication cycle have been initially identified through genome wide screens in C. elegans and they present functional homologs in the fruit fly Drosophila melanogaster (D) and in humans (Homo sapiens, Hs). Their recruitment to the centrosome and their activity are sequential [17,18,20]. Throughout evolution the three major steps required for centriole duplication have been conserved [16, 17] (Fig. Fig. 2.2). They consist of:

Proteins required for centrosome duplication . The core machinery required for centrosome duplication is well conserved [16, 17]. The proteins sequentially required for centriole duplication in C. elegans, D. melanogaster and H. sapiens are listed. They act timely to induce recruitment of kinase activity to the centrosome (1), formation of a centriole-primordium (2) and incorporation of MTs at the newly formed centriole (3)
-
1.
Recruitment of kinase activity to the centrosome (proteins involved: SPD-2/HsCEP192/DSPD-2 or HsCEP152/DAsl and ZYG-1/HsPLK4/DSak)
-
2.
Formation of a procentriole-primordium (proteins involved: SAS-6/HsSAS6/DSas-6 and SAS-5/HsSTIL/DAna2)
-
3.
Incorporation of MTs at the newly formed procentriole (proteins involved: SAS-4/HsCPAP/DSas4, HsCP110, Hsγ-tubulin, HsCEP135/DBld10). Of note, other members of the tubulin superfamily (zeta-, epsilon- and delta-tubulin) are required to form centrioles in certain cells (for a review, see [25])
For simplicity, throughout this chapter, we will refer to these genes with their Hs name.
2.2 The Centrosome and the Mitotic Spindle
The mitotic spindle supports chromosome separation during mitosis. It consists of a bipolar, antiparallel array of MTs [11, 26, 27]. In animal cells it is composed by MTs nucleated mainly by the centrosome. Astral MTs are typical of centrosomal-spindles: they emanate from the centrosome into the cytosol and—contacting the cortex—play important roles in spindle orientation [23,24,25,31]. The mitotic spindle is composed by different populations of coexisting MTs. These can be distinguished based on their orientation, function and stability. Kinetochore MTs connect the chromosomes to the spindle machinery while MTs that emanate from opposite poles and interact in an antiparallel fashion are named interpolar MTs [27] (Fig. 2.1).
MTs can also be assembled by the Augmin complex through branching from pre-existing MTs [32]. Additionally, MTs are nucleated at the level of the chromatin by the establishment of a RanGTP gradient after nuclear envelope breakdown [28,29,30,31,32,38]. Chromatin-dependent spindle assembly occurs physiologically in different systems. For instance, this occurs frequently during female meiosis, even in human oocytes [39]. However, this process can also support spindle assembly when centrosomes are experimentally removed (e.g. [35,36,37,43]).
Importantly, while bipolar mitotic spindle assembly can occur in the absence of centrosomes, these organelles are normally required to ensure correct spindle orientation. Spindle orientation determines the position of daughter cells at the end of mitosis and contributes to the differential inheritance of cytoplasmic and cortical factors [40, 39,40,41,42,48].
2.3 The Centrosome and the Cilium
One important role of the centrosome in interphase is to function as a basal body to template the assembly of cilia and flagella. In light of human brain anatomy and function, two kinds of cilia need to be described: motile cilia and primary cilia.
Motile cilia normally present 9 MT doublets plus a central pair of MTs and are required to mediate fluids movement in the human body. In the brain, ependymal cells are responsible for the flux of the cerebrospinal fluid (CSF) . These cells are multiciliated: they present hundreds of cilia, which are formed by sequential, multiple non-canonical centriole duplication cycles [49, 50].
Primary cilia slightly differ from motile cilia in their structure, as they lack the central pair of MTs [51]. These cilia, also known as non-motile cilia, mainly function as signalling hubs. In the brain, they are important to sense signalling molecules transported in the CSF , including growth factors, sonic hedgehog (Shh) and Wnt ligands [47,48,49,50,51,52,58].
2.4 General Principles Governing the Effects of Centrosome Dysfunction
Centrosomes play important roles in determining the outcome of cell division, both in terms of cell fate and cell survival. We conceive that centrosome defects can impair this process by affecting at least four different mechanisms: orientation of the mitotic spindle [28, 30, 40, 45, 48, 59, 60], correct chromosome segregation [56,57,58,64] and assembly of a primary cilium [53]. In addition, centrosome loss can affect cell proliferation: it has been recently shown that centrosome removal triggers p53 activation and arrests vertebrate cell in G1 [65, 66].
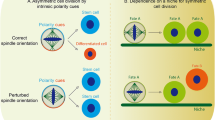
The position of the spindle defines where daughter cells will be positioned at the end of mitosis as well as the cortical/cytoplasmic inheritance they will receive. During symmetric cell division , the factors inherited by daughter cells are equivalent and the daughter cells will have similar fates. During asymmetric cell division , daughters are unequal and will differ in fate [46]. In Drosophila neural stem cells, cell fate-determinants are anchored to the membrane and directly transmitted to daughter cells by cortical inheritance, while in mammalian NSCs structural elements such as junctions, the apical membrane and the basal lamina are proposed to be responsible for cell fate determination [46, 57, 62,63,69]. Interestingly, the centrosome itself can be inherited asymmetrically by stem cells and differentiating cells [65,66,67,73]. The role of centrosome asymmetry during development or in maintaining tissue integrity is still not understood. A plausible explanation comes from a study performed in vertebrate cells in culture where it has been shown that, after mitosis, primary cilia grow asynchronously in the two daughter cells [74]. The cell that inherits the older centrosome will initiates ciliogenesis earlier than its sister, putting forward the concept that centrosome asymmetry might influence the capacity to sense environmental signals [74].
Centrosome defects can generate abnormal cilia, both in terms of number or structure. For instance, the nucleation of extra primary cilia can impact on the ability of a cell to transduce Sonic Hedgehog (Shh) and Wnt signaling and might lead to alterations in cell fate [53, 58].
In addition, defects in centrosome number (mainly centrosome amplification) and function can impair spindle activity during chromosome segregation. This can lead to alterations in the karyotype of daughter cells, a condition known as aneuploidy [75]. The pathological implications of an abnormal number of chromosomes are extremely broad and can be associated with both over proliferation (e.g. cancer) [76] or defective growth (e.g. microcephaly) [62], which will be further discussed in the following paragraphs.
Aneuploidy per se can be at the basis of premature differentiation or cell death [62, 72,73,74,80]. Additionally, lengthening of the G1 phase of the cell cycle has been shown to cause exhaustion of proliferative divisions and favour differentiation, probably by allowing the cell extra time to sense differentiation signals [76,77,83].
2.5 Neocortex Development: Evolutionary Insights
Drosophila is an invertebrate organism commonly used to understand the genetic bases of developmental processes. Brain development in Drosophila is quite stereotyped and the deep knowledge of its anatomy and of the cell types composing it render it an ideal model to explore the role of different factors in neurogenesis. During embryonic development, a population of neural stem cells called neuroblasts (NBs) delaminates from the neuroectoderm and give rise to the larval brain, composed by two lobes and a ventral nerve cord [84]. After a period of dormancy, proliferation of the larval NBs resumes. NBs divide in an asymmetric fashion and generate two daughter cells with distinct fates: a new NB, which retains the stem-cell potential, and a ganglion mother cell, which undergoes a single additional division. At the end, each NB gives rise to a reasonably invariant set of neurons and glial cells [85].
The vertebrate brain (including Zebrafish, mouse and human brain) can roughly be subdivided in three parts : the forebrain, the midbrain, and the hindbrain [86].
The mammalian brain is characterized by the development of the neocortex, composed of six layers of neurons. It is the part of cerebral cortex which underwent the biggest and most recent phylogenetic expansion, mainly by growth in the lateral and radial dimension [57, 87, 88]. It originates from the divisions of neuroepithelial cells, which give rise to a set of intermediate progenitors that will undergo additional divisions. Taverna et al. have proposed a classification of these populations based on the localization of progenitor mitosis with respect to the ventricular zone (VZ) (apical—AP, basal—BP and subapical progenitors—SAP), the extent of cell polarity and their proliferative capacity [57].
The evolution of different types of progenitor cells in the primate and human brain has contributed to the expansion of the cerebral cortex [88]. In particular, a novel type of non-epithelial progenitors has been described in the outer subventricular zone (OSVZ) , which are proposed to have contributed to the evolutionary expansion of the human brain [89].
The human brain strongly differs from that of other mammals by its degree of corticalization, which accompanies an increase in cognitive functions [90, 91]. This impressive growth in brain size has been accommodated in the skull thanks to gyrification. Interestingly, it has been reported that the formation of gyri in the otherwise unfolded mouse cortex (lissencephalic) can be induced by modifying the expression pattern of a single protein or by forcing the expression of a human gene [92, 93].
2.5.1 Centrosome and Brain Development: Lessons from Drosophila
In order to explain the effects of centrosomal defects on brain development, we will start by presenting two extreme cases: what happen when centrosomes are absent and when they are present in excess (while for others—more specific—models, we address the readers to the corresponding paragraphs). Both scenarios can be obtained by mutating or overexpressing proteins involved in the centrosome duplication cycle. For instance, mutations in Plk4 result in centrosome loss, while Plk4 overexpression causes centrosome amplification [59, 94].
In the absence of centrosomes, mitotic spindles can be assembled from the vicinity of chromatin or from pre-existing MTs [32, 36, 37, 95]. However, centrioles are indispensible to nucleate sensory cilia and sperm flagella [91,92,98]. Flies without centrioles can develop in viable adults, but they die shortly after eclosion because their sensory neurons lack cilia, affecting vital function such as movement, smell and prioception [59, 94, 94,95,101]. Acentrosomal mitoses do not generate aneuploidy in dividing NSCs. However, due to lack of astral MTs, spindle orientation and asymmetric cell division are perturbed, leading to an expansion of the stem cell pool, which is tumorigenic in allogeneic transplantations [40, 44, 47, 102].
When extra centrosomes are present, their efficient clustering at the spindles poles allows the formation of bipolar spindles and ensures correct chromosome segregation. Flies with centrosome amplification do not present gross cilia defects. However, asymmetric cell division is again perturbed, and the brain holds tumorigenic potential [59].
Overall, we think that centrosome defects mostly affect spindle positioning in Drosophila and that this is the main route by which cell proliferation is affected. However, the mammalian brain can respond differently to the same kind of perturbations (see paragraph, see below for further details).
Of note, not all tissues have the same way of responding to centrosome loss and amplification. While centrosomes number in the brain is not a variable influencing faithful chromosome segregation, it can generate aneuploidy in the wing disc [63, 103]. These results suggests that, when centrosomes are perturbed, mitotic fidelity will rely on the strength of alternative mechanisms for spindle assembly or on the capacity to achieve centrosome clustering.
2.5.2 Centrosome and Brain Development: Lessons from Zebrafish
Small head size phenotypes were obtained by Novorol and colleagues in Zebrafish after knockdown (KD) of four centrosomal genes: stil, aspm wdr62 and odf2 [104] (see also [105]), generating abnormal centrosome number and localization. In these mutants the microcephalic brain results mainly from mitotic defects - namely prometaphase delay [104], while the contribution of p53-dependent apoptosis is minor. In addition, the work from Pfaff, K.L. and colleagues [106], also reported a strong mitotic phenotype with highly disorganized spindles in Zebrafish in the absence of Stil. An additional Zebrafish model with non-functional centrosomes was obtained by targeting NEDD1, which is required to recruit γ-tubulin at the centrosome [107]. NEDD1 knockdown (KD) resulted in mitotic arrest and apoptosis. Depending on the intensity of the KD, the phenotype was spanning from embryonic lethality to severe defects in the brain [107].
Depletion of Plk4 causes a strong size reduction in Zebrafish, mainly due to abnormal spindles and mitotic defects, including a substantial delay in mitotic progression [108]. Moreover, the impairment of centriole duplication due to Plk4 knockdown results in a dilution of basal bodies and causes a dose-dependent ciliary phenotype [108]: while a mild reduction in Plk4 levels mainly affect mitosis by reducing the number of centrioles and thus altering the bipolar configuration of the spindle, a stronger KD resulting in complete centriole/basal body loss impairs the ability of cells of growing cilia [108].
The consequences of centrosome amplification in the Zebrafish brain have been studied recently [109]. In this system, extra centrosomes do not cluster and induce the formation of multipolar spindles. Multipolar divisions can lead to the presence of multiple nuclei in one of the daughter cells. When occurring in the neuroepithelial progenitors, this leads to apoptosis, tissue degeneration and death, affecting mostly retinal neuronal layering [109].
2.5.3 Centrosome and Brain Development: Lessons from Mouse
Similarly to Drosophila, centrosome removal and amplification in mouse can be achieved genetically by manipulating genes involved in the centrosome duplication cycle [62, 110].
Since most vertebrate cells are ciliated, lack of centrioles would be expected to cause lethality due to the absence of cilia. However, CPAP mutant mouse embryos die earlier than mutants lacking cilia. In rodents, centriole presence becomes essential from embryonic day 9 [105,106,107,108,114] and experimental removal of centrosomes causes mitotic delay and p53-dependent cell death in the embryo [110, 115, 116]. Bazzi and Anderson recently showed that a null mutation in CPAP results in embryonic lethality at midgestation. They also observed a prometaphase delay and demonstrated its involvement in p53 activation and p53-dependent apoptosis [110].
When progressive loss of centrioles is taking place in the neuronal precursors, mice develop microcephaly [45, 110]. p53 removal rescues cell death and the reduced brain size phenotype. However, it does not rescue the defects in tissue architecture due to abnormal spindle orientation that leads to misplacement of neural progenitors [45]. Very recently it has been shown that—in addition to randomizing spindle orientation—CPAP silencing in post-mitotic neurons leads to abnormal morphology and slower neuronal migration [117]. The defective neuronal migration described here is ascribable to the function that CPAP exert on interphase rather than mitotic MTs, and open new possible roles for centrosomal genes in contributing to growth disorders. Supporting this view, Gabriel, E. and colleagues have shown that CPAP promotes neural progenitors fate by promoting cilia disassembly, rather than by a centrosomal function [118].
As centrosome loss, centrosome amplification in the mouse central nervous system causes microcephaly [62]. Compared to Drosophila, mouse neural stem cells have less efficient clustering mechanisms. Multipolar spindles cause errors in chromosome segregation that leads to aneuploidy and p53-dependent cell death . Interestingly, p53 inhibition rescues cell death, but prompts premature differentiation of progenitor cells, mirroring results obtained in Drosophila aneuploid brains [119]. However, while in flies premature differentiation is a primary response to aneuploidy, in mouse it is probably a secondary mechanism taking over only when the p53 primary response is not efficient. These observations seem paradoxical, since aneuploidy has long been regarded uniquely as conferring proliferative advantage. However, recent studies showing a negative effect of aneuploidy on proliferation put forward the novel concept that chromosome imbalance mostly hinders proliferation , and only specific gain or losses might favor malignant transformation (which might not occur through proliferative advantage—e.g. see [115,116,122]).
2.6 The Centrosome and Its Role in Primordial Microcephalic Disorders
Primordial microcephalic disorders include a spectrum of diseases characterized by severe growth retardation. Autosomal recessive primary microcephaly (MCPH) (2.6.1), microcephalic osteodysplastic primordial dwarfism type II (MOPD-II) (2.6.2) and primordial dwarfism disease Seckel syndrome (SCKS) (2.6.3) are all primordial microcephalic disorders that share phenotypic and genetic traits. In the following paragraphs, we will present a summary of clinical description and molecular insights for each of these syndromes. These disorders can be linked to premature exhaustion of proliferative division of stem cells, due to premature differentiation or cell death . The causes underlying cell death/differentiation can be multiple and include defects in spindle robustness [123], in spindle positioning/orientation [124], in cell cycle progression or DNA damage repair [125] or in chromosome segregation [62].
2.6.1 Autosomal Recessive Primary Microcephaly (MCPH: Microcephaly Primary Hereditary)
MCPH is characterized by a reduction of the occipito-frontal circumference, which can be nearly normal at birth (−2) but is inevitably worsen in the first year of life (−3) [126]. It is a rare genetic disease, found in about 100 families [127]. Brain size reduction is proportionate, albeit affecting particularly the cerebral cortex [128]. Other clinical features are mental retardation, mild seizures and particular neuronal migration defects [121,122,123,129].
MCPH causal mutations are found in a large number of genes and can all cause premature differentiation, cell death and displacement of neural progenitors. Thirteen MCPH loci (1–13) have so far been identified in human patients, encoding for 13 different genes. MCPH genes can be grouped in 3 (partially overlapping) categories :
-
1.
genes with a role in centrosome and spindle function (CEP152, CEP63, SAS-6, STIL, CPAP/CENPJ, CEP135, CDK5RAP2, CDK6, ASPM, WDR62 and STIL; see next paragraphs for a detailed description);
-
2.
genes with a role in chromosome dynamics. This include the kinetochore gene CASC5, encoding for KNL1 [130] and CENPE. CENPE is encoded in an MCPH locus, but generates a more severe phenotype, similar to MOPD-II [131];
-
3.
genes with a role in DNA-damage related pathways. In this category we can include Microcephalin, PHC1, ZNF335 [127,128,129,135]. Of note, CDK6, ASPM, STIL are involved in both cell cycle regulation and centrosome function.
In light of recent results an additional, fourth class of genes involved in cell cycle regulation (Microcephalin and ASPM), should be considered [81, 133].
For the purposes of this chapter, we will focus on the genes with a role in centrosome and spindle function (category 1) and we will describe their identification in MCPH, MOPD-II and SCKS. For the other genes , we address the reader to [127, 136, 137]. However, a small digression should be made on Microcephalin (Mcph1). Microcephalin is mostly known for its role in DNA-related processes (DNA damage response, chromosome condensation) [129, 138, 139] but was also involved in the etiology of microcephaly through its role in coupling the centrosome cycle with mitosis [140]. Indeed, mutations in Drosophila MCPH1 are early embryonic lethal. Mutant embryos exhibit asynchronous nuclear and centrosome cycle, abnormal centrosomes and spindles, chromatin bridging due to premature chromosome condensation and mitotic arrest [141, 142].
Genes with a role in centrosome and spindle function that are at the origin of MCPH were previously classified in centriole duplication genes, genes encoding PCM proteins and genes that encode spindle-pole associated proteins [143].
2.6.1.1 Centriole Duplication Genes : CEP152, STIL, CEP135, CEP63, SAS-6 and CPAP/CENPJ
CEP152 underwent positive selection in humans [144]. A non-conservative amino acid change in CEP152 has been identified by SNP genotyping in three MCPH families. Only one of the cases reported was heterozygous for the missense mutation, with the second allele characterized by a premature stop codon [144].
Mutations in STIL that results in truncation of the protein has been reported in 4 MCPH families [145]. In 2014, Arquint and Nigg [146] described two truncating mutations in STIL found in MCPH patients which perturbs its ubiquitination, thus compromising its degradation and causing centrosome amplification.
MCPH mutations in CPAP/CENPJ have fist been described by Bond and colleagues, [147]. The authors found a homozygous single-base deletion and a substitution resulting in an amino acid change in a very conserved residue of the protein.
A truncated form of the protein encoded by CEP135 has been described in MCPH patients by Hussain et al. [148] and Farooq et al. [149]. So far, reported MCPH mutations in CEP135 are a single base deletion in exon 8 and a splice site mutation leading to complete skipping of exon 11 with loss of the C-terminus domain of CEP135 necessary for the interaction with SAS-6 [149].
The protein encoded by CEP63 interacts with CEP152 and plays an important role in regulating centrosome number. Homozygous mutations in this gene generating a premature stop codon have been found to cause MCPH [150]. Further analysis demonstrated that the protein is normally localized in a discrete ring around the parental centriole and that this localization is lost in patient-derived cells. Centrosome maturation and separation were not found to be perturbed, however, the presence of abnormal spindles—probably due to delayed procentriole assembly and erroneous centriole engagement—highlighted possible defects in centrosome duplication [150]. Surprisingly, the mitotic phenotype observed in CEP63KD cells was rescued by exogenous targeting of CEP152 to the centrosome, suggesting that the microcephalic phenotype could be ascribed to the role of CEP63 in recruiting CEP152 in rapidly proliferating cells. Interestingly, CEP63 deficient mice recapitulate SCKS. In this model, mitotic errors due to centrosomal defects cause p53-dependent cell death, resulting in a reduced brain size. In addition, CEP63 loss impairs male meiotic recombination [151].
A missense mutation within a highly conserved region of SAS-6 has been found by homozygosity mapping in individuals from a Pakistani consanguineous family. Tissue culture analysis of the Ile62Thr SAS-6 mutant revealed that this form of the protein is less efficient in sustaining centriole formation . Further analysis by protein KD revealed the presence of monopolar spindles [152].
2.6.1.2 Genes Encoding PCM Proteins: CDK5RAP2 and CDK6
The formation of a functional mitotic centrosome requires the expansion of PCM around the centrioles, in a process known as centrosome maturation [153, 154].
CDK5RAP2 is a pericentriolar protein required for γ-tubulin recruitment and MT nucleation at the centrosome [155, 156]. It is a MCPH protein highly expressed in the neuronal progenitor pool. Its loss causes depletion of apical progenitors due to cell cycle exit and premature differentiation [157]. CDK5RAP2 mutations were initially identified in two MCPH families. The mutations found were in coding and non-coding regions of the gene respectively, causing an amino acid substitution in the first case and aberrant splicing (generating a truncated protein) in the second [147]. Strikingly, mutations in CDK5RAP2 gene in mice (153) or in human pluripotent stem cell-derived 3D organoid culture system (cerebral organoids) can recapitulate microcephaly, probably due to premature neurogenic non-proliferative divisions [159]. Interestingly, CDK5RAP2 mutations most likely contribute to MCPH onset through defects in several processes. Characterization of a Hertwig’s anemia mutant mouse model revealed the presence of multipolar spindles, spindle mispositioning and increased apoptosis due to an inversion in CDK5RAP2 [158]. Cerebral organoids derived from reprogramming of patient skin fibroblasts, mainly displayed misoriented spindles [159]. Finally, in the avian B cell line DT40, CDK5RAP2 function has been linked to the cohesion between centrioles and has been shown to promote cell cycle arrest in cells that underwent DNA-damage [160]. In flies, CDK5RAP2 plays several roles: for instance, it is required for centrosome maturation [161], it regulates centrosome size [162], establishes centrosome asymmetry [163] and represses dendrite branching [164], sustaining the view that mutations in this gene might contribute to MCPH in different ways.
CDK6 is a cyclin dependent kinase required for cell cycle progression and it was found to be mutated in a Pakistani family with MCPH [165]. The mutation—Alanine to Threonine conversion—occurs at the level of a very conserved residue. In the same study, the protein was found to localize at the mitotic centrosomes, but this localization was lost in patient primary fibroblast, leading to the conclusion that CDK6 could play a role in organizing centrosomal-MTs and in centrosome positioning.
2.6.1.3 Genes Encoding Spindle-Pole Associated Proteins: ASPM and WDR62
ASPM is a protein required for spindle integrity, as it plays a role in focusing the poles of the mitotic spindle [166]. It is normally down regulated during the switch from proliferative to neurogenic division of neural progenitors and is required to maintain spindle orientation [167]. Mutations in ASPM are the most common cause of MCPH [168] and strong evidence favor a role for ASPM in neurogenesis: Pulvers and colleagues [169] showed that in mice, ASPM mutations similar to those causing microcephaly in humans, cause abnormal protein localization during mitosis. They also report the appearance of mild microcephaly that can be rescued by the human transgene. A very recent paper proposed a role for ASPM in the etiology of MCPH by regulating cell cycle progression through G1 instead of spindle orientation. Capecchi and Pozner [81] generated a new mouse model and demonstrated that ASPM can tune cyclin E ubiquitination and—thus—mitotic progression through G1. Interestingly, mutations in the Drosophila functional homolog of ASPM can also cause severe defects in brain size and neuroepithelium morphogenesis. The absence of ASPM results in abnormal mitosis and increased apoptosis. In addition, ASPM mutants present abnormal spindle positioning and abnormal interkinetic nuclear migration, which compromises tissue architecture [170].
Missense and frame-shifting mutations in WDR62 have been identified in seven MCPH families, making this gene the second most commonly mutated in MCPH after ASPM. WDR62 is specifically expressed in neuronal precursors undergoing mitosis and, similarly to what has been observed for CDK6, the centrosomal localization of the protein is lost in the mutant form [171]. More recently, the study of a WDR62 mouse model established that mutant progenitor cells show spindle instability (including multipolar spindle formation) leading to mitotic arrest, cell death and microcephaly [172]. Interestingly, mutations in the Drosophila functional homolog of WDR62 affect the asymmetry that normally characterizes apical and basal MTOCs during interphase. This is due to a lack of Plk1/Polo recruitment on the apical centrosome , which mediates MTOC activity [173].
2.6.2 Majewski/Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPD-II)
MOPD-II patients are characterized by severe pre-natal and postnatal growth failure with proportionate microcephaly at birth, that evolves in disproportionate microcephaly [169,170,176]. MOPD can be clinically distinguished from Seckel syndrome mainly from the radiologic finding of skeletal dysplasia [175], as well as by less severe mental retardation but more pronounced growth defects [177]. Mutations in the PCNT gene (encoding a PCM protein) were described to be at the origin of both SCKS and MOPD-II, with the diagnosis often revised when evidence of skeletal dysplasia appear [178]. Absence of PCNT causes defects in spindle structure, which leads to chromosome missegregation [179]. In [177] MOPDII was defined as a “genetically homogeneous condition due to loss of function of pericentrin”. Analyzing the current knowledge, Delaval A. and Doxsey J. proposed three mechanisms to explain the implication of pericentrin in a phenotype of reduced growth: (1) through its role as a DNA-damage checkpoint protein, (2) because of its function in MT nucleation and (3) in light of its role in spindle orientation and organization [180].
-
1.
Role of pericentrin as a checkpoint protein. Mutations in PCNT were reported in individuals with SCKS and cells from these patients displayed defects in the ATR-dependent checkpoint signaling for DNA damage [181]. In addition, pericentrin plays a role in anchoring Chk1 at the centrosome, thus regulating the activation of centrosomal cyclin B-Cdk1. Its mutation would then be responsible for premature mitotic entry, even in the presence of DNA damage [182]
-
2.
Function of pericentrin in MT nucleation. Localization of Pericentrin at the centrosome is required to sustain MT nucleation during mitosis [183] and thus to ensure mitotic spindle organization [123]. Interestingly, pericentrin loss phenocopies CDK5RAP2 loss in mice and causes a reduced recruitment of CDK5RAP2 at the centrosome [157].
-
3.
Role of pericentrin in spindle organization. PCNT depletion in human cells causes γ-tubulin loss at the centrosome and disrupts astral MT nucleation, [123], spindle positioning [184] and organization, impairing chromosome segregation [210].
Of note, mutations in mouse PCNT and in the Drosophila functional homolog cause ciliary phenotypes. A mouse model with hypomorphic PCNT mutation displayed malformed cilia in the olfactory chemosensory neurons [185]. Similarly, adult flies present defects in sensory neuron cilia and sperm flagella function. However, in this system , PCNT is dispensable for mitosis and spindle formation [101].
2.6.3 Seckel Syndrome (SCKS)
Seckel syndrome was described by Seckel in 1960 as a severe form of dwarfism accompanied by microcephaly, a distinctive facies and mental retardation [186]. It is a rare and heterogeneous type of primordial dwarfism, very similar to MOPD and firstly distinguished from it by Majewski and Goecke [175]. Mutated genes identified so far in SCKS play a role in centriole duplication and are CPAP/CENPJ and CEP152 [144, 183,184,189], initially associated with MCPH (see MCPH section for further details on CEP152), and PCNT (see MOPD-II section for further details). Interestingly, in mouse, a hypomorphic allele of CPAP/CENPJ recapitulates several features of Seckel syndrome. Those arise from defective spindle formation and genetic defects such as polyploidy, aneuploidy and apoptosis [190].
2.7 Centrosomal Genes and Other Growth Syndromes
Genes with a role in centrosome and spindle function that are at the origin of other forms of primordial microcephalic disorders other than MCPH, SKCS and MOPD-II include CPAP, CEP152, PCNT (see previous sections for detailed descriptions), the centriole duplication gene PLK4 and the PCM gene TUBGCP4.
Plk4
PLK4 is the master regulator of centriole duplication [94, 191]. In 2014, two different groups identified mutation in the Plk4 genes in individuals with microcephalic primordial dwarfism [108, 192]. Martin and colleagues analyzed a Zebrafish model for Plk4 loss of function and found that centriole biogenesis is compromised, causing both longer, abnormal mitosis—which lead to impaired growth—and ciliary phenotypes. Interestingly, the growth and ciliary phenotypes were dependent on Plk4 dosage. Indeed, cilia loss correlates with complete absence of basal bodies, while mitosis can proceed and being perturbed even when centrioles are present, but their number is reduced. This suggests that—in this system—mitosis is more sensitive to centriole depletion than ciliogenesis [108].
TUBGCP4
TUBGCP4 (tubulin gamma complex associated protein 4) is a component of the γ-tubulin ring complex. Compound heterozygous mutations in TUBGCP4 have recently been reported in individuals with autosomal recessive microcephaly and patient-derived fibroblasts presented abnormal MT organization and aneuploidy [193]. Moreover, striking nuclear defects were reported: enlarged nuclei of abnormal shapes, chromatin bridges and multinucleation were detected in patient-derived fibroblasts [193].
In addition, mutations in POC1 centriolar protein A (POC1A) have been reported in patients affected by primordial dwarfism. Patient’s fibroblasts displayed abnormal spindles and impaired ciliogenesis [194] and have been shown to contain centrosome amplification [195].
2.8 Other Centrosome-Related Developmental Syndromes
Oral-Facial-Digital Syndrome Type I (OFD1)
OFD1 is a complex syndrome, characterized by polycystic kidney disease and malformations of the mouth, face, brain and digits. It represents an interesting case related to defective primary cilium signaling, although it results from mutations in a basal body gene rather than a ciliary gene. The syndrome is mainly caused by caused by mutations in the CXORF5/OFD1 [196], which colocalizes with γ-tubulin, suggesting an association not only with the cilium—but also with the centrosome [197]. Mutations in CXORF5/OFD1 cause dysfunctions of the primary cilium, abnormal proliferation, abnormal Hedgehog and Wnt signalling and defects in planar cell polarity [198]. In normal human embryos, the gene product localizes in the organs affected by the syndrome, including the brain.
Meier Gorlin Syndrome
Meier-Gorlin syndrome is a form of microcephalic primordial dwarfism often due to mutations in DNA-replication proteins, like Orc1 [199]. Orc1 depletion in Zebrafish was proposed to cause reduced body size due to an impairment of replication licensing and cell cycle lengthening [199]. However, in addition to controlling DNA replication by interacting with Cyclin A–CDK2, a different domain of Orc1 controls Cyclin E–CDK2-dependent centriole and centrosome copy number [200, 201]. Analysis of Meier-Gorlin causing mutations in Orc1, revealed that they can cause centrosome amplification [201], putting forward the idea that extra centrosomes might contribute to the growth-defective phenotype observed.
2.9 Concluding Remarks and Current Opinions on Microcephaly
The genetic background of animal models used to study human diseases can dramatically influence the phenotypes observed. A representative example can be found in CDK5RAP2 mouse models. While the one used in [158] could recapitulate microcephaly, the system used in [202] did not reveal significant defects in brain size. The high degree of human brain corticalization, its complexity and the presence of specific progenitor cell populations are unique features, difficult to recapitulate with rodent model systems. Thus, the introduction of cerebral organoids represents a great opportunity to study the etiology of human diseases.
Centrosome defects are very often at the origin of microcephaly and reduced growth. However, other genetic or environmental phenomena can generate similar effects. This is for example the case of fetal alcohol syndrome [203]. In addition, a recent outbreak of microcephaly in Brazil with the number of cases increased of 20-fold in the last months [204, 205], indeed suggests that the developing human brain is more vulnerable in terms of size than other organs. Why does the Zika virus, a flavivirus that in adults causes relatively mild syndromes and is transmitted by mosquitoes [206], affects specifically the developing brain remains to be understood.
Zika tropism to the brain was reported in two studies in 1952 and 1971 [207, 208], but only recently it was shown to infect human neural progenitor cells, causing cell death and cell-cycle alterations [209] and providing a possible explanation for the role of Zika in establishing brain size reduction.
Based on the current knowledge, we propose that the main mechanisms by which abnormal centrosomal components can induce defective brain growth are aneuploidy and alteration of cell cycle length. These can cause cell cycle exit or cell death, leading to a premature exhaustion of neural progenitors. The recent Zika outbreak raised awareness on microcephaly, which is normally a rare condition. Despite great advances in elucidating its causes, the reasons why the development of a normal-sized, well-organized brain is more susceptible to centrosome defects than the rest of the body in vertebrates and mammals remains to be understood.
Importantly, the zygotic centrosome mutations found in MCPH mostly generate architecturally normal but smaller brains, without affecting body size. Why is the brain so vulnerable to centrosome mutations is an important question that remains unanswered. One possibility is that neural progenitors are particularly susceptible to centrosome mutations when compared to other progenitors in the body. Alternatively, establishment of brain size might rely more than other organs on cell divisions that occur during developmental stages . However, other possibilities should not be discarded and this remains—in our view—the major open question in the field.
References
Bornens M (2002) Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol 14:25–34
Bobinnec Y, Khodjakov A, Mir LM, Rieder CL, Edde B, Bornens M (1998) Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J Cell Biol 143:1575–1589
Mennella V, Keszthelyi B, McDonald KL, Chhun B, Kan F, Rogers GC, Huang B, Agard DA (2012) Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Biol 14:1159–1168
Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139:663–678
Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA (2012) 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open 1:965–976
Karsenti E, Newport J, Hubble R, Kirschner M (1984b) Interconversion of metaphase and interphase microtubule arrays, as studied by the injection of centrosomes and nuclei into Xenopus eggs. J Cell Biol 98:1730–1745
Kellogg DR, Moritz M, Alberts BM (1994) The centrosome and cellular organization. Annu Rev Biochem 63:639–674
Ishikawa H, Marshall WF (2011) Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 12:222–234
Moritz M, Braunfeld MB, Guenebaut V, Heuser J, Agard DA (2000) Structure of the gamma-tubulin ring complex: a template for microtubule nucleation. Nat Cell Biol 2:365–370
Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13:83–117
Mitchison TJ (1993) Localization of an exchangeable GTP binding site at the plus end of microtubules. Science 261:1044–1047
Anderson RG (1972) The three-dimensional structure of the basal body from the rhesus monkey oviduct. J Cell Biol 54:246–265
Karsenti E, Kobayashi S, Mitchison T, Kirschner M (1984a) Role of the centrosome in organizing the interphase microtubule array: properties of cytoplasts containing or lacking centrosomes. J Cell Biol 98:1763–1776
Stearns T, Evans L, Kirschner M (1991) Gamma-tubulin is a highly conserved component of the centrosome. Cell 65(5):825–836
Tsou MF, Stearns T (2006) Mechanism limiting centrosome duplication to once per cell cycle. Nature 442:947–951
Carvalho-Santos Z, Machado P, Branco P, Tavares-Cadete F, Rodrigues-Martins A, Pereira-Leal JB, Bettencourt-Dias M (2010) Stepwise evolution of the centriole-assembly pathway. J Cell Sci 123:1414–1426
Hodges ME, Scheumann N, Wickstead B, Langdale JA, Gull K (2010) Reconstructing the evolutionary history of the centriole from protein components. J Cell Sci 123:1407–1413
Delattre M, Canard C, Gonczy P (2006) Sequential protein recruitment in C. elegans centriole formation. Curr Biol 16:1844–1849
Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA (2007) Plk4-induced centriole biogenesis in human cells. Dev Cell 13:190–202
Pelletier L, O'Toole E, Schwager A, Hyman AA, Muller-Reichert T (2006) Centriole assembly in Caenorhabditis elegans. Nature 444:619–623
Kohlmaier G et al (2009) Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr Biol 19(12):1012–1018
Dobbelaere J et al (2008) A genome-wide RNAi screen to dissect centriole duplication and centrosome maturation in Drosophila. PLoS Biol 6(9):e224
Faragher AJ, Fry AM (2003) Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol Biol Cell 14(7):2876–2889
Glover DM et al (1995) Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81(1):95–105
McKean PG, Vaughan S, Gull K (2001) The extended tubulin superfamily. J Cell Sci 114:2723–2733
Reber S, Hyman AA (2015) Emergent properties of the metaphase spindle. Cold Spring Harb Perspect Biol 7:a015784
Wittmann T, Hyman A, Desai A (2001) The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol 3:E28–E34
Gonzalez C (2007) Spindle orientation, asymmetric division and tumour suppression in Drosophila stem cells. Nat Rev Genet 8:462–472
Laan L, Pavin N, Husson J, Romet-Lemonne G, van Duijn M, Lopez MP, Vale RD, Julicher F, Reck-Peterson SL, Dogterom M (2012) Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 148:502–514
Morin X, Bellaiche Y (2011) Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell 21:102–119
Siller KH, Doe CQ (2009) Spindle orientation during asymmetric cell division. Nat Cell Biol 11:365–374
Goshima G, Mayer M, Zhang N, Stuurman N, Vale RD (2008) Augmin: a protein complex required for centrosome-independent microtubule generation within the spindle. J Cell Biol 181:421–429
Carazo-Salas RE, Guarguaglini G, Gruss OJ, Segref A, Karsenti E, Mattaj IW (1999) Generation of GTP-bound ran by RCC1 is required for chromatin-induced mitotic spindle formation. Nature 400:178–181
Kalab P, Pu RT, Dasso M (1999) The ran GTPase regulates mitotic spindle assembly. Curr Biol 9:481–484
Khodjakov A, Cole RW, Oakley BR, Rieder CL (2000) Centrosome-independent mitotic spindle formation in vertebrates. Curr Biol 10:59–67
Khodjakov A, Copenagle L, Gordon MB, Compton DA, Kapoor TM (2003) Minus-end capture of preformed kinetochore fibers contributes to spindle morphogenesis. J Cell Biol 160:671–683
Maiato H, Rieder CL, Khodjakov A (2004) Kinetochore-driven formation of kinetochore fibers contributes to spindle assembly during animal mitosis. J Cell Biol 167:831–840
Ohba T, Nakamura M, Nishitani H, Nishimoto T (1999) Self-organization of microtubule asters induced in Xenopus egg extracts by GTP-bound ran. Science 284:1356–1358
Holubcova Z, Blayney M, Elder K, Schuh M (2015) Human oocytes. Error-prone chromosome-mediated spindle assembly favors chromosome segregation defects in human oocytes. Science 348:1143–1147
Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW (2006) Flies without centrioles. Cell 125:1375–1386
Heald R, Tournebize R, Habermann A, Karsenti E, Hyman A (1997) Spindle assembly in Xenopus egg extracts: respective roles of centrosomes and microtubule self-organization. J Cell Biol 138:615–628
Nahaboo W, Zouak M, Askjaer P, Delattre M (2015) Chromatids segregate without centrosomes during Caenorhabditis elegans Mitosis in a ran- and CLASP-dependent manner. Mol Biol Cell 26:2020–2029
Toya M, Terasawa M, Nagata K, Iida Y, Sugimoto A (2011) A kinase-independent role for aurora a in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat Cell Biol 13:708–714
Giansanti MG, Gatti M, Bonaccorsi S (2001) The role of centrosomes and astral microtubules during asymmetric division of Drosophila neuroblasts. Development 128:1137–1145
Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH (2014) Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17:1528–1535
Knoblich JA (2010) Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol 11:849–860
Megraw TL, Kao LR, Kaufman TC (2001) Zygotic development without functional mitotic centrosomes. Curr Biol 11:116–120
Mora-Bermudez F, Matsuzaki F, Huttner WB (2014) Specific polar subpopulations of astral microtubules control spindle orientation and symmetric neural stem cell division. eLife 3:e02875
Al Jord A, Lemaitre AI, Delgehyr N, Faucourt M, Spassky N, Meunier A (2014) Centriole amplification by mother and daughter centrioles differs in multiciliated cells. Nature 516:104–107
Klos Dehring DA, Vladar EK, Werner ME, Mitchell JW, Hwang P, Mitchell BJ (2013) Deuterosome-mediated centriole biogenesis. Dev Cell 27:103–112
Satir P, Christensen ST (2007) Overview of structure and function of mammalian cilia. Annu Rev Physiol 69:377–400
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF (2005) Vertebrate smoothened functions at the primary cilium. Nature 437:1018–1021
Mahjoub MR, Stearns T (2012) Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr Biol 22:1628–1634
Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317:372–376
Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA (2004) Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131:5639–5647
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A et al (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between wnt signaling pathways. Nat Genet 37:537–543
Taverna E, Gotz M, Huttner WB (2014) The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 30:465–502
Wallingford JB, Mitchell B (2011) Strange as it may seem: the many links between wnt signaling, planar cell polarity, and cilia. Genes Dev 25:201–213
Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133:1032–1042
Yamashita YM, Jones DL, Fuller MT (2003) Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 301:1547–1550
Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460:278–282
Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R (2013) Centrosome amplification causes microcephaly. Nat Cell Biol 15:731–740
Sabino D, Gogendeau D, Gambarotto D, Nano M, Pennetier C, Dingli F, Arras G, Loew D, Basto R (2015) Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol 25:879–889
Silkworth WT, Nardi IK, Scholl LM, Cimini D (2009) Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 4:e6564
Lambrus BG, Uetake Y, Clutario KM, Daggubati V, Snyder M, Sluder G, Holland AJ (2015) p53 protects against genome instability following centriole duplication failure. J Cell Biol 210:63–77
Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Cell biology. Reversible centriole depletion with an inhibitor of polo-like kinase 4. Science 348:1155–1160
Jan YN, Jan LY (1998) Asymmetric cell division. Nature 392:775–778
Liu X, Hashimoto-Torii K, Torii M, Ding C, Rakic P (2010) Gap junctions/hemichannels modulate interkinetic nuclear migration in the forebrain precursors. J Neurosci Off J Soc Neurosci 30:4197–4209
Rhyu MS, Jan LY, Jan YN (1994) Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell 76:477–491
Paridaen JT, Wilsch-Brauninger M, Huttner WB (2013) Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 155:333–344
Rebollo E, Sampaio P, Januschke J, Llamazares S, Varmark H, Gonzalez C (2007) Functionally unequal centrosomes drive spindle orientation in asymmetrically dividing Drosophila neural stem cells. Dev Cell 12:467–474
Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH (2009) Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature 461:947–955
Yamashita YM, Mahowald AP, Perlin JR, Fuller MT (2007) Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 315:518–521
Anderson CT, Stearns T (2009) Centriole age underlies asynchronous primary cilium growth in mammalian cells. Curr Biol 19:1498–1502
Ricke RM, van Deursen JM (2013) Aneuploidy in health, disease, and aging. J Cell Biol 201:11–21
Gordon DJ, Resio B, Pellman D (2012) Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13:189–203
Gogendeau D, Siudeja K, Gambarotto D, Pennetier C, Bardin AJ, Basto R (2015) Aneuploidy causes premature differentiation of neural and intestinal stem cells. Nat Commun 6:8894
Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P (2010) The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A 107:14188–14193
McNamee LM, Brodsky MH (2009) p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics 182:423–435
Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, Kawamoto T, Miyamoto M, Hirayama T, Okaniwa M et al (2015) Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun 6:7668
Capecchi MR, Pozner A (2015) ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination. Nat Commun 6:8763
Fluckiger AC, Marcy G, Marchand M, Negre D, Cosset FL, Mitalipov S, Wolf D, Savatier P, Dehay C (2006) Cell cycle features of primate embryonic stem cells. Stem Cells 24:547–556
Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S (2002) Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin a/E and E2F activities. Oncogene 21:8320–8333
Hartenstein V, Spindler S, Pereanu W, Fung S (2008) The development of the Drosophila larval brain. Adv Exp Med Biol 628:1–31
Homem CC, Knoblich JA (2012) Drosophila neuroblasts: a model for stem cell biology. Development 139:4297–4310
Northcutt RG 2002 Understanding vertebrate brain evolution. From the symposium recent advances in neurobiology presented at the annual meeting of the society for integrative and comparative biology, 2–6 January 2002, at Anaheim, California
Borrell V, Reillo I (2012) Emerging roles of neural stem cells in cerebral cortex development and evolution. Dev Neurobiol 72:955–971
Fish JL, Dehay C, Kennedy H, Huttner WB (2008) Making bigger brains-the evolution of neural-progenitor-cell division. J Cell Sci 121:2783–2793
Hansen DV, Lui JH, Parker PR, Kriegstein AR (2010) Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464:554–561
Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A et al (2006) An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443:167–172
Roth G, Dicke U (2005) Evolution of the brain and intelligence. Trends Cogn Sci 9:250–257
Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, Haffner C, Sykes A, Wong FK, Peters J et al (2015) Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347:1465–1470
Stahl R, Walcher T, De Juan RC, Pilz GA, Cappello S, Irmler M, Sanz-Aquela JM, Beckers J, Blum R, Borrell V et al (2013) Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153:535–549
Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, Carmo N, Balloux F, Callaini G, Glover DM (2005) SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 15:2199–2207
Dinarina A, Pugieux C, Corral MM, Loose M, Spatz J, Karsenti E, Nedelec F (2009) Chromatin shapes the mitotic spindle. Cell 138:502–513
Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS (2004) Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117:527–539
Bate M, Martinez-Arias A (1993) The development of Drosophila melanogaster. Cold Spring Harbor Laboratory Press, New York
Riparbelli MG, Callaini G (2011) Male gametogenesis without centrioles. Dev Biol 349:427–439
Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P et al (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36:744–749
Blachon S, Gopalakrishnan J, Omori Y, Polyanovsky A, Church A, Nicastro D, Malicki J, Avidor-Reiss T (2008) Drosophila asterless and vertebrate Cep152 are orthologs essential for centriole duplication. Genetics 180:2081–2094
Martinez-Campos M, Basto R, Baker J, Kernan M, Raff JW (2004) The Drosophila pericentrin-like protein is essential for cilia/flagella function, but appears to be dispensable for mitosis. J Cell Biol 165:673–683
Castellanos E, Dominguez P, Gonzalez C (2008) Centrosome dysfunction in Drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol 18:1209–1214
Poulton JS, Cuningham JC, Peifer M (2014) Acentrosomal Drosophila epithelial cells exhibit abnormal cell division, leading to cell death and compensatory proliferation. Dev Cell 30:731–745
Novorol C, Burkhardt J, Wood KJ, Iqbal A, Roque C, Coutts N, Almeida AD, He J, Wilkinson CJ, Harris WA (2013) Microcephaly models in the developing zebrafish retinal neuroepithelium point to an underlying defect in metaphase progression. Open Biol 3:130065
Kim HT, Lee MS, Choi JH, Jung JY, Ahn DG, Yeo SY, Choi DK, Kim CH (2011) The microcephaly gene aspm is involved in brain development in zebrafish. Biochem Biophys Res Commun 409:640–644
Pfaff KL, Straub CT, Chiang K, Bear DM, Zhou Y, Zon LI (2007) The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol Cell Biol 27:5887–5897
Manning JA, Lewis M, Koblar SA, Kumar S (2010) An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ 17:1302–1314
Martin CA, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, Leitch A, Nurnberg G, Toliat MR, Murray JE, Hunt D et al (2014) Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet 46:1283–1292
Dzafic E, Strzyz PJ, Wilsch-Brauninger M, Norden C (2015) Centriole amplification in zebrafish affects proliferation and survival but not differentiation of neural progenitor cells. Cell Rep 13:168–182
Bazzi H, Anderson KV (2014) Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci U S A 111:E1491–E1500
Calarco-Gillam PD, Siebert MC, Hubble R, Mitchison T, Kirschner M (1983) Centrosome development in early mouse embryos as defined by an autoantibody against pericentriolar material. Cell 35:621–629
David A, Liu F, Tibelius A, Vulprecht J, Wald D, Rothermel U, Ohana R, Seitel A, Metzger J, Ashery-Padan R et al (2014) Lack of centrioles and primary cilia in STIL(−/−) mouse embryos. Cell Cycle 13:2859–2868
Gueth-Hallonet C, Antony C, Aghion J, Santa-Maria A, Lajoie-Mazenc I, Wright M, Maro B (1993) Gamma-tubulin is present in acentriolar MTOCs during early mouse development. J Cell Sci 105(Pt 1):157–166
Szollosi D, Calarco P, Donahue RP (1972) Absence of centrioles in the first and second meiotic spindles of mouse oocytes. J Cell Sci 11:521–541
Hudson JW, Kozarova A, Cheung P, Macmillan JC, Swallow CJ, Cross JC, Dennis JW (2001) Late mitotic failure in mice lacking sak, a polo-like kinase. Curr Biol 11:441–446
Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, Kuehn MR (1999) The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 399:691–694
Garcez PP, Diaz-Alonso J, Crespo-Enriquez I, Castro D, Bell D, Guillemot F (2015) Cenpj/CPAP regulates progenitor divisions and neuronal migration in the cerebral cortex downstream of Ascl1. Nat Commun 6:6474
Gabriel E, Wason A, Ramani A, Gooi LM, Keller P, Pozniakovsky A, Poser I, Noack F, Telugu NS, Calegari F et al (2016) CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J 35:803–819
Gogendeau D, Siudeja K, Gambarotto D, Pennetier C, Bardin AJ, Basto R (2015) Aneuploidy causes premature differentiation of neural and intestinal stem cells. Nat Commun 6:8894
Pfau SJ, Silberman RE, Knouse KA, Amon A (2016) Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev 30:1395–1408
Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z (2012) Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 8:608
Thorburn RR, Gonzalez C, Brar GA, Christen S, Carlile TM, Ingolia NT, Sauer U, Weissman JS, Amon A (2013) Aneuploid yeast strains exhibit defects in cell growth and passage through START. Mol Biol Cell 24:1274–1289
Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ (2004) Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell 15:3642–3657
Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Fluckiger I, Gonczy P (2011) Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J Cell Sci 124:3884–3893
Klingseisen A, Jackson AP (2011) Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev 25:2011–2024
Passemard S, Titomanlio L, Elmaleh M, Afenjar A, Alessandri JL, Andria G, de Villemeur TB, Boespflug-Tanguy O, Burglen L, Del Giudice E et al (2009) Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 73:962–969
Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P (2010) Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol 90:363–383
Woods CG, Bond J, Enard W (2005) Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet 76:717–728
Trimborn M, Bell SM, Felix C, Rashid Y, Jafri H, Griffiths PD, Neumann LM, Krebs A, Reis A, Sperling K et al (2004) Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet 75:261–266
Genin A, Desir J, Lambert N, Biervliet M, Van Der Aa N, Pierquin G, Killian A, Tosi M, Urbina M, Lefort A et al (2012) Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum Mol Genet 21:5306–5317
Mirzaa GM, Vitre B, Carpenter G, Abramowicz I, Gleeson JG, Paciorkowski AR, Cleveland DW, Dobyns WB, O’Driscoll M (2014) Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum Genet 133:1023–1039
Awad S, Al-Dosari MS, Al-Yacoub N, Colak D, Salih MA, Alkuraya FS, Poizat C (2013) Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum Mol Genet 22:2200–2213
Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ et al (2002) Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 71:136–142
Kiyomitsu T, Obuse C, Yanagida M (2007) Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell 13:663–676
Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J et al (2012) Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151:1097–1112
Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al-Qahtani MH et al 2015 Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 8(Suppl 1):S4
Mahmood S, Ahmad W, Hassan MJ (2011) Autosomal recessive primary microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis 6:39
Trimborn M, Ghani M, Walther DJ, Dopatka M, Dutrannoy V, Busche A, Meyer F, Nowak S, Nowak J, Zabel C et al (2010) Establishment of a mouse model with misregulated chromosome condensation due to defective Mcph1 function. PLoS One 5:e9242
Zhou ZW, Tapias A, Bruhn C, Gruber R, Sukchev M, Wang ZQ (2013) DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 12:645–655
Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ (2011) MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat Cell Biol 13:1325–1334
Brunk K, Vernay B, Griffith E, Reynolds NL, Strutt D, Ingham PW, Jackson AP (2007) Microcephalin coordinates mitosis in the syncytial Drosophila embryo. J Cell Sci 120:3578–3588
Rickmyre JL, Dasgupta S, Ooi DL, Keel J, Lee E, Kirschner MW, Waddell S, Lee LA (2007) The Drosophila homolog of MCPH1, a human microcephaly gene, is required for genomic stability in the early embryo. J Cell Sci 120:3565–3577
Nano M, Basto R (2015) The Janus soul of centrosomes: a paradoxical role in disease? Chromosome Res 24(1):127–144
Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC et al (2010) Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet 87:40–51
Kumar A, Girimaji SC, Duvvari MR, Blanton SH (2009) Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet 84:286–290
Arquint C, Nigg EA (2014) STIL microcephaly mutations interfere with APC/C-mediated degradation and cause centriole amplification. Curr Biol 24:351–360
Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO et al (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37:353–355
Hussain MS, Baig SM, Neumann S, Nurnberg G, Farooq M, Ahmad I, Alef T, Hennies HC, Technau M, Altmuller J et al (2012) A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am J Hum Genet 90:871–878
Farooq M, Fatima A, Mang Y, Hansen L, Kjaer KW, Baig SM, Larsen LA, Tommerup N (2016) A novel splice site mutation in CEP135 is associated with primary microcephaly in a Pakistani family. J Hum Genet 61:271–273
Sir JH, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D'Santos C, Woods CG, Gergely F (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43:1147–1153
Marjanovic M, Sanchez-Huertas C, Terre B, Gomez R, Scheel JF, Pacheco S, Knobel PA, Martinez-Marchal A, Aivio S, Palenzuela L et al (2015) CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat Commun 6:7676
Khan MA, Rupp VM, Orpinell M, Hussain MS, Altmuller J, Steinmetz MO, Enzinger C, Thiele H, Hohne W, Nurnberg G et al (2014) A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family. Hum Mol Genet 23:5940–5949
Fu J, Glover DM (2012) Structured illumination of the interface between centriole and peri-centriolar material. Open Biol 2:120104
Conduit PT, Richens JH, Wainman A, Holder J, Vicente CC, Pratt MB, Dix CI, Novak ZA, Dobbie IM, Schermelleh L et al (2014b) A molecular mechanism of mitotic centrosome assembly in Drosophila. elife 3:e03399
Choi YK, Liu P, Sze SK, Dai C, Qi RZ (2010) CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J Cell Biol 191:1089–1095
Fong KW, Choi YK, Rattner JB, Qi RZ (2008) CDK5RAP2 is a pericentriolar protein that functions in centrosomal attachment of the gamma-tubulin ring complex. Mol Biol Cell 19:115–125
Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH (2010) Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron 66:386–402
Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD (2010) Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137:1907–1917
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501:373–379
Barr AR, Kilmartin JV, Gergely F (2010) CDK5RAP2 functions in centrosome to spindle pole attachment and DNA damage response. J Cell Biol 189:23–39
Conduit PT, Feng Z, Richens JH, Baumbach J, Wainman A, Bakshi SD, Dobbelaere J, Johnson S, Lea SM, Raff JW (2014a) The centrosome-specific phosphorylation of Cnn by Polo/Plk1 drives Cnn scaffold assembly and centrosome maturation. Dev Cell 28:659–669
Conduit PT, Brunk K, Dobbelaere J, Dix CI, Lucas EP, Raff JW (2010) Centrioles regulate centrosome size by controlling the rate of Cnn incorporation into the PCM. Curr Biol 20:2178–2186
Conduit PT, Raff JW (2010) Cnn dynamics drive centrosome size asymmetry to ensure daughter centriole retention in Drosophila neuroblasts. Curr Biol 20:2187–2192
Yalgin C, Ebrahimi S, Delandre C, Yoong LF, Akimoto S, Tran H, Amikura R, Spokony R, Torben-Nielsen B, White KP et al (2015) Centrosomin represses dendrite branching by orienting microtubule nucleation. Nat Neurosci 18:1437–1445
Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nurnberg G, Tariq M, Jameel M, Khan TN, Fatima A et al (2013) CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 22:5199–5214
do Carmo Avides M, Glover DM (1999) Abnormal spindle protein, asp, and the integrity of mitotic centrosomal microtubule organizing centers. Science 283:1733–1735
Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB (2006) Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci U S A 103:10438–10443
Gul A, Hassan MJ, Mahmood S, Chen W, Rahmani S, Naseer MI, Dellefave L, Muhammad N, Rafiq MA, Ansar M et al (2006) Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: novel sequence variants in ASPM gene. Neurogenetics 7:105–110
Pulvers JN, Bryk J, Fish JL, Wilsch-Brauninger M, Arai Y, Schreier D, Naumann R, Helppi J, Habermann B, Vogt J et al (2010) Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc Natl Acad Sci U S A 107:16595–16600
Rujano MA, Sanchez-Pulido L, Pennetier C, le Dez G, Basto R (2013) The microcephaly protein asp regulates neuroepithelium morphogenesis by controlling the spatial distribution of myosin II. Nat Cell Biol 15:1294–1306
Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A et al (2010) WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 42:1010–1014
Chen JF, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L (2014) Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat Commun 5:3885
Ramdas Nair A, Singh P, Salvador Garcia D, Rodriguez-Crespo D, Egger B, Cabernard C (2016) The microcephaly-associated protein Wdr62/CG7337 is required to maintain centrosome asymmetry in Drosophila neuroblasts. Cell Rep 14:1100–1113
Hall JG, Flora C, Scott CI Jr, Pauli RM, Tanaka KI (2004) Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet A 130A:55–72
Majewski F, Goecke T (1982) Studies of microcephalic primordial dwarfism I: approach to a delineation of the Seckel syndrome. Am J Med Genet 12:7–21
Majewski F, Goecke TO (1998) Microcephalic osteodysplastic primordial dwarfism type II: report of three cases and review. Am J Med Genet 80:25–31
Willems M, Genevieve D, Borck G, Baumann C, Baujat G, Bieth E, Edery P, Farra C, Gerard M, Heron D et al (2010) Molecular analysis of pericentrin gene (PCNT) in a series of 24 Seckel/microcephalic osteodysplastic primordial dwarfism type II (MOPD II) families. J Med Genet 47:797–802
Piane M, Della Monica M, Piatelli G, Lulli P, Lonardo F, Chessa L, Scarano G (2009) Majewski osteodysplastic primordial dwarfism type II (MOPD II) syndrome previously diagnosed as Seckel syndrome: report of a novel mutation of the PCNT gene. Am J Med Genet A 149A:2452–2456
Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH et al (2008) Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 319:816–819
Delaval B, Doxsey SJ (2010) Pericentrin in cellular function and disease. J Cell Biol 188:181–190
Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC et al (2008) Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet 40:232–236
Tibelius A, Marhold J, Zentgraf H, Heilig CE, Neitzel H, Ducommun B, Rauch A, Ho AD, Bartek J, Kramer A (2009) Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J Cell Biol 185:1149–1157
Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M (1994) Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 76:639–650
Purohit A, Tynan SH, Vallee R, Doxsey SJ (1999) Direct interaction of pericentrin with cytoplasmic dynein light intermediate chain contributes to mitotic spindle organization. J Cell Biol 147:481–492
Miyoshi K, Kasahara K, Miyazaki I, Shimizu S, Taniguchi M, Matsuzaki S, Tohyama M, Asanuma M (2009) Pericentrin, a centrosomal protein related to microcephalic primordial dwarfism, is required for olfactory cilia assembly in mice. FASEB J 23:3289–3297
Seckel HPG (1960) Bird-headed Dwarfs: studies in developmental anthropology including human proportions. Charles C Thomas, Springfield
Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF et al (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32:316–320
Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS (2010) Novel CENPJ mutation causes Seckel syndrome. J Med Genet 47:411–414
Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tuysuz B, Nurnberg G et al (2011) CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 43:23–26
McIntyre RE, Lakshminarasimhan Chavali P, Ismail O, Carragher DM, Sanchez-Andrade G, Forment JV, Fu B, Del Castillo V-HM, Edwards A, van der Weyden L et al (2012) Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS Genet 8:e1003022
Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA (2005) The polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 7:1140–1146
Shaheen R, Al Tala S, Almoisheer A, Alkuraya FS (2014) Mutation in PLK4, encoding a master regulator of centriole formation, defines a novel locus for primordial dwarfism. J Med Genet 51:814–816
Scheidecker S, Etard C, Haren L, Stoetzel C, Hull S, Arno G, Plagnol V, Drunat S, Passemard S, Toutain A et al (2015) Mutations in TUBGCP4 alter microtubule organization via the gamma-tubulin ring complex in autosomal-recessive microcephaly with chorioretinopathy. Am J Hum Genet 96:666–674
Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, Alshammari MJ, Al-Sheddi T, Adly N, Al-Dosari MS, Megason SG et al (2012) POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am J Hum Genet 91:330–336
Koparir A, Karatas OF, Yuceturk B, Yuksel B, Bayrak AO, Gerdan OF, Sagiroglu MS, Gezdirici A, Kirimtay K, Selcuk E et al (2015) Novel POC1A mutation in primordial dwarfism reveals new insights for centriole biogenesis. Hum Mol Genet 24:5378–5387
Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS et al (2001) Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet 68:569–576
Romio L, Wright V, Price K, Winyard PJ, Donnai D, Porteous ME, Franco B, Giorgio G, Malcolm S, Woolf AS et al (2003) OFD1, the gene mutated in oral-facial-digital syndrome type 1, is expressed in the metanephros and in human embryonic renal mesenchymal cells. J Am Soc Nephrol: JASN 14:680–689
AlKattan WM, Al-Qattan MM, Bafaqeeh SA (2015) The pathogenesis of the clinical features of oral-facial-digital syndrome type I. Saudi Med J 36:1277–1284
Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A, Kerzendorfer C, Martin CA, Yeyati P, Al Sanna N, Bober M et al (2011) Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet 43:350–355
Hemerly AS, Prasanth SG, Siddiqui K, Stillman B (2009) Orc1 controls centriole and centrosome copy number in human cells. Science 323:789–793
Hossain M, Stillman B (2012) Meier-Gorlin syndrome mutations disrupt an Orc1 CDK inhibitory domain and cause centrosome reduplication. Genes Dev 26:1797–1810
Barrera JA, Kao LR, Hammer RE, Seemann J, Fuchs JL, Megraw TL (2010) CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev Cell 18:913–926
Niccols A (2007) Fetal alcohol syndrome and the developing socio-emotional brain. Brain Cogn 65:135–142
Pan American Health Organization (2015) Epidemiological alert. Neurological syndrome, congenital malformations, and Zika virus infection. Implications for public health in the Americas. Pan American Health Organization, Washington. Available from: http://www.paho.org/hq/index.phpoption=com_docman&task=doc_download&Itemid=&gid=32405 [cited 2016 Apr 13]
WHO (2015) Zika virus outbreaks in the Americas. Releve epidemiologique hebdomadaire/Section d’hygiene du Secretariat de la Societe des Nations = Weekly epidemiological record/Health Section of the Secretariat of the League of Nations 90:609–610
Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, Doriqui MJ, Neri JI, Neto JM, Wanderley HY et al (2016) Possible association between Zika virus infection and microcephaly – Brazil, 2015. MMWR Morb Mortal Wkly Rep 65:59–62
Bell TM, Field EJ, Narang HK (1971) Zika virus infection of the central nervous system of mice. Archiv fur die gesamte Virusforschung 35:183–193
Dick GW, Kitchen SF, Haddow AJ (1952) Zika virus. I Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520
Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM et al (2016) Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. doi:10.1016/j.stem.2016.02.016
Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, Boddaert N, Picard C, Sbiti A, Asermouh A et al (2007) Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet 39:454–456
Acknowledgements
We thank V. Marthiens for helpful comments on the manuscript. Work in our lab is supported by an ANR (ANR DIVACEN), an INCa (AAP INCa 2015/118) grants, the CNRS, I. Curie, La Ligue contre le Cancer and FRM (M.N.) Our lab is a member of the CelTisPhyBio labex.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Nano, M., Basto, R. (2017). Consequences of Centrosome Dysfunction During Brain Development. In: Gotta, M., Meraldi, P. (eds) Cell Division Machinery and Disease. Advances in Experimental Medicine and Biology, vol 1002. Springer, Cham. https://doi.org/10.1007/978-3-319-57127-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-57127-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57125-6
Online ISBN: 978-3-319-57127-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)