
Abstract
Nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) belongs to one of the best described and most intensively studied transcription factors in biochemistry in the last 30 years. The NF-κB signaling cascade exists in two variants, the canonical and noncanonical pathway, and its transcription factors are key regulators of several biochemical processes like immune responses, inflammation, survival, and cellular development and growth.
Examination of various transgenic mouse models targeting NF-κB itself or signaling members discovered the implication of NF-κB in chronic inflammatory diseases and cancer development in different organs as in the skin, intestine, and liver.
In this review the focus lies on the central organ of metabolic and inflammatory processes: the liver. It seems that NF-κB is pivotal for the homeostasis in the different hepatic cell types concerning hepatic failure, fibrosis, and HCC progression. NF-κB has the ability to be a potential target in the attempt to circumvent or medicate liver fibrosis and HCC.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Liver Fibrosis
- Liver Fibrosis Progression
- Liver Cell Type
- Enhance Hepatocyte Proliferation
- Linear Ubiquitin Chain Assembly Complex
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
6.1 Introduction
Nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) is one of the best and most intensive studied transcription factors in the field of biomedicine. It was discovered and first described 30 years ago by David Baltimore and colleagues [1]. Since that time many studies revealed the outstanding meaning of NF-κB in the development of inflammatory diseases such as arthritis and psoriasis, inflammatory bowel diseases, asthma, and neurodegenerative heart diseases and its contribution to cancer development [2,3,4]. It is expressed in most mammalian cell types and tissues and controls the transcription of genes involved in immune responses, cell survival, proliferation, and differentiation [5].
Next to in vitro approaches, examination of several transgenic mouse models with different NF-κB targets gave the opportunity to raise our understanding of the complex mechanisms behind inflammation-driven diseases in vivo. Moreover, the work with conditional murine knockout models in different organs uncovered the central role of NF-κB in mediating innate immune responses and cytokine expression in order to react on pathological outcomes of inflammation like chronical skin inflammation (e.g. psoriasis) and hepatocarcinogenesis [6,7,8,9,10]. The severe affection of organs or tissues during disbalanced NF-κB activation gains importance and constitutes a therapeutic challenge. Further investigations will support the development of clinical trials targeting certain molecules of the NF-κB signaling cascade.
6.2 Members of the NF-κB/Rel Family
The NF-κB signaling pathway is evolutionarily highly conserved and is, besides mammalians, also found in the fruit fly Drosophila melanogaster, cnidarians, porifera, viruses, and mollusks [11,12,13,14]. Caenorhabditis elegans and yeast are the main exceptions [15, 16].
Mammalian NF-κB itself is composed of different types of dimers, appearing as homo- or heterodimers. The single compounds of these dimers are p50 (NFKB1), p52 (NFKB2), c-Rel (REL), p65/RelA (RELA), and RelB (RELB). All five transcription factors are characterized by an N-terminal Rel homology domain (RHD-NTD) which is needed to mediate DNA binding, homo- and heterodimerization, and nuclear translocation. In the nucleus NF-κB dimers bind at κB sites inside enhancer/promoter regions of target genes where they control transcription by recruiting coactivators and corepressors [17,18,19]. Furthermore, only p65, RelB, and c-Rel comprise a C-terminal transactivation domain (TAD) which is required for gene transcription. The other two members, p50 and p52, lacking TAD and are processed from the precursor proteins p105 (p50) and p100 (p52) (Fig. 6.1). Both are not directly involved in gene transcription except in combination with p65, RelB, c-Rel, or other proteins which are able to recruit coactivators. After successfully entering into the nucleus, it binds at the following consensus sequence 5′-GGGRNYYYCC-3′ (R, purine; Y, pyrimidine; N, any nucleotide) of DNA κB sites [19]. The TAD and RHD act in each case autonomously and underlay posttranscriptional modifications which might have an influence on NF-κB activation at the level of transcription and/or DNA binding [20].

Schematic diagram of the domain structures of each individual NF-κB, IκB, and IKK complex protein family members. All three family groups have a typical domain structure as the Rel homology domain (RHD) for the NF-κB family members, the ankyrin repeat domains (ARD) for the IκB family members, and the leucine-zipper (LZ) motif for the IKK complex members. On the basis of their function, p100 and p105 are also associated with the NF-κB and IκB family. CC coiled coil, DD death domain, GRR glycine-rich region, HLH helix-loop-helix, NBD NEMO-binding domain, PEST proline-, glutamic acid-, serine-, and threonine-rich region, TAD transactivation domain. Adapted from Oeckinghaus et al.: The NF-κB Family of Transcription Factors and its Regulation, Cold Spring Harb Perspect Biol. 2009, 1(4): 1–14
The most abundant combination of NF-κB dimers are p50/p65 and p50/50, whereas, in contrast, the homodimer p50/p50 can act as a transcriptional repressor [21, 22].
6.3 The Negative Controllers of NF-κB: IκBs
NF-κB dimers are located in the cytoplasm, and translocation from the cytoplasm through the nucleus is regulated by another group of proteins named nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor (IκBs) [15]. These proteins are tightly associated with the NF-κB dimers, preventing NF-κB activation by hindering NF-κB translocation through the nuclear membrane. The IκB family includes six members: IκBα (NFKBIA), IκBβ (NFKBIB), IκBε (NFKBIE), IκBγ (NFKB1), IκBζ (NFKBIZ), and Bcl-3 (BCL3) and the NF-κB precursors p100 and p105. All of them share several ankyrin repeat domains (ARD), which are necessary to interact with the RHD of NF-κB [23]. Crystal structure analysis of the IκBα/NF-κB (p50/p65) heterodimer and IκBβ/NF-κB (p50/p50) homodimer allowed a closer look through the binding conditions of each complex and revealed a binding ratio of 1:1. Inside the IκBα/NF-κB complex, the ankyrin repeat six and the C-terminal PEST sequence of IκBα are associated with the p65 RHD-NTD, impeding binding to the DNA κB site (Fig. 6.1). Additionally, p65 undergoes such a conformational change, which strongly supports the linkage to IκBα, holding NF-κB in its inactive state [24,25,26].
Every IκB member has its own favorite NF-κB dimer. IκBα/β/ε binds to NF-κB dimers, which contain a minimum of one p65 or c-Rel subunit. p100 and p102 are connected to all NF-κB subunits. IκBζ and Bcl-3 have a preference for p50 and p52 homodimers [27, 28].
Here, IκBα is investigated at best and is a central regulatory factor in the canonical NF-κB signaling pathway as described in the next chapter.
6.4 The Canonical NF-κB Pathway
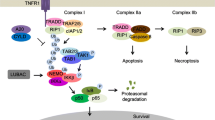
Various cellular stress stimuli and certain endogenous and exogenous ligands lead to NF-κB activation. The stimulus decides if the canonical or noncanonical NF-κB pathway is activated. Both cascades lead to NF-κB nuclear translocation but are regulated by different checkpoints within the cascade. In the last few years, extensive studies have been performed to characterize both variants of NF-κB activation and to identify important key players. It was shown that the canonical pathway is primarily activated during physiological stress conditions like inflammation, exposure to bacterial products like lipopolysaccharide (LPS), and oxidative stress [29, 30]. The canonical NF-κB signaling cascade is generally activated by the following receptors: tumor necrosis factor receptor (TNFR), interleukin 1 receptor (IL-1R), Toll-like receptor (TLR), B-cell receptor (BCR), and T-cell receptor (TCR) [31]. Receptor ligation leads to the recruitment of certain adapter proteins to TNF receptor-associated factor (TRAF) and receptor-interacting protein kinase 1 (RIPK1). RIPK1 is connected to the TGFβ-activated kinase 1 (TAK1)-binding protein (TAB2-TAB3-TAK1) complex and the NF-κB essential modulator (NEMO) via ubiquitin chains, bringing TAK1 into close vicinity to NEMO (IKKγ), the regulatory subunit of the IκB kinase (IKK) complex. This complex is composed of two more members, the catalytic subunits IKKα (IKK1) and IKKβ (IKK2), and represents the crucial step in NF-κB nuclear translocation by controlling proteasomal degradation of IκBα. More precisely, phosphorylation of TAK1 at Thr178 and Thr184 permits a direct phosphorylation of IKKβ inside its activation loop at Ser177 and Ser181 leading straightly to IκBα phosphorylation at Ser32 and Ser36. Phosphorylation of IκBα follows activation of the IKK complex, the second essential regulatory step in NF-κB activation, because IκBα undergoes K48-linked polyubiquitination by the SCFβTrCP ubiquitin ligase complex, which induces its fast degradation by the 26S proteasome. Finally, IκBα degradation exposes the nuclear localization site (NLS) of NF-κB, which is needed for nuclear access, DNA binding, and transcription of target genes (Fig. 6.2) [32,33,34,35,36,37].

The canonical and noncanonical NF-κB signaling cascade. After activation of the respective receptors, which are able to induce the canonical NF-κB pathway, the TAB-TAK1 complex and NEMO, the regulatory subunit of the IKK complex, get ubiquitinated by RIPK1. These ubiquitin chains bring both complexes into closer vicinity to each other, whereby TAK1 phosphorylates IKKβ, one of the catalytic subunits of the IKK complex. The activated IKK complex phosphorylates two serine residues of the NF-κB inhibitor IκBα, thereby initiating its proteasomal degradation. After IκBα degradation, NF-κB translocates through the nucleus to induce gene expression. The noncanonical NF-κB signaling cascade is controlled at the upper part by NIK. NIK mediates phosphorylation leading to the activation of the homodimer IKKα, another catalytic subunit of the IKK complex. IKKα cleaves the inactive precursor p100 into the active NF-κB subunit p52. In combination with RelB, it forms a heterodimeric NF-κB molecule which enters the nucleus to induce target gene expression. BCR B-cell receptor, BAFF-R B-cell-activating factor receptor, FADD Fas-associated death domain, IκBα inhibitor of NF-κB, IKKα/β IκB kinase α/β, LT-βR lymphotoxin-β receptor, NEMO NF-κB essential modulator, NIK NF-κB-inducing kinase, RANK receptor activator of NF-κB, RIPK1 receptor-interacting protein 1, TAB2/3 TGFβ-activated kinase 1 (TAK1)-binding protein, TAK1 TGFβ-activated kinase 1, TCR T-cell receptor, TLR Toll-like receptor, TRADD TNF receptor-associated death domain, TRAF2/3 TNF receptor-associated factor 2/3, TNFR tumor necrosis factor receptor. Adapted from Luedde et al.: The role of NF-κB in hepatic disease models, Translational Research in Chronic Liver Disease, Falk Workshop, Shaker Verlag Aachen 2009: 57–89
6.5 The Noncanonical or Alternative NF-κB Pathway
Next to the extensively studied canonical pathway, an alternative NF-κB activation exists. The so-called noncanonical NF-κB pathway activates NF-κB not by degradation of IκBα/β/ε and p105, but via processing the inactive p100/RelB NF-κB heterodimer through the active p52/RelB heterodimer [38, 39]. This pathway seems to be crucial in lymphoid organogenesis, B-cell maturation and survival, dendritic cell activation, and osteoclastogenesis. The major ligands which initiate these variants of NF-κB activation are the lymphotoxin β receptor (LTβR), B-cell-activating factor belonging to the TNF family receptor (BAFF-R), CD40, and receptor activator of NF-κB (RANK). Moreover, uncontrolled stimulation of the noncanonical NF-κB signaling cascade leads to severe diseases such as B-cell lymphomas, ulcerative colitis, and rheumatoid arthritis [40].
Processing of p100 depends on the NF-κB-inducing kinase (NIK), IKKα homodimers, and βTrCP, a subunit of the SCFβTrCP ubiquitin ligase. NIK mediates phosphorylation of p100 at Ser866 and Ser870 in its NIK-responsive domain (NRD) and IKKα activation. Activated IKKα phosphorylates p100 at Ser99, Ser108, Ser115, Ser123, and Ser872 which is needed for the recruitment of βTrCP. The SCFβTrCP ubiquitin ligase complex facilitates ubiquitination of p100 and thereby its 26S proteasomal degradation to p52. Interestingly, canonical and noncanonical pathways exhibit some similarities regarding regulatory mechanisms as shown by equivalent phosphorylation sites of p100 and IκBα or ubiquitin-mediated 26S proteasomal degradation of NF-κB inhibitors (Fig. 6.2) [41,42,43]. In contrast to the canonical pathway, the noncanonical cascade is characterized by a slow and persistent signaling and protein synthesis [44, 45].
Next to ubiquitin, SUMOylation is another regulatory mechanism to shape NF-κB signaling in both pathways. SUMOylation induces posttranslational modifications like ubiquitination and phosphorylation and influences protein-protein interaction and gene transcription [46]. It was shown in different studies that the interplay of SUMO, SUMO proteases, and NF-κB signaling members like NEMO, IκBα, or p100 represent another important level of signal transduction [47, 48].
6.6 NF-κB-Associated Human Diseases and Genetic Mouse Models
Due to the immense effort taken in examining NF-κB and its biological function, the crucial role of NF-κB in cellular homeostasis was uncovered. Several human diseases like psoriasis, colitis ulcerosa, Crohn’s disease, rheumatoid arthritis, or cancer development are a result of dysregulated NF-κB activation [2,3,4].
For a better understanding of the mechanisms behind NF-κB dysregulation in vivo, numerous mouse models were developed and examined. These comprise constitutive, tissue-specific conditional knockouts using the cre/loxP technology, gene knock-ins, and reporter mice of single or more IKK subunits or in combination with other members of the NF-κB signaling cascade [6,7,8, 49,50,51,52,53].
Caused by the tremendous information on the different genetic mouse models and their implication in broadening our understanding of NF-κB-associated human diseases, the next paragraph will focus particularly on one organ which has been extensively studied over the last few years and is a prime example for the importance of NF-κB homeostasis: the liver.
6.7 NF-κB and Its Critical Role for Liver Homeostasis
The liver is the biggest organ in the human body with a weight of 1.2–1.8 kg and makes approximately 2–3% of the whole body weight. The liver is of great importance because it is responsible for different metabolic processes like synthesis of vitally essential proteins (e.g. albumin, blood coagulation factors, hormones), utilization of food residues (e.g. conversion of glucose to amylum), detoxification of intermediate catabolic metabolites (e.g. from medicine), and bile production. Additionally, it is a storage organ for important macromolecules such as hormones or amylum. Besides the wide metabolic functions, the liver has the capability of reacting immunologically as well [54]. During all these biochemical and immunological processes, the different cell types forming the liver are faced with degradation products which might be harmful, such as oxygen radicals, or are attacked by bacterial components like lipopolysaccharides (LPS) or viruses, such as hepatitis viruses. NF-κB activation protects the cells against apoptosis and supports pro-inflammatory responses. Hepatocytes are the dominant hepatic cell type and stress factors, such as cytokines like TNFα or IL-1, and initiate the NF-κB signaling cascade to protect them against cell death [31].
Despite the bad reputation of inflammatory processes, it plays in the liver a central role for wound healing induced by injury processes, such as alcohol abuse or medication intake, which bear a high risk for liver fibrosis and cirrhosis progression and, finally, hepatocellular carcinoma (HCC) development. However, also viral infections such as hepatitis B virus (HBV) and HCV are potent inducers of liver fibrosis and HCC development. During fibrosis, the liver tissue undergoes a perennial process of inflammation, apoptotic and necroptotic events, and compensatory renewal. These chronical processes cause severe side effects, such as the production of highly reactive molecules like reactive oxygen species (ROS), chromosomal aberrations, and possibly malignant alterations of proliferating hepatocytes. Other diseases and reasons which can also trigger HCC formation are nonalcoholic steatohepatitis (NASH), obesity, diabetes, aflatoxin-contaminated nutrients, exposure to toxic compounds like vinyl chloride, and genetic predisposition such as hemochromatosis [55, 56].
6.8 A Deeper View on TNF and NF-κB
Only two possibilities remain if a cell is faced with strong stress: survival or death. TNFα and IL-1 are classical stress inducers and belong to the TNF superfamily. Next to these prominent members, Fas (Apo-1) and TRAIL are also well known and are currently intensively studied proteins. These signaling cascades simultaneously induce expression of pro-survival and proapoptotic proteins and, according to the stimulation strength, determine if the cell dies or survives [57,58,59].
The balance between life and death is absolutely essential to keep the liver in a healthy status. Apoptosis and necroptosis are different ways of cellular death but are strictly coordinated processes with common molecular characteristics, but with different cellular fragmentation processes into several small pieces [60].
TNFα acts on different biological processes facilitated by TNF-R1 and TNF-R2. Soluble TNFα initiates TNF-R1 signaling, whereas activation of TNF-R2 signaling needs binding of membrane-bound TNFα [61]. Activation of the TNF-R1 by TNFα leads to its trimerization and initiates recruitment of different adaptor proteins building the membrane-bound TNFR-complex I. The complex I comprises TRADD, RIPK1, cIAP1/2, and TRAF2/5. All seven TRAF family members have a C-terminal coiled coil domain which supports protein-protein interaction. Only TRAF2-7 exhibits an N-terminal RING domain which transfers K63-linked ubiquitin to target proteins, acting as E3 ligases. Nevertheless, it is not clarified till now if TRAF proteins mainly act as E3 ligases or as adaptors [20, 62]. It was shown that TRAFs are involved in both, canonical and noncanonical NF-κB pathway, and those TRAFs are also needed for activation of other signaling cascades, therefore acting as a distributor platform for several pathways. RIPK1 is, differently to the TRAFs, exclusively engaged in the canonical NF-κB pathway. RIPK1 and TRAF2/5 seem to interact with each other by TRAF2/5-mediated K63-linked polyubiquitination of RIPK1 (Lys377) [63]. Alternatively, it is also discussed that cIAP1/2 is responsible for RIPK1 polyubiquitination and that TRAF2 only recruits them to the receptor complex. Despite intensive research the exact function of TRAF2/5 and cIAP1/2 concerning RIPK1 ubiquitination could not have been solved satisfactorily to date [64]. Besides K63-linked ubiquitination, RIPK1 undergoes a second, linear Met1-linked ubiquitination simultaneously. This is mediated by the linear ubiquitin chain assembly complex (LUBAC), another E3 ligase complex [65, 66]. RIPK1 is linked with the linear chain to NEMO and with the K63-linked chain to the TAB2/3-TAK1 complex and brings the IKK complex and TAK1 in closer vicinity to each other, leading to the phosphorylation of IKKβ and finally NF-κB activation as described above.
6.9 NF-κB and Its Function in Hepatogenesis
A deeper understanding of NF-κB importance and its components was achieved by using genetically modified mouse models. These models include a setup of diverse genetic approaches such as constitutive knockout models, dominant-negative expression or overexpression of single or double IKK subunits or IκB proteins, tissue-specific conditional knockouts by using cre/loxP recombination, reporter systems, and gene knock-ins [50].
Knockout mice deficient in Rela (p65) die in the uterus between embryonic days 15 (E15) and E16 because of hepatocyte apoptosis. The primary cause of this event is the failure of a TNFα-mediated IκBα induction and granulocyte/macrophage colony-stimulating factor (GM-CSF) as shown in murine embryonic fibroblasts (MEFs), revealing that RelA has a protective function against TNFα [67, 68]. These findings were approved by generating a double knockout of Tnf and Rela, which led to a normal embryonic development and a full rescue from lethality [69]. However, another study reported similar results, but these double knockout mice died 10 days after birth from acute hepatitis and neutrophil infiltration [70]. Both studies indicate that RelA and TNF-R1 are not essential for liver development in mice, but seem to sensitize these animals to infections leading to death within a very short time frame. Further investigations of other NF-κB subunits showed that the genetic loss of both c-Rel (Rel) and Rela causes liver failure as well [71]. Moreover, genetic ablation of both transcription factors triggers impaired maturation of B cells, T cells, and macrophages, denoting important roles in controlling genes relevant for immune responses. Genetic manipulation of murine livers by using adenoviral technique allowed transcription of an IκB superrepressor (Ad5IkappaB) which abolished NF-κB linkage to the respective DNA binding sites. After partial hepatectomy the adenoviral infected livers displayed increased apoptosis rates of hepatocytes, proving the importance of NF-κB [72]. For the remaining NF-κB subunits RelB, NF-κB1, and NF-κB2, they were shown to play important roles in the differentiation and proper function of hemopoietic cells [73].
Next to the studies on NF-κB subunits, some studies carried out to define the functional impact of the single IKK members. Constitutive deletion of Ikkβ (Ikk2 −/−) in mice leads to embryonic lethality at day E12.5 as a cause of enhanced liver damage. Cell culture experiments of primarily isolated MEFs from these animals revealed impaired NF-κB activation in response to TNFα and IL-1. These results revealed a central function for IKKβ in controlling liver development and NF-κB activity, whereas loss of IKKβ cannot be fully compensated by IKKα (IKK1) [74,75,76]. Ablation of the catalytic subunit Nemo (Ikkγ −/−) has a similar phenotypical effect as the embryos die between day E12.5 and E13.0 from massive apoptotic liver failure, and experiments with isolated Ikkγ −/− MEFs also showed disturbed NF-κB activity after treatment with TNFα, IL-1, LPS, and Poly(I:C), leading to high susceptibility to apoptosis [77]. As stated above, IKKα does not seem to have an impact on liver development and NF-κB induction triggered by pro-inflammatory substances. Ikkα −/− mice die perinatally and develop severe skeletal and skin-related defects during embryogenesis [78, 79].
6.10 NF-κB and Its Pivotal Role for Liver Integrity
Generation of tissue-specific knockouts by using the cre/loxP recombination system gives the opportunity of a much more detailed view on NF-κB in particular with regard to protection against cytokine-induced hepatitis. Another great advantage of the cre/loxP technology is the time-dependent loss of target genes determined by activation of the respective promoter during later embryonic developmental stages or postnatally, circumventing embryonic lethality effects. This was demonstrated by a conditional cre-driven hepatocyte knockout of Rela/p65. These mice are viable, and isolated primary hepatocytes treated with TNFα were highly sensitive to apoptosis with concurrently enhanced c-Jun N-terminal kinase (JNK) expression and degradation of the anti-apoptotic protein cellular FLICE inhibitory protein long (c-FLIPL) [80]. Investigation of hepatocyte-specific Ikkβ deletion in adult mice revealed an unexpected slight sensitivity in response to TNFα or LPS administration contrary to the murine embryonical state [52, 80, 81]. However, treatment with concanavalin A (ConA) promotes severe liver failure in adult Ikkβ-deficient animals, which is mainly supported by increased activation of JNK, a key mediator of ConA-induced liver failure. Next to JNK activation, ConA is also a potent activator of T cells, indicating that the anti-apoptotic function of IKKβ is the prevention of T cell-mediated cell death associated with decreased JNK activity [81].
Metabolic diseases represent another potential risk factor in mediating inflammatory processes like type 2 diabetes and obesity. Liver-specific Ikkβ ablation abolished insulin sensitivity, while these animals showed insulin resistance in muscle and fat induced by a high-fat diet and upon aging [82].
Double knockout of Ikkα and Ikkβ (IKKα/βLPC-KO) in liver parenchymal cells (hepatocytes and cholangiocytes) supported increased susceptibility of hepatocytes to LPS in vivo, which was not detected in the single knockout conditions, claiming for a more redundant function for both IKKs in canonical NF-κB activation. Of note, simultaneous deficiency of IKKα and IKKβ or the combined ablation of Ikkα and Nemo, but not Nemo alone, led to spontaneous development of cholangitis with disturbed portal bile ducts accompanied by severe jaundice, revealing the importance of both catalytic NF-κB subunits in controlling liver immunology and bile duct integrity [83]. Additional pivotal discoveries concerning the physiological impact of the IKK complex were achieved by examining adult mouse livers lacking the regulatory subunit NEMO. These livers are highly sensitive against TNF- and LPS-mediated inflammation and subsequently cell death in vivo and in vitro [8, 52].
Further studies regarding genetic ablation of other NF-κB signaling members alone or in combination with different IKKs revealed a great impact on cellular homeostasis as well. Hepatocyte-specific deletion of the mitogen-activated kinase kinase kinase (MAP3K) TGF-β-activated kinase 1 (TAK1LPC-KO; TAK1Δhep) leads to a comparable phenotype as seen for IKKα/βLPC-KO with considerable cholangitis, early HCC development, and lethal jaundice at younger age [6, 51]. Deletion of death receptor-associated adaptor proteins like Fas-associated protein with death domain (Fadd) or Tnfr1 in combination with Nemo (NEMO/FADDLPC-KO) or Tak1 (TAK1/TNFR1∆hep) showed strongly reduced signs of inflammation, fibrosis, and cell death, raising evidence for a pro-apoptotic trigger driving these phenotypes [8, 51]. A look more downstream of the death receptor pathways, regarding casapse-8, highlighted a rescue of Caspase8/NEMO∆hep mice from steatosis and HCC development, but these animals developed a severe spontaneous phenotype of liver necrosis, cholestasis, and biliary lesions, most likely caused by a FasR-induced RIPK1-RIPK3-mediated necroptosis [84]. As stated above, the hepatoprotective function of NF-κB is also influenced by a sustained reduced expression level of JNK upon TNFα administration [85,86,87]. JNK belongs to the MAPK family and is activated via TRAF2, RIPK1, and MKK4/7. JNK is a major mediator of cell death, triggered not only by pro-inflammatory cytokines such as TNFα and IL-1β but also by cellular stressors such as UV radiation, osmotic, oxidative, hypoxic, and genotoxic events [88, 89]. The connective bridge between NF-κB and JNK is TAK1, which is able to phosphorylate IKKβ and MKK4/7, depending on stimulus strength. Next to JNK, TAK1 is also able to phosphorylate MKK3/6, which are activators of p38, another MAPK involved in cell proliferation, differentiation, and cell death [90, 91]. Studies with p38α/IKK2LPC-KO mice showed increased hepatocyte sensitivity against TNFα and LPS administration in vivo, which was not the case for the single knockout conditions. Moreover, significantly increased JNK expression levels could be detected, but were not strong enough to induce liver failure [92]. The results revealed impressively that the NF-κB signaling cascade is not an isolated pathway and is embedded in a broad network of stress-related signaling pathways which are tightly regulated in protecting the liver against harmful events.
6.11 NF-κB and Its Implication for HCC Development
Chronic inflammatory liver diseases belong to the main preconditions for generation and progression of liver cirrhosis and subsequently HCC development, affecting 80–90% of patients with liver cirrhosis [55]. The formation of cancer is generally characterized by a disbalance between cell death and survival/proliferation [93]. In most HCCs, NF-κB is constantly active, which drives a continuous burst of pro-inflammatory and anti-apoptotic signals [94, 95]. In the last two decades, murine genetic studies had a great impact on the understanding of molecular mechanisms driving HCC development [6,7,8,9, 96]. It has been shown that tumor formation is a process in which different liver cell types are involved and where NF-κB activation is time-dependent altered, particularly at early and late stages of cancer development.
One of the first studies in this direction was done on Multidrug resistance protein 2 (Mdr2 −/−) mice (human homologue MDR3), lacking a permeability (P)-glycoprotein which is located in the bile canalicular membrane of hepatocytes with a function as a phospholipid export pump. Disruption of the functionality of the pump causes a spontaneous phenotype described by cholangitis with a dysfunctional biliary delivery, ending in HCC development at 4–6 months of age [96]. In this phenotype, enhanced NF-κB activity leads to a higher TNFα expression, and impairment of NF-κB activity with a hepatocyte-specific inducible IκB superrepressor transgene negatively affected tumor progression and hepatocyte cell death at later stages of tumor development, whereas NF-κB blockage at the initial stage of tumor formation has no inhibitory effect [97]. Besides this tumor-promoting effect of NF-κB, other studies argue for a tumor-suppressive function. Hepatocyte-specific knockout of Nemo promotes hepatitis in these animals at 2 months of age and spontaneous HCC development 12 months after birth, triggered by cytokines and a constant low intrinsic dosage of LPS coming from the commensal gut bacteria [8]. Further studies with different combined hepatocyte-specific knockouts of Nemo and Tnfr1, Trail-r, or Fas or quadruple knockout showed no rescue or improvement of this phenotype. Still the combination of NEMO/FADDLPC-KO caused a much milder progress of liver failure and inflammation and abolished HCC development. Examination of liver sections from 1-year-old NEMOLPC-KO/Tnf−/− mice with developed liver tumors revealed that TNF is not important for the development of spontaneous cell death, hepatitis, and HCC in NEMOLPC-KO livers. Neither depletion of natural killer cells nor an intercrossing with Rag-1 ablated mice (NEMOLPC-KO/Rag1−/−) prevented liver damage [49]. All these results demonstrated that the spontaneously developed hepatocyte-specific NEMO phenotype is neither the result of death receptor-mediated signaling cascades nor an immune response triggered once, and additional studies are needed to identify the mechanism behind. Interestingly, double knockout of Nemo and Tak1 in hepatocytes rescued the massive phenotype detected in the Tak1 single knockout condition (as stated above), claiming for a NF-κB-independent tumor formation in the TAK1LPC-KO livers [6, 8]. Generation of knock-in mice endogenously expressing catalytically inactive RIPK1 D138N (Ripk1 D138N/D138N) are alive after birth unlike mice conditionally lacking Ripk1. Moreover, these mice are protected against TNFα treatment and poly (I:C)-induced necroptosis in vitro and TNFα administration in vivo, indicating that the kinase activity of RIPK1 is not a prerequisite for cell survival but is crucial for TNFα-induced necroptosis [53]. Moreover, this result is supported by the finding that hepatocyte death and HCC development in NEMOLPC-KO mice is triggered by RIPK1’s kinase activity, independent of NF-κB activity and RIPK1’s scaffolding function. A complete NF-κB blockage induced by hepatocyte-specific single or combined knockout of Rela, c-rel, or Relb did not affect the liver, whereas constitutively active IKKβ prevented hepatocarcinogenesis in NEMOLPC-KO animals. These results revealed a NEMO protective function against HCC development. Hepatocyte-specific ablation of RIPK1 activated a TRADD-related apoptosis and HCC development, showing two different functions of RIPK1 [98].
Hepatocarcinogenesis chemically induced by a single injection of diethylnitrosamine (DEN) in 15-day-old IKKβΔhep mice causes in 2-month-old mice massive liver tumor development which is not seen in untreated IKKβΔhep livers [9, 81]. Compared to the untreated mice, DEN treatment supports enhanced ROS production correlating with increased JNK expression levels, hepatocyte death, and compensatory hepatocyte proliferation, which is similar to the results of the ConA-treated IKKβΔhep mice [81]. Moreover, experiments with IKKβ−/− fibroblasts showed that antioxidants like manganese superoxide dismutase (MnSOD) or MAPK phosphatases are needed to avoid ROS-mediated sustained JNK activity [99]. Besides ROS, nitric oxide (NO•) is another kind of agent radically synthesized by inducible nitric oxide synthase (iNOS), which is able to induce chronic inflammation and might influence tumor formation by controlling cell proliferation, angiogenesis, survival, medical resistance, and DNA repair [100,101,102]. Examination of iNos knockout mice showed significantly reduced NF-κB activities, and a higher concentration of iNOS is related to tumor proliferation, genomic instability microvascularization, and worse diagnosis for HCC patients. Treatment with iNOS inhibitors, like aminoguanidine, has a negative effect on HCC progression and NF-κB activity and a positive influence on apoptosis in vivo and in vitro. Moreover, interruption of the NF-κB cascade with sulfasalazine or siRNA led to decreased iNOS expression in different HCC cell lines [103].
Next to the components of the canonical NF-κB pathway, HCC development could also be triggered via the LTβR, one of the activators of the noncanonical NF-κB pathway. Enhanced levels of LTα, LTβ, and LTβR were detected in human HBV-/HCV-infected livers and in HCC. Examination of the transgenic mouse models of LTα and LTβ showed that hepatocyte-specific overexpression promotes fibrosis and HCC development. Ablation of IKKβ, specifically in hepatocytes, rescued mice from HCC development [7].
These results indicate a carcinogenesis promoting function of NF-κB, which is strongly supported by pro-inflammatory mediators.
6.12 NF-κBs Contribution on Liver Fibrosis and Cirrhosis
Liver fibrosis is a disease that develops as a consequence of recurrent “out-of-control” wound healing processes. Persistent removal of damaged and/or inflamed tissue with compensatory renewal leads over time to composition of fibrillar collagen in the affected areas, ending up in scarring liver tissue and loss of metabolic functional areas. Hepatic stellate cells (HSCs) are mainly involved in this process, whereas after certain stimuli this cell population is activated and undergoes a transformation to hepatic myofibroblasts which are built by decomposition of extracellular matrix proteins the collagen scars in the tissue. HBV and HCV infections, autoimmune hepatitis, alcohol abuse, NASH, and cholestasis trigger liver fibrosis and finally cirrhosis. For a long period, it was assumed that activated HSCs/hepatic myofibroblasts are the main source of liver fibrosis, but it seems to be a multifaceted process in which different signaling pathways and other liver cell types such as hepatocytes and Kupffer cells might be of importance [104]. Examination of intercrossed mice with a constitutively active human IKKβ (CAIKK2) allele in postnatal livers, controlled by a tetracycline promoter system, revealed modest liver injury, infiltration of immune cells, enhanced hepatocyte proliferation, and spontaneous liver fibrosis progression. Deeper analysis detected significantly enhanced levels of chemokines and certain chemokine receptors, while interruption of CAIKK2 expression led to declined expression levels in hepatocytes. Moreover, disruption of CAIKK2 expression for few weeks also reduced HSC activation but without significant improvement of fibrosis reduction. Only macrophage removal with liposomal clodronate positively affected liver fibrosis development caused by a reduced NF-κB activation. This indicates that next to transformed HSCs, recruitment of pro-inflammatory immune cells such as macrophages, mediated by prolonged hepatocellular NF-κB activation, are involved in promoting liver fibrosis [105]. Previous studies detected in activated HSCs enhanced levels of NF-κB, correlating not only with increased expression of pro-inflammatory and adhesion molecules, but also with upregulation of anti-apoptotic proteins like TRAF1/2, cIAP1/2, Bcl-XL, and GADD45β [106]. Moreover, it seems that the CD95/Fas pathway is responsible for HSC death, whereas TGF-β and TNFα induce pro-survival signaling. Disruption of NF-κB activity by overexpression of an IκB superrepressor led to decreased expression levels of anti-apoptotic proteins like Bcl-XL and enhanced proapoptotic proteins as caspase-3 during TGF-β and TNFα stimulation [107, 108]. Another study revealed a constitutive phosphorylation of p65 at Ser536 mediated by IKKβ and the autocrine renin-angiotensin system in human hepatic myofibroblasts. IKKβ-mediated phosphorylation of p65 allows nuclear translocation, whereas angiotensin II promotes myofibroblast survival in an autocrine and paracrine way [109]. Besides increased NF-κB activation, enhanced p-JNK levels could be detected in human- and murine-activated HSCs, indicating that JNK has an important function in liver fibrosis progression [110].
In addition to the death receptor family, two members of the Toll-like receptor (TLR) family, TLR4 and TLR9, were uncovered to be involved in mediating NF-κB activation during liver fibrosis progression as their activation could be a consequence of gut bacterial components [111, 112].
6.13 NF-κB in HVB/HCV Infections
Approximately 3% of the total world population have been infected with HCV, whereas more than 170 million people are suffering from chronic hepatitis, cirrhosis, and HCC. The annual rate of HCC development is strongly increased in cirrhotic livers [113]. The HCV core, the envelop protein E2, and HCV subgenomic replicons have been shown to enhance p38 and extracellular signal-regulated kinase (ERK) phosphorylation and initiate NF-κB [114, 115]. Moreover, it seems that oxidative stress (ROS) and TGF-β1 are important mediators of fibrogenesis in HCV infection. Silencing of p38, JNK, ERK1/2, and NF-κB (p65) in hepatocytes in vitro revealed their implication in enhanced TGF-β1 expression [116]. Experiments using different inhibitors for ROS (diphenyliodonium [DPI]), JNK (SP600125), IRE1 (Irestatin 9389), and NF-κB (6-amino-4-(4-phenoxyphenylethylamino [AQ]), have blocked significantly HCV-induced NF-κB and TGF-β1-mediated SMAD signaling. Silencing of JNK and IRE1 using siRNA inhibited efficiently ER stress, ROS, NF-κB, and TGF-β1 activity [117]. Moreover, it was shown in vitro that the hepatitis B virus X (HBx) protein, in addition to HCV infection, maintains enhanced NF-κB and AP-1 activation which might indicate a supportive role of HBx in HCC development [118]. Patients suffering from HBV have a much higher potential to develop HCC confirmed by studies which detected in nearly all examined HCCs of HBV patients chromosomally integrated viral DNA. Next to the HBx protein, the HBV surface antigen PreS2 is able to activate NF-κB and AP-1 via PKC-mediated induction of the c-Raf-1/MAPK signaling pathway, which further leads to increased hepatocyte proliferation, indicating a tumor-promoting function for PreS2 [119].
Stimulation of HCV core protein infected HeLa and HuH-7 cells with TNFα or LTα1/β2 mediates increased or sustained IκBβ degradation, whereas degradation of IκBα was only detected in LTα1/β2-stimulated HeLa cells. Besides cytokine treatment, higher levels of NF-κB activity were also detected in untreated HeLa and HuH-7 cells only harboring the HCV core protein. This finding shows that the HCV core protein is able to positively alter NF-κB initiation and, in combination with cytokine treatment, markedly increased this effect, which might have a direct influence on a stable and continuous NF-κB activation in HCV-infected cells [120].
After viral infection, the immune system reacts with the production of different NF-κB-related cytokines, especially IFNs to defend the infection [121,122,123]. NF-κB and IFN regulatory factor-3 (IRF-3) initiate alone or together the antiviral Janus kinases/signal transducer and activator (JAK-STAT) signaling cascade [124]. Despite the protective host defense response to eliminate viral infection accompanied by removing and restoring damaged tissue, continuous NF-κB activation also has negative side effects, such as activation of quiescent HSCs or proliferation of hepatocytes with oncogenic mutations, leading to liver fibrogenesis and HCC development. Therefore, therapeutics are needed which keep virus-induced oxidative stress or mediators of fibrosis at a minimum [125, 126].
6.14 NF-κB and Its Meaning for Obesity
Overweight and metabolic diseases have strongly increased all over the world, ranging from childhood to adult stage. The WHO estimated for 2015 that around 2.3 billion adults would be overweighted and approximately 700 million people would be obese [127]. Obesity is a gate opener not only for several diseases, such as insulin resistance, type 2 diabetes, cardiovascular disease, and atherosclerosis, but also for dementia, airway disease, and cancer [128, 129]. Genetic examination of murine and human adipose tissue, as well as murine hepatic tissue, uncovered strongly increased activation of an inflammatory and immune response gene network triggered by a multifaceted genetic loci and environmental issues [130,131,132]. However, the molecular mechanism of how obesity influences macrophage response is not yet fully understood. Currently, different theories are trying to explain how macrophages and obesity are connected to each other, e.g. by TLR4 induction through a high content of saturated fats, by inflammation of the central nervous system, or by the commensal gut microbiota [133,134,135,136].
Adipocytes are the cellular components of the adipose tissue and next to lipid storage they are also able to generate and release pro-inflammatory cytokines and adipokines which attract monocytes and T cells to infiltrate the adipose tissue, where monocytes differentiate to M1 macrophages. It is discussed that one major source triggering this process is metabolic stress caused by overnutrition, which leads to high levels of non-metabolized free fatty acids and ER stress inducing inflammatory responses. However, also removal of apoptotic adipocytes is another potent inducer of macrophage recruitment [128]. Besides macrophage recruitment, activation of TLRs is considered to initiate NF-κB-mediated generation of pro-inflammatory molecules. The NF-κB target IKKε was detected to be needed for high-fat diet (HFD)-induced obesity. Depletion of IKKε revealed a positive influence on high-fat diet fed mice, because these animals were protected from insulin resistance and hepatic steatosis, and they did not show any sign of chronic inflammation in liver or adipose tissue or induction of inflammatory pathways [137]. Similar results were obtained by HFD-treated mice with IKKβ deficiency in hepatocytes (Ikbkb Δhep) or in myeloid (Ikbkb Δmye) cells. These animals showed liver-specific insulin sensitivity, but revealed insulin resistance in muscle and adipose tissue, which was also examined in obese or aged mice. The Ikbkb Δmye mice had preserved total insulin sensitivity with protection against insulin resistance [82]. Constitutively expressed IKKβ in hepatocytes caused increased NF-κB activity comparable to values detected in HFD-treated or obese mice. These animals also developed type 2 diabetes (T2D) with characteristic features such as hyperglycemia, severe hepatic insulin resistance and mild systemic insulin resistance, also affecting muscle tissue. Liver-specific expression of the IκBα superrepressor rescued this phenotype [138]. These results indicate a potential NF-κB function in obesity-related inflammation and T2D locally in the liver and myeloid cells, which seem to be crucial for improving systemic insulin resistance. Sustained excessive food intake also leads to NF-κB initiation via different pro-inflammatory pathways as in adipocytes and macrophages; TLR4 was discovered to be activated by free fatty acids. Ablation of TLR4 protected these mice from insulin resistance in muscle tissue, disturbed glucose metabolism, and inflammatory signaling in liver and adipose tissue [139]. Additional studies showed that loss of function mutation in TLR4 also hindered the development of insulin resistance in adipocytes and in diet-initiated obesity [135, 140, 141]. Next to TLR4, TLR2 seems to have a comparable role in mediating NF-κB activation during HFD-induced obesity and the formation of insulin resistance [142, 143]. Metabolic stress signals attract monocytes to remove apoptotic pancreatic β-cells, thereby secreting TNFα, IL-6, and IL-1 to support β-cell dying. Increased TNFα levels in blood and peripheral tissues were detected in insulin-resistant rodents as well, and HFD promotes enhanced secretion of TNFα, IL-6, and IL-1 in hepatocytes and adipocytes. Inactivation of TNFα in different genetic rodent models mediated a significant peripheral glucose uptake during insulin secretion. Moreover, hepatocyte deficiency of IL-6 receptor (IL-6RαL-KO) impaired obesity-related insulin resistance and glucose tolerance. Depletion of IL-6Rα induced a massive inflammatory response indicated by enhanced levels of TNFα, IL-6, and IL-10 and IκBα phosphorylation. Glucose tolerance in these animals was reinstated by TNFα neutralization or Kupffer cell deficiency [144,145,146]. All these results imply a critical role for NF-κB in obesity-mediated insulin resistance and T2D progression and that a controlled release of inflammatory inducers is necessary for a healthy hepatic metabolism.
6.15 NF-κB and Its Function in Hepatic Ischemia/Reperfusion (I/R) Injury
During and after liver transplantation or hepatic resection, ischemia/reperfusion (I/R) is a main reason for liver failure caused by hypoxia or anoxia if oxygen supply and tissue pH are reinstated after clamping of the hepatic blood flow. The pathobiochemical mechanisms behind I/R are versatile and affect all liver cell types [147, 148].
Several studies were done to clarify if I/R induces apoptosis or necroptosis [149,150,151,152,153,154]. During I/R, hepatocytes express damage-associated molecular patterns (DAMPs)/pathogen-associated molecular patterns (PAMPs) on their surface, and macrophages, Kupffer cells, and dendritic cells bind these DAMPs/PAMPS by TLRs, thereby initiating the immune system. Adaptive and innate immune cells are recruited to the affected tissue sections where they secrete additional inflammatory mediators as TNFα, IL-1, IL-12, and INFγ which fuel these processes [148, 155]. Hepatic adenoviral overexpression of an IκB superrepressor in rats impeded NF-κB activation by decreased TNFα expression, preventing negative effects of hepatic I/R [156]. Furthermore, it was shown that heat shock preconditioning of rat livers significantly impaired I/R-induced NF-κB activation and expression of several inflammatory mediators [157]. Conditional ablation of Ikkβ in murine hepatocytes led to impaired liver necrosis and inflammation during I/R compared to the wild-type condition. Also administration of the chemical IKKβ inhibitor AS602868 failed to induce I/R-mediated liver failure without induction of pro-apoptotic side effects, making this inhibitor potentially useful for therapy [52]. A20 is another critical NF-κB target gene activated during inflammatory processes in hepatocytes and is involved in blocking apoptosis, but is also part of a negative feedback loop to control NF-κB activation [158,159,160]. Murine hepatic recombinant adenoviral overexpression of A20 revealed a significantly increased survival rate after I/R in these animals, indicated by considerably reduced bilirubin and transaminase levels, reduced hemorrhagic necrosis and steatosis, and enhanced hepatocyte proliferation. Moreover, A20 induced the release of peroxisome proliferator-activated receptor alpha (PPARα), an important controller of lipid homeostasis and oxidative damage, which prevented oxidative induced necrosis [161].
These results revealed that interference of IKKβ activation by pharmacological treatment is a putative instrument to protect livers from I/R injury. A20, another crucial NF-κB target, seems to be an additional promising candidate which might have the capacity as a therapeutic target which efficiently blocks I/R [86, 161]. In contrast, mice given recombinant receptor activator of NF-κB ligand (RANKL) before or during hepatic I/R showed enhanced NF-κB activation accompanied by less liver damage [162].
6.16 NF-κB as Therapeutic Target
Liver diseases are characterized by multifaceted biochemical processes, whereas the balance between apoptotic and survival signaling pathways is of great importance for liver homeostasis. Impaired homeostasis leads to chronic inflammation and compensatory proliferation ending up in liver cirrhosis and HCC development. Tremendous work was done in these fields, and particularly the use of genetic modified mouse models showed that NF-κB is one of the most important key players in preserving liver integrity. However, the function of NF-κB in the development of liver diseases is not black and white nor is it equal for all liver cell types. This makes it extremely challenging to design cell type and disease-specific drugs which only affect the cells of interest and not interfering with NF-κB activity or other signaling cascades in healthy cells.
Several biological and chemical compounds have been tested to modify NF-κB activity as steroids, selective estrogen receptor modulators, antioxidants, proteasome inhibitors, and IKK inhibitors [126].
The administration of hormones is critical to assess because of the increased risk of negative side effect induction in the liver or other organs or their prospective impact on the respective gender [126, 163]. Phytochemicals from tea or other plants have a great antioxidant potential which seems to have a significant positive influence on liver health, but further investigations are needed for refining substance purification and antioxidant composition [164,165,166,167]. The development of chemical compounds, such as proteasome inhibitors like Bortezomib, mediates reduced NF-κB activation. Treatment of human hepatoma cells with MG132, another protease inhibitor, resulted in apoptosis induction but affected the β-catenin pathway. Currently, the use of proteasome inhibitors as a therapeutic target needs more scientific research because of the diversity of bad side effects induced next to decreased NF-κB activation [168,169,170,171]. Regarding specific NF-κB blockage, the IKK complex member IKKβ seems to be a putative target. It has been shown that aspirin and sulfasalazine are able to impair the catalytic activity of the IKK complex. Furthermore, the development of a new class of chemical compounds, referred to as “small molecules,” is able to impede NF-κB activation by binding to the ATP-binding pocket of IKKβ, thereby inducing conformational changes. Despite the higher binding specificity of small molecules for their targets and effectiveness compared to other compounds, a potential risk remains for the development of side effects, such as induction of inflammatory responses [126, 172, 173].
In addition to biological and chemical compounds, therapeutics on the DNA/RNA level have also been developed. Different studies were done with small interfering RNAs (siRNAs), microRNAs (miRNAs), and antisense oligodeoxynucleotides (ODNs), placing emphasis on liver fibrosis. Several studies have obtained promising results, but more genetic and clinical trial studies are needed to gain a better mechanistic understanding and to improve application effectiveness of small RNA and DNA products to enhance therapeutic success in the treatment of liver diseases [174].
In summary, further scientific and clinical studies are needed to develop effective therapeutics that are fine-tuned for the control of hepatic NF-κB activation to prevent progression of liver diseases, but without inducing harmful side effects.
References
Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi:10.1016/0092-8674(86)90346-6.
Barnes PJ, Karin M. Nuclear factor-κB—a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi:10.1056/NEJM199704103361506.
Camandola S, Mattson MP. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets. 2007;11:123–32. doi:10.1517/14728222.11.2.123.
MacLellan WR, Schneider MD. Death by design programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–44. doi:10.1161/01.RES.81.2.137.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi:10.1016/j.cell.2010.01.025.
Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, Weber A, Weiskirchen R, Liedtke C, Gassler N, Müller M, de Vos R, Wolf MJ, Boege Y, Seleznik GM, Zeller N, Erny D, Fuchs T, Zoller S, Cairo S, Buendia M-A, Prinz M, Akira S, Tacke F, Heikenwalder M, Trautwein C, Luedde T. TAK1 suppresses a NEMO-dependent but NF-κB-independent pathway to liver cancer. Cancer Cell. 2010;17:481–96. doi:10.1016/j.ccr.2010.03.021.
Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien P-A, Thimme R, Blum H, Nedospasov SA, Zatloukal K, Ramzan M, Ciesek S, Pietschmann T, Marche PN, Karin M, Kopf M, Browning JL, Aguzzi A, Heikenwalder M. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi:10.1016/j.ccr.2009.08.021.
Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi:10.1016/j.ccr.2006.12.016.
Maeda S, Kamata H, Luo J-L, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi:10.1016/j.cell.2005.04.014.
Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. 2014;14:289–301. doi:10.1038/nri3646.
Augustin R, Fraune S, Bosch TCG. How Hydra senses and destroys microbes. Semin Immunol. 2010;22:54–8. doi:10.1016/j.smim.2009.11.002.
Hemmrich G, Miller DJ, Bosch TCG. The evolution of immunity: a low-life perspective. Trends Immunol. 2007;28:449–54. doi:10.1016/j.it.2007.08.003.
Lange C, Hemmrich G, Klostermeier UC, López-Quintero JA, Miller DJ, Rahn T, Weiss Y, Bosch TCG, Rosenstiel P. Defining the origins of the NOD-like receptor system at the base of animal evolution. Mol Biol Evol. 2011;28:1687–702. doi:10.1093/molbev/msq349.
Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier MEA, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, Larroux C, Putnam NH, Stanke M, Adamska M, Darling A, Degnan SM, Oakley TH, Plachetzki DC, Zhai Y, Adamski M, Calcino A, Cummins SF, Goodstein DM, Harris C, Jackson DJ, Leys SP, Shu S, Woodcroft BJ, Vervoort M, Kosik KS, Manning G, Degnan BM, Rokhsar DS. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466:720–6. doi:10.1038/nature09201.
Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi:10.1038/sj.onc.1209954.
Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from C. elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58. doi:10.1038/nri2689.
Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi:10.1146/annurev.iy.12.040194.001041.
Blank V, Kourilsky P, Israël A. NF-κB and related proteins: rel/dorsal homologies meet ankyrin-like repeats. Trends Biochem Sci. 1992;17:135–40. doi:10.1016/0968-0004(92)90321-Y.
Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: structural views. Oncogene. 1999;18:6845–52. doi:10.1038/sj.onc.1203224.
Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–62. doi:10.1016/j.cell.2008.01.020.
Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of κB DNA binding by Rel/NF-κB dimers. J Biol Chem. 2000;275:24392–9. doi:10.1074/jbc.M003784200.
Saha A, Hammond CE, Trojanowska M, Smolka AJ. Helicobacter pylori-induced H,K-ATPase alpha-subunit gene repression is mediated by NF-kappaB p50 homodimer promoter binding. Am J Physiol Gastrointest Liver Physiol. 2008;294:G795–807. doi:10.1152/ajpgi.00431.2007.
Inoue J, Kerr LD, Rashid D, Davis N, Bose HR, Verma IM. Direct association of pp40/I kappa B beta with rel/NF-kappa B transcription factors: role of ankyrin repeats in the inhibition of DNA binding activity. Proc Natl Acad Sci U S A. 1992;89:4333–7.
Huxford T, Huang D-B, Malek S, Ghosh G. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell. 1998;95:759–70. doi:10.1016/S0092-8674(00)81699-2.
Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–58.
Malek S, Huang D-B, Huxford T, Ghosh S, Ghosh G. X-ray crystal structure of an IκBβ·NF-κB p65 homodimer complex. J Biol Chem. 2003;278:23094–100. doi:10.1074/jbc.M301022200.
Huxford T, Ghosh G. A structural guide to proteins of the NF-κB signaling module. Cold Spring Harb Perspect Biol. 2009;1:a000075. doi:10.1101/cshperspect.a000075.
Michel F. Crystal structure of the ankyrin repeat domain of Bcl-3: a unique member of the IkappaB protein family. EMBO J. 2001;20:6180–90. doi:10.1093/emboj/20.22.6180.
Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–23. doi:10.1038/ni.2060.
Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–15. doi:10.1038/cr.2010.178.
Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi:10.1038/ni.2065.
Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–13.
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–54. doi:10.1038/41493.
Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, Chiu Y-H, Deng L, Chen ZJ. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–48. doi:10.1016/j.molcel.2004.08.008.
Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. 2014;49:185–94. doi:10.1007/s00535-013-0931-x.
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–51. doi:10.1038/35085597.
Yu Y, Ge N, Xie M, Sun W, Burlingame S, Pass AK, Nuchtern JG, Zhang D, Fu S, Schneider MD, Fan J, Yang J. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFκB and AP-1 activation as well as IL-6 gene expression. J Biol Chem. 2008;283:24497–505. doi:10.1074/jbc.M802825200.
Coope HJ, Atkinson PGP, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GGB, Johnston LH, Ley SC. CD40 regulates the processing of NF‐κB2 p100 to p52. EMBO J. 2002;21:5375–85. doi:10.1093/emboj/cdf542.
Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem. 2003;278:23278–84. doi:10.1074/jbc.M300106200.
Dejardin E. The alternative NF-κB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–79. doi:10.1016/j.bcp.2006.08.007.
Fong A, Sun S-C. Genetic evidence for the essential role of β-transducin repeat-containing protein in the inducible processing of NF-κB2/p100. J Biol Chem. 2002;277:22111–4.
Shih VF-S, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi:10.1038/cr.2010.161.
Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–9.
Liang C, Zhang M, Sun S-C. beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. 2006;18:1309–17. doi:10.1016/j.cellsig.2005.10.011.
Sun S-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21:71–85. doi:10.1038/cr.2010.177.
Bettermann K, Benesch M, Weis S, Haybaeck J. SUMOylation in carcinogenesis. Cancer Lett. 2012;316:113–25. doi:10.1016/j.canlet.2011.10.036.
Gao C, Huang W, Kanasaki K, Xu Y, Gao C, Huang W, Kanasaki K, Xu Y. The role of ubiquitination and sumoylation in diabetic nephropathy, the role of ubiquitination and sumoylation in diabetic nephropathy. BioMed Res Int. 2014;2014:e160692. doi:10.1155/2014/160692.
Mabb AM, Miyamoto S. SUMO and NF-kappaB ties. Cell Mol Life Sci. 2007;64:1979–96. doi:10.1007/s00018-007-7005-2.
Ehlken H, Krishna-Subramanian S, Ochoa-Callejero L, Kondylis V, Nadi NE, Straub BK, Schirmacher P, Walczak H, Kollias G, Pasparakis M. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014;21:1721–32. doi:10.1038/cdd.2014.83.
Gerondakis S, Grumont R, Gugasyan R, Wong L, Isomura I, Ho W, Banerjee A. Unravelling the complexities of the NF-κB signalling pathway using mouse knockout and transgenic models. Oncogene. 2006;25:6781–99. doi:10.1038/sj.onc.1209944.
Inokuchi S, Aoyama T, Miura K, Österreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A. 2010;107:844–9. doi:10.1073/pnas.0909781107.
Luedde T, Assmus U, Wüstefeld T, Meyer zu Vilsendorf A, Roskams T, Schmidt-Supprian M, Rajewsky K, Brenner DA, Manns MP, Pasparakis M, Trautwein C. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest. 2005;115:849–59. doi:10.1172/JCI200523493.
Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Lee TH, Chan FKM, Pasparakis M, Kelliher MA. RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–43. doi:10.4049/jimmunol.1400590.
Martini FH, Timmons MJ, Tallitsch RB. Human anatomy. 8th ed. Boston, MA: Pearson; 2014.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27. doi:10.1056/NEJMra1001683.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. doi:10.1053/j.gastro.2007.04.061.
Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41. doi:10.1038/cdd.2011.155.
Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350–64. doi:10.1038/cdd.2014.81.
Schleich K, Warnken U, Fricker N, Öztürk S, Richter P, Kammerer K, Schnölzer M, Krammer PH, Lavrik IN. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell. 2012;47:306–19. doi:10.1016/j.molcel.2012.05.006.
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–59. doi:10.1016/j.bbamcr.2013.06.001.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi:10.1038/sj.cdd.4401189.
Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8:7. doi:10.1186/1750-2187-8-7.
Israël A. The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi:10.1101/cshperspect.a000158.
Wertz IE, Dixit VM. Signaling to NF-κB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2:a003350. doi:10.1101/cshperspect.a003350.
Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–44. doi:10.1016/j.molcel.2009.10.013.
Walczak H, Iwai K, Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. doi:10.1186/1741-7007-10-23.
Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–4.
Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–70. doi:10.1038/376167a0.
Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci U S A. 1999;96:2994–9.
Rosenfeld ME, Prichard L, Shiojiri N, Fausto N. Prevention of hepatic apoptosis and embryonic lethality in RelA/TNFR-1 double knockout mice. Am J Pathol. 2000;156:997–1007.
Grossmann M, Metcalf D, Merryfull J, Beg A, Baltimore D, Gerondakis S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc Natl Acad Sci U S A. 1999;96:11848–53.
Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest. 1998;101:802–11. doi:10.1172/JCI483.
Gerondakis S, Grumont R, Rourke I, Grossmann M. The regulation and roles of Rel/NF-κB transcription factors during lymphocyte activation. Curr Opin Immunol. 1998;10:353–9. doi:10.1016/S0952-7915(98)80175-1.
Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999a;284:321–5.
Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999b;189:1839–45.
Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–9.
Rudolph D, Yeh W-C, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 2000;14:854–62.
Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–20.
Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–6.
Geisler F, Algül H, Paxian S, Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–503. doi:10.1053/j.gastro.2007.03.033.
Maeda S, Chang L, Li Z-W, Luo J-L, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–37.
Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–8. doi:10.1038/nm1185.
Luedde T, Heinrichsdorff J, de Lorenzi R, De Vos R, Roskams T, Pasparakis M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc Natl Acad Sci U S A. 2008;105:9733–8. doi:10.1073/pnas.0800198105.
Liedtke C, Bangen J-M, Freimuth J, Beraza N, Lambertz D, Cubero FJ, Hatting M, Karlmark KR, Streetz KL, Krombach GA, Tacke F, Gassler N, Riethmacher D, Trautwein C. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology. 2011;141:2176–87. doi:10.1053/j.gastro.2011.08.037.
Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: the crosstalk between the NF-κB and JNK pathways. Biol Chem. 2009;390:965–76. doi:10.1515/BC.2009.111.
Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–9. doi:10.1152/ajpgi.00422.2005.
Wullaert A, Heyninck K, Beyaert R. Mechanisms of crosstalk between TNF-induced NF-κB and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–101. doi:10.1016/j.bcp.2006.07.003.
Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52.
Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–95. doi:10.1080/15216540500097111.
Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–60. doi:10.1016/S0968-0004(00)01595-4.
Schieven GL. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem. 2005;5:921–8. doi:10.2174/1568026054985902.
Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 2008;9:1048–54. doi:10.1038/embor.2008.149.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi:10.1016/j.cell.2011.02.013.
Li W, Tan D, Zenali MJ, Brown RE. Constitutive activation of nuclear factor-kappa B (NF-kB) signaling pathway in fibrolamellar hepatocellular carcinoma. Int J Clin Exp Pathol. 2010;3:238–43.
Tai DI, Tsai SL, Chang YH, Huang SN, Chen TC, Chang KS, Liaw YF. Constitutive activation of nuclear factor kappaB in hepatocellular carcinoma. Cancer. 2000;89:2274–81.
Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, van den Bergh Weerman MA, Verkruisen RP, Groen AK, Oude Elferink RP. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145:1237–45.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi:10.1038/nature02924.
Kondylis V, Polykratis A, Ehlken H, Ochoa-Callejero L, Straub BK, Krishna-Subramanian S, Van T-M, Curth H-M, Heise N, Weih F, Klein U, Schirmacher P, Kelliher M, Pasparakis M. NEMO prevents steatohepatitis and hepatocellular carcinoma by inhibiting RIPK1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell. 2015;28:582–98. doi:10.1016/j.ccell.2015.10.001.
Kamata H, Honda S-I, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi:10.1016/j.cell.2004.12.041.
Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–80. doi:10.1002/ijc.23173.
Lasagna N, Fantappiè O, Solazzo M, Morbidelli L, Marchetti S, Cipriani G, Ziche M, Mazzanti R. Hepatocyte growth factor and inducible nitric oxide synthase are involved in multidrug resistance-induced angiogenesis in hepatocellular carcinoma cell lines. Cancer Res. 2006;66:2673–82. doi:10.1158/0008-5472.CAN-05-2290.
Ying L, Hofseth AB, Browning DD, Nagarkatti M, Nagarkatti PS, Hofseth LJ. Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res. 2007;67:9286–93. doi:10.1158/0008-5472.CAN-07-2238.
Calvisi DF, Pinna F, Ladu S, Pellegrino R, Muroni MR, Simile MM, Frau M, Tomasi ML, Miglio MRD, Seddaiu MA, Daino L, Sanna V, Feo F, Pascale RM. Aberrant iNOS signaling is under genetic control in rodent liver cancer and potentially prognostic for the human disease. Carcinogenesis. 2008;29:1639–47. doi:10.1093/carcin/bgn155.
Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. 2015;22:512–8. doi:10.1002/jhbp.245.
Sunami Y, Leithäuser F, Gul S, Fiedler K, Güldiken N, Espenlaub S, Holzmann K-H, Hipp N, Sindrilaru A, Luedde T, Baumann B, Wissel S, Kreppel F, Schneider M, Scharffetter-Kochanek K, Kochanek S, Strnad P, Wirth T. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatol. Baltim. Md. 2012;56:1117–28. doi:10.1002/hep.25711.
Elsharkawy AM, Wright MC, Hay RT, Arthur MJ, Hughes T, Bahr MJ, Degitz K, Mann DA. Persistent activation of nuclear factor-κB in cultured rat hepatic stellate cells involves the induction of potentially novel rel-like factors and prolonged changes in the expression of IκB family proteins. Hepatology. 1999;30:761–9. doi:10.1002/hep.510300327.
Oakley F, Trim N, Constandinou CM, Ye W, Gray AM, Frantz G, Hillan K, Kendall T, Benyon RC, Mann DA, Iredale JP. Hepatocytes express nerve growth factor during liver injury: evidence for paracrine regulation of hepatic stellate cell apoptosis. Am J Pathol. 2003;163:1849–58. doi:10.1016/S0002-9440(10)63544-4.
Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The bcl, NFκB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-β or TNF-α on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554–61. doi:10.1078/0171-9335-00182.
Oakley F, Teoh V, Ching-A-Sue G, Bataller R, Colmenero J, Jonsson JR, Eliopoulos AG, Watson MR, Manas D, Mann DA. Angiotensin II activates IκB kinase phosphorylation of RelA at Ser536 to promote myofibroblast survival and liver fibrosis. Gastroenterology. 2009;136:2334–2344.e1. doi:10.1053/j.gastro.2009.02.081.
Kluwe J, Pradere J-P, Gwak G-Y, Mencin A, Minicis SD, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–59. doi:10.1053/j.gastro.2009.09.015.
Gäbele E, Mühlbauer M, Dorn C, Weiss TS, Froh M, Schnabl B, Wiest R, Schölmerich J, Obermeier F, Hellerbrand C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun. 2008;376:271–6. doi:10.1016/j.bbrc.2008.08.096.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. doi:10.1038/nm1663.
Goossens N, Hoshida Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin Mol Hepatol. 2015;21:105–14. doi:10.3350/cmh.2015.21.2.105.
Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–24.
Zhao L-J, Zhao P, Chen Q-L, Ren H, Pan W, Qi Z-T. Mitogen-activated protein kinase signalling pathways triggered by the hepatitis C virus envelope protein E2: implications for the prevention of infection. Cell Prolif. 2007;40:508–21. doi:10.1111/j.1365-2184.2007.00453.x.
Lin W, Tsai W-L, Shao R-X, Wu G, Peng LF, Barlow LL, Chung WJ, Zhang L, Zhao H, Jang J-Y, Chung RT. HCV regulates TGF-β1 production through the generation of reactive oxygen species in an NFκB-dependent manner. Gastroenterology. 2010;138:2509–2518.e1. doi:10.1053/j.gastro.2010.03.008.
Chusri P, Kumthip K, Hong J, Zhu C, Duan X, Jilg N, Fusco DN, Brisac C, Schaefer EA, Cai D, Peng LF, Maneekarn N, Lin W, Chung RT. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci Rep. 2016;6:22487. doi:10.1038/srep22487.
Kanda T, Yokosuka O, Nagao K, Saisho H. State of hepatitis C viral replication enhances activation of NF-kB- and AP-1-signaling induced by hepatitis B virus X. Cancer Lett. 2006;234:143–8. doi:10.1016/j.canlet.2005.03.030.
Hildt E, Hofschneider PH. The PreS2 activators of the hepatitis B virus: activators of tumour promoter pathways. Recent Results Cancer Res. 1998;154:315–29.
You LR, Chen CM, Lee YH. Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J Virol. 1999;73:1672–81.
Bose S, Banerjee AK. Innate immune response against nonsegmented negative strand RNA viruses. J Interferon Cytokine Res. 2003;23:401–12. doi:10.1089/107999003322277810.
Hiscott J, Grandvaux N, Sharma S, Tenoever BR, Servant MJ, Lin R. Convergence of the NF-κB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann N Y Acad Sci. 2003;1010:237–48. doi:10.1196/annals.1299.042.
Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–54. doi:10.1089/1079990041689665.
Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: cellular executioner or white knight? Curr Med Chem. 2007;14:1279–89.
Sasaki R, Kanda T, Nakamura M, Nakamoto S, Haga Y, Wu S, Shirasawa H, Yokosuka O. Possible involvement of hepatitis B virus infection of hepatocytes in the attenuation of apoptosis in hepatic stellate cells. PLoS One. 2016;11:e0146314. doi:10.1371/journal.pone.0146314.
Sun B, Karin M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene. 2008;27:6228–44. doi:10.1038/onc.2008.300.
Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am. 2010;39:1–7. doi:10.1016/j.gtc.2009.12.014.
Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi:10.1016/j.cmet.2010.12.008.
Poloz Y, Stambolic V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 2015;6:e2037. doi:10.1038/cddis.2015.381.
Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy M-F, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake TA, Lusis AJ, Schadt EE. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008b;452:429–35. doi:10.1038/nature06757.
Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, Mouy M, Steinthorsdottir V, Eiriksdottir GH, Bjornsdottir G, Reynisdottir I, Gudbjartsson D, Helgadottir A, Jonasdottir A, Jonasdottir A, Styrkarsdottir U, Gretarsdottir S, Magnusson KP, Stefansson H, Fossdal R, Kristjansson K, Gislason HG, Stefansson T, Leifsson BG, Thorsteinsdottir U, Lamb JR, Gulcher JR, Reitman ML, Kong A, Schadt EE, Stefansson K. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–8. doi:10.1038/nature06758.
Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, Torosyan G, Majid S, Falkard B, Kleinhanz RR, Karlsson J, Castellani LW, Mumick S, Wang K, Xie T, Coon M, Zhang C, Estrada-Smith D, Farber CR, Wang SS, van Nas A, Ghazalpour A, Zhang B, Macneil DJ, Lamb JR, Dipple KM, Reitman ML, Mehrabian M, Lum PY, Schadt EE, Lusis AJ, Drake TA. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–23. doi:10.1038/ng.325.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3. doi:10.1038/4441022a.
Nguyen SD, Sok D-E. Effect of 3,4-dihydroxyphenylalanine on Cu(2+)-induced inactivation of HDL-associated paraoxonasel and oxidation of HDL; inactivation of paraoxonasel activity independent of HDL lipid oxidation. Free Radic Res. 2004;38:969–76. doi:10.1080/10715760400000943.
Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC, Prada PO, Hirabara SM, Schenka AA, Araújo EP, Vassallo J, Curi R, Velloso LA, Saad MJA. Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–98. doi:10.2337/db06-1595.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–31. doi:10.1126/science.1179721.
Chiang S-H, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma J-T, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR. The protein kinase IKKɛ regulates energy balance in obese mice. Cell. 2009;138:961–75. doi:10.1016/j.cell.2009.06.046.
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–90. doi:10.1038/nm1166.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi:10.1172/JCI28898.
Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity. 2008;16:1248–55. doi:10.1038/oby.2008.210.
Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, Burcelin R, Alessi MC. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue vin response to a high-fat diet. Diabetologia. 2007;50:1267–76. doi:10.1007/s00125-007-0654-8.
Davis JE, Braucher DR, Walker-Daniels J, Spurlock ME. Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J Nutr Biochem. 2011;22:136–41. doi:10.1016/j.jnutbio.2009.12.008.
Nguyen MTA, Favelyukis S, Nguyen A-K, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–92. doi:10.1074/jbc.M706762200.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi:10.1126/science.7678183.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389:610–4. doi:10.1038/39335.
Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, Juntti-Berggren L, Li L-S, van Rooijen N, Libert C, Berggren P-O, Brüning JC. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12:237–49. doi:10.1016/j.cmet.2010.06.011.
Mendes-Braz M, Elias-Miró M, Jiménez-Castro MB, Casillas-Ramírez A, Ramalho FS, Peralta C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J Biomed Biotechnol. 2012;2012:298657. doi:10.1155/2012/298657.
Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59:1094–106. doi:10.1016/j.jhep.2013.06.017.
Gao W, Bentley RC, Madden JF, Clavien P-A. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology. 1998;27:1652–60. doi:10.1002/hep.510270626.
Kohli V, Selzner M, Madden JF, Bentley RC, Clavien PA. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation. 1999;67:1099–105.
Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13:2797–804. doi:10.1111/ajt.12448.
Massip-Salcedo M, Roselló-Catafau J, Prieto J, Avíla MA, Peralta C. The response of the hepatocyte to ischemia. Liver Int. 2007;27:6–16. doi:10.1111/j.1478-3231.2006.01390.x.
Rauen U, Kerkweg U, Weisheit D, Petrat F, Sustmann R, de Groot H. Cold-induced apoptosis of hepatocytes: mitochondrial permeability transition triggered by nonmitochondrial chelatable iron. Free Radic Biol Med. 2003;35:1664–78.
Theruvath TP, Czerny C, Ramshesh VK, Zhong Z, Chavin KD, Lemasters JJ. C-Jun N-terminal kinase 2 promotes graft injury via the mitochondrial permeability transition after mouse liver transplantation. Am J Transplant. 2008;8:1819–28. doi:10.1111/j.1600-6143.2008.02336.x.
Shuh M, Bohorquez H, Loss GE, Cohen AJ. Tumor necrosis factor-α: life and death of hepatocytes during liver ischemia/reperfusion injury. Ochsner J. 2013;13:119–30.
Suetsugu H, Iimuro Y, Uehara T, Nishio T, Harada N, Yoshida M, Hatano E, Son G, Fujimoto J, Yamaoka Y. Nuclear factor κB inactivation in the rat liver ameliorates short term total warm ischaemia/reperfusion injury. Gut. 2005;54:835–42. doi:10.1136/gut.2004.043034.
Uchinami H, Yamamoto Y, Kume M, Yonezawa K, Ishikawa Y, Taura K, Nakajima A, Hata K, Yamaoka Y. Effect of heat shock preconditioning on NF-κB/I-κB pathway during I/R injury of the rat liver. Am J Physiol Gastrointest Liver Physiol. 2002;282:G962–71. doi:10.1152/ajpgi.00466.2001.
Ruland J. Return to homeostasis: downregulation of NF-κB responses. Nat Immunol. 2011;12:709–14. doi:10.1038/ni.2055.
Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi:10.1038/nature02794.
Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–11.
Ramsey HE, Da Silva CG, Longo CR, Csizmadia E, Studer P, Patel VI, Damrauer SM, Siracuse JJ, Daniel S, Ferran C. A20 protects mice from lethal liver ischemia reperfusion injury by increasing peroxisome proliferator-activated receptor-α expression. Liver Transpl. 2009;15:1613–21. doi:10.1002/lt.21879.
Sakai N, Van Sweringen HL, Schuster R, Blanchard J, Burns JM, Tevar AD, Edwards MJ, Lentsch AB. Receptor activator of nuclear factor-κB ligand (RANKL) protects against hepatic ischemia/reperfusion injury in mice. Hepatol. Baltim. Md. 2012;55:888–97. doi:10.1002/hep.24756.
Marinò M, Morabito E, Altea MA, Ambrogini E, Oliveri F, Brunetto MR, Pollina LE, Campani D, Vitti P, Bartalena L, Pincheral A, Marcocci C. Autoimmune hepatitis during intravenous glucocorticoid pulse therapy for Graves’ ophthalmopathy treated successfully with glucocorticoids themselves. J Endocrinol Invest. 2005;28:280–4.
Bae M-K, Kim S-H, Jeong J-W, Lee YM, Kim H-S, Kim S-R, Yun I, Bae S-K, Kim K-W. Curcumin inhibits hypoxia-induced angiogenesis via down-regulation of HIF-1. Oncol Rep. 2006;15:1557–62.
Chen D, Milacic V, Chen MS, Wan SB, Lam WH, Huo C, Landis-Piwowar KR, Cui QC, Wali A, Chan TH, Dou QP. Tea polyphenols, their biological effects and potential molecular targets. Histol Histopathol. 2008a;23:487–96.
Toledo LP, Ong TP, Pinho ALG, Jordão A, Vanucchi H, Moreno FS. Inhibitory effects of lutein and lycopene on placental glutathione S-transferase-positive preneoplastic lesions and DNA strand breakage induced in Wistar rats by the resistant hepatocyte model of hepatocarcinogenesis. Nutr Cancer. 2003;47:62–9. doi:10.1207/s15327914nc4701_8.
Umemura T, Kai S, Hasegawa R, Kanki K, Kitamura Y, Nishikawa A, Hirose M. Prevention of dual promoting effects of pentachlorophenol, an environmental pollutant, on diethylnitrosamine-induced hepato- and cholangiocarcinogenesis in mice by green tea infusion. Carcinogenesis. 2003;24:1105–9. doi:10.1093/carcin/bgg053.
Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–44. doi:10.1002/hep.21036.
Cervello M, Giannitrapani L, La Rosa M, Notarbartolo M, Labbozzetta M, Poma P, Montalto G, D’Alessandro N. Induction of apoptosis by the proteasome inhibitor MG132 in human HCC cells: possible correlation with specific caspase-dependent cleavage of beta-catenin and inhibition of beta-catenin-mediated transactivation. Int J Mol Med. 2004;13:741–8.
Hegewisch-Becker S, Sterneck M, Schubert U, Rogiers X, Guerciolini R, Pierce JE, Hossfeld DK. Phase I/II trial of bortezomib in patients with unresectable hepatocellular carcinoma (HCC). J Clin Oncol. 2004;22:335S.
Joshi-Barve S, Barve SS, Butt W, Klein J, McClain CJ. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatol. Baltim. Md. 2003;38:1178–87. doi:10.1053/jhep.2003.50470.
Greten FR, Arkan MC, Bollrath J, Hsu L-C, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–31. doi:10.1016/j.cell.2007.07.009.
Liang M-C, Bardhan S, Li C, Pace EA, Porco JA, Gilmore TD. Jesterone dimer, a synthetic derivative of the fungal metabolite jesterone, blocks activation of transcription factor nuclear factor kappaB by inhibiting the inhibitor of kappaB kinase. Mol Pharmacol. 2003;64:123–31. doi:10.1124/mol.64.1.123.
Kim K-H, Park K-K. Small RNA- and DNA-based gene therapy for the treatment of liver cirrhosis, where we are? World J Gastroenterol. 2014;20:14696–705. doi:10.3748/wjg.v20.i40.14696.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Bettermann, K. (2017). NF-κB and Its Implication in Liver Health and Cancer Development. In: Haybaeck, J. (eds) Mechanisms of Molecular Carcinogenesis – Volume 1. Springer, Cham. https://doi.org/10.1007/978-3-319-53659-0_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-53659-0_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53657-6
Online ISBN: 978-3-319-53659-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)