
Abstract
Enterohemorrhagic Escherichia coli (EHEC) is a significant foodborne attaching and effacing (A/E) pathogen that causes diarrhea, hemorrhagic colitis and the hemolytic-uremic syndrome (HUS) in humans. EHEC is closely related to enteropathogenic E. coli (EPEC) and both induce characteristic A/E lesions on the gut mucosal surface. During EHEC and EPEC infection, host innate immune responses, such as inflammation and cell death are rapidly activated, upon the detection of bacterial components and virulence factor activity. To promote A/E lesion formation and dissemination of the pathogen in the body, EHEC and EPEC deliver a repertoire of effector proteins, including Tir, NleA/EspI and NleB to -H, to the host cell cytosol via a type III secretion system (T3SS). These interfere with a range of host cell processes, including host defense mechanisms. Several T3SS effector proteins specifically modify or cleave host proteins involved in inflammation and cell death, thereby inactivating these pathways. The identification of the host targets and the characterization of the biochemical function of T3SS effectors have greatly contributed to understanding the pathogenesis of EHEC and EPEC infections.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Enterohemorrhagic Escherichia coli (EHEC) is a major threat to human health in both low-income and high-income countries . EHEC was first recognized as a human pathogen in 1983 during two outbreaks of hemorrhagic colitis in the USA which were caused by contaminated undercooked hamburger meat (Riley et al. 1983). Early studies revealed, approximately 75% of O157:H7 EHEC outbreaks in the US were attributed to the consumption of contaminated beef products (Callaway et al. 2009), although agricultural plants, pork products, freshly consumed fruits and vegetables and water, have also been reported as a source of EHEC infection due to contamination with animal feces (From the Centers for Disease Control and Prevention 1995; Feng and Reddy 2013). Apart from food contamination, other identified routes of EHEC transmission include person-to-person contact, animal-to-person contact and airborne transmission (Durso et al. 2005; Spika et al. 1986; Varma et al. 2003).
Among EHEC strains, serotype O157:H7 is the most prominent clinical serotype and is responsible for a large number of cases linked to food and waterborne outbreaks worldwide. EHEC O157:H7 has a very low infectious dose, with as few as 10–100 microorganisms able to cause disease in humans (Boyce et al. 1995). Upon ingestion of O157:H7, diarrhea begins after 1–8 days (average onset 3–4 days) and rapidly progresses into bloody diarrhea in more than 70% of EHEC-infected people (Mead and Griffin 1998). After ca. 7 days of diarrhea, approximately 5–10% of patients develop hemolytic uremic syndrome (HUS) , with children and the elderly particularly at risk of this life-threatening complication (Manning et al. 2008; Karmali et al. 2010).
A feature of the interaction of EHEC with the intestinal epithelium is the ability of the bacteria to form attaching and effacing (A/E) lesions and to produce Shiga toxins (Stx) . A/E lesions are histopathological changes on the intestinal mucosa characterized by extracellular attachment of the bacteria to the cell surface, the formation of pedestal-like structures underneath the adherent bacteria and the local effacement of microvilli (Frankel et al. 1998). A/E lesion formation by EHEC is dependent on a pathogenicity island known as the locus of enterocyte effacement (LEE) , which is conserved across all A/E pathogens, including closely related enteropathogenic E. coli (EPEC). The LEE encodes more than 40 genes, including regulators, the adhesin, intimin, translocators, chaperones, a type III secretion system (T3SS) and translocated effector proteins that play an essential role in EHEC pathogenesis (McDaniel et al. 1995; Elliott et al. 2000). For example, the LEE-encoded T3SS effector , termed the translocated intimin receptor (Tir) , binds the bacterial outer membrane adhesin, intimin, and promotes actin polymerization at the site of bacterial attachment by recruiting host proteins associated with cytoskeletal actin dynamics (Wong et al. 2011). Although EPEC and EHEC share a similar mechanism of host cell attachment, Stx additionally influences the interaction of EHEC with the intestinal mucosa. Bacteriophage-encoded Stx comprise two subgroups, Stx1 and Stx2, and individual EHEC isolates may carry one or both Stx1 and Stx2 (Kaper et al. 2004; Muthing et al. 2009). Stx is toxic for human cells and mediates the inhibition of protein synthesis resulting in large-scale cell death through apoptosis and the severe pathologies of hemorrhagic colitis and HUS (Endo et al. 1988; Karmali et al. 1983).
Aside from A/E lesion formation, EHEC and EPEC employ T3SS effectors, including LEE-encoded and non-LEE-encoded effectors, to promote bacterial colonization and survival within the host. In recent years, T3SS effectors from EHEC and other A/E pathogens have emerged as novel enzymes that specifically modify or directly degrade target host proteins, thereby interfering with fundamental biological processes such as cytoskeleton organization , inflammatory responses and apoptosis signaling pathways (Wong et al. 2011). The inhibition of pro-inflammatory responses and apoptosis that are involved in eliminating the pathogen or restricting the severity of disease, is an essential virulence mechanism that allows EHEC to persist in the host. Here, we will focus on manipulation of the innate immune response by EHEC that is mediated through the interaction of bacterial effectors with host proteins. The closely related human A/E pathogen, EPEC, and the mouse pathogen, Citrobacter rodentium, will also be considered.
The Host Response to EHEC Infection
Upon pathogen infection, the mammalian innate immune system recognizes microbial molecules , including lipopolysaccharide (LPS) , peptidoglycan , flagellin and nucleic acid , otherwise known as pathogen-associated molecular patterns (PAMPs) (Akira et al. 2006). The detection of PAMPs via a suite of pattern recognition receptors (PRRs) leads to activation of the innate immune response that provides immediate defense against infection. Based on their localization, PRRs are classified into cell surface receptors , such as Toll-like receptors (TLR) and C-type lectin receptors (CLR) , and cytoplasmic receptors , including NOD-like receptors (NLR) and RIG-I-like receptors (RLR) (Takeuchi and Akira 2010). The initiation of innate immune signaling by PRRs leads to the activation of pro-inflammatory gene expression, which results in the production of cytokines, chemokines and antimicrobial peptides that ultimately facilitate elimination of the pathogens either by direct killing or phagocytosis.
Following ingestion, EHEC initiates infection of the surface epithelium of the intestinal mucosa. The intestinal epithelium primarily functions to separate host tissues from the external environment but also plays an essential role in activating the immune response to pathogenic microorganisms (Wong Fok Lung et al. 2014). Infected epithelial cells generally express increased secretion of cytokines, chemokines and antimicrobial peptides, which results from the activation of NF-κB and MAPK signaling pathways . During EHEC and EPEC infection, high levels of Interleukin 8 (IL-8 ) expression and secretion in epithelial cells accompanied by the infiltration of neutrophils at infected sites have been demonstrated in a number of in vivo studies (Tzipori et al. 1985, 1989). This phenotype has been mainly attributed to the recognition of bacterial virulence factors through PRRs . Stx has also been implicated in triggering a marked increase of IL-8 production in polarized T84 human intestinal epithelial cells and Caco2 cells (Thorpe et al. 1999, 2001). However, depletion of H7 flagellin rather than Stx from the EHEC culture supernatant fluid abolished IL-8 induction in epithelial cells, suggesting flagellin is the principal factor that contributes to the EHEC-induced inflammatory response (Zhou et al. 2003; Berin et al. 2002). Consistently, EHEC H7 flagellin significantly enhanced transcription of IL-8 and Chemokine Ligand 20 (CCL20) in colonic epithelial cells and induced neutrophil infiltration in an in vivo study using human intestinal xenografts (Miyamoto et al. 2006). In addition to flagellin, EHEC LPS is associated with the stimulation of immune responses. In vivo studies using the mouse A/E pathogen C. rodentium show that Tlr4 −/− mice, which are insensitive to bacterial LPS, produce lower levels of cytokine and have less neutrophil and macrophage infiltration at the infection site (Khan et al. 2006).
Although Stx and LPS are not essential to induce pro-inflammatory cytokine secretion in intestinal epithelial cells, their roles in inducing inflammation systemically are recognized as important. In children with HUS, EHEC LPS binds and activates platelets, resulting in the expression of CD40 Ligand (CD40L) which can in turn increase the amount of Toll-like Receptor (TLR4) on neutrophils and monocytes, and induce the production of inflammatory cytokines in vascular endothelial cells (Stahl et al. 2006). In addition, translocation of Stx across the intestinal epithelium is promoted by neutrophil transmigration, although the mechanism is not fully characterized. Hence Stx can access the host circulatory system and induce pro-inflammatory reactions (Hurley et al. 2001; Walters et al. 1989). Stx also binds to monocytes through Globotriascylcermide 3 (Gb3) receptors in patients with HUS, while in vitro studies have revealed that stimulation of monocytes with Stx leads to a significant increase in the production of IL-6, IL-8 , TNF and IL-1β (van Setten et al. 1996).
In addition to inflammation, immune detection of bacterial pathogens can trigger apoptotic signaling cascades that are associated with the clearance of infected cells. Stx appears to be the major virulence factor responsible for inducing apoptosis of epithelial cells during EHEC infection in vitro (Smith et al. 2003; Schuller et al. 2004; Barnett Foster et al. 2000; Kashiwamura et al. 2009), and evidence from in vivo studies in rabbits suggests that loss of microvilli is associated with Stx-induced apoptosis (Smith et al. 2003; Schuller et al. 2004; Kashiwamura et al. 2009). Additionally, inflammatory cytokines , such as Tumor Necrosis Factor (TNF) , can activate cell death signaling by binding death receptors and the activity of some T3SS effector proteins; namely EspF, Map and Cif, are associated with apoptosis induction.
Inflammation and Cell Death Signaling
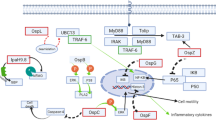
NF-κB signaling is a crucial part of the host immune response that induces pro-inflammatory gene expression upon stimulation (Mukaida et al. 1990). The NF-κB proteins comprise a family of transcription factors that includes p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), c-Rel and RelB which regulate gene expression by binding to promoters/enhancers of target genes (Ghosh et al. 1998). All NF-κB transcription factors possess an N-terminal Rel homology domain that mediates homo- and hetero-dimerization with other NF-κB proteins, nuclear translocation and DNA-binding. The C-terminal transcription activation domains (TADs) of p65, c-Rel and RelB control the activation of target gene transcription (Vallabhapurapu and Karin 2009), whereas p50 and p52 lack TADs and can either down-regulate gene expression by directly binding with NF-κB-responsive promoters or activate transcription through recruitment of p65, c-Rel, RelB or other TAD-containing proteins (Vallabhapurapu and Karin 2009).
Although NF-κB may be activated by various pathways, most converge at IKK (IκB Kinase) complex activation, which is comprised of two kinase subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit NEMO (Liu and Chen 2011). Upon stimulation by pathogen-derived molecules, PRRs recruit various adaptor proteins to form signaling complexes that can activate IKK kinase and direct IKK phosphorylation. Activation of the IKK complex mediates phosphorylation and ubiquitination of IκB proteins that serve as NF-κB inhibitors by retaining NF-κB dimers in the cytoplasm of inactive cells. Ubiquitinated IκB proteins are targeted for degradation by the host 26S proteasome, following which NF-κB subunits are released and translocated into the host cell nucleus where they enhance transcription of inflammatory genes (Hayden and Ghosh 2008).
MAPK signaling functions to coordinate cell responses to numerous physical and chemical stimuli and also contributes to IL-8 production in epithelial cells during EHEC and EPEC infection. MAPK represents a group of conserved proteins that are subdivided into three classes of serine/threonine kinases, ERK, p38 and JNK. The activation of JNK, which involves phosphorylation at threonine residue (Thr183) and serine residue (Ser185), is induced through MAP3Ks in response to TLR, IL-1R (IL-1 receptor) or TNFR (TNF receptor) stimulation (Johnson and Lapadat 2002). Previous studies have shown that A/E pathogen infection triggers JNK activation which in turn activates the transcription factor c-Jun, a member of the activation protein-1 (AP-1) family that has similar functions to NF-κB in regulating gene transcription during inflammation (Mukaida et al. 1994).
Apoptosis in the epithelium has recently emerged as an important host immune defense mechanism that combats intestinal bacterial infection. Killing of infected cells helps to control and eliminate pathogens, clear damaged cells and induce an adaptive immune response. Intrinsic signals such as DNA damage as well as extrinsic signals such as the stimulation of death receptors can trigger caspase-dependent cell death. Cell death receptors , including TNFR1 , TRAIL (TNF-associated apoptosis-inducing ligand) receptor and FAS (TNFRSF6), are stimulated upon infection with their respective ligands TNF, TRAIL and FasL. These receptors activate intracellular cell death signaling by recruiting a range of adaptor proteins through their conserved cytoplasmic death domains (DD) (Park et al. 2007). Activation of death receptors can also lead to non-apoptotic signaling, namely activation of inflammatory signaling, indicating that there is substantial crosstalk between the components of these pathways.
Pathogen Response to the Host: Inflammation
Inflammation pathways involving NF-κB and MAPK signaling and inflammatory cytokine production are strongly influenced by EHEC virulence factors in the early stages of infection. The activity of some T3SS effectors, such as EspT and NleF, leads to enhanced NF-κB activation (Raymond et al. 2011; Pallett et al. 2014), whereas other T3SS effectors suppress the inflammatory response through specific targeting of host signaling proteins.
Inhibition of NF-κB Signaling
EHEC and other A/E pathogens employ multiple T3SS effectors to inhibit IκB degradation and nuclear translocation of NF-κB. One of these is NleE , a highly conserved T3SS effector encoded on the virulence-associated O-island (OI) 122 in EHEC O157:H7 EDL933. NleE from EPEC O127:H6 E2348/69 is encoded by integrative element 6 (IE6) (Nadler et al. 2010; Iguchi et al. 2009). NleE is highly conserved, with NleE proteins from EPEC, EHEC and C. rodentium displaying more than 85% amino acid identity (Zurawski et al. 2008). NleE has homologues in Shigella spp., known as OspZ, and amino acid similarity between NleE and OspZ is also high (ca. 86% similarity). Notably, Shigella flexneri serogroup 2a carries a truncated OspZ that lacks 36 amino acids at the C-terminus and is non-functional for NF-κB inhibition (Newton et al. 2010).
Early studies revealed that ectopically-expressed NleE from EHEC, EPEC and C. rodentium and full length OspZ from Shigella spp. can block IκB degradation and p65 nuclear translocation in response to either TNF (tumor necrosis factor) or IL-1β stimulation, ultimately resulting in a decrease in the expression and production of inflammatory cytokines such as IL-8 (Newton et al. 2010). Consistently, the presence of NleE during EPEC infection significantly reduces the level of pro-inflammatory cytokine production from infected epithelial cells. In addition, the ability of NleE to control NF-κB activation can also be seen in human dendritic cells (DCs) , where DCs infected with the nleE mutant of EPEC E2348/69 secreted markedly higher levels of IL-8, TNF and IL-6 compared to wild type E2348/69 (Vossenkamper et al. 2010).
Recently, NleE was characterized as a S-adenosyl-L-methionine (SAM) dependent methyltransferase that specifically modifies the zinc coordinating cysteine in the Npl4 Zinc figure (NZF) domain of human TAB2 and TAB3 (TAK1-binding proteins 2 and 3 ) (Zhang et al. 2012). TAB2 and TAB3 are adaptor proteins with redundant functions that play an essential role in signaling pathways via Toll-like, IL-1 and TNF receptors. Upon activation, the NZF domains of TAB2/3 recognize and bind to K63-linked polyubiquitin chains on upstream receptor-associated proteins TRAF6 and TRAF2 (Takaesu et al. 2000; Sato et al. 2009). Normally, upstream signals, transduced via the interaction of TAB2/3 with ubiquitinated TRAF proteins, activate TAK1 and subsequently induce IKKβ phosphorylation and IκB degradation (Takaesu et al. 2000). Cysteine-methylated TAB2-NZF and TAB3-NZF lose their polyubiquitin chain binding activity, indicating that NZF-domain methylation abolishes signal transduction by interrupting the interaction between TAB2/3 and TRAF2/6 (Zhang et al. 2012). The methyltransferase activity of NleE is dependent on a conserved C-terminal motif 209IDSYMK214, which is explained by the crystal structure of NleE where tyrosine (Tyr212), together with arginine (Arg107) and glutamine (Glu191), form the binding pocket for SAM (Yao et al. 2014). Despite the potent in vitro activity of NleE, in vivo studies with C. rodentium revealed no significant differences in bacterial load between wild type C. rodentium and an nleE null mutant strain, probably due to redundant functions with other T3SS effectors that target NF-κB activation (Kelly et al. 2006).
The non-LEE encoded effectors NleH1 and NleH2 have also been associated with the inhibition of NF-κB activation. NleH1 and NleH2 share 84% amino acid sequence identity and both are present in EHEC and EPEC. This is in contrast to C. rodentium, which carries only one copy of nleH (Gao et al. 2009; Feuerbacher and Hardwidge 2014). Although NleH1 and NleH2 are closely related effectors that both contain a C-terminal kinase domain, they interfere with NF-κB activation through different mechanisms. Recent studies revealed that the host target of NleH1 is ribosomal protein 3 (RPS3) , a known co-activator of NF-κB that can be phosphorylated by IKKβ on serine (Ser209). Upon phosphorylation, RPS3 translocates to the nucleus and activates gene transcription (Gao et al. 2009). Upon binding to NleH1, not NleH2, phosphorylation of RPS3 by IKKβ is inhibited, and, even though the N-terminal region of NleH1 is sufficient for the binding, the C-terminal kinase activity of NleH1 appears to be important for this inhibition (Pham et al. 2012). NleH1 and NleH2 share 30% and 27% amino acid identity, respectively, with the Shigella effector OspG (Kim et al. 2005). OspG is a Ser/Thr protein kinase that subverts NF-κB by preventing ubiquitin-conjugating enzyme-mediated ubiquitination and degradation of phospho-IκBα (Kim et al. 2005). Similar to NleH1, the kinase activity of OspG is critical for modulating NF-κB activation (Gao et al. 2009; Kim et al. 2005). NleH2 suppresses NF-κB activation in cultured epithelial cells when IKKβ is overexpressed, and appears to not to have any influence on RPS3 nuclear translocation (Wan et al. 2011). While NleH1/NleH2 -induced NF-κB inhibition has been attributed to the subversion of IκBα phosphorylation/degradation, another study revealed that neither NleH1 nor NleH2 was able to modulate the phosphorylation and degradation of IκBα individually (Pham et al. 2012). This study further demonstrated that NleH1 and NleH2 bound to each other, and that this interaction regulated their ability to inhibit NF-κB signaling through an unknown mechanism (Pham et al. 2012).
In vivo studies using gnotobiotic piglets have revealed that deletion of nleH1 results in reduced bacterial load, more severe inflammatory responses and higher levels of phosphorylated RPS3 in the colon of infected piglets than those infected with wild type EHEC O157:H7. Infection with the nleH2 mutant strain, however, only showed diminished colonization (Wan et al. 2011). Similarly, C. rodentium infection of mice has revealed that nleH contributes to full intestinal colonization, and that this phenotype can also be mediated by EHEC nleH1 but not nleH2 (Feuerbacher and Hardwidge 2014). Another study revealed that a C. rodentium nleH mutant was attenuated for colonization in the early stages of infection and induced less activation of NF-κB activation than wild type C. rodentium (Hemrajani et al. 2008).
NF-κB signaling is further influenced by the zinc metalloprotease T3SS effector NleC that directly degrades NF-κB subunits, p65 and p50 (Baruch et al. 2011; Muhlen et al. 2011; Pearson et al. 2011; Sham et al. 2011). NleC is encoded by OI-36 in EHEC EDL933 and PP4 in EPEC E2348/69, and is highly conserved among all A/E pathogens. The amino acid sequences of NleC from EHEC and EPEC are 100% identical and share 95% similarity with NleC from C. rodentium (Iguchi et al. 2009; Perna et al. 2001). The conserved zinc metalloprotease motif 183HEIIH187 is essential for the proteolytic activity of NleC, as NleC, which carries a mutated metalloprotease motif, loses its ability to cleave the NF-κB subunits p65 and p50 (Marches et al. 2005).
NleC-mediated cleavage of p65 can be observed in cultured epithelial cells by ectopic expression or during EPEC infection. Although different cleavage sites of p65 were originally proposed, namely between proline (Pro10) and alanine (Ala11) or between cysteine (Cys38) and glutamic (Glu39), both sites are located within the N-terminal Rel homology domain that mediates the binding of p65 to promoters or enhancers of target genes (Pearson et al. 2011; Baruch et al. 2011; Yen et al. 2010). Subsequent studies confirmed the cleavage site as Cys38/Glu39 and a recent molecular analysis of NleC revealed that two motifs in the Rel homology domain of p65, 22EIIE25 and 177PVLS180, were critical as the substrate recognition and binding sites for NleC (Giogha et al. 2015). In addition, other NF-κB subunits, such as p50, c-Rel, IκBα, and coactivator of transcription regulators p300, have been reported as substrates of NleC ; however, some of these targets remain a topic of debate among certain research groups (Muhlen et al. 2011; Pearson et al. 2011; Shames et al. 2011a).
The role of NleC in pathogenesis has been examined in vivo using an EHEC O157:H7 infection model in lambs and calves. However, an EHEC strain lacking NleC did not show any significant differences in colonization when compared to wild type EHEC O157:H7 (Marches et al. 2005). Similarly, C57BL/6 mice infected with either wild type C. rodentium or an nleC mutant had equivalent numbers of the pathogen in the colon, however, nleC deletion resulted in increased tissue damage in mice, suggesting that NleC functions in vivo to reduce tissue pathology (Kelly et al. 2006; Sham et al. 2011; Marches et al. 2005).
The LEE-encoded effector , Tir, which is critical for A/E lesion formation, has also been implicated in inhibiting host innate signaling. The C-terminal region of Tir shares sequence similarity with host cellular immunoreceptor tyrosine-based inhibitory motifs (ITIM) , which are on the cytoplasmic tail of many immunoreceptors. Phosphorylation of a conserved tyrosine in ITIM-containing proteins allows the receptor to bind to protein tyrosine phosphatases, such as SHP-1 and SHP-2. These phosphatases, in turn, down-regulate host immune responses by decreasing the phosphorylation of a variety of immune signaling-related proteins (Zhang et al. 2000; Neel et al. 2003). The phosphorylation of Tir on amino acid residues Tyr483 and Tyr511 leads to an interaction between Tir and SHP-1, resulting in suppression of cytokine production including TNF and IL-6 (Yan et al. 2012, 2013). Apart from ITIM similarity, Tir also binds to the NF-κB adaptor protein TRAF2 and consequently inhibits TNF-induced NF-κB signaling (Ruchaud-Sparagano et al. 2011).
Inhibition of MAPK Signaling
In addition to NleC, A/E pathogens encode another conserved zinc metalloprotease, termed NleD. NleD is a 30-kD T3SS effector encoded on OI-36 in EHEC EDL933 and PP4 in EPEC E2348/69, respectively, and the amino acid sequence similarity between them is 99% (Pearson and Hartland 2014). NleD contains a conserved zinc metalloprotease motif, 141HELLH145, which is crucial for proteolytic activity (Baruch et al. 2011). In contrast to NleC, NleD targets MAPK signaling through direct cleavage of the conserved activation loop in the MAPKs, JNK and p38 (Baruch et al. 2011). Targeting of MAPK by NleD may be a mechanism that contributes to the profound decrease in IL-8 secretion during EPEC infection, since an nleBECD mutant induced higher levels of IL-8 in infected epithelial cells than an nleBEC mutant (Baruch et al. 2011). NleD may also contribute to promoting cell survival, as the cleavage of JNK can arrest JNK-induced apoptosis (Shaulian and Karin 2001).
Pathogen Response to the Host: Apoptosis
Effectors that Induce Apoptosis
Programmed cell death pathways can be induced directly upon the secretion of T3SS effectors by EHEC during infection. All of EspF, Map and Cif have been associated with induction of cell death. Due to carriage of a mitochondrial targeting sequence (MTS) located at the N-terminus, EspF localizes to host mitochondria and induces disruption of mitochondrial membrane potential, which in turn initiates apoptosis signaling by stimulating cytochrome c release and caspase-9 cleavage (Nougayrede et al. 2007; Dean et al. 2010). EspF also binds Abcf2 in mitochondria which may explain the induction of caspase-9 cleavage since lower Abcf2 levels in mitochondria correlate with caspase-9 cleavage (Nougayrede et al. 2007). The T3SS effector Map also harbors an MTS in the N-terminal region and Map is predicted to activate intrinsic cell death during A/E pathogen infection (Kenny and Jepson 2000). In vitro studies have revealed that Map leads to mitochondrial membrane disruption; however, the precise function of Map in vivo is still unclear (Ma et al. 2006). Finally, the T3SS effector Cif can arrest cell-cycle progression at G1/S and G2/M in host cells by inhibiting the ubiquitination and degradation of proteins that regulate cell cycle. This induces a delayed form of apoptosis (Marches et al. 2003; Taieb et al. 2006; Samba-Louaka et al. 2008, 2009). Cif inactivates Cullin Ring E3 ligases (CRL ) by deamidation of NEDD8 (Crow et al. 2012; Toro et al. 2013; Cui et al. 2010). Neddylation is usually required to activate CRLs by inducing a conformational change that allows the E3 ligase to ubiquitinate target proteins. In addition, Cif prevents perforin-2 ubiquitination and hence its activation and bactericidal activity (McCormack et al. 2015).
Another conserved T3SS effector , NleB1 , is encoded directly upstream of nleE in OI-122 of EHEC EDL933 or IE6 of EPEC E2348/69 and a close homologue, NleB2, is encoded in OI-36 (EHEC EDL933) or PP4 (EPEC E2348/69). When expressed ectopically in cultured epithelial cells, NleB1 can dampen NF-κB signaling in response to TNF but not IL-1β (Newton et al. 2010). However, during infection, NleB1 has little impact on the inhibition of pro-inflammatory cytokine production (Pearson et al. 2013; Li et al. 2013). Instead, NleB1 has a key role in blocking extrinsic apoptosis signaling stimulated by death receptor activation through FAS or TNFR1. NleB1 possesses N-acetylglucosamine (GlcNAc) transferase activity that specifically modifies the death domains (DD) of death receptor adaptor proteins by transferring GlcNAc to a conserved arginine in FADD (Arg117), TRADD and RIPK1 (Pearson et al. 2013; Li et al. 2013). This activity blocks caspase-8 activation and subsequent cell death by preventing formation of the death-inducing signaling complex (DISC) . These findings are consistent with previous discoveries revealing that Arg117 of FADD is an essential residue that mediates death domain interactions between FAS-FADD and FADD-TRADD (Imtiyaz et al. 2005; Wang et al. 2010). TRADD is an adaptor protein involved in the NF-κB pathway where it forms a component of the TNFR1 complex I with RIPK1 and TRAF2. Thus the modification of Arg235 in TRADD may explain the role of NleB1 in suppressing NF-κB activation under certain experimental conditions (Newton et al. 2010).
The GlcNAc transferase activity of NleB1 depends on a catalytic motif, DxD, that is conserved in NleB1 and NleB2, and also the SseK T3SS effectors from Salmonella spp. (Gao et al. 2013). In vivo studies using C. rodentium, which carries only one copy of nleB revealed, that either deletion of nleB or mutation of the DxD motif results in attenuation of colonization and increased caspase-8 cleavage in the colon of infected mice (Kelly et al. 2006; Pearson et al. 2013). Currently, the host targets and functions of NleB2 and the SseK effectors are not clear, although some activity against DD containing proteins has been observed (Kelly et al. 2006; Pearson et al. 2013).
NleF is also a conserved T3SS effector that is encoded on OI-71 in EHEC EDL933, and PP6 in EPEC E2348/69. The amino acid sequences of NleF from EPEC and EHEC are 100% identical (Iguchi et al. 2009; Deng et al. 2004). NleF inhibits intrinsic and extrinsic apoptosis in vitro by binding caspase-4, caspase-8 and caspase-9 (Blasche et al. 2013). The inhibition appears to occur through the binding of NleF to the active site of these caspases. However, during infection, deletion of nleF from EPEC E2348/69 does not lead to a significant increase in caspase-3/7 activity in cultured epithelial cells, although this experiment did not induce apoptosis in infected cells using an exogenous signal (Blasche et al. 2013). NleF also promotes NF-κB nuclear translocation and IL-8 production, although the mechanism by which NleF activates NF-κB is not clear (Pallett et al. 2014).
NleH1 and NleH2 also modulate intrinsic cell death signaling pathways. The function of NleH in inhibiting apoptosis was initially determined in vitro where a higher number of cultured epithelial cells died when infected with an ΔnleH1/nleH2 double mutant compared to cells infected with wild type EPEC E2348/69 (Hemrajani et al. 2010). NleH was then implicated in the inhibition of caspase-3 cleavage which was observed in EPEC-infected epithelial cells and C. rodentium-infected mice, which may arise from an interaction between NleH and the apoptosis inhibitor protein, Bax inhibitor-1 (BI-1) (Hemrajani et al. 2010; Robinson et al. 2010).
Additionally, the LEE-encoded effectors , EspZ and EspO, have been implicated in maintaining epithelial integrity during A/E pathogen colonization. EspZ (SepZ) is a conserved T3SS effector carried by all A/E pathogens and the amino acid sequences of EspZ from EPEC and EHEC are 71.7% identical (Kanack et al. 2005). Initial research revealed that deletion of espZ from C. rodentium significantly reduced the colonization of C. rodentium in vivo (Deng et al. 2004) and in vitro studies further revealed that EspZ protects epithelial cells from cell death during EPEC infection. The mechanism of EspZ activity in enhancing cell survival was originally attributed to its ability to bind CD98 and upregulate integrin signaling and focal adhesion kinase (FAK) signaling (Shames et al. 2010). More recently, EspZ has been observed to localize to mitochondria and may reduce cell cytotoxicity through binding the inner mitochondrial membrane 17b (TIM17b), thereby maintaining mitochondrial membrane potential (Shames et al. 2011b). Rabbit enteropathogenic E. coli (REPEC) ΔespZ mutants also exhibit attenuated virulence during infection and induce markedly higher levels of host cell death (Wilbur et al. 2015). EspZ assists in regulating T3SS effector translocation from a plasma membrane location which may contribute to its virulence function (Berger et al. 2012), but, to date, the biochemical basis of EspZ activity is unknown.
Another LEE-encoded effector , EspO , which is homologous to OspE from Shigella flexneri, enhances host cell-cell adherence during infection. Previous studies have revealed that OspE promotes the stability of focal adhesions (FA) by interacting with integrin-linked kinase (ILK) , and also by binding to the PDZ-containing protein PDLIM7 to modulate the activity of protein kinase C (PKC), which is associated with cell spreading and adherence (Kim et al. 2009; Yi et al. 2014). EspO carries the Trp68 equivalent that is involved in OspE-ILK interaction and has also been observed to localize with FA during EHEC infection in cultured epithelial cells, indicating that EspO may have a similar role to OspE in bacterial gut infection (Kim et al. 2009; Morita-Ishihara et al. 2013).
Inflammasomes and Immunity to A/E Pathogens
Mice deficient in certain inflammasome components, including Nlrc4, Nlrp3, Asc and Caspase-1 (but not Caspase-11), are more susceptible to C. rodentium infection, suggesting that inflammasome sensing plays a role in defense against A/E pathogens (Lupfer et al. 2014; Nordlander et al. 2014). Inflammasomes of the intestinal epithelium play a general protective role during infection by removing damaged enterocytes and producing inflammasome dependent cytokines (Sellin et al. 2015). Findings from a recent study suggest that A/E pathogens counter this using the T3SS effector, NleA/EspI, which binds directly to Nlrp3 and blocks de-ubiquitination, thereby preventing inflammasome activation (Yen et al. 2015).
Conclusions
The manipulation of host signaling pathways, especially those associated with host immunity, is a key strategy utilized by EHEC and other A/E pathogens for successful infection. A growing number of host proteins have been identified as targets of T3SS effectors that are inactivated through specific post-translational modifications. These discoveries have not only expanded our understanding of pathogen-host interactions, but, in some cases, have also helped to define the function of host proteins in signaling pathways. Indeed, identification of the host proteins modified by T3SS effectors during infection has greatly informed us about the immune responses important for combating infection, as these are precisely the pathways targeted by the pathogen for inhibition. Overall, there is still much to learn about roles of T3SS effectors during infection; in particular, those where no enzymatic activity has been identified (Table 1). Given this, some reassessment of T3SS biology in EHEC and other A/E pathogens is needed. Future research must also be cautious about inferring the function of homologous bacterial effectors from different pathogens as these may display different activities and target distinct host signaling pathways.
Abbreviations
- A/E pathogen:
-
Attaching/effacing pathogen
- AP-1:
-
Activation protein-1
- BI-1:
-
Bax inhibitor-1
- CLR:
-
C-type lection receptor
- CRL:
-
Cullin Ring E3 ligases
- DC:
-
Dendritic cell
- DD:
-
Death domains
- DISC:
-
Death-inducing signaling complex
- EHEC:
-
Enterohemorrhagic Escherichia coli
- FA:
-
Focal adhesion
- GlcNAc:
-
N-acetylglucosamine
- HUS:
-
Hemolytic uremic syndrome
- IE:
-
Integrative element
- ILK:
-
Integrin-linked kinase
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motifs
- LEE:
-
Locus of enterocyte effacement
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MTS:
-
Mitochondrial targeting sequence
- NF-κB:
-
Nuclear-factor κB
- NLR:
-
NOD-like receptor
- NZF:
-
Npl4 Zinc figure
- OI:
-
O-island
- PAMPs:
-
Pathogen-associated molecular patterns
- PKC:
-
Protein kinase C
- PRRs:
-
Pattern recognition receptors
- RLR:
-
RIG-I-like receptor
- SAM:
-
S-adenosyl-L-methionine
- Stx:
-
Shiga toxins
- T3SS:
-
Type III secretion system
- TAB2 and TAB3:
-
TAK1-binding proteins 2 and 3
- TAD:
-
Transcription activation domain
- TIM17b:
-
Translocase of inner mitochondrial membrane 17b
- Tir:
-
Translocated intimin receptor
- TLR:
-
Toll-like receptor
- TNFR1:
-
Tumor necrosis factor receptor 1
- TRAIL:
-
TNF-associated apoptosis-inducing ligand
References
Akira, S., Uematsu, S., & Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell, 124(4), 783–801.
Barnett Foster, D., et al. (2000). Enterohemorrhagic Escherichia coli induces apoptosis which augments bacterial binding and phosphatidylethanolamine exposure on the plasma membrane outer leaflet. Infection and Immunity, 68(6), 3108–3115.
Baruch, K., et al. (2011). Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. The EMBO Journal, 30(1), 221–231.
Berger, C. N., et al. (2012). EspZ of enteropathogenic and enterohemorrhagic Escherichia coli regulates type III secretion system protein translocation. MBio, 3(5), e00317–e00312.
Berin, M. C., et al. (2002). Role of EHEC O157:H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cellular Microbiology, 4(10), 635–648.
Blasche, S., et al. (2013). The E. coli effector protein NleF is a caspase inhibitor. PloS One, 8(3), e58937.
Boyce, T. G., Swerdlow, D. L., & Griffin, P. M. (1995). Escherichia coli O157:H7 and the hemolytic-uremic syndrome. The New England Journal of Medicine, 333(6), 364–368.
Callaway, T. R., et al. (2009). Diet, Escherichia coli O157:H7, and cattle: A review after 10 years. Current Issues in Molecular Biology, 11(2), 67–79.
Crow, A., et al. (2012). The molecular basis of ubiquitin-like protein NEDD8 deamidation by the bacterial effector protein Cif. Proceedings of the National Academy of Sciences of the United States of America, 109(27), E1830–E1838.
Cui, J., et al. (2010). Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science, 329(5996), 1215–1218.
Dean, P., et al. (2010). The enteropathogenic E. coli effector EspF targets and disrupts the nucleolus by a process regulated by mitochondrial dysfunction. PLoS Pathogens, 6(6), e1000961.
Deng, W., et al. (2004). Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proceedings of the National Academy of Sciences of the United States of America, 101(10), 3597–3602.
Durso, L. M., et al. (2005). Shiga-toxigenic Escherichia coli O157:H7 infections among livestock exhibitors and visitors at a Texas County fair. Vector Borne and Zoonotic Diseases, 5(2), 193–201.
Elliott, S. J., et al. (2000). The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infection and Immunity, 68(11), 6115–6126.
Endo, Y., et al. (1988). Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. European Journal of Biochemistry, 171(1–2), 45–50.
Feng, P. C., & Reddy, S. (2013). Prevalences of Shiga toxin subtypes and selected other virulence factors among Shiga-toxigenic Escherichia coli strains isolated from fresh produce. Applied and Environmental Microbiology, 79(22), 6917–6923.
Feuerbacher, L. A., & Hardwidge, P. R. (2014). Influence of NleH effector expression, host genetics, and inflammation on Citrobacter rodentium colonization of mice. Microbes and Infection, 16(5), 429–433.
Frankel, G., et al. (1998). Enteropathogenic and enterohaemorrhagic Escherichia coli: More subversive elements. Molecular Microbiology, 30(5), 911–921.
From the Centers for Disease Control and Prevention. (1995). Escherichia coli O157:H7 outbreak linked to commercially distributed dry-cured salami – Washington and California, 1994. JAMA, 273(13), 985–986.
Gao, X., et al. (2009). Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathogens, 5(12), e1000708.
Gao, X., et al. (2013). NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-kappaB activation. Cell Host & Microbe, 13(1), 87–99.
Ghosh, S., May, M. J., & Kopp, E. B. (1998). NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annual Review of Immunology, 16, 225–260.
Giogha, C., et al. (2015). Substrate recognition by the zinc metalloprotease effector NleC from enteropathogenic Escherichia coli. Cellular Microbiology, 17, 1766–1778.
Hayden, M. S., & Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell, 132(3), 344–362.
Hemrajani, C., et al. (2008). Role of NleH, a type III secreted effector from attaching and effacing pathogens, in colonization of the bovine, ovine, and murine gut. Infection and Immunity, 76(11), 4804–4813.
Hemrajani, C., et al. (2010). NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proceedings of the National Academy of Sciences of the United States of America, 107(7), 3129–3134.
Hurley, B. P., Thorpe, C. M., & Acheson, D. W. (2001). Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infection and Immunity, 69(10), 6148–6155.
Iguchi, A., et al. (2009). Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. Journal of Bacteriology, 191(1), 347–354.
Imtiyaz, H. Z., Zhang, Y., & Zhang, J. (2005). Structural requirements for signal-induced target binding of FADD determined by functional reconstitution of FADD deficiency. The Journal of Biological Chemistry, 280(36), 31360–31367.
Johnson, G. L., & Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science, 298(5600), 1911–1912.
Kanack, K. J., et al. (2005). SepZ/EspZ is secreted and translocated into HeLa cells by the enteropathogenic Escherichia coli type III secretion system. Infection and Immunity, 73(7), 4327–4337.
Kaper, J. B., Nataro, J. P., & Mobley, H. L. (2004). Pathogenic Escherichia coli. Nature Reviews. Microbiology, 2(2), 123–140.
Karmali, M. A., et al. (1983). Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet, 1(8325), 619–620.
Karmali, M. A., Gannon, V., & Sargeant, J. M. (2010). Verocytotoxin-producing Escherichia coli (VTEC). Veterinary Microbiology, 140(3–4), 360–370.
Kashiwamura, M., et al. (2009). Shiga toxin kills epithelial cells isolated from distal but not proximal part of mouse colon. Biological & Pharmaceutical Bulletin, 32(9), 1614–1617.
Kelly, M., et al. (2006). Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infection and Immunity, 74(4), 2328–2337.
Kenny, B., & Jepson, M. (2000). Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cellular Microbiology, 2(6), 579–590.
Khan, M. A., et al. (2006). Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infection and Immunity, 74(5), 2522–2536.
Kim, D. W., et al. (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proceedings of the National Academy of Sciences of the United States of America, 102(39), 14046–14051.
Kim, M., et al. (2009). Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature, 459(7246), 578–582.
Li, S., et al. (2013). Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature, 501(7466), 242–246.
Liu, S., & Chen, Z. J. (2011). Expanding role of ubiquitination in NF-kappaB signaling. Cell Research, 21(1), 6–21.
Lupfer, C. R., et al. (2014). Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathogens, 10(9), e1004410.
Ma, C., et al. (2006). Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: Role of mitochondrial associated protein (Map). Cellular Microbiology, 8(10), 1669–1686.
Manning, S. D., et al. (2008). Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proceedings of the National Academy of Sciences of the United States of America, 105(12), 4868–4873.
Marches, O., et al. (2003). Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Molecular Microbiology, 50(5), 1553–1567.
Marches, O., et al. (2005). Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infection and Immunity, 73(12), 8411–8417.
McCormack, R. M., et al. (2015). Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of Perforin-2. eLife, 4, e06505.
McDaniel, T. K., et al. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proceedings of the National Academy of Sciences of the United States of America, 92(5), 1664–1668.
Mead, P. S., & Griffin, P. M. (1998). Escherichia coli O157:H7. Lancet, 352(9135), 1207–1212.
Miyamoto, Y., et al. (2006). Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cellular Microbiology, 8(5), 869–879.
Morita-Ishihara, T., et al. (2013). EspO1-2 regulates EspM2-mediated RhoA activity to s(43): p. 30101-13. Tabilize formation of focal adhesions in enterohemorrhagic Escherichia coli-infected host cells. PloS One, 8(2), e55960.
Muhlen, S., Ruchaud-Sparagano, M. H., & Kenny, B. (2011). Proteasome-independent degradation of canonical NFkappaB complex components by the NleC protein of pathogenic Escherichia coli. The Journal of Biological Chemistry, 286(7), 5100–5107.
Mukaida, N., Mahe, Y., & Matsushima, K. (1990). Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. The Journal of Biological Chemistry, 265(34), 21128–21133.
Mukaida, N., et al. (1994). Molecular mechanism of interleukin-8 gene expression. Journal of Leukocyte Biology, 56(5), 554–558.
Muthing, J., et al. (2009). Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thrombosis and Haemostasis, 101(2), 252–264.
Nadler, C., et al. (2010). The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathogens, 6(1), e1000743.
Neel, B. G., Gu, H., & Pao, L. (2003). The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in Biochemical Sciences, 28(6), 284–293.
Newton, H. J., et al. (2010). The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathogens, 6(5), e1000898.
Nordlander, S., Pott, J., & Maloy, K. J. (2014). NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunology, 7(4), 775–785.
Nougayrede, J. P., Foster, G. H., & Donnenberg, M. S. (2007). Enteropathogenic Escherichia coli effector EspF interacts with host protein Abcf2. Cellular Microbiology, 9(3), 680–693.
Orchard, R. C., et al. (2012). Identification of F-actin as the dynamic hub in a microbial-induced GTPase polarity circuit. Cell, 148(4), 803–815.
Pallett, M. A., et al. (2014). The type III secretion effector NleF of enteropathogenic Escherichia coli activates NF-kappaB early during infection. Infection and Immunity, 82(11), 4878–4888.
Park, H. H., et al. (2007). The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annual Review of Immunology, 25, 561–586.
Pearson, J. S., & Hartland, E. L. (2014). The inflammatory response during Enterohemorrhagic Escherichia coli infection. Microbiology Spectrum, 2(4), 321–339.
Pearson, J. S., et al. (2011). A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-kappaB for degradation. Molecular Microbiology, 80(1), 219–230.
Pearson, J. S., et al. (2013). A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature, 501(7466), 247–251.
Perna, N. T., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature, 409(6819), 529–533.
Pham, T. H., et al. (2012). Functional differences and interactions between the Escherichia coli type III secretion system effectors NleH1 and NleH2. Infection and Immunity, 80(6), 2133–2140.
Raymond, B., et al. (2011). The WxxxE effector EspT triggers expression of immune mediators in an Erk/JNK and NF-kappaB-dependent manner. Cellular Microbiology, 13(12), 1881–1893.
Riley, L. W., et al. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. The New England Journal of Medicine, 308(12), 681–685.
Robinson, K. S., et al. (2010). The enteropathogenic Escherichia coli effector NleH inhibits apoptosis induced by Clostridium difficile toxin B. Microbiology, 156(Pt 6), 1815–1823.
Ruchaud-Sparagano, M. H., et al. (2011). The enteropathogenic E. coli (EPEC) Tir effector inhibits NF-kappaB activity by targeting TNFalpha receptor-associated factors. PLoS Pathogens, 7(12), e1002414.
Samba-Louaka, A., et al. (2008). Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cellular Microbiology, 10(12), 2496–2508.
Samba-Louaka, A., et al. (2009). The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infection and Immunity, 77(12), 5471–5477.
Sato, Y., et al. (2009). Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by NZF domains of TAB2 and TAB3. The EMBO Journal, 28(24), 3903–3909.
Schuller, S., Frankel, G., & Phillips, A. D. (2004). Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cellular Microbiology, 6(3), 289–301.
Sellin, M. E., et al. (2015). Inflammasomes of the intestinal epithelium. Trends in Immunology, 36(8), 442–450.
Sham, H. P., et al. (2011). Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-kappaB and p38 mitogen-activated protein kinase activation. Infection and Immunity, 79(9), 3552–3562.
Shames, S. R., et al. (2010). The pathogenic E. coli type III effector EspZ interacts with host CD98 and facilitates host cell prosurvival signalling. Cellular Microbiology, 12(9), 1322–1339.
Shames, S. R., et al. (2011a). The pathogenic Escherichia coli type III secreted protease NleC degrades the host acetyltransferase p300. Cellular Microbiology, 13(10), 1542–1557.
Shames, S. R., et al. (2011b). The type III system-secreted effector EspZ localizes to host mitochondria and interacts with the translocase of inner mitochondrial membrane 17b. Infection and Immunity, 79(12), 4784–4790.
Shaulian, E., & Karin, M. (2001). AP-1 in cell proliferation and survival. Oncogene, 20(19), 2390–2400.
Smith, W. E., et al. (2003). Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infection and Immunity, 71(3), 1497–1504.
Spika, J. S., et al. (1986). Hemolytic uremic syndrome and diarrhea associated with Escherichia coli O157:H7 in a day care center. The Journal of Pediatrics, 109(2), 287–291.
Stahl, A. L., et al. (2006). Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood, 108(1), 167–176.
Taieb, F., et al. (2006). Escherichia coli Cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. Cellular Microbiology, 8(12), 1910–1921.
Takaesu, G., et al. (2000). TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Molecular Cell, 5(4), 649–658.
Takeuchi, O., & Akira, S. (2010). Pattern recognition receptors and inflammation. Cell, 140(6), 805–820.
Thorpe, C. M., et al. (1999). Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infection and Immunity, 67(11), 5985–5993.
Thorpe, C. M., et al. (2001). Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infection and Immunity, 69(10), 6140–6147.
Toro, T. B., Toth, J. I., & Petroski, M. D. (2013). The cyclomodulin cycle inhibiting factor (CIF) alters cullin neddylation dynamics. The Journal of Biological Chemistry, 288(21), 14716–14726.
Tzipori, S., et al. (1985). Enteropathogenic Escherichia coli enteritis: Evaluation of the gnotobiotic piglet as a model of human infection. Gut, 26(6), 570–578.
Tzipori, S., Gibson, R., & Montanaro, J. (1989). Nature and distribution of mucosal lesions associated with enteropathogenic and enterohemorrhagic Escherichia coli in piglets and the role of plasmid-mediated factors. Infection and Immunity, 57(4), 1142–1150.
van Setten, P. A., et al. (1996). Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood, 88(1), 174–183.
Vallabhapurapu, S., & Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annual Review of Immunology, 27, 693–733.
Varma, J. K., et al. (2003). An outbreak of Escherichia coli O157 infection following exposure to a contaminated building. JAMA, 290(20), 2709–2712.
Vossenkamper, A., et al. (2010). Inhibition of NF-kappaB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. Journal of Immunology, 185(7), 4118–4127.
Walters, M. D., et al. (1989). The polymorphonuclear leucocyte count in childhood haemolytic uraemic syndrome. Pediatric Nephrology, 3(2), 130–134.
Wan, F., et al. (2011). IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nature Immunology, 12(4), 335–343.
Wang, L., et al. (2010). The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nature Structural & Molecular Biology, 17(11), 1324–1329.
Wilbur, J. S., et al. (2015). The secreted effector protein EspZ is essential for virulence of rabbit enteropathogenic Escherichia coli. Infection and Immunity, 83(3), 1139–1149.
Wong Fok Lung, T., et al. (2014). The cell death response to enteropathogenic Escherichia coli infection. Cellular Microbiology, 16(12), 1736–1745.
Wong, A. R., et al. (2011). Enteropathogenic and enterohaemorrhagic Escherichia coli: Even more subversive elements. Molecular Microbiology, 80(6), 1420–1438.
Yan, D., et al. (2012). Inhibition of TLR signaling by a bacterial protein containing immunoreceptor tyrosine-based inhibitory motifs. Nature Immunology, 13(11), 1063–1071.
Yan, D., et al. (2013). Enteropathogenic Escherichia coli Tir recruits cellular SHP-2 through ITIM motifs to suppress host immune response. Cellular Signalling, 25(9), 1887–1894.
Yao, Q., et al. (2014). Structure and specificity of the bacterial cysteine methyltransferase effector NleE suggests a novel substrate in human DNA repair pathway. PLoS Pathogens, 10(11), e1004522.
Yen, H., et al. (2010). NleC, a type III secretion protease, compromises NF-kappaB activation by targeting p65/RelA. PLoS Pathogens, 6(12), e1001231.
Yen, H., Sugimoto, N., & Tobe, T. (2015). Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PLoS Pathogens, 11(9), e1005121.
Yi, C. R., et al. (2014). Systematic analysis of bacterial effector-postsynaptic density 95/disc large/zonula occludens-1 (PDZ) domain interactions demonstrates Shigella OspE protein promotes protein kinase C activation via PDLIM proteins. The Journal of Biological Chemistry, 289, 30101–30113.
Zhang, Q., et al. (2000). Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promoter. The American Journal of Pathology, 157(4), 1137–1146.
Zhang, L., et al. (2012). Cysteine methylation disrupts ubiquitin-chain sensing in NF-kappaB activation. Nature, 481(7380), 204–208.
Zhou, X., et al. (2003). Flagellin of enteropathogenic Escherichia coli stimulates interleukin-8 production in T84 cells. Infection and Immunity, 71(4), 2120–2129.
Zurawski, D. V., et al. (2008). The NleE/OspZ family of effector proteins is required for polymorphonuclear transepithelial migration, a characteristic shared by enteropathogenic Escherichia coli and Shigella flexneri infections. Infection and Immunity, 76(1), 369–379.
Acknowledgements
This work was supported by grants to ELH from the Australian National Health and Medical Research Council (APP1044061). JSP is the recipient of an NHMRC Early Career Fellowship. YZ is the recipient of a University of Melbourne International Research Scholarship (MIRS).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zhang, Y., Pearson, J.S., Hartland, E.L. (2017). Host Innate Immune Factors Influencing Enterohemorrhagic Escherichia coli Pathogenicity. In: Gurtler, J., Doyle, M., Kornacki, J. (eds) Foodborne Pathogens. Food Microbiology and Food Safety(). Springer, Cham. https://doi.org/10.1007/978-3-319-56836-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-56836-2_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56834-8
Online ISBN: 978-3-319-56836-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)