
Abstract
Myasthenia gravis (MG) is an acquired autoimmune disease of the neuromuscular junction characterized by fluctuating weakness and exhaustion of the striated skeletal muscle [1, 2].
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


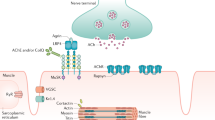
Myasthenia gravis (MG) is an acquired autoimmune disease of the neuromuscular junction characterized by fluctuating weakness and fatigability of the striated skeletal muscle [1, 2].
Myasthenia is part of the group of autoimmune channelopathies, diseases characterized by the presence of antibodies directed against the receptor structures of the neuromuscular junction. In addition to myasthenia gravis, they include Lambert-Eaton myasthenic syndrome and neuromyotonia.
Most patients present IgG1 antibodies directed against the acetylcholine receptor (AChRs) located on the postsynaptic membrane [3]. A variable portion of patients, defined as seronegative, do not have antiAchRr antibodies but have antibodies against a muscle-specific tyrosine kinase (MuSK) [4, 5]. Other antibodies directed against different antigens targets, such as LRP4 (low-density lipoprotein receptor 4) [6] and agrin [7], have been recently identified in patients who tested negative for Ab antiAchR and Ab anti-MuSK.
1 Epidemiology
Myasthenia gravis is a rare disease. According to a review of 55 studies [8] conducted between 1950 and 2007 it has an estimated incidence of 5.3 cases per million person-years and a prevalence of 77.7 cases per million. The prevalence of the disease has increased since the middle of last century: this is due to an improvement in diagnostic accuracy, an aging population and increased longevity of the patients affected.
The incidence of the disease in boys and girls before puberty is similar, whereas after puberty the male:female ratio is 4:6. The disease can start at any age: the incidence in female patients peaks in the third decade and in the sixth to seventh decades in male patients [9].
2 Pathophysiology
Myasthenia gravis is a disease that meets the criteria of autoimmune diseases mediated by autoantibodies [1, 2], more specifically:
-
1.
Most patients have pathogenic antibodies which react against a specific antigen;
-
2.
There are animal models of the disease created by passive transfer of antibodies;
-
3.
The disease responds to immunomodulatory/immunosuppressive therapies.
Eighty to 90% of patients with generalized MG have antibodies against the acetylcholine receptor (Ab antiAchR). The receptor is a protein, consisting of five different subunits, which assemble to form a ion channel [10]. The antibodies interact with different antigenic epitopes of the receptor and belong to the class of immunoglobulins IgG1. These antibodies have the ability to functionally inhibit the AchR, accelerate degradation and promote lysis by complement activation [11]. The factor which causes the immune system to loose tolerance against AchR is still unknown; however it is believed that the thymus plays an important role, given the presence in its context of myoid cells, which expresses the receptor and can act as antigen presenting cells [12].
Ten percent of patients with generalized myasthenia gravis don’t have Ab-antiAchR and are defined as seronegative. In 40–70% of seronegative Caucasian patients, antibodies against a muscle-specific tyrosine kinase receptor (MuSK) have been identified. [4, 5, 13] Similar values have been reported in the Japanese case series [14]. This tyrosine kinase is located at the neuromuscular junction and is involved in the stabilization processes of AchR at the level of post-synaptic membrane [15]. MUSK antibodies positive myasthenia gravis might recognize pathophysiological mechanisms different from Ab antiAchR positive myasthenia. The anti-MuSK antibodies belong mainly to the IgG4 subclass which does not activate complement, however IgG1 subclasses are present in low concentrations and are able to activate complement when they bind to MuSK [16].
In patients who test negative for Ab antiAchR and antiMusK, the presence of antibodies directed against protein 4 related to lipoprotein (LRP4) has been recently reported. In larger series [17,18,19] antibodies against LRP4 have been reported in 2.09% of double seronegative cases.
These antibodies are IgG1, which are able to activate the complement and thus they are potentially pathogenic. However, more research is needed to confirm this hypothesis.
3 Clinical Features
The basic clinical features of the disease are fluctuating weakness and fatigability of the striated voluntary muscles. Two main forms can be distinguished with reference to the districts involved: ocular MG, which affects only the extraocular muscles, and generalized MG involving the striated skeletal muscles with possible involvement of the bulbar district [9].
Typically, muscle weakness fluctuates during the day: it tends to be worse in the afternoon or evening, increases during physical work and is relieved by rest. In the early stages of the disease, symptoms may be absent upon awakening, whereas they tend to be constantly present with the progression of the disease, even if moderate to severe fluctuations are possible [20]. Despite the fact that myasthenia gravis can produce weakness in any muscle group, there are some manifestations that are quite characteristic [20]:
-
More than 50% of patients show ocular symptoms (ptosis and diplopia): about half of these will develop generalized symptoms within 2 years;
-
Approximately 15% of patients present bulbar symptoms (dysarthria, dysphagia, fatigability when chewing);
-
Less than 5% of patients present isolated proximal muscle weakness.
Less commonly presenting patterns include: isolated neck weakness (dropped head); isolated weakness of the respiratory muscles; distal muscle weakness.
Ocular muscles. Weakness of the eyelid muscles can cause ptosis. Ptosis can occur bilaterally and then improve in one of the eyes causing unilateral ptosis, or it may be unilateral at the beginning and then become bilateral. Involvement of the extraocular muscles causes binocular diplopia which can be horizontal or vertical, in more severe cases it can lead to ophthalmoparesis/ophthalmoplegia. Pupils are always spared.
Bulbar muscles. The mandibular musculature is often involved causing weakness during chewing. The patient often reports that this occurs halfway through the meal. When mandibular weakness is present at rest, patients hold their fingers under the jaw to keep the mouth closed. Weakness of the oropharyngeal muscles causes dysarthria and dysphagia. The patient is affected by rhinolalia because of the weakness of palatal muscles. The symptoms worsen with prolonged speech. Dysphagia may be relevant, and there is often nasal regurgitation.
Facial muscles. Facial muscles are often involved, causing reduced expression, weak eye closure, difficulty in whistling, puffing out cheeks and a typical smile called a “snarl smile”: the central part of the lips is lifted while the corners of the mouth can not be lifted
-
Axial musculature. The flexors and extensors of the neck are commonly affected and this can lead to the head falling forwards.
-
Limb muscles. Weakness predominantly affects the proximal muscles (deltoids, quadriceps and psoas) of the upper and lower limbs. In addition to the proximal muscles, the extensor muscles of the fingers and wrist are also commonly involved.
Respiratory musculature. Involvement of respiratory muscles (diaphragm, intercostal) can cause respiratory failure.
The natural history of the disease is characterized by exacerbations and remissions. The most active phase of the disease usually coincides with the first 5–7 years. Symptoms usually peak in the first 2–3 years of the disease. In a U.S. case study carried out on 1976 patients, the symptoms reached maximum severity within 2 years in 82% of the cases [21].
In an Italian retrospective study carried out on 1152 patients, the symptoms of 77% of the patients reached their nadir in the first 3 years [22]. Spontaneous remissions occur in 10–15% of cases in the first 10 years of illness. About 50% of patients presenting with ocular symptoms developed generalized symptoms within 2–3 years. There are no predictive factors; in particular the presence of Ab antiAchR, a positive response to the decremental repetitive stimulation or a positive single fiber elctromyography are not predictive of generalization.
The life expectancy of a myasthenic patient is currently comparable to the life expectancy of the general population. The mortality rate is 20–30% at 10 years in untreated cases. Most clinicians believe that the disease involves three phases, although these are modified and influenced by the current immunotherapies:
-
An active phase characterized by large fluctuations and severity of symptoms, which occurs in the first 5–7 years of onset; most myasthenic crises occur in this period;
-
A period of stability in which symptoms persist but are stable enough and can be worsened by intervening factors such as infections, reduction of drug dose, surgical stress;
-
A third phase in which remission of the disease is possible in the course of immunotherapy or after its suspension.
In literature there are different MG classifications, more precisely Osserman and Jenkins’ classification [23] and the classification of the Myasthenia Gravis Foundation of America (MGFA) [24]. The Osserman and Jenkins’ classification [23] distinguishes the following forms:
-
Group I: pure ocular MG
-
Group IIA: mild generalized MG
-
Group IIB: generalized MG with bulbar disorders
-
Group III: “fulminant” MG (rapidly evolving MG reaching maximum severity within 6 months and involving respiratory muscles)
-
Group IV: chronic MG (severe evolution of patients with modest disease for 2 or more years).
The Myasthenia Gravis Foundation of America (MGFA) stressed the importance of a grading system and standardized clinical evaluation, which is why in 2000 it proposed some standard recommendations in clinical research on myasthenia, recommendations drawn up by an ad hoc Task Force (Task Force of the MGFA, 2000) [24]. Here, in summary, is the classification of MG currently proposed by MGFA:
-
Class I: ocular MG;
-
Class II: mild generalized Myasthenia:
-
IIA: with predominant involvement of the limb muscles
-
IIB: with predominant involvement of the limb and prevalent involvement of bulbar-respiratory muscles
-
-
Class III: mild generalized Myasthenia:
-
IIIA: with prevalent involvement of limb muscles
-
IIIB: with involvement of the limb and predominant impairment of bulbar-respiratory muscles
-
-
Class IV: severe generalized Myasthenia:
-
IVA: with predominant involvement of the limb muscles;
-
IVB: with involvement of the limbs and prevalent impaired bulbar muscles.
-
-
Class V: Defined by the need for intubation, with or without mechanical ventilation, with the exception of intubation used during the routine postoperative period; the nasogastric tube falls into the IVB category.
4 Diagnostic Tests
The diagnostic tests are intended to confirm the clinical diagnosis formulated on the basis of clinical history and physical examination. We distinguish:
-
1.
Tests at the patient’s bedside (ice test and tensilon test);
-
2.
Serological tests;
-
3.
Neurophysiological tests;
-
4.
Radiological examinations;
-
1.
The ice test and the Tensilon test (edrophonium chloride) can be considered as clinical examination extensions, rather than as true laboratory tests.
-
(a)
The ice test is used in patients with ptosis. It is based on the physiological principle that neuromuscular transmission improves by cooling the muscle. In practice, a bag containing ice is placed on the eyelids, which are kept closed for 2 min. The bag is then removed and it is immediately assessed whether there has been an improvement in the ptosis. The sensitivity of the test is about 80% in patients with severe ptosis [25, 26].
-
(b)
Edrophonium chloride (Tensilon), available in 10 mg vials, is an acetilcolinesterasis inhibitor that has a rapid and short duration of action (5–10 min). The test should be given to patients with evidence of ptosis or ophthalmoparesis, in whom it is easy to observe improvement after administration of the drug. Ten milligrams of the drug are intravenously administered while the heart rhythm is checked for the possible onset of a slowdown in atrio-ventricular conduction. The drug is administered as follows: a first dose of 2 mg followed by another dose of 2 mg every 60 s up to a total dose of 10 mg. The clinical response is then assessed. This test has 80–90% sensitivity, but its specificity is rather low [3].
-
(a)
-
2.
Serological test:
-
(a)
Antireceptor acetylcholine antibody dosage: the dosage of anti-AChR antibodies is performed in serum according to the radioimmunoassay (RIA) method using human acetylcholine receptor as antigen [27]. These antibodies are present in about 85% of patients with generalized disease [3]; furthermore almost all patients with myasthenia gravis and thymoma are seropositive for these antibodies [28]. However, they are present only in 40–55% of patients with ocular myasthenia [3]. These antibodies are highly specific for myasthenia gravis, rare cases of false positives low titer have been reported in the Lambert Eaton syndrome (5%), in motor neuron disease (3–5%) and in polymyositis (<1%)
-
(b)
Anti-MuSK antibodies dosage. Antibodies against specific muscle receptor tyrosine kinase (MuSK) have been reported in 38–50% of patients with generalized MG who tested negative for Ab antiAchR [4, 5, 14, 17, 29]. These antibodies are not usually present in patients who only have ocular myasthenia, although rare cases have been described [30].
Typically, patients with generalized forms positive for Ab antiAchR do not have anti-MuSK antibodies, even if one case study reported that 11% of the patients showed double positivity [14]. The analysis of available clinical case studies regarding MuSK positive patients showed that these antibodies define a category of patients with some special features. They are usually female patients with prevalent involvement of the ocular and bulbar areas (oculo-bulbar form) and a high incidence of respiratory failure. Furthermore, there is a low incidence of thymic pathology.
It is also reported that these patients display a poor response to inhibitors of acetylcholinesterase but a good response to plasmapheresis and immunosuppressive therapies [4, 5, 14, 17, 29].
-
(c)
Anti-titin antibodies and ryanodine antibodies: in addition to the anti-AchR antibodies, patients with MG may have antibodies directed against the striated muscle components, in particular against the titin protein and against the ryanodine receptor (component of the sarcoplasmic reticulum involved in calcium release).
The pathogenic role of these antibodies has not yet been defined. A correlation has been observed between positivity of these antibodies and the presence of thymoma. Their presence can be considered as a tumour marker. Therefore, they are useful in patients with uncertain evidence of thymic enlargement or when recurrence of a thymic tumor is suspected [31, 32].
-
(d)
Lastly, in cases of seronegative myasthenia, specialized laboratories can search for the presence of low affinity antiAchR IgG or anti LPR4 antibodies.
-
(a)
-
3.
Neurophysiological tests.
The neurophysiological tests (repetitive nerve stimulation and single fiber EMG) can show a deficit of neuromuscular transmission and thus significantly contribute to the diagnosis. In generalized myasthenia gravis, the sensitivity of repetitive nerve stimulation (RNS) and single-fiber electromyography (SF-EMG) is respectively 75% and 95% (34–35). In the ocular forms RS can be negative in up to 50% of cases while the SF-EMG has a sensitivity of 85–95%. (34–35)
-
(a)
Repetitive stimulation (RNS). This is the neurophysiological test which is most commonly used when myasthenia gravis is suspected. The examination is performed placing a recording electrode on the muscle and stimulating the corresponding motor nerve.
The nerve is repeatedly stimulated at low frequency (2–5 Hz) and then the corresponding compound muscle action potential (CMAP) is recorded. Myasthenia patients typically show a reduction of the CMAP amplitude between the I and IV/V response (decremental response). The test is considered positive if there is a decrease of at least 10%.
The study can be carried out on different muscles; it is generally useful to test symptomatic districts. In addition, sensitivity appears to be greater in the proximal muscles than in the distal muscles (e.g. trapezoid more sensitive than abductor V), even if stimulation is better tolerated on distal muscles.
-
(b)
Single fiber electromyography (SF-EMG). The SF-EMG is technically more challenging than the RNS. The method allows the simultaneous recording of the action potential of two muscle fibers innervated by the same motor axon.
The time interval between the two potentials is defined as ‘jitter’. In myasthenia gravis, the loss of the safety factor of neuromuscular transmission determines an increase in jitter. The increase in jitter is not specific to myasthenia gravis, because it can also be found in other conditions such as motor neuron disease, polymyositis, peripheral neuropathies, Eaton-Lambert. However, these diseases have other associated electroneurographic and electromyographic findings, such as to allow an adequate neurophysiological differential diagnosis.
To maximize SFEMG sensitivity, a facial district muscle and a limb are typically tested.
We report the recommendations regarding neurophysiological tests proposed by the AAM (American Association of Electrodiagnostic Medicine) [34] based on a review of the literature:
-
The RNS must be carried out on a nerve corresponding to a symptomatic district and the resut is considered positive when a reproducible decrease of at least 10% of the amplitude of the potential between the I and IV/V response is detected in at least one district.
Please respect the following conditions:
-
Discontinue anticholinesterase therapy at least 12 h before the test;
-
Immobilize the limb if possible;
-
Adjust stimulating rate between 2 and 5 Hz;
-
Adjust basal stimulation, post-tetanic stimulation or post-exercise at 2–5 Hz followed by stimulation at regular intervals from 30 s to 1 min, for 5 min;
-
Maintain skin temperature as close as possible to 35 °C.
-
-
If RNS is normal, but there is a strong suspicion of MG, an examination by SF-EMG must be carried out, at least in one symptomatic region; if this is negative but there is a strong clinical suspicion, the investigation must be performed on a second region. The results are considered pathological if more than 10% of the pairs of potential have a higher jitter than normal, the average jitter exceed the standard limits or impulse blocking is present.
-
-
(a)
-
4.
Radiological investigations. Radiological investigations refer to the radiological assessment of the mediastinum. More than 75% of generalized MG patients who test positive for Ab antiAchR show abnormalities of the thymus. In about 85% of cases thymic hyperplasia and in 15% of cases thymic tumors, mainly thymoma [35].
A chest CT scan with contrast or magnetic resonance imaging for mediastinal evaluation is part of the diagnostic workout in patients with myasthenia gravis. There are no controlled litterature study comparing the two methods, in order to determine which is the most suitable for the diagnosis of thymoma and thymic hyperplasia.
In case of doubts in radiological results antitina/ryanodine antibody testing may be usefull, as they are associated with the presence of thymoma.
-
5.
Collateral investigations: autoimmune pathologies of the thyroid are quite frequently associated with MG (3–8% of cases), dosing TSH and antithyroid antibodies is therefore useful, regardless of the presence of symptoms attributable to thyroid dysfunction. Furthermore, it can be associated with many other autoimmune diseases, but the need for further investigation in the field of autoimmunity will be dictated by the clinical features of each patient.
Recommendations on the use of diagnostic tests:
-
Clinical data is of great importance, particularly the association between weakness, fatigability, and fluctuation of symptoms (in the absence of muscle atrophy and preserved tendon reflexes) are a crucial aspect in guiding diagnostic suspicion;
-
The dosage of specific autoantibodies has the highest sensitivity and specificity in generalized MG; their positivity can make the neurophysiological assessment unnecessary;
-
In patients showing generalized symptoms and negativity for Ab antiAchR dosage, repetitive stimulation should be performed, and antiMuSK antibodies should be dosed; in patients with seronegative ocular MG and a proceed with a SFEMG test, which has the highest sensitivity;
-
Dosage of acetylcholine antireceptor antibodies has a purely diagnostic use, and variations in antibody titer is not usefull for therapeutic approach.
-
5 Differential Diagnosis
The main differential diagnoses in Myasthenia Gravis, keeping in consideration both the purely ocular form and the generalized form, include:
-
Thyroid ophtalmopathy
-
Eaton-lambert myasthenic syndrome
-
Myopathies with involvement of the ocular area (oculopharyngeal dystrophy, myotonic dystrophy, mitochondrial myopathies with or without progressive ophthalmoplegia);
-
Acute poliradicolonevritis;
-
Motor neuron diseases;
-
Alteration of one or more cranial nerves;
-
Encephalic trunk diseaes;
-
Organophosfate poisoning, botulism;
-
Congenital myasthenic syndrome;
-
Myasthenia induced by penicillamine.
6 Therapy
There are several treatment stategies in the management of patients with Myasthenia Gravis. The therapeutic approach follows some basic principles, but it is largely individualized according to the clinical characteristics of the patient. Therapy is generally decided on the basis of age, severity of illness, and possible involvement of the bulbar and/or respiratory system.
Neither the autoantibody titers, nor the entity of decremental response of RNS influence the therapeutic approach, that is clinically determined.
The available therapeutic strategies are:
-
1.
Anti-acetylcholinesterase drugs (AntiAChE);
-
2.
Immunosuppressive therapy;
-
3.
Immunomodulatory therapies;
-
4.
Surgical treatment (thymectomy).
-
1.
AntiAChE drugs play a purely symptomatic role; they inhibit the metabolism of acetylcholine thus increasing its availability at the neuromuscular junction, facilitating the link with the specific receptor, and favoring muscle contraction [36].
There are no randomized controlled trials on the use of antiAChEs, but individual case studies and clinical experience have demonstrated a proven effectiveness. The duration of action is maximal within 2–3 h, for this reason they must be administered repeatedly during the day. AntiAChEs usually represent the first line of treatment, also because of their relative safety and ease of use.
Pyridostigmine (Mestinon) is the usual choice; neostigmine is commercially available but it is not generally used.
It is important to note that:
-
Most of the patients who suffer from generalized MG, at least in the initial phase, show good/excellent clinical response to AntiAChEs;
-
In purely ocular MG forms, the patients’ response is often unsatisfactory or completely absent;
-
The response to AntiAChEs can vary in different muscle groups; the dosage must therefore be adjusted according to the relative importance of the most compromised and functionally relevant regions;
-
The failure to find a significant clinical effect does not justify a progressive increase of dosage, but suggest that an immunosuppressive treatment must be started.
The dosages used range from 60 to 120 mg in four administrations per day (every 3.5–4 h). There is also a 180 mg modified-release form which can be administered before going to bed when it is necessary to reduce fatigue on awakening.
Side effects: muscarinic (diarrhoea, upset stomach, increased bronchial secretions and saliva) occur most frequently. Particular attention must be paid to the increase in bronchial secretions and drooling in patients who already have difficulty swallowing and wheezing. Increasing the dose in an attempt to reduce muscle deficiency is not recommended for these patients. Nicotinic-type side effects can include muscle cramps and twitching and more rarely an accentuation of muscle weakness (cholinergic crisis, difficult to observe at commonly used doses).
-
-
2.
Immunosuppressive therapy.
Immunosuppressive drugs are necessary for patients who are symptomatic despite treatment with pyridostigmine, or whose symptoms return after a temporary response to pyridostigmine. The choice of immunosuppressive drug type is based on considerations that take into account the clinical picture, the speed of the drug’s action, its side effects, the patient’s comorbidities. Here below we report the levels of evidence and recommendation regarding each form of treatment, followed by some general considerations.
-
(a)
Corticosteroids:
Remission or improvement of the clinical picture is reported in 70–80% of myasthenic patients treated with corticosteroids. Observational studies and clinical experience support the efficacy of glucocorticoids in the treatment of myasthenia gravis. Limited evidence from randomized, controlled trials likewise suggests that glucocorticoid treatment offers significant short-term benefit in MG compared with placebo. [36,37,38,39].
Indications:
-
Patients with generalized or bulbar myasthenia;
-
Patients with purely ocular disabling forms;
-
Myasthenic crisis.
Prednisone (0.75–1 mg/kg daily) is administered in a single dose in the morning; this dosage is maintained up to the maximum clinical improvement achievable (average within 2 months) [38]. In purely ocular forms, a starting dose of 25–50 mg/day is quickly effective in most cases.
Glucocorticoid tapering can be done with the final goal of achieving either a daily or alternate-day regimen. We usually reduce the dose of 10% every 6–8 weeks. The same diagram also applies to purely ocular forms. It is important to note that the start of steroid treatment in patients with generalized MG—especially in those with bulbar impairment—requires hospitalization, given the possible clinical deterioration that could significantly impair chewing and swallowing and could worsen respiratory failure; in these patients it is useful to associate treatment with plasmapheresis or immunoglobulin (see below).
Ocular MG does not require hospitalization. The most frequent side effects related to steroid therapy are cushingoid appearance, cataracts, weight gain, metasteroid diabetes, hypertension and osteoporosis. The patients rarely develop mental disorders. If the glucocorticoids cannot be tapered below a reasonably acceptable level without recurrence of symptoms, or if the patient does not respond satisfactorily, then other immunotherapeutic agents are usually needed, either to supplant the glucocorticoids or as a “glucocorticoid-sparing” agent.
Some recommendations are important for patients treated with steroids, in particular:
-
Follow a diet poor in sodium and carbohydrates;
-
Check bood pressure periodically;
-
Check glycemic balance periodically;
-
Perodically check ocular tension and transparency of the lens;
-
Undergo an annual Computed Bone Mineralometry (in the spine) in order to assess bone mineralization
-
Establish a preventive therapy for osteoporosis with calcium, bisphosphonates and Vitamin D.
-
-
(b)
Immunosuppressive drugs:
Indications:
-
Poor effectiveness of steroids, frequent clinical relapses;
-
Need for a drastic reduction of the steroid dosage because of major side effects or in patients who have contraindications to high-dose steroids;
Azathioprine:
Azathioprine is the most frequently immunosuppressant used in the treatment of MG; the drug is metabolized at 6-mercaptopurine, which inhibits the synthesis of DNA and RNA and interferes with the function of T-lymphocytes. A randomized controlled trial has demonstrated the efficacy of azathioprine as a steroid-sparing agent [40] and clinical studies support its efficacy [41, 42]. It is important to highlight the slowness of the drug’s action, which should be administered for at least 1 year before establishing effectivness. Therefore this drug cannot be considered useful for quickly reducing neurological deficits. Azathioprine is administered at a dosage of 2.5–3 mg/kg per day, in 2–3 doses. The treatment must be started gradually with 50 mg daily for the first week and increased to 50 mg every week until the required dose is achieved, according to the patient’s weight. The drug should be administered on a full stomach to prevent gastric intolerance. It is important to check haematology and liver function every week at the beginning of treatment, then once a month when the required dose has been reached; blood tests should be performed periodically throughout the duration of the treatment in order to promptly detect the onset of toxicity.
The most frequent side effects are: gastric intolerance, myelosuppression and hepatotoxicity. There are no studies that provide clear information on duration of treatment with azathioprine after obtaining clinical remission or a satisfactory clinical improvement; discontinuation of the drug is therefore a decision to be assessed case by case, bearing in mind that it is possible to observe reactivation of the disease after discontinuation [43].
Cyclophosphamide
Cyclophosphamide is an alkylating agent, with a strong immunosuppressive action on T and B lymphocytes. Evidence of its effectiveness derives from a controlled study of 23 myasthenic patients [44]. Cyclophosphamide is an alternative to azathioprine in the following cases: when azathioprine is ineffective after an adequate evaluation period (at least 1 year); when there is azathioprine intolerance; when it is necessary to establish an immunosuppressive treatment because of serious contraindications to the use of high-dose prednisone or the appearance of serious side effects from steroid such as to require a rapid reduction; when there is prolonged refractoriness to combined treatment (steroid+another immunosuppressive therapy).
An important limitation of the drug derives from its major side effects, especially from its effect on the reproductive system in terms of infertility.
This drug can be administered orally at a dose of 2.5–3 mg/kg/day (split into two to three doses), or alternatively it can be administered in bolus i.v. monthly, at a dose of 0.750–1 g/m2.
Similarly to what happens with azathioprine, treatments with this drug should be delivered gradually, blood and urine tests should be carried out regularly in order to check possible occurrence of hemorrhagic cystitis: in this respect it is necessary to force diuresis by increasing the daily fluid intake and associating the administration of acetylcysteine two to three times a day in order to protect the bladder mucosa.
Side effects include alopecia, nausea and vomiting; hemorrhagic cystitis, infertility, amenorrhea. As far as haematology is concerned, the same considerations made for azathioprine are valid.
Cyclosporine
Cyclosporine is an immunosuppressant that reduces the production of IL-2, inhibits the function of T-helper lymphocytes and dampens T lymphocyte-dependent immune responses. A randomized controlled study on 20 MG patients showed that cyclosporine was effective in improving clinical score compared to placebo [45–46]. These conclusions were also reached by open and retrospective studies [47].
However, the incidence of significant side effects should be emphasized, in particular nephrotoxicity and blood hypertension. Cyclosporine is considered a third line drug and its administration is limited to patients who have not responded to treatment with azathioprine, mycophenolate, and cannot be treated with cyclophosphamide. The dosage is 3 mg/kg/day (minimum dose, taking into account that with higher doses of 5–6 mg/kg/day there is an increased risk of renal toxicity). There should be periodic monitoring of blood urea nitrogen, creatinine, creatinine clearance and blood pressure.
Mycophenolate Mofetil
Mycophenolate mofetil with its active metabolite (mycophenolic acid) is an inhibitor of purine nucleotide synthesis and has a selective effect on proliferating lymphocytes.
Some open label clinical studies [48, 49] and two retrospective studies [50, 51] suggest the potential efficacy and steroid-sparing effect of mycophenolate. However, these results were not confirmed by three randomized clinical trials [52,53,54]. Mycophenolate is indicated in patients who have not responded to treatment with azathioprine.
The recommended dosage is 2 g/day, divided into two doses. The drug is generally well tolerated and the most common adverse side effects are gastrointestinal, mostly nausea or diarrhea. It is necessary to wait at least 5 months before a clinical benefit can be observed.
Methotrexate
Methotrexate is an immunosuppressive agent that reduces the synthesis of purines and pyrimidines and interferes with DNA synthesis. A recent single-blind trial [55] proposed methotrexate as an alternative to azathioprine; further prospective studies are needed to confirm this finding.
Tacrolimus (FK506)
Tacrolimus is is an immunosuppressive macrolide molecule similar in action to cyclosporine; indeed it acts on T lymphocyte proliferation by inhibiting the activated pathway mediated by calcineurin. Tacrolimus is less neprotoxic than cyclosporine. In a number of uncontrolled studies, tacrolimus has been used successfully to treat MG at low doses (generally 3 to 8 mg/day) [56, 57]. It should be considered when there is no response to azathioprine, or as an alternative to mycophenolate or cyclophosphamide. The most common side effects include hyperglycemia, hypomagnesemia, tremors and numbness.
Rituximab
Rituximab is a monoclonal antibody against CD20 positive B cells. There is no randomized trial evidence regarding the effectiveness of rituximab in MG. However, a large and growing number of case series support its use in patients with refractory myasthenia gravis [58, 59]. Some of these studies suggest its efficacy in MuSK positive patients [60, 61].
Etanercept
Etanercept is a recombinant protein consisting of the receptor for tumour necrosis factor (TNF) joined to the Fc portion of human IgG1. This drug binds TNF and blocks its interaction with the receptor on the cell surface. Experimental models of autoimmune myasthenia have shown that blocking TNF-alpha, a proinflammatory cytokine, suppresses the disease. A pilot study lasting various months used the drug in 8 MG patients. Seven of them improved and one of them deteriorated [62]. Further studies are needed to better define the safety and potential effectiveness of the drug in MG.
-
-
(a)
-
3.
Immunomodulatory therapy. The aim of immunomodulatory treatments used in MG is to obtain rapid clinical improvement, especially in patients with bulbar involvement.
These treatments include: (a) plasmapheresis, and (b) high-dose intravenous immunoglobulin.
Indications:
-
Serious bulbar or generalized forms, especially in rapid clinical deterioration;
-
Treatment of myasthenic crises;
-
Insufficient response to ongoing immunosuppressive therapy;
-
Clinical deterioration at the start of steroid therapy;
-
Period of non-effectiveness of immunosuppressive therapy;
-
Preparation for thymectomy (in patients with severe or generalized bulbar forms).
Therapeutic Apheresis
Plasmapheresis: The rationale of plasmapheresis lies in the rapid removal of circulating antibodies (by centrifugation or membrane filtration), with the aim of obtaining rapid clinical improvement. The clinical response occurs over a period of days and continues for 4–6 weeks. There are no adequate RCT studies on the effectiveness of plasmapheresis, but many case series have documented the short-term efficacy in MG, particularly in patients with myasthenic crisis [63, 64].
Experts believe that the proposal of controlled trials is unethical in MG and thus plasmapheresis is recommended as a short-term treatment. As shown in the Cochrane review [64], there is no uniformity in the apheretic protocols adopted in the available studies, particularly regarding the number of sessions, the characteristics of patients included in the studies, and evaluation methods. It is important to note that:
-
There are no clinical parameters predictive of clinical efficacy of plasmapheresis in the individual patient;
-
There is no correlation between autoantibody titer and effectiveness of plasmapheresis;
-
Positivity of the acetylcholine antireceptor antibody titer is not a necessary requirement to indicate the procedure;
-
Plasmapheresis should be considered, in most cases, as an “acute” treatment which is useful for temporarily resolving the neurological deficit;
-
The minority of patients who do not respond to immunosuppressive therapy (assessed for an appropriate period) may benefit from apheretic treatments repeated at regular intervals. In this regard the Cochrane review points out that there are no RCT trials regarding long-term outcome of MG patients treated with plasmapheresis [64].
Assessment and preparation of patient for apheresis treatment:
-
1.
Verification of clinical indication according to strictly neurological criteria (medium to severe myasthenic patients, especially with a deficit of bulbar muscles, respiratory failure patients, intensive care patients);
-
2.
Internist’s evaluation in order to rule out any contraindications, in particular cardiovascular diseases, coagulation deficit, ongoing anticoagulation therapy, concomitant infectious processes;
-
3.
Evaluation of vascular access in order to predict the need for central access
-
4.
Informing the patient about the procedure and obtaining an informed consens.
-
Side effects: Hypotension and/or bradycardia, bleeding complications, possible onset of general perioral numbness or cramping due to temporary hypocalcemia induced by citrate. Multiple plasmapheresis cycles can lead to inadequate peripheral venous access leading to the need for central access; chronic catheter-related complications such as infection and thrombosis may occur.
IgG selective immunoadsorption is a plasma treatment technique which can selectively remove IgG immunoglobulins (and thus autoantibodies which are relevant from a pathogenic viewpoint). This method involves the use of a) filters containing protein A derived from staphylococcal wall or b) filters containing sheep polyclonal anti-human IgG.
High-dose intravenous immunoglobulin. Several open studies have supported the short-term effectiveness of immunoglobulin administered intravenously at high doses; the main ongoing studies are also re-evaluated in the Cochrane review on the topic [65]. Two randomized studies comparing immunoglobulin and plasmapheresis did not show a significant difference in the effectiveness of the procedures within 15 days of their administration [63, 66]. Immunoglobulins have the same rationale and indications mentioned above for therapeutic plasmapheresis. They represent a useful alternative where there are inadequate vascular accesses and where there are cardiovascular-related contraindications to plasmapheresis.
Administration schedule: 2 g/kg total dose, administered in 2–5 days. It is worth pointing out that, especially in patients with severe bulbar disorders, plasmapheresis seems to provide faster clinical improvement compared to immunoglobulins.
Side effects: immunoglobulins are generally well tolerated. Patients frequently report headaches, which tend to reduce by slowing the rate of administration. The most serious effects, though rare, include the possible occurrence of aseptic meningitis, thrombotic phenomena, anaphylaxis, blood hyperviscosity. Particular attention should be paid to the administration of high doses of immunoglobulins in patients with pre-existing cardiovascular diseases, kidney failure, paraproteinemia. As already noted for plasmapheresis, in selected patients refractory to conventional therapy, an approach with periodic administration of immunoglobulins should be offered; however, this practice has not been validated by controlled studies.
-
4.
Surgical therapy: thymectomy
Thymectomy aims at removing the potential source of origin and/or maintenance of the autoimmune process underlying the disease. Pending the results of the MGMTX trial [67], there are no RCT studies that unequivocally demonstrate the effectiveness of thymectomy. This is a randomized, multicenter comparative study between thymectomy versus no surgery in severe generalized Ab antiAchR positive patients treated with steroid therapy.The role of thymectomy and its impact on the natural history of the disease was reviewed by an ad hoc Task Force sponsored by the Myasthenia Gravis Foundation of America (MGFA) [68]. The literature taken into consideration was not homogeneous in regard to clinical evaluation methods, associated therapies and especially the definition clinical remission. This systematic analysis led to the following conclusions:
-
Detection of a higher relative median remission rate for operated patients compared to non-operated patients;
-
Presence of bias in all studies, with different prognostic variables regarding the basic characteristics of the patients in the study populations;
-
Positive association between thymectomy and improved outcome after univariate analysis (taking into account individual variables such as gender, age and severity of disease;
-
Absence of clearcut evidence of improvement after thymectomy inmultivariate analysis.
-
Hence, the authors “are not able to determine from available studies whether the association found between thymectomy and clinical improvement is due to thymectomy or if it is simply the result of differences in the basic characteristics between operated and non-operated patients”. On this basis, the authors conclude that for patients with autoimmune myasthenia without thymoma, thymectomy can be recommended as an option to increase the likelihood of improvement or remission.
Thymectomy currently has the following indications:
-
(a)
Patients with radiologic evidence of thymic enlargement (especially in case of thymomas), regardless of age at onset of the disease;
-
(b)
Patients suffering from generalized and/or bulbar forms, even without radiological signs of thymic enlargement with onset in middle age (<60 years) or at a young age;
-
(c)
There is yet insufficient evidence to justify thymectomy in myasthenia patients with late onset and no signs of thymic enlargement;
-
(d)
Data concerning thymectomy in seronegative patients with antiMuSK antibodies are still limited; usually these patients did not present thymic pathology, however some cases associated with thymic hyperplasia have been reported in literature [69].
It is important to remember that thymectomy is not an urgent therapeutic procedure and the patient should therefore undergo surgery in the best clinical conditions to avoid any possible post-surgical deterioration.
The objective of thymectomy is to ensure the greatest possible removal of thymic tissue: in addition to the thymus, the adipose mediastinal and cervical tissue must be removed as it may contain thymic tissue islands.
There are four different surgical procedures: transcervical thymectomy, transternal thymectomy, combined thymectomy (transternal + transcervical) and minimally invasive thymectomy (video-assisted or robotic-assisted).
All methods ensure the removal of the thymus; the difference lies in the amount of adipose perithymic tissue removed.
The video-thoracoscopic enlarged thymectomy (VATET) was introduced some years ago. This technique enables wide visualization and exploration of the mediastinal space and the removal, in addition to the thymus, of the adipose tissue from the pericardium to the thyroid, without sternotomy. This method is much less invasive and better tolerated by the patient than the traditional technique [70].
A minimally invasive thymectomy can be proposed as the method of choice in all patients with a radiologically normal thymus, or with an enlarged thymus due to hyperplasia; in cases of thymoma the methodology should be discussed case by case considering the size of the lesion and its radiological features [70].
7 Myasthenic Crisis
7.1 Definition
Myasthenic crisis is a life-threatening condition that involves a rapid deterioration of neuromuscular function with respiratory failure and severe impairment of bulbar innervated muscles. It is a critical condition that requires hospitalization in an intensive care unit (ICU) and may lead to intubation and mechanical ventilation.
7.2 Epidemiology
Approximately 10–20% of myasthenic patients experience at least one myasthenic crisis during the course of the disease [71], the annual risk of developing a crisis being approximately 2–3% [72]. In 15–20% of patients a myasthenic crisis may be the first manifestation of the disease [72]. Most myasthenic crises occur in the first years after diagnosis, when the disease is in its most active phase.
7.2.1 Clinical Picture
Patients who develop a myasthenic crisis generally experience an exacerbation of weakness and fatigability of bulbar and limb muscles before the crisis. In some cases however, patients may show respiratory failure which is disproportionate in comparison with bulbar/generalized symptoms and more rarely, respiratory failure may be the only clinical manifestation [73].
A myasthenic crisis can be determined by several precipitating factors including inflammatory/infectious processes, surgery, pregnancy, breastfeeding, reduction of immunosuppressive therapy. Several drugs may also interfere with neuromuscular transmission and are considered precipitating factors. Finally, in some casesa myasthenic crisis occurs spontaneously as part of the natural history of the disease.
7.3 Management of Myasthenic Crisis
The approach to myasthenic crisis involves:
-
Admission to ICU;
-
Evaluation of swallowing and placement of nasogastric tube;
-
Evaluation of respiratory function and elective intubation if the clinical evaluation or pulmonary function tests indicate respiratory insufficiency;
-
Immunomodulatory treatment (plasmapheresis or immunoglobulin) which will be associated (or
modified if already in progress) with an immunosuppressive treatment. The severity of the clinical picture generally imposes the start of steroid treatment or its increase up to full dose as described above.
-
Identification and treatment of any precipitating factors (e.g. intercurrent infectious processes, contraindicated drugs …)
-
Start weaning off mechanical ventilation when lung function has improved and only after starting treatment with plasmapheresis or immunoglobulin.
Evaluation of respiratory function is based both on clinical signs/symptoms and on respiratory muscle function tests [74,75,76]. Clinical signs or symptoms of respiratory failure are breathlessness, hypophonia, increased respiratory rate, involvement of accessory respiratory muscles, abdominal flail chest.
Vital capacity (VC) and maximum inspiratory pressure (MIP) are the main parameters used to monitor the strength of respiratory muscles. VC reflects the mechanical function of both the inspiratory and expiratory muscles; it is assessed by asking the patient to take a big breath and then exhale forcefully into a spirometer.
On the other hand MIP provides information on the inspiratory force; it is assessed by asking the patient to inhale against a closed valve and measuring the pressure that is generated at mouth level.
Typically, intubation is recommended when VC drops below 15–20 mL/kg and MIP is less than—30 cm H2O; in the presence of clinical signs of respiratory distress; in the case of metabolic acidosis or ineffective removal of secretions. [74,75,76].
In intubated patients it is preferable to suspend anticholinesterases medications in order to avoid excessive secretions.Plasmapheresis and intravenous immunoglobulins are used in the treatment of myasthenic crisis in order to achieve rapid improvement of the clinical picture: the effectiveness of these treatments is however limited in time (3–4 weeks); it is therefore necessary to add appropriate steroid treatment.
The rationale for plasmapheresis lies in the rapid removal (by centrifugation or membrane filtration) of circulating antibodies. As shown in the Cochrane review on the subject [64] there is no uniformity in apheretic protocols. One classic scheme provides five exchanges (3–5L of plasma each) in 7 or 14 days. Although done daily in some circumstances, exchanges done every other day are probably more effective in reducing the antibody levels due to the time it takes for the extravascular immunoglobulin to re-equilibrate after each plasma exchange. Intravenous immunoglobulins, like plasmapheresis, allow rapid clinical improvement. The total administered dose is 2 g/kg in 2–5 days.
There are no randomized studies that have compared plasmapheresis or intravenous immunoglobulins with placebo in the treatment of myasthenic crisis. On the contrary a randomized controlled trial [66, 77] showed a comparable efficacy for plasmapheresis and intravenous immunoglobulins in case of myastenic crisis. After 2 weeks a similar number of patients improved in both groups. Although at 2 weeks a larger number of patients in the immunoglobulin group (17.5%) had a QMGS score (quantitative myasthenia gravis score) which was worse than patients treated with plasmapheresis (2%), this difference was not significant. This data is also confirmed by a systematic review on the subject [65].
Many experts, however, prefer plasmapheresis as first line treatment, since it is more rapid in determining clinical improvement. Others, by contrast, prefer intravenous immunoglobulins because they are easier to administer and with lower incidence of severe side effects. The decision to start weaning off mechanical ventilation should be individualized for each patient. Generally, in the myasthenic patient, some spontaneous breathing trials should be attempted:
-
After the patient begins treatment with plasmapheresis/;
-
When there is evidence of improvement in respiratory muscle strength (CV > 15–20 mL/kg, MIP more negative than—30 cm H2O);
-
When the secretions are manageable;
-
With adequate cough.
The complications most commonly associated with myasthenic crisis are fever, infections (pneumonia, bronchitis, urinary tract infection, sepsis), deep venous thrombosis, heart failure, cardiac arrhythmias and cardiac arrest.
Improved therapies and intensive treatments have dramatically improved the prognosis of myasthenic crisis, whose mortality rate has decreased from 75% in the 1950s to 5% in the 1990s [78].
8 General Scheme for Treating the Myasthenic Patient
-
1.
The initial treatment involves the use of antiAChE; in case of poor response, especially in cases with bulbar signs, it is worth starting a steroid treatment; antiAChE is very quickly effective, so if it does not have an effect, steroid/immunosuppressive therapy must be prescribed prematurely;
-
2.
If there is a clinical relapse, if the steroid is ineffective, or if major side effects appear, consider the combined therapy of steroid + immunosuppressant (first choice: azathioprine);
-
3.
The presence of thymoma represents an absolute indication to thymectomy; stabilize the patient with proper treatment using antiAChE and/or immunosuppressive drugs before surgery; do not perform urgent surgery.
-
4.
Except for patients with thymoma, thymectomy is recommended for patients with disease onset in middle age or at a young age; there are no data to support the efficacy of thymectomy in patients with exclusively bulbar myasthenia; if possible, consider a minimally invasive technique;
-
5.
Plasmapheresis and immunoglobulins represent an emergency therapeutic options for patients with serious generalized and/or bulbar forms (including patients already on assisted ventilation, for whom the time spent in intensive care can be reduced); in rare cases refractory to drug therapy, a chronic treatment with plasmapheresis, intravenous immunoglobulin or selective immunoadsorption should be used.
-
6.
In patients with partial or total respiratory failure consider, apart from mechanical ventilation, the early use of immunomodulatory therapy (plasmapheresis and high-dose immunoglobulins) in conjunction with immunosuppressive therapy at full doses;
-
7.
Instruct the patient to come to regular follow-up outpatient visits, especially patients receiving chronic steroid treatment, so as not to prolong every single current dose;
8.1 Drugs Contraindicated in Myasthenic Patients
Aminoglycoside antibiotics, antiarrhythmic drugs belonging to quinidines and beta-blockers, curare drugs and others releasing non-depolarizing substances, can directly interfere with neuromuscular transmission and thus cause deterioration of the disease.
Because of their muscle relaxant action, benzodiazepines can accentuate an existing ventilatory defect. Clinical deterioration was also reported during treatment with chloroquine and penicillamine, which can induce the synthesis of specific antibodies as well as the disease in susceptible individuals (symptomatology ceases several weeks after discontinuation of the drug). There is no contraindication to local anesthesia.

Algorithm for the early management of myasthenia gravis exacerbation.
References
Drachman DB (1994) Myasthenia gravis. N Engl J Med 330:1797–1810
Vincent A (2002) Unraveling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2:797–804
Meriggioli MN, Sanders DB (2009) Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 8(5):475–490
Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A (2001) Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7:365–368
McConville J, Farrugia ME, Beeson D et al (2004) Detection and characterization of musk antibodies in seronegative myasthenia gravis. Ann Neurol 55(4):580–584
Zhang B, Tzartos JS, Belimezi M et al (2012) Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 69:445–451
Cossins J, Belaya K, Zolowska K et al (2012) The search for new antigenic targets in myasthenia gravis. Ann N Y Acad Sci 1275:123–128
Carr AS, Cr C, McCarron PO et al (2010) A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol 18(10):46
Engel AG (1994) Acquired autoimmune myasthenia gravis. In: Engel AG, Franzini-Armstrong C (eds) Myology. McGraw-Hill, New York, pp 1769–1797
Karlin A, Akabas MH (1995) Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron 15(6):1231
Drachman DB, Adams RN, Josifek LF, Self SG (1982) Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 307:769
Hohlfeld R, Wekerle H (2008) Reflections on the “intrathymic pathogenesis” of myasthenia gravis. Neuroimmunology 201–202:21–27
Liyanage Y, Hoch W, Beeson D, Vincent A (2002) The agrin/muscle-specific kinase pathway: new targets for autoimmune and genetic disorders at the neuromuscular junction. Muscle Nerve 25(1):4
Ohta K, Shigemoto K, Kubo S et al (2004) MuSK antibodies in AChR Ab-seropositive MG vs AChR Ab-seronegative MG. Neurology 62:2132
Ghazanfari N, Fernandez KJ, Murata Y, Morsch M, Ngo ST, Reddel SW, Noakes PG, Phillips WD (2011) Muscle specific kinase: organiser of synaptic membrane domains. Int J Biochem Cell Biol 43(3):295–298
Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, Beeson D, Willcox N, Vincent A (2008) IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 131:1940
Deymeer F, Gungor-Tuncer O, Yilmaz V, Parman Y, Serdaroglu P, Ozdemir C, Vincent A, Saruhan-Direskeneli G (2007) Clinical comparison of anti-MuSK- vs anti-AChR-positive and seronegative myasthenia gravis. Neurology 68(8):609
Higuchi O, Hamuro J, Motomura M, Yamanashi Y (2011) Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69(2):418–422
Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, Melms A, Kröger S (2012) Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 259(3):427
Hoosterhuis HJGH (1997) Clinical aspects and epidemiology. In: Myasthenia gravis. Groningen Neurological Press, Groningen, pp 17–48
Grob D, Brunner N, Namba T, Pagala M (2008) Lifetime course of myasthenia gravis. Muscle Nerve 37(2):141
Mantegazza R, Beghi E, Pareyson D, Antozzi C, Peluchetti D, Sghirlanzoni A et al (1990) A multicenter follow-up study of 1152 patients with myasthenia gravis in Italy. J Neurol 237:339–344
Osserman KE, Jenkins G (1971) Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med 38:497–537
Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America, Jaretzki A III, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, Sanders DB (2000) Myasthenia gravis. Recommendations for clinical research standards. Neurology 55:16–23
Sethi KD, Rivner MH (1987) Swift TR Ice pack test for myasthenia gravis. Neurology 37(8):1383
Golnik KC, Pena R, Lee AG, Eggenberger ER (1999) An ice test for the diagnosis of myasthenia gravis. Ophthalmology 106(7):1282
Vincent A, Newsom-Davis J (1985) Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry 48:1246–1252
Choi Decroos E, Hobson-Webb LD, Juel VC, Massey JM, Sanders DB (2014) Do acetylcholine receptor and striated muscle antibodies predict the presence of thymoma in patients with myasthenia gravis? Muscle Nerve 49(1):30–34
Vincent A, McConville J, Farrugia ME, Newsom-Davis J (2004) Seronegative myasthenia gravis. Semin Neurol 24(1):125
Caress JB, Hunt CH, Batish SD (2005) Anti-MuSK myasthenia gravis presenting with purely ocular findings. Arch Neurol 62(6):1002
Baggi F, Andreetta F, Antozzi C, Simoncini O, Confalonieri P, Labeit S, Cornelio F, Mantegazza R (1998) Anti-titin and antiryanodine receptor antibodies in myasthenia gravis patients with thymoma. Ann N Y Acad Sci 841:538–541
Yamamoto AM, Gajdos P, Eymard B, Tranchant C, Warter JM, Gomez L et al (2001) Anti-Titin antibodies in myasthenia gravis. Right association with thymoma and heterogeneity of nonthymomatous patients. Arch Neurol 58:885–890
Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D (1992) Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 15(6):720
AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine (2001) Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve 24(9):1239
Castleman B (1966) The pathology of the thymus gland in myasthenia gravis. Ann N Y Acad Sci 135(1):496
Rowland LP (1980) Controversies about the treatment of myasthenia gravis. J Neurol Neurosurg Psychiatry 43:644–659
Pascuzzi RM, Coslett HB, Johns TR (1984) Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 15:291–298
Sghirlanzoni A, Peluchetti D, Mantegazza R, Fiacchino F, Cornelio F (1984) Myasthenia gravis: prolonged treatment with steroids. Neurology 34:170–174
Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR (2005) Coticosteroids for myasthenia gravis. Cochrane Database Syst Rev (2):CD002828
Palace J, Newsom-Davis J, Lecky B, The Myasthenia Gravis Study Group (1998) A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 50:1778–1783
Witte AS, Cornblath DR, Parry GJ, Lisak RP, Schatz NJ (1984) Azathioprine in the treatment of myasthenia gravis. Ann Neurol 15:602–605
Mantegazza R, Antozzi C, Peluchetti D, Sghirlanzoni A, Cornelio F (1988) Azathioprine as a single drug or in combination with steroids in the treatment of myasthenia gravis. J Neurol 235:449–453
Hohlfeld R, Toyka K, Besinger UA, Gerhold B, Heininger K (1985) Myasthenia gravis: reactivation of clinical disease and of autoimmune factors after discontinuation of long term azathioprine. Ann Neurol 17:238–242
De Feo LG, Schottlender J, Martelli NA, Molfino NA (2002) Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravis. Muscle Nerve (1):31–36
Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G (1987) Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 316:719–724
Tindall RSA, Phillips JT, Rollins JA, Wells L, Hall K (1993) A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci 681:539–551
Ciafaloni E, Nikhar NK, Massey JM, Sanders DB (2000) Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology 55:448–450
Ciafaloni E, Massey JM, Tucker-Lipscomb B, Sanders DB (2001) Mycophenale mofetil for myasthenia gravis. Neurology 56:97–99
Chaudhry V, Cornblath DR, Griffin JW, O’Brien R, Drachman DB (2001) Mycophonelate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology 56:94–96
Meriggioli MN, Ciafaloni E, Al-Hayk KA, Rowin J, Tucker-Lipscomb B, Massey JM, Sanders DB (2003) Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 61:1438–1440
Hehir MK, Burns TM, Alpers J et al (2010) Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 41:593
Sanders DB, Hart IK, Mantegazza R et al (2008) An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 71:400
Muscle Study Group (2008) A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 71:394
Meriggioli MN, Rowin J, Richman JG, Leurgans S (2003) Mycophenolate mofetil for myasthenia gravis: a double-blind, placebo-controlled pilot study. Ann N Y Acad Sci 998:494
Heckmann JM, Rawoot A, Bateman K et al (2011) A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 11:97
Konishi T, Yoshiyama Y, Takamori M, Yagi K, Mukai E, Saida T, Japanese FK506 MG Study Group (2003) Clinical study of FK506 in patients with myasthenia gravis. Muscle Nerve 28:570–574
Yoshikawa H, Mabuchi K, Yasukawa Y, Takamori M, Yamada M (2002) Low-dose tacrolimus for intractable myasthenia gravis. J Clin Neurosci 9(6):627–628
Lebrun C, Bourg V, Tieulie N, Thomas P (2009) Successful treatment of refractory generalized myasthenia gravis with rituximab. Eur J Neurol 16:246
Zebardast N, Patwa HS, Novella SP, Goldstein JM (2010) Rituximab in the management of refractory myasthenia gravis. Muscle Nerve 41:375
Stein B, Bird SJ (2011) Rituximab in the treatment of MuSK antibody-positive myasthenia gravis. J Clin Neuromuscul Dis 12:163
Díaz-Manera J, Martínez-Hernández E, Querol L et al (2012) Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 78:189
Rowin J, Meriggioli MN, Tüzün E et al (2004) Etanercept treatment in corticosteroid-dependent myasthenia gravis. Neurology 63:2390
Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C, for the Myasthenia Gravis Clinical Study Group (1997) Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol 41:789–796
Gajdos P, Chevret S, Toyka K (2002) Plasma exchange for myasthenia gravis. Cochrane Database Syst Rev (4):CD002275
Gajdos P, Chevret S, Toyka K (2012) Intravenous immunoglobulins for myasthenia gravis. Cochrane Database Syst Rev (12):CD002277
Barth D, Nouri MN, Ng E et al (2011) Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 76(23):2017–2023
Wolfe GI, Kaminski HJ, Jaretzki A et al (1998) Development of a thymectomy trial in nonthymomatous myasthenia gravis patients receiving immunosuppressive therapy. Ann N Y Acad Sci 998:473–480
Gronseth GS, Barohn RJ (2000) Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 55:7–15
Lauriola L, Ranelletti F, Maggiano N, Guerriero M, Punzi C, Marsili F, Bartoccioni E, Evoli A (2005) Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology 64:536–538
Mantegazza R, Baggi F, Bernasconi P, Antozzi C, Confalonieri P, Novellino L, Spinelli L, Ferrò MT, Beghi E, Cornelio F (2003) Video-assisted thoracoscopic extended thymectomy and extended transternal thymectomy (T-3b) in non-thymomatous myasthenia gravis patients: remission after 6 years of follow-up. J Neurol Sci 212:31–36
Wendell LC, Levine JM (2011) Myasthenic crisis. Neurohospitalist 1(1):16–22
Berrouschot J, Baumann I, Kalischewski P, Sterker M, Schneider D (1997) Therapy of myasthenic crisis. Crit Care Med 25(7):1228–1235
Mier A, Laroche C, Green M (1990) Unsuspected myasthenia gravis presenting as respiratory failure. Thorax 45(5):422
Juel VC (2004) Myasthenia gravis: management of myasthenic crisis and perioperative care. Semin Neurol 24(1):75
Jani-Acsadi A, Lisak RP (2007) Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 261(1-2):127–133
Chaudhuri A, Behan PO (2009) Myasthenic crisis. QJM 102(2):97–107
Bril V, Barnett-Tapia C, Barth D et al (2012) IVIg and PLEX in the treatment of myasthenia gravis. Ann N Y Acad Sci 1275:1–6
Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ (2009) Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 72(18):1548
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rigamonti, A. (2017). Myasthenia Gravis. In: Agostoni, E. (eds) Emergencies in Neuromuscular Disease. Emergency Management in Neurology. Springer, Cham. https://doi.org/10.1007/978-3-319-56654-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-56654-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56653-5
Online ISBN: 978-3-319-56654-2
eBook Packages: MedicineMedicine (R0)