
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia among older individuals, with genetics and aging being two of the most potent risk factors. The course of AD dementia is characterized by early memory impairment, along with progressive decline in other cognitive functions, often with associated behavioral changes. The diagnosis of AD dementia can be made with the basic elements of a good clinical history of illness, neurological examination and laboratory/imaging testing. Several in vivo biomarkers of AD are available and while primarily focused on recognition of earlier stages of AD, may have potential roles in the setting of the demented patient. The treatment of AD addresses cognitive and behavioral symptoms with symptomatic treatments. Although disease modifying strategies are under study, the current therapies have not been shown to slow progression of the disease. Several lifestyle interventions that may have a pro-cognitive effect should be encouraged.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Alzheimer’s disease
- Dementia
- Amyloid imaging
- Positron emission tomography
- Cholinesterase inhibitors
- Memantine
Clinical Pearls:
-
The absence of memory symptoms early in the symptomatic course should cast doubt on the diagnosis of AD.
-
Hallucinations occurring early in the clinical course should raise the possibility of an alternative diagnosis such as Lewy Body Disease (see Chap. 8) or presence of an intercurrent illness or drug effect.
-
PET amyloid imaging has been correlated with plaque burden post-mortem. Thus, a negative PET amyloid study is inconsistent with a clinical diagnosis of AD dementia.
-
Atrophy on MRI scan is not specific for AD; hippocampal volume loss can be seen in hippocampal sclerosis and other neurodegenerative diseases.
-
Most individuals with dementia will have Alzheimer’s disease, either by itself or with other brain pathologies; be alert to the presence of those other conditions.
-
Regarding diagnosis, go low tech—most of the focus should be on obtaining a thorough history from an informant and probing for features that are not typical of Alzheimer’s (early hallucinations, REM sleep behavior, early physical symptoms such as incontinence, gait or balance problems).
-
Regarding treatment, subtract before you add—before adding an agent for AD, be sure to eliminate any drugs that may have an anti-cognitive effect if possible.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia among older individuals. While AD is now recognized to be a disease process with a long prodromal phase (see Chap. 3), this section will focus on AD dementia , the point most likely to present to the physician and the phase of illness which demands the most resources from caregivers and the medical system.
Though the absolute percentage of the population with AD is hard to ascertain, several epidemiological characteristics are worth considering when evaluating the dementia patient [1]:
-
Among individuals with dementia, AD pathology (either solely or in combination with other pathologies) is present in up to 80% of cases.
-
Incidence and prevalence of AD roughly doubles every 5 years after the age of 65; the frequency of the disease at or below 65 years old is low, 1–2%, but in some estimates, reaches close to 50% of individuals over the age of 85.
Genetics has a significant role in determining risk of AD [2]:
-
There is an increased risk of AD among first degree relatives of cases.
-
Young onset forms of familial AD that present in the third through sixth decade have been linked with mutations in three genes: the Amyloid Precursor Protein (Chromosome 21), presenilin 1 (chromosome 14) and presenilin 2 (chromosome 1).
-
Apolipoprotein (ApoE) polymorphism E4 is associated with an increased risk of AD. Individuals homozygous for E4 carry a tenfold increased risk of AD, with some estimates suggesting a 90% risk by their mid to late 70s, whereas those heterozygous for E4 are at four times the risk of non-carriers and are at 50% risk of developing AD by their mid to late 70s.
Clinical Manifestations
The symptomatic hallmark of AD is impaired memory function—initially involving recall of new information such as conversations, names, object locations—with remote memories relatively preserved [3]. However, over time, even distant memories become effected, either forgotten or jumbled.
Higher levels of cognitive processing (“executive” functions) are also affected relatively early in AD. Difficulty with tasks involving planning, reasoning, decision making or judgment become apparent. Impairment in these spheres may manifest by problems performing more complex tasks such as home repairs or other projects around the house, coordinating large family meals, or making prudent financial decisions.
Eventually, the entire gamut of learned processes recede. Concerns about word finding may occur early in AD but generally language abilities are preserved. As the disease progresses, patients may become more obviously aphasic, either producing language without much meaning or more commonly, loss of fluency and inability to converse. Impaired visuospatial abilities lead to getting lost in familiar places, including, when advanced, one’s home. In moderate to severe stages of AD, the development of apraxia results in increased dependence on others as hygiene and grooming (brushing teeth for example), and eventually feeding and toileting are affected. Patients also may lose the ability to recognize objects (agnosia).
Often behavioral changes are present as an early feature of AD. Apathy and loss of initiative/motivation are common and not infrequently mistaken as depression. Depressive symptoms can occur in AD as well, with estimates suggesting occurrence in up to 30–50% during the course of disease [4]. Differentiating apathy from depression is assisted by the absence of other depressive symptoms such as anhedonia, sad mood, or vegetative symptoms in apathetic patients. And even when depression is present, it is often influenced by environmental manipulation with major depression being rare in AD.
Irritability or poor impulse control can be observed early in the disease process in some individuals. Caregivers may report that the patient’s premorbid personality has been accentuated, though some individuals seem to have a distinct change in their patience level. Unfortunately, some behavioral problems are amplified by the caregiver’s approach to the patient.
As AD progresses, other behavioral symptoms may emerge including delusions and hallucinations . The former may take the form as delusions of theft, infidelity, misidentification of family members, or that their house is not their home. Hallucinations are less common and usually visual.
There are certain symptoms or clinical signs that are not typical of AD and include the presence of: gait disturbance, motor impairment, incontinence or seizures early in the course (though all of these symptoms can occur later in the disease). Table 4.1 provides clinical features that aid in diagnosis.
While episodic memory loss is considered the hallmark symptom of AD, several non-amnestic presentations have been recognized and discussed separately (see Chap. 5). These have as the primary clinical features:
-
Progressive language impairment (logopenic aphasia).
-
Progressive impairment of visual processing (posterior cortical atrophy).
-
Dysexecutive and/or behavioral impairment (“frontal” AD).
Diagnosis
Specific criterion for the diagnosis of AD have been developed by the International Work Group (IWG; see Chap. 3) and the National Institute of Aging and the Alzheimer’s Association (NIA/AA) (Table 4.2). These criteria shares with others in the literature the requirement for the presence of dementia, a gradually progressive course and absence of other specific causes for the dementia [5]. Using the NIA/AA criteria, the positive predictive value for the diagnosis of AD has generally exceeded 80–85%. However, the specificity of the diagnostic criteria tends to be lower, closer to 60% [6].
The diagnosis of AD dementia can be made with the basic elements of a good clinical history of illness, neurological examination and laboratory/imaging testing (Fig. 4.1).

Amyloid PET . (a) Normal; (b) abnormal with amyloid burden typical of AD
Possible AD dementia (Table 4.3) is diagnosed when patients with features of AD have atypical or mixed presentations.
Examination
Evaluation of the dementia patient begins with a thorough history, but generally not from the patient. Though some AD patients do have insight into their illness, it is imperative to obtain information from a reliable collateral source. Obtaining a history of short term memory impairment of gradual onset and progression with other cognitive functions declining, in the absence of any marked physical changes early, is a necessary start to the diagnosis.
The neurological examination is focused on both confirming the reported cognitive changes via mental status testing and surveillance for any neurological signs that may point to an alternative diagnosis. Several standardized cognitive screening tests are available with the Mini Mental Status Examination (MMSE) and Montreal Cognitive Assessment (MoCA) being two of the most commonly used. While both are easily administered, their limitations are worth keeping in mind. The MMSE lacks any assessment of “executive” function, is heavily weighted toward orientation, has a ceiling effect and can be influenced by education, ethnicity, and other factors. Alternatively, the MoCA is useful in detecting milder degrees of impairment and does include tests that reflect executive functioning [8].
Until the later stages of the disease, AD progresses in the absence of any marked physical changes. Thus presence of focal neurological signs or extrapyramidal features directs one to an alternative diagnosis (e.g. vascular dementia or Lewy body disease ).
Certain laboratory tests have become a routine part of the workup of AD with the purpose of eliminating other potentially reversible causes of dementia. The basic tests are listed in Fig. 4.1 with other studies that may be chosen as indicated by the clinical picture [9]. While most individuals with gradually progressive dementia will have a neurodegenerative or vascular etiology, a not insignificant proportion (about 15%) will have a comorbid condition that can be treated.
The diagnostic role of neuroimaging is to rule out structural lesions that can cause dementia. Non-contrasted Magnetic Resonance Imaging (MRI) or Computerized Tomography (CT) of the brain are often adequate for that purpose. Of the two methods, MRI imaging can better visualize posterior fossa lesions or small vascular lesions, as well as patterns of focal atrophy.
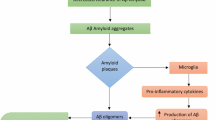
Several in vivo biomarkers of AD are available and primarily focused on recognition of earlier stages of AD. While it is unclear yet how the clinical application of these markers improves diagnosis of AD dementia, there may be several potential roles of these biomarkers in the setting of the demented patient. It is known from clinicopathological studies that older individuals with dementia often have multiple pathologies at autopsy that may include amyloid plaques and tau protein tangles indicative of AD, along with cerebrovascular disease, Lewy bodies and other aggregated proteins. Having tests indicative of AD may assist in differential diagnosis and inform therapeutic decisions. Biomarkers for AD fall into two categories: those that reflect β amyloid accumulation and those that denote neuronal degeneration. Markers of amyloid deposition include decreased levels of β amyloid in the cerebrospinal fluid (CSF) (presumably due to β amyloid aggregation into fibrillar plaques) and elevated uptake of tracers that bind to amyloid on Positron Emission Tomography (PET) (Fig. 4.1). Studies to date suggest a relatively close correlation between these two markers [10]. Moreover, PET amyloid imaging has been correlated with plaque burden post-mortem. Thus, a negative PET amyloid study is inconsistent with a clinical diagnosis of AD dementia.
Several biomarkers of neuronal damage complement the amyloid markers. Rise in CSF total tau or phosphorylated tau is considered a marker of cellular injury and when coupled with low CSF β amyloid provides a diagnostic signature of AD.
Synaptic dysfunction may be reflected through 18F- fluorodeoxyglucose (FDG) PET imaging. In AD, lower FDG uptake (lower glucose metabolism) is characteristically seen in the temporoparietal regions with involvement of precuneus and posterior cingulate (Fig. 4.2).

FDG PET. (a) Normal; (b) abnormal with reduced temporo-parietal metabolism characteristic of AD
Regional atrophy on structural MRI imaging is also thought to indicate neurodegeneration. Volumes of medial temporal lobe structures, particularly hippocampus, are reduced in AD and can be measured by automated techniques.
While all of these biomarkers are commercially available, their cost effectiveness in AD diagnosis has yet to be demonstrated. The combination of volumetric MRI and PET markers provides useful findings supportive of a diagnosis of AD.
Though there are many possible causes of cognitive impairment in the elderly, several stand out to consider in the patient that presents with a history of chronic progressive cognitive decline. Cerebrovascular disease should be suspected in the patient with a history of vascular risk factors, stuttering/stepwise course, or neurological signs on examination such as focal findings, gait disturbance, hyperreflexia, or incontinence. To be confident of that diagnosis, one should see evidence of multiple infarcts or extensive white matter disease on structural imaging. Lewy body disease comes into the differential diagnosis with a history of visual hallucinations early in the course, fluctuating levels of alertness/daytime fatigue, rapid eye movement (REM) sleep behavioral disorder, and mild extrapyramidal signs on examination. Those with more acute changes in cognitive function or behavior should be vigorously evaluated for inappropriate medications or other systemic illnesses such as metabolic/infectious etiologies.
Treatment
The treatment of AD addresses cognitive and behavioral symptoms with symptomatic treatments. Although disease modifying strategies are under study, the current therapies have not been shown to slow progression of the disease.
The principal treatments for all severities of AD dementia are cholinesterase inhibitors (CEI) . Three agents are currently approved in the United States, donepezil (Aricept™), rivastigmine (Exelon™) and galantamine (Razadyne™). Rivastigmine is available in patch form; donepezil and galantamine are given orally. These drugs putatively work by blocking acetylcholinesterase, the enzyme that degrades acetylcholine, thus increasing the cholinergic tone that is compromised as cholinergic neurons degenerate with disease progression.
The CEIs have repeatedly demonstrated a modest benefit on measures of cognitive function and Activities of Daily Living (ADLs) . Notable improvement is seen in 20% of treated patients, with a larger proportion realizing a temporary lessening of symptomatic decline [11]. Given the less than dramatic response from CEIs, patients and caregivers need to be counseled on realistic expectations from these drugs. Yet it is worth remembering that delaying a symptomatic decline in ADLs may translate to being able to stay at more independent levels of home or residential care.
All CEIs are equally efficacious. On the other hand, the degree of cholinergic side effects may differ from agent to agent. The most common adverse effects are nausea and vomiting, bowel urgency or frank diarrhea (though other cholinergic effects can be seen such as vivid dreams, muscle cramps and bradycardia). The likelihood of these (or other side effects occurring) may be mitigated by slow titration of dose. If patients do not tolerate one CEI, it may be worth switching to at least one other.
Once an AD patient progresses to moderate to severe dementia, the addition of memantine (Namenda™) should be considered. An NMDA receptor antagonist, memantine is thought to reduce glutaminergic tone which has been shown to be increased in AD. This mechanism may improve signal transmission. In patients with moderate to severe AD, memantine slightly improves cognitive function and ADLs, and may have a favorable effect on behavior. Memantine has been shown to have efficacy either as monotherapy or in combination with a CEI. Dizziness has been noted as a possible side effect of memantine and it is available in immediate release and delayed release form, as well as in a combination capsule with donepezil (Namzaric™). Because the CEIs and memantine are thought to be only symptomatic drugs, it is worth discussing withdrawal of these medications if the patient has lost all ADLs and has no meaningful interaction with the family or caregivers.
Medical foods are a class of agents that are safe and address a metabolic defect but are not tested with the same rigor as drugs approved by the Food and Drug Administration (FDA). Axona™ is a medium chain triglyceride (caprylic triglyceride) proposed to address the metabolic defect of AD. Cerefolin™ is a B vitamin combination that treats hyperhomocysteinemia observed in some AD patients. Souvenaid™ is available in many countries and is composed of elements involved in supporting synaptic function.
There is an active industry promoting agents that are over the counter and said to improve cognitive health. Currently, there is no consistent evidence to recommend any of these agents. On the other hand, when feasible, lifestyle interventions that may have a pro-cognitive effect should be encouraged. Epidemiological studies have suggested that regular exercise, social engagement, good control of vascular risk factors and diets that include foods rich in antioxidants may have a positive effect on brain health in general. Helpful guidance can be found on healthybrains.org.
Behavioral disturbances are common in AD and can significantly complicate caregiving. Table 4.4 summarizes an initial approach to behavioral problems. While no therapeutic agents have a specific indication for behavioral disorders in AD, pharmacologic management of behavior is often based on how similar non-dementia diagnoses in psychiatry are treated. However, certain caveats need to be considered in administering psychotropics to an elderly, dementia population including [12]:
-
Antipsychotic medications carry a FDA “black box” warning of increased risk of stroke and death in older demented patients and should be considered if the psychosis is very disturbing for the patient or results in the patient being a danger to themselves or others.
-
Benzodiazepines have been associated with increased confusion, sedation and falls in the elderly.
Figure 4.3 provides guides for pharmacotherapy of AD.

Management of AD
ICD-10 Codes
Alzheimer’s disease, unspecified | G30.9 |
Alzheimer’s disease with early onset | G30.0 |
Alzheimer’s disease with late onset | G30.1 |
Focal onset Alzheimer’s disease | G30.8 |
References
Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–52.
Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72.
Farlow MR. Alzheimer’s disease. Continuum. 2007;13(2):39–68.
Aarsland D, Sharp S, Ballard C. Psychiatric and behavioral symptoms in Alzheimer’s disease and other dementias: etiology and management. Curr Neurol Neurosci Rep. 2005;5:345–54.
Dubois B, Feldman H, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29.
McKhann G, Drachmann D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–44.
McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Finney GR, Minagar A, Heilman KM. Assessment of mental status. Neurol Clin. 2016;34(1):1–16.
Arevalo-Rodriguez I, Pedraza OL, Rodríguez A, et al. Alzheimer's disease dementia guidelines for diagnostic testing: a systematic review. Am J Alzheimers Dis Other Demen. 2013;28(2):111–9.
Scheltens P, Blennow K, Breteler MM, et al. Alzheimer’s disease. Lancet. 2016;388(10043):505–17.
Farlow MR, Cummings JL. Effective pharmacologic management of Alzheimer’s disease. Am J Med. 2007;120(5):388–97.
Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. BMJ. 2015;350:h369.
Disclosures
Dr. Bernick is a speaker for Allergan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Bernick, C. (2017). Alzheimer’s Disease. In: Tousi, B., Cummings, J. (eds) Neuro-Geriatrics. Springer, Cham. https://doi.org/10.1007/978-3-319-56484-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-56484-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56483-8
Online ISBN: 978-3-319-56484-5
eBook Packages: MedicineMedicine (R0)