
Abstract
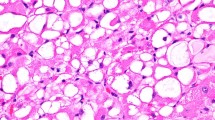
Histologically, skeletal muscle cells are distinguished by eosinophilic cytoplasm containing light and dark striations that correspond to the alternating arrangement of thin and thick myofilaments (Fig. 8. 1). These cells can be seen in varying degrees in soft tissue tumors with skeletal muscle differentiation. However, in tumors that are poorly differentiated, rhabdomyoblastic differentiation must sometimes be confirmed by immunohistochemical stains for skeletal muscle markers (e.g., myogenin) [1].
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Alveolar rhabdomyosarcoma
- Embryonal rhabdomyosarcoma
- Pleomorphic rhabdomyosarcoma
- Rhabdomyoma
- Skeletal muscle tumor
Histologically, skeletal muscle cells are distinguished by eosinophilic cytoplasm containing light and dark striations that correspond to the alternating arrangement of thin and thick myofilaments (Fig. 8.1). These cells can be seen in varying degrees in soft tissue tumors with skeletal muscle differentiation. However, in tumors that are poorly differentiated, rhabdomyoblastic differentiation must sometimes be confirmed by immunohistochemical stains for skeletal muscle markers (e.g., myogenin) [1].

Normal skeletal muscle cells with striated and eosinophilic cytoplasm
8.1 Rhabdomyoma
Rhabdomyomas are rare and benign neoplasms of skeletal muscle differentiation. These are divided into cardiac, adult, fetal, and genital types. Cardiac rhabdomyomas typically arise as single or multiple tumors in the right or left heart ventricles and are often associated with tuberous sclerosis. Patients can present with symptoms of arrhythmia or cardiac dysfunction [2, 3]. Adult rhabdomyomas arise as slow-growing lesions in the head and neck in adults after the fifth decade of life. Patients can present with problems swallowing or experience vocal cord dysfunction [4,5,6]. Fetal rhabdomyomas predominantly occur in the head and neck in children and have been reported in patients with nevoid basal cell carcinoma syndrome [7, 8]. Genital rhabdomyoma usually arises as a slowly growing polypoid mass in the vagina or vulva in middle-aged women (average age of 45 years) [9].
Pathology
The histologic appearance of rhabdomyomas depends on the subtype. Cardiac rhabdomyomas contain sheets of polygonal cells that are highly vacuolated, otherwise known as “spider cells” (Fig. 8.2). Adult rhabdomyomas show polygonal cells with eosinophilic and granular cytoplasm. Scattered vacuolated cells can also be seen. Fetal rhabdomyomas demonstrate spindle cells with thin and elongated nuclei (Figs. 8.3 and 8.4). Depending on the morphologic maturity of the tumor, these tumor cells may contain cytoplasmic cross striations. Genital rhabdomyomas demonstrate an aggregate of muscle cells with scattered cross striations (Fig. 8.5) [6].

Cardiac rhabdomyoma with “spider cells”

Fetal rhabdomyoma with spindle cells

Fetal rhabdomyoma with spindle cells with very focal cross striations (arrow). These cells can sometimes be difficult to distinguish from a rhabdomyosarcoma

Genital rhabdomyoma with skeletal muscle cells containing cross striations
Ancillary Studies
-
Consistent with their myogenic nature, rhabdomyomas stain with desmin.
Differential Diagnosis
-
Hibernoma
-
Embryonal rhabdomyosarcoma
Comment
-
1.
While hibernomas can have cells with eosinophilic cytoplasm, they usually contain scattered adipocytes that indicate their lipogenic differentiation. Unlike rhabdomyomas, hibernomas are negative for muscle markers such as desmin.
-
2.
Rhabdomyomas with particularly immature cells, such as fetal rhabdomyoma, can be difficult to discern from embryonal rhabdomyosarcoma.
-
3.
Embryonal rhabdomyosarcoma is typically more infiltrative and has increased mitosis and necrosis compared to a rhabdomyoma [6].
-
4.
Adult rhabdomyomas are treated by simple excision. Up to 40% of lesions can recur, but these are typically treated with re-excision [4].
-
5.
Given that cardiac rhabdomyomas often spontaneously regress, surgical excision is only performed in patients who are experiencing arrhythmias or hemodynamic compromise.
-
6.
Patients who are diagnosed with cardiac rhabdomyoma should be assessed for tuberous sclerosis [6, 10].
-
7.
Fetal rhabdomyomas are typically cured by simple excision. Less than 10% recur [7].
8.2 Embryonal Rhabdomyosarcoma
Embryonal rhabdomyosarcoma is a malignant soft tissue sarcoma of skeletal muscle differentiation that predominantly occurs in young children, often under 10 years of age [11]. Frequent sites of occurrence include the head and neck and hollow organs, such as the nasopharynx, bladder, and vagina [6, 12].
Pathology
Grossly these tumors have a white and friable appearance [9]. Microscopically, they are composed of highly immature cells in a variably myxoid background (Fig. 8.6). The neoplastic cells have scant cytoplasm, and only occasionally contain skeletal muscle type striations, otherwise known as rhabdomyoblasts (Fig. 8.7). These rhabdomyoblasts are increased in tumors that have been previously treated with chemotherapy (Fig. 8.8) [13]. In the botryoid variant of embryonal rhabdomyosarcoma, the tumor cells approach but do not overrun the overlying squamous or urothelial mucosa (Fig. 8.9).

Embryonal rhabdomyosarcoma with highly immature tumor cells with limited cytoplasm

Rare rhabdomyoblast demonstrating eosinophilic cytoplasm and cross striations (arrow) in an embryonal rhabdomyosarcoma

Rhabdomyosarcoma previously treated with chemotherapy showing multiple rhabdomyoblasts

The tumor cells in this botryoid variant of embryonal rhabdomyosarcoma condense around the adjacent urothelium
Ancillary Studies
-
The tumor cells are positive for desmin, consistent with their myogenic nature.
-
Additionally, the neoplastic cells show nuclear expression of myogenin and MyoD1.
Differential Diagnosis
-
Alveolar rhabdomyosarcoma
-
Ewing sarcoma
-
Pleomorphic rhabdomyosarcoma
-
Fetal rhabdomyoma
Comment
-
Embryonal rhabdomyosarcoma lacks the characteristic translocations involving the EWSR1 gene or FOXO1 gene seen in Ewing sarcoma or alveolar rhabdomyosarcoma, respectively.
-
Although embryonal rhabdomyosarcoma can exhibit focal areas of pleomorphism, pleomorphic rhabdomyosarcoma is diffusely anaplastic and typically arises in the extremities of adults.
-
Although fetal rhabdomyoma can have immature cells, these tumors lack the mitosis and necrosis seen in embryonal rhabdomyosarcoma.
-
Botryoid rhabdomyosarcoma has an overall 5-year survival rate of approximately 95% [14].
-
Non-botryoid embryonal rhabdomyosarcoma has an overall 5-year survival rate of approximately 67% [14].
-
Treatment consists of a combination of chemotherapy and surgical resection. Radiation therapy can be used in cases of nodal metastasis, positive margins, or poor response to chemotherapy [6].
8.3 Alveolar Rhabdomyosarcoma
Alveolar rhabdomyosarcoma is a malignant soft tissue tumor that frequently arises during the second and third decades of life. Common sites of involvement include the extremities, head, neck, and trunk [14].
Pathology
Histologically, alveolar rhabdomyosarcoma shows highly immature tumor cells with very little cytoplasm that are partitioned by fibrous septae (Fig. 8.10). Although tumor cells adhere to these fibrous bands, the more centrally located neoplastic cells drop out and give the tumor an “alveolar” appearance similar to lung parenchyma (Figs. 8.11 and 8.12). Scattered neoplastic giant cells can also be seen (Fig. 8.13). Only rare if any rhabdomyoblasts can be identified (Fig. 8.14) [15].

Alveolar rhabdomyosarcoma showing poorly differentiated tumor cells partitioned by fibrous septa

The tumor cells in this alveolar rhabdomyosarcoma are adherent to the fibrous septa

Central dropout of tumor cells (arrows) among the fibrous septa creates an “alveolar”-like appearance in the tumor

Scattered neoplastic giant cells (arrow) are often present in an alveolar rhabdomyosarcoma

Scattered rhabdomyoblasts are rarely found in alveolar rhabdomyosarcoma
Ancillary Studies
-
Alveolar rhabdomyosarcomas are positive for desmin, myogenin, and MyoD1 immunohistochemical stains.
-
Approximately 60% of alveolar rhabdomyosarcomas manifest a t(2;13) translocation that results in a PAX3-FOX01A fusion transcript.
-
Approximately 20% of alveolar rhabdomyosarcomas demonstrate a t(1;13) translocation that results in a PAX7-FOX01A fusion transcript [16].
Differential Diagnosis
-
Ewing sarcoma
-
Desmoplastic round cell tumor
-
Embryonal rhabdomyosarcoma
Comment
-
Alveolar rhabdomyosarcoma can be difficult to distinguish from other poorly differentiated round cell tumors such as Ewing sarcoma or desmoplastic round cell tumor.
-
Ewing sarcoma and desmoplastic round cell tumor characteristically manifest mutations involving the EWSR1 gene.
-
Although alveolar rhabdomyosarcoma and embryonal rhabdomyosarcoma can have the same immunohistochemical staining pattern, alveolar rhabdomyosarcoma can be diagnosed by detection of the PAX3-FOX01A or PAX7-FOX01A fusion transcripts.
-
The distinction of alveolar rhabdomyosarcoma from embryonal rhabdomyosarcoma is relevant as alveolar rhabdomyosarcoma has a poorer survival (5-year survival of 54%) [14].
-
The prognosis is better in alveolar rhabdomyosarcomas with a PAX7-FOX01A fusion transcript than a PAX3-FOX01A fusion transcript [17].
-
Frequent sites of metastasis include the lung and lymph nodes.
-
Treatment usually includes chemotherapy and radiation therapy. Second-line chemotherapy is administered for tumors that do not respond to initial treatment [9].
8.4 Pleomorphic Rhabdomyosarcoma
Pleomorphic rhabdomyosarcoma is a malignant soft tissue neoplasm that arises later than other rhabdomyosarcomas, typically after the fourth decade of life. These most commonly arise in the deep extremities, such as the thigh, but can arise in other sites. When diagnosed, these tumors are usually greater than 10 cm in size [6, 18].
Pathology
In contrast to embryonal and alveolar rhabdomyosarcomas which are predominantly composed of immature cells with scant cytoplasm, pleomorphic rhabdomyosarcomas are composed of cells with abundant eosinophilic cytoplasm and bizarre nuclei (Fig. 8.15).

Pleomorphic rhabdomyosarcoma with highly atypical tumor cells
Ancillary Studies
-
The tumor cells are positive for desmin, myogenin, and MyoD1 (Fig. 8.16).
Fig. 8.16 
The tumor cells in this pleomorphic rhabdomyosarcoma are positive for MyoD1, consistent with skeletal muscle differentiation

Differential Diagnosis
-
Embryonal or alveolar rhabdomyosarcoma
-
Undifferentiated pleomorphic sarcoma
-
Pleomorphic leiomyosarcoma
Comment
-
Embryonal and alveolar rhabdomyosarcoma can focally have pleomorphic areas, but these are limited to only a portion of the tumor. Also, pleomorphic rhabdomyosarcoma usually occurs in older adults.
-
Pleomorphic rhabdomyosarcomas can look similar to other pleomorphic sarcomas, such as undifferentiated pleomorphic sarcoma or pleomorphic leiomyosarcoma. By definition, these other tumors lack the morphologic or immunohistochemical features of skeletal muscle differentiation.
-
Pleomorphic rhabdomyosarcoma is a highly aggressive tumor. The majority will metastasize and the median survival is approximately 20 months [6, 15].
-
Treatment usually consists of wide excision and adjuvant radiation therapy [9].
Facts to Remember
-
1.
Rhabdomyomas are rare soft tissue tumors that manifest skeletal muscle differentiation. Different subtypes of rhabdomyomas have specific clinical and morphologic features.
-
2.
Embryonal rhabdomyosarcoma often arises in infants and children in the head and neck and genitourinary area. The botryoid variant of rhabdomyosarcoma has a particularly good prognosis.
-
3.
Alveolar rhabdomyosarcoma occurs in young adults, frequently in the extremities. This type of rhabdomyosarcoma is more aggressive than embryonal rhabdomyosarcoma and contains either PAX3-FOX01A or PAX7-FOX01A fusion transcripts.
-
4.
Pleomorphic rhabdomyosarcomas arise in older adults and contain bizarre tumor cells. These tumors are highly aggressive and carry a poor prognosis.
References
Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, et al. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol. 2001;25(9):1150–7.
De Rosa G, De Carolis MP, Pardeo M, Bersani I, Tempera A, De Nisco A, et al. Neonatal emergencies associated with cardiac rhabdomyomas: an 8-year experience. Fetal Diagn Ther. 2011;29(2):169–77.
Burke AP, Virmani R. Cardiac rhabdomyoma: a clinicopathologic study. Mod Pathol. 1991;4(1):70–4.
Kapadia SB, Meis JM, Frisman DM, Ellis GL, Heffner DK, Hyams VJ. Adult rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study. Hum Pathol. 1993;24(6):608–17.
Pichi B, Manciocco V, Marchesi P, Pellini R, Ruscito P, Vidiri A, et al. Rhabdomyoma of the parapharyngeal space presenting with dysphagia. Dysphagia. 2008;23(2):202–4.
Goldblum J, Weiss S, Folpe AL, editors. Enzinger and Weiss’s soft tissue tumors. 6th ed. Philadelphia: Elsevier; 2013.
Kapadia SB, Meis JM, Frisman DM, Ellis GL, Heffner DK. Fetal rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study of 24 cases. Hum Pathol. 1993;24(7):754–65.
DiSanto S, Abt AB, Boal DK, Krummel TM. Fetal rhabdomyoma and nevoid basal cell carcinoma syndrome. Pediatr Pathol. 1992;12(3):441–7.
Hornick JL, editor. Practical soft tissue pathology: a diagnostic approach. 1st ed. Philadelphia: Saunders; 2013.
Fesslova V, Villa L, Rizzuti T, Mastrangelo M, Mosca F. Natural history and long-term outcome of cardiac rhabdomyomas detected prenatally. Prenat Diagn. 2004;24(4):241–8.
Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer. 2009;115(18):4218–26.
Newton Jr WA, Soule EH, Hamoudi AB, Reiman HM, Shimada H, Beltangady M, et al. Histopathology of childhood sarcomas, intergroup rhabdomyosarcoma studies I and II: clinicopathologic correlation. J Clin Oncol. 1988;6(1):67–75.
Miettinen M, editor. Modern soft tisssue pathology: tumors and non-neoplastic conditions. 1st ed. New York: Cambridge University Press; 2010.
Newton Jr WA, Gehan EA, Webber BL, Marsden HB, van Unnik AJ, Hamoudi AB, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification – an intergroup rhabdomyosarcoma study. Cancer. 1995;76(6):1073–85.
Fletcher CDM, Bridge JA, Hogendoorn PC, Mertens F, editors. Pathology and genetics of tumours of soft tissue and bone. 4th ed. Lyon: World Health Organization; 2013.
Parham DM, Qualman SJ, Teot L, Barr FG, Morotti R, Sorensen PH, et al. Correlation between histology and PAX/FKHR fusion status in alveolar rhabdomyosarcoma: a report from the Children's oncology group. Am J Surg Pathol. 2007;31(6):895–901.
Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20(11):2672–9.
Gaffney EF, Dervan PA, Fletcher CD. Pleomorphic rhabdomyosarcoma in adulthood. Analysis of 11 cases with definition of diagnostic criteria. Am J Surg Pathol. 1993;17(6):601–9.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Perry, K. (2017). Skeletal Muscle Tumors. In: Soft Tissue Pathology for Clinicians. Pathology for Clinicians. Springer, Cham. https://doi.org/10.1007/978-3-319-55654-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-55654-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55653-6
Online ISBN: 978-3-319-55654-3
eBook Packages: MedicineMedicine (R0)