
Abstract
Mutant cells that are defective for certain components of the mammalian DNA damage response (DDR) have been shown to display hypersensitivity to killing by ionizing radiations; these findings have prompted the idea that drugs that emulate these DDR deficiencies might serve as clinically useful radiosensitizers for improving results in cancer therapy. In this chapter, the ways in which several agents now established as radiosensitizers do in fact function by inhibiting parts of the DDR are first presented. The various subsystems of the DDR are next reviewed, and several potential molecular targets for discovery or design of chemical modifiers that could lead novel radiosensitizing drugs are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- DNA Damage Response (DDR)
- DNA Repair
- Double Strand Break (DSB)
- Homologous Recombination Repair (HR)
- Non-homologous End Joining Repair (NHEJ)
- Radiation therapy (XRT)
- Radiosensitizer
- Therapeutic Ratio (TR)
1 The Challenge of Continuing to Improve the Therapeutic Ratio for Radiation Therapy in Human Oncology
Radiation therapy (XRT) continues to be a key modality in modern cancer medicine for both curative and palliative management of a variety of malignant diseases. It has been suggested that the role radiation therapy plays in obtaining local-regional control of tumors might in fact become increasingly important in the coming years as innovative systemic therapies, such as molecularly targeted agents and immune checkpoint modulators, improve our ability to eradicate microscopic metastatic disease for some patients (Citrin and Mitchell 2014). The recent 1–2 decades have witnessed impressive technical advances in the planning and delivery of XRT including multiple imaging modality simulation, 4-dimensional treatment planning and delivery, practical multibeam intensity modulated XRT, and daily image guidance for target positioning verification; several of these have supported development of effective new XRT approaches such as stereotactic body radiotherapy. Improving the conformality of XRT dose delivery in both space and time through these approaches is expected to improve the therapeutic ratio (TR) for XRT by minimizing normal tissue toxicity (Citrin and Mitchell 2014).
An improved TR for XRT can also be achieved by selectively enhancing the lethal effects of ionizing radiation (IR) on tumor versus normal tissues (Citrin and Mitchell 2014; Jekimovs et al. 2014; Gavande et al. 2016). Such an outcome might be obtainable by targeting certain features of malignant disease such as intratumoral hypoxia or host antitumor immune responses for example, but much current interest is centered upon manipulating biological IR responses at the level of individual malignant cells (Citrin and Mitchell 2014; Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013). The term radiosensitizer properly refers to a chemical that increases cell death in response to a given dose of IR while being completely innocuous to cells in the absence of IR treatment (Citrin and Mitchell 2014; Higgins et al. 2015), although the term is often informally applied to agents such a chemotherapeutic drugs that can themselves be toxic to cells at sufficient doses; the latter are instead properly called chemical modifiers of cellular radiation response (Citrin and Mitchell 2014; Higgins et al. 2015). Drugs that usefully enhance tumor responses to IR are already extremely important in the clinic. Recent large gains in our understanding of the molecular genetics of cancer, along with ever-improving approaches to protein structure-based drug design, point to the strong likelihood that new, potent chemical modifiers for use with XRT will contribute to advances in human cancer medicine. This brief chapter will update and expand upon several recent excellent reviews of this topic (Citrin and Mitchell 2014; Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013; Begg et al. 2011).
2 Radiosensitizing Agents Targeting the DNA Damage Response: Established Radiosensitizing Agents
2.1 Overview
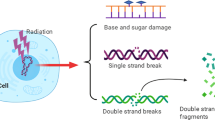
Formation of DNA double strand breaks (DSB) at sufficient levels in human and other mammalian genomes, as by IR or certain chemotherapeutic agents, activates a complex signal transduction cascade—the DNA Damage Response (DDR)—that culminates in a range of cellular outcomes including repair of the DNA damage, cell cycle arrest at specific checkpoints, and programmed cell death; the DDR and these various endpoints have been reviewed extensively (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Jeggo and Lobrich 2015; Waters et al. 2014; Jackson 2009) and will be covered here only briefly. Natural and engineered mammalian cells and animals that have gene mutations causing functional defects for components of the DDR are commonly IR hypersensitive to varying degrees, along with displaying “genomic instability” (see later). Human patients and mutant mouse strains with germline DDR gene defects often display increased susceptibility to various malignancies; some also have immunodeficiency related to a failure to repair the programmed DSB formed during the course of immunoglobulin and T-cell receptor gene maturation (Goodarzi and Jeggo 2013; Jackson 2009; Curtin 2012; Weinberg and Hanahan 2011). The finding that DDR gene mutations can cause mammalian cell IR hypersensitivity has given the impetus in recent years for discovery of chemical compounds that could inhibit the functions of various DDR proteins and emulate the effects of such gene defects (sometimes called “pharmacological phenocopy”); these agents would be expected to be candidate preclinical cellular radio- and chemo-sensitizers that might ultimately lead to useful drugs for human cancer medicine (Citrin and Mitchell 2014; Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013). The finding that inhibition of different facets of the DDR is in fact the molecular mechanism of action for some radiosensitizers now used clinically, as detailed next, serves to validate this strategy.
2.2 Targeting DNA Damage Sensing/Signaling Pathways
Following induction of DSB in human nuclear DNA, their presence is sensed by the Mre11/Rad50/NBS1 (MRN) protein complex, which is recruited to the break ends by the human single stranded binding protein 1 (hSSB1); MRN then promotes association of the key damage signaling kinase Ataxia Telangiectasia Mutated (ATM) with the DSB (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). ATM kinase is thereby activated toward phosphorylation of itself, of the components of the MRN complex, and of the variant histone H2AX (thereby generating γH2AX) (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). γH2AX formation then propagates for many thousands of base pairs from the DSB ends into the DNA strands, and initiates assembly of an array of additional DDR proteins to form a molecular macrostructure, the ionizing radiation induced focus (IRIF) (Goodarzi and Jeggo 2013). ATM signaling is amplified by interactions within the IRIF and then transduced to downstream effectors such as the CHK2 kinase-p53 axis to initiate cell cycle arrest or apoptosis (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). Cells with deficient MRN function show severely impaired ATM signaling (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). The MRN complex (in particular, the Mre11 nuclease) plays an additional key role in the cellular “choice” between utilization of homologous recombination (HR) versus non-homologous end joining (NHEJ) mechanisms for the repair of DSB in the late-S and G2 phases of the cell cycle ((Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016) and see below). The observed IR sensitivity of MRN function-deficient cells thus makes sense.
Heat treatment of sufficient temperature and duration very potently sensitizes malignant human cells to killing by IR, a phenomenon termed hyperthermic radiosensitization (HtRs) (Dewey 2009). Given the magnitude of this effect, clinical application of HtRs has been of interest for decades, but implementation of this has been hindered previously by the engineering challenges connected with heating deep tumors in situ; this obstacle might now be overcome, however, with the development of MR-guided focused ultrasound technology (see Chapter XX of this text). The molecular basis of HtRs has been elucidated using a genetic approach termed epistasis analysis; this strategy is predicated on the fact that two separate functional defects (“hits”) in the same mechanistic pathway should have no more consequence than either single hit alone does. In contrast, hits to separate pathways that function in parallel in a complementary fashion (for example, the NHEJ and HR mechanisms for DSB repair; see below) typically have a greater impact on cell physiology than either hit alone. Previous work had implicated one or more of the MRN complex protein components in HtRs, possibly via active export of these proteins out of the nucleus in response to heat treatment (Seno and Dynlacht 2004). The effect heating on clonogenic inactivation by IR was next investigated using cells having natural (cells derived from Mre11- or Nbs1-defective patients) or engineered (siRNA knockdown) deficiencies of Mre11, Nbs1, or Rad50 function (Dynlacht et al. 2011). For Nbs1 and Rad50, unheated cells were hypersensitive to killing by IR compared to normal cells, but that sensitivity was increased still further by the heat treatment. In contrast, heating of the Mre11 cells did not alter their already marked sensitivity to IR (Dynlacht et al. 2011). Purified Mre11 protein was also found to be unusually sensitive to heat denaturation in vitro (Dynlacht et al. 2011). The interpretation of these findings is that the Mre11-mutant cells are already maximally deficient for MRN-ATM signaling after DNA damage so that heat denaturation of any residual Mre11 protein has no consequence. In contrast, low levels of Nbs1 or Rad50 activity in the respective mutant cells did support some MRN-ATM signaling after IR, and this was abolished by total heat inactivation of cellular Mre11. In keeping with these findings, recently developed small molecule inhibitors of the Mre11 endo- and exonuclease activities are radiosensitizing agents in vitro (Shibata et al. 2014).
2.3 Targeting DSB Repair Pathways
Repair of DNA DSB in eukaryotic cell relies on two fundamentally distinct mechanisms, nonhomologous end joining (NHEJ) and homologous recombination (HR) repair (sometimes called homology directed repair) (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Waters et al. 2014; Pannunzio et al. 2014; Moynahan and Jasin 2010). NHEJ entails the nucleolytic processing and direct ligation of DSB ends that may approximately restore the original configuration of a stretch of DNA (typically with loss of some of the nucleotides adjacent to the strand breaks). Alternatively, DNA ends from remote parts of the genome might be brought together by NHEJ repair, forming a chromosomal translocation or inversion (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). The kinetics of end joining by this mechanism is determined by the chemistry of the DNA termini and by the chromatin configuration of the DNA within which the DSB occurs; chemically complex ends produced by relatively high LET radiations, and the compacted chromatin associated with heterochromatic chromosomal regions lead to slower end rejoining (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). Quite recently, it has been possible to distinguish between so-called canonical NHEJ (the mechanism identified first) and one or more “alternative” NHEJ pathways (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Pannunzio et al. 2014); these differ with respect to the specific proteins involved and the precise molecular details of DNA end synapsis during DSB end rejoining (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Pannunzio et al. 2014). How these various NHEJ processes might be differently targeted for achieving radiosensitization remains to be seen.
HR repair of DSB is initiated by quite extensive exonucleolytic degradation of DNA, starting at the break ends and proceeding in the 5′ → 3′ direction, thereby forming long single strands having 3′ hydroxyl termini (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Moynahan and Jasin 2010). Those strands invade nearby intact homologous DNA (this is typically afforded by the adjacent sister chromatid following replication) and prime the synthesis of new DNA corresponding to the regions surrounding the DSB, using the intact complementary strands as templates. The new DNA strands are then extracted from the sister chromatid and reannealed; ligation of the new DNA 3′ ends to 5′ ends of the broken chromatid completes repair of the DSB (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Moynahan and Jasin 2010).
HR contrasts with NHEJ in that the latter has no mechanistic requirement for the presence intact homologous DNA nearby (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). For this reason, NHEJ is practicable throughout the cell cycle, while HR is confined to the G2 phase and to those genomic regions that have already undergone replication during S phase (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016). NHEJ appears to be responsible for the majority of DSB repair throughout the cell cycle in mammalian cells, this despite it being much more “error prone” than HR, given the likelihood of small DNA sequence changes in the vicinity of the DSB and the potential to form gross chromosomal aberrations (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Pannunzio et al. 2014). The mechanistic basis for the cellular “decision” between use of NHEJ versus HR for the repair of a given DSB during G2 or S phase is at present poorly understood and is the subject of much research activity, although the MRN complex appears to be intimately involved (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Shibata et al. 2014).
2.3.1 Targeting the NHEJ Pathway
Curative-intent treatment programs combining XRT and a platinum-containing chemotherapeutic drug such as cisplatin (CDDP) are widely used in human oncology, and have led to improved clinical outcomes for different forms of lung cancer, head and neck cancer, and carcinoma of the uterine cervix among other malignancies (Sears et al. 2016). The mechanistic basis for radiosensitization by platinum-containing drugs has been investigated in vitro. An epistasis analysis showed that CDDP treatment did not sensitize the killing of NHEJ-deficient cells by IR, while HR-defective were markedly sensitized (Raaphorst et al. 2005); this result indicates that NHEJ does not function properly in CDDP-treated cells. NHEJ can be assayed in vitro in mammalian cells extracts by following ligation of linear substrate DNA molecules (Sears and Turchi 2012; Diggle et al. 2005). The presence of CDDP damage in the substrate molecules inhibited NHEJ in vitro (Sears and Turchi 2012; Diggle et al. 2005), while extracts prepared from CDDP-treated cells were NHEJ-competent for repair of undamaged substrate DNA molecules (Sears and Turchi 2012); these results support the model that it is CDDP adducts in DNA near DSB are a block to repair by NHEJ. In keeping with this model, so-call host reactivation of transfected substrate DNA molecules by NHEJ is proficient for undamaged substrates transfected into CDDP-treated cells, but deficient for CDDP-damaged substrates transfected into untreated cells (Sears and Turchi 2012).
2.3.2 Targeting the HR Pathway
The HR pathway for DSB repair depends upon synthesis of long stretches of new DNA using an undamaged sister chromatid as the template (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Moynahan and Jasin 2010); it might thus be anticipated that antimetabolite chemotherapy drugs which deprive mammalian cells of DNA synthetic precursors might serve as effective HR repair inhibitors. Gemcitabine is an antimetabolite chemotherapeutic agent that inhibits ribonucleotide reductase and the de novo synthesis of deoxynucleotide DNA precursors. Gemcitabine is also a potent radiosensitizers (Shewach and Lawrence 1995; Van Putten et al. 2001; Wachters et al. 2001). Using epistasis analysis, the mechanism of gemcitabine radiosensitization was investigated in mammalian cells deficient in NHEJ and HR (Van Putten et al. 2001; Wachters et al. 2001). Following treatment with gemcitabine, radiosensitization was observed in the NHEJ-deficient cells, but this was markedly decreased in the HR-deficient cell, demonstrating that this drug blocks the latter pathway (Van Putten et al. 2001; Wachters et al. 2001). Investigation of the mechanism of radiosensitization by various fluoropyrimidine family drugs, another class of clinically useful antimetabolite chemotherapeutic radiosensitizers, has proven to be more complex (Canman et al. 1994). This may be because, while these agents inhibit thymidine (TdR) DNA precursor synthesis, the simultaneously promote incorporation of deoxyuridine (UdR) into nascent DNA in place of TdR (Canman et al. 1994).
2.4 Targeting Prosurvival/Anti-apoptotic Signaling Pathways
Following DNA damage and several other cellular stresses, many normal cells and certain malignant cells undergo a process of programmed cell death termed apoptosis (Balcer-Kubiczek 2012). For cells exposed to IR, DDR signaling through ATM ultimately impinges upon the tumor suppressor protein p53 and activates it as a transcription factor (Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Balcer-Kubiczek 2012). Depending upon cellular context, p53 activation promotes either apoptosis or cell cycle arrest in G1 phase by transcriptional regulation of specific genes (Balcer-Kubiczek 2012). In the checks and balances of cellular governance, pro-apoptotic influences are countered by pro-survival signaling; the Akt serine/threonine kinases are key mediators of the latter process (Balcer-Kubiczek 2012; Toulany and Roderman 2015). Loss of proper apoptotic response to cellular stress appears to be an essential component of carcinogenesis for many cell types (Weinberg and Hanahan 2011); enhanced Akt signaling by several different mechanisms has been found to be one means by which malignant cells can achieve this abrogation of apoptosis (Cengel et al. 2007; Mckenna et al. 2003; Cheung and Testa 2013). Akt activation in tumors is associated with chemotherapy and radiotherapy treatment resistance (Cengel et al. 2007; Mckenna et al. 2003; Cheung and Testa 2013; Sekhar et al. 2011; Kao et al. 2007; Misale et al. 2012; Garrido-Laguna et al. 2012), and down regulation of Akt signaling by dominant negative inhibition or drug targeting of its upstream signaling partner PI3 kinase (PI3K) has been shown in some cases to cause radiosensitization (Tanno et al. 2004). These finding are believed to reflect, at least in some cells, mitigation of the killing effects of IR by pro-survival Akt signaling by these inhibitory interventions (Toulany and Roderman 2015; Tanno et al. 2004; Brognard et al. 2001). Importantly, however, more recent results indicate that activated Akt also plays significant, directs role in cellular responses to IR including DNA DSB sensing/signaling and regulation of NHEJ; targeting activated Akt may thus instead (or in addition) radiosensitize some cells by inhibiting DSB repair (Toulany et al. 2008, 2012; Park et al. 2009).
Akt is a downstream effector of several mitogenic (pro-growth) signaling cascades that are frequently found to be corrupted in tumor cells (Toulany and Roderman 2015; Cheung and Testa 2013); prominent among these are activating mutations and amplification of genes encoding cell surface receptor tyrosine kinases (RTKs) such as EGFR (Weinberg and Hanahan 2011; Toulany and Roderman 2015; Cheung and Testa 2013). One target of deregulated EGFR signaling in tumors, via PI3K, is Akt (Toulany and Roderman 2015; Cheung and Testa 2013). Significant correlations have been found for many tumors between EGFR activation and Akt activation (Cheung and Testa 2013; Nijkamp et al. 2011), and between EGFR activation and chemo/radiation resistance (Ang et al. 2002; Nakamura 2007); findings such as these have made EGFR signaling an attractive target for cancer therapy (Seshacharyulu et al. 2012; Dassonville et al. 2007). Cetuximab, a recombinant monoclonal antibody therapeutic that down regulates EGFR signaling has been investigated in combination with XRT patients with colon cancer, non-small cell lung cancer, and head and neck cancers (Seshacharyulu et al. 2012), and is now in routine use for the latter given its efficacy as a radiosensitizer (Bobber et al. 2010). Erlotinib, a small molecule EGFR TK inhibitor also shows radiosensitizing activity for some malignant cells, and has shown this activity in the clinic in a promising way for patients with brain metastases of non-small cell lung cancer (Zheng et al. 2016).
3 DNA Damage Response: Recent Results for Radiosensitizing Agents
3.1 Targeting DNA Damage Sensing/Signaling Pathways
The ataxia telangiectasia-mutated (ATM) protein is defective in that disease and is a pivotal player in the early steps of DSB detection (Jekimovs et al. 2014; Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Jeggo and Lobrich 2015). A-T patients inherit two defective germline copies of the ATM gene, and are characteristically both cancer-prone and hypersensitive to IR (Jekimovs et al. 2014; Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Jeggo and Lobrich 2015). ATM inactivation by somatic mutation is a common finding in many tumors, and is thought to be a mechanism for the genomic instability that is believed to be required of carcinogenesis (Jekimovs et al. 2014; Jeggo and Lobrich 2015). ATM is a member of the PI3K-like kinase (PIKK) family that also includes ATR (for ATM-mutated and Rad3-related), DNA-PKcs (DNA-dependent protein kinase catalytic subunit, a core component of the canonical NHEJ pathway), and PI3K itself, along with multiple other proteins (Jeggo and Lobrich 2015). The kinase ATP binding pocket has been a common target for discovery of numerous kinase-inhibiting drugs such as erlotinib; this approach has been problematic for the PIKK family proteins due to difficulty in achieving sufficient target specificity to allow clinical use, however (Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013). Sufficient success in this regard has been obtained to date for the ATR kinase (Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013; Jeggo and Lobrich 2015; Sanjiv et al. 2016) to allow inhibitory drugs to enter clinical trials, some in combination with XRT.
As is the case for ATM, ATR contributes to maintaining genomic integrity after various genotoxic insults. ATR appears to be activated primarily by single stranded DNA associated with replication forks during periods of replicative stress (Jeggo and Lobrich 2015; Waters et al. 2014; Jackson 2009), such as DNA synthesis using damaged template strands following IR exposure (Jeggo and Lobrich 2015; Waters et al. 2014; Jackson 2009). Like for ATM, ATR activation leads to cell cycle arrest at a specific checkpoint, in this case S/G2 phase. It has been hypothesized that the loss of G1 checkpoint in ATM-deficient tumor cells renders them more dependent upon integrity of the ATR-mediated S/G2 checkpoint after genotoxic insults such as IR exposure (Sanjiv et al. 2016). The specific ATR kinase inhibitor VX-970 has been shown to sensitize adenocarcinoma cells to IR and to several chemotherapeutic drugs that cause replication stress (Sanjiv et al. 2016). Based on these findings, VX-970 is currently being investigated in combination with whole brain XRT in patients with brain metastases from non-small cell lung cancer (NCI trial designation NCT02589522).
3.2 Targeting DSB Repair Pathways
3.2.1 Targeting the NHEJ Pathway
The Ku70 protein is a core component, along with Ku80 protein and DNA-PKcs, of the DNA-PK complex that mediates the initial steps of canonical NHEJ (Goodarzi and Jeggo 2013; Waters et al. 2014). Ku70 protein was found to interact with the androgen receptor in prostate carcinoma (CaP) cells (Al-Ubaidi et al. 2013), an observation that motivated study of Ku70 levels and endogenous NHEJ activity in biopsies of CaP in patients, both before and after surgical or pharmacological (GnRH treatment) castration (Al-Ubaidi et al. 2013). Ku70 levels were found to be reduced, and spontaneous γH2AX foci (this is a measure of persisting DSB formed during replication, an indication of reduced NHEJ) were increased following castration (Al-Ubaidi et al. 2013). The remarkable result of these findings was the prediction that initial (“neoadjuvant”) androgen deprivation, via surgical or pharmacological means, might be a radiosensitizer for subsequent prostate XRT. To test this hypothesis, 48 patients in a small pilot trial were randomly assigned to neoadjuvant androgen deprivation or not prior to receiving XRT as 2 Gy × 5 fractions; biopsies were obtained prior to any intervention or after the 10 Gy XRT (Tarish et al. 2015). Autophosphorylation of DNA-PKcs, a measure of canonical NHEJ activity (Jeggo and Lobrich 2015; Toulany and Roderman 2015), was strongly upregulated following XRT without prior androgen deprivation, but completely abolished with the androgen deprivation (Tarish et al. 2015). An increased number of persisting nuclear γH2AX foci were observed in the trial castration arm, a finding also consistent with failure of DSB repair in the setting of neoadjuvant androgen deprivation (Tarish et al. 2015). Studies with long term follow up and larger patient numbers will be required to determine whether the putative NHEJ inhibition and radiosensitization in this setting leads to superior local control of CaP.
3.2.2 Targeting the HR Pathway
Protein acetylation on lysine residues is a post-translational protein modification that is increasingly appreciated to be a mode of intracellular signaling comparable to protein phosphorylation (Ceccacci and Minucci 2016; West and Johnstone 2014). This modification was first described for chromatin histone proteins, and the enzymes that add and remove these acetyl groups are called histone acetyl transferases (HATs) and histone deacetylases (HDACs), respectively; many non-histone proteins are clearly substrates for HATs and HDACs as well, however (Ceccacci and Minucci 2016; West and Johnstone 2014; Elia et al. 2015). Signaling via protein acetylation/deacetylation has recently been found to play an important role in the mammalian cellular DNA damage response (Elia et al. 2015). A number of HDAC-inhibiting drugs are showing interesting activity as anti-cancer agents, particularly for hematolymphoid malignancies (Ceccacci and Minucci 2016; West and Johnstone 2014). Several HDAC inhibitors have also been shown to act as radiosensitizers (Camphausen et al. 2004; Chinnaiyan et al. 2008; Chen et al. 2012). Although effects on histone modification and chromatin configuration were suggested to be the mechanistic basis for these findings, persisting acetylation of non-histone proteins clearly also plays a role. For example, we and others have shown that HDAC inhibitors reduce DSB repair via the HR pathway (Chen et al. 2012; Adimoolam et al. 2007). In the case of potent and clinically approved drug SAHA, treatment of multiple myeloma cells at low, non-toxic concentrations led to significant radiosensitization, specifically by inhibiting the HR pathway for DSB repair (Chen et al. 2012). This effect is apparently caused by blocking upregulation of the key HR pathway protein Rad51, and inhibiting normal Rad51 association with chromatin, following IR exposure (Chen et al. 2012; Adimoolam et al. 2007). It is not yet known what protein(s) experiences persistent acetylation in the presence of the drug to mediate these events. Concurrent SAHA treatment would thus be a rational approach to enhancing efficacy of XRT for this disease.
The findings noted here support the ideas that small molecule inhibitors of the NHEJ and HR repair pathways may be useful clinically as cytotoxins and radiosensitizers. As regards the former, an inhibitor of DNA-PKcs and the PIKK m-TOR (mammalian target of rapamycin), CC-115 has shown activity as a single agent in chronic lymphocytic leukemia (Thijssen et al. 2016), and is being characterized with respect to dosing and tolerability in a Phase I trial of advanced solid malignancies (NCI trial designation NCT01353625). Use of this agent along with XRT in patients has not yet been reported. Small molecule inhibitors of HR have been reported, and are in the preclinical phase of development (Huang et al. 2011; Budke et al. 2013; Zhu et al. 2013).
3.3 Targeting Pro-apoptotic Signaling
In addition to transcriptional activation of pro-apoptotic nuclear genes (Balcer-Kubiczek 2012), the p53 tumor suppressor protein can also activate apoptosis as a DDR endpoint through processes mediated at the outer mitochondrial membrane (Chipuk et al. 2002; Chen et al. 2011; Sykes et al. 2006). Treatment with the HDAC inhibitor drug Valproic Acid (VPA) was shown to promote radiosensitization via apoptosis after IR exposure—a response not typically displayed–in two CaP cell lines, but not a third one (Chen et al. 2011). The CaP line showing no radiosensitization did not contain any p53 protein, while the other two lines did, perhaps implicating p53 in the radiosensitization mechanism. However, one of the two CaP lines that did display radiosensitization contained only mutant forms of p53 protein having no activity as a transcription factor (Chen et al. 2011); this fact would argue against the model for p53 involvement in the radiosensitization (at least with respect to its best known function as an regulator of nuclear gene expression).
p53 is a substrate for the HAT Tip60, which acetylates a specific p53 lysine residue in response to DNA damage and other pro-apoptotic stresses (Sykes et al. 2006); this modification activates p53 transcription factor activity toward pro-apoptotic gene targets specifically (Sykes et al. 2006). We and others showed that this modification also activates p53 toward its pro-apoptotic interactions at the mitochondrial membrane, and that some mutant forms of p53 that are inactive as transcription factors are proficient for this process (Chen et al. 2011; Mellert et al. 2011). Following IR, p53 in untreated CaP cells is acetylated by Tip60, but it is then promptly deacetylated (by HDAC1 (Mellert et al. 2011)) and an apoptotic response does not ensue. With VPA treatment, p53 acetylation persists and a sufficient quantity of this modified form of the protein accumulates at the mitochondrial membrane to trigger apoptosis (Chen et al. 2011; Mellert et al. 2011). This adds to amount of cell killing produced by a given dose of IR—the definition of radiosensitization. This mechanism appears to be operative in colorectal carcinoma cells as well (Chen et al. 2009).
4 Future Directions and Promise
Statistical analyses and in vitro studies have led to the conclusion that multiple independent mutational genetic changes are required to convert a fully normal human cell into a fully malignant one (Hahn et al. 1999). Loeb was first to recognized that the fidelity of mammalian genomic replication is sufficiently good that this number of mutations could never accumulate in a human cell during a human lifetime and cause a malignant tumor, a conclusion clearly inconsistent with the clinical problem of cancer. Based on this, Loeb proposed the Mutator Phenotype hypothesis (Loeb 2016), which states the premalignant cells must somehow lose mechanisms of replicative fidelity early in the course of neoplastic transformation and thereby become able to acquire the required mutation burden. A number of observations have shown this idea to be correct, notably the findings of recurrent DNA repair pathway defects in a wide range of human tumor types (Jeggo and Lobrich 2015; Waters et al. 2014; Jackson 2009; Curtin 2012; Weinberg and Hanahan 2011). Malignant cells having a mutator phenotype are often said to have developed “genomic instability”.
Mutational inactivation of components of the DNA Damage Response is a frequent cause of genomic instability in tumors (Jeggo and Lobrich 2015; Waters et al. 2014; Jackson 2009; Curtin 2012; Weinberg and Hanahan 2011). It has become clear that, in some cases, such mutational events create in malignant cells an absolute dependence upon the integrity of other DDR components for cell survival, a situation termed “synthetic lethality” (Gavande et al. 2016; Sanjiv et al. 2016; Morgan and Lawrecne 2015). The now classic example of this is the dependence of HR pathway-deficient BRCA1/2 tumor cells on integrity of the DNA Base Excision Repair (BER) system (Jackson 2009; Morgan and Lawrecne 2015). The BER system deals with DNA single strand breaks (SSB) that arise in nascent DNA during replication (Jackson 2009; Morgan and Lawrecne 2015) and it is regulated by poly-ADP ribose polymerase (PARP). If nascent strand SSB are left unrepaired, they will be present in some of the template DNA strands during the next round of replication, and cause replication fork collapse. Rescue of collapsed replication forks depends in turn upon function of the HR pathway. If BER is blocked in BRCA1/2 cells by inhibiting PARP with the drug olaparib, cell death results from breakdown of DNA replication (Jackson 2009; Morgan and Lawrecne 2015). Conversely, malignant cells having BER defects resulting from loss of DNA polymerase β (Morgan and Lawrecne 2015) would be expected to experience synthetic lethality with HR pathway inhibition.
As noted before, IR exposure is capable of provoking synthetic lethal interactions: ATR inhibition with VX-970 in malignant cells is tolerated in unstressed cells, but it is lethal in combination with replicative stress provoked by certain chemotherapeutic agents and IR (Sanjiv et al. 2016). It seems likely that other cases of synthetic lethality in tumor cells, in the context of combined IR and DDR inhibitor treatment, will be found, given the enormous range of genotoxic damages that IR causes (Citrin and Mitchell 2014; Jekimovs et al. 2014; Gavande et al. 2016; Higgins et al. 2015; Raleigh and Haas-Kogan 2013; Begg et al. 2011; Goodarzi and Jeggo 2013; Ceccaldi et al. 2016; Jeggo and Lobrich 2015; Waters et al. 2014). This prospect is an exciting one, given the remarkable ability to localize IR dose deposition with modern radiation therapy technology.
References
Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ (2007) HDAC inhibitor PCI-24781 decreased RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA 104:19482–19487
Al-Ubaidi FL, Schultz N, Loseva O, Egevad L, Granfors T, Helleday T (2013) Castration therapy results in decreased Ku70 levels in prostate cancer. Clin Cancer Res 19:1547–1556
Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH et al (2002) Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 62:7350–7356
Balcer-Kubiczek EK (2012) Apoptosis in radiation therapy: a double-edged sword. Exp Oncol 34:277–285
Begg AC, Stewart FA, Vens C (2011) Strategies to improve radiotherapy with targeted drugs. Nature Rev Cancer 11:239–253
Bobber JA, Harari PM, Giralt J, Cohen RB, Jones CU et al (2010) Radiotherapy plus Cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomized trial, and relation between Cetuximab-induced rash and survival. Lancet Oncol 11:21–28
Brognard J, Clark AS, Ni Y, Dennis PA (2001) Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res 61:3986–3997
Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M et al (2013) An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J Med Chem 56:254–263
Camphausen K, Burgan W, Cerra M, Oswald KA, Trepel JB et al (2004) Enhanced radiation enhanced-induced killing and prolongation of gammaH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res 64:316–321
Canman CE, Radany EH, Parsels LA, Davis MA, Lawrence TS, Maybaum J (1994) Cancer Res 54:2296–2298
Ceccacci E, Minucci S (2016) Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer 114:605–611
Ceccaldi R, Rondinelli B, D’Andrea AD (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26:52–63
Cengel KA, Voong KR, Chandrasekaran S, Maggiorella L, Brunner TB, Stanbridge E et al (2007) Oncogenic K-Ras signals through epidermal growth factor receptor and wild-type H-Ras to promote radiation survival in pancreatic and colorectal carcinoma cells. Neoplasia 9:341–348
Chen X, Wong P, Radany E, Wong JY (2009) HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm 24:689–699
Chen X, Wong JYC, Wong P, Radany EH (2011) Low-dose valproic acid enhances radiosensitivity of prostate cancer through acetylated p53-dependent modulation of mitochondrial membrane potential and apoptosis. Mol Cancer Res 9:448–461
Chen X, Wong P, Radany EH, Stark JM, Laulier C, Wong JY (2012) Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res 10:1052–1064
Cheung M, Testa JR (2013) Diverse mechanisms of AKT pathway activation inhuman malignancy. Curr Cancer Drug Targets 13:234–244
Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES et al (2008) Postradiation sensitization by the histone deacetylase inhibitor valproic acid. Clin Cancer Res 14:5410–5415
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD et al (2002) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014
Citrin DE, Mitchell JB (2014) Altering the response to radiation: sensitizers and protectors. Semin Oncol 41:848–859
Curtin N (2012) J DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12:801–817
Dassonville O, Bozec A, Fischel JL, Milano G (2007) EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors. Similarities and differences. Crit Rev Oncol Hematol 62:53–61
Dewey WC (2009) Arrhenius relationships from the molecule and cell to the clinic. Int J Hyperthermia 23:3–20
Diggle CP, Bentley J, Knowles MA, Kiltie AE (2005) Inhibition of double-strand break non-homologous end joining by cisplatin adducts in human cell extracts. Nucleic Acids Res 33:2531–2539
Dynlacht JR, Batuello CN, Lopez JT, Kim KK, Turchi JJ (2011) Identification of Mre11 as a target for heat radiosensitization. Radiat Res 176:323–332
Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA et al (2015) Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell 59:867–881
Garrido-Laguna I, Hong DS, Janku F, Nguyen LM, Falchook GS, Fu S et al (2012) KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS ONE 7:e38033
Gavande NS, Vandervere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR et al (2016) DNA repair targeted therapy: the past or future of cancer treatment? Pharmcol Ther 160:65–83
Goodarzi AA, Jeggo PA (2013) The repair and signaling responses to DNA double-strand breks. AnvGenet 82:1–45
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumor cells with defined genetic elements. Nature 400:464–468
Higgins GS, O’Cathail SM, Muschel RJ, McKenna WG (2015) Drug radiotherapy combinations: review of previous failures and reasons for future optimism. Cancer Treat Rev 41:105–113
Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV (2011) Identification of specific inhibitors of human RAD51 recombinase using high throughput screening. ACS Chem Biol 6:628–635
Jackson SP (2009) Bartek J The DNA damage response in human biology and disease. Nature 461:1071–1078
Jeggo PA, Lobrich M (2015) How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem J 471:1–11
Jekimovs C, Bolderson E, Suraweera A, Adams M, O’Byrne KJ, Richard DJ (2014) Chemotherapeutic compounds targeting the DNA double strand break repair pathways. Front Oncol 4:1–18
Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A (2007) Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem 282:21206–21212
Loeb LA (2016) Human cancers express a mutator phenotype: hypothesis, origin, and consequences. Cancer Res 76:2057–2059
Mckenna WG, Muchel RJ, Gupta AK, Hahn SM, Bernhard EJ (2003) The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 22:5866–5875
Mellert HS, Stanek TJ, Sykes SM, Rauscher FJ, Schultz DC, McMahon SB (2011) Deacetylation of the DNA-binding domain regulates p53-mediated apoptosis. J Biol Chem 286:4264–4270
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D et al (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486:532–536
Morgan MA, Lawrecne TS (2015) Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin Cancer Res 21:2898–2904
Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11:196–207
Nakamura JL (2007) The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications. Expert Opin Ther Targets 11:463–472
Nijkamp MM, Hoogsteen IJ, Span PN, Takes RP, Lok J, Rijken PF et al (2011) Spatial relationship of phosphorylated epidermal growth factor receptor and activated AKT in head and neck squamous cell carcinoma. Radiother Oncol 101:165–170
Pannunzio NR, Li S, Watanabe G, Lieber MR (2014) NHEJ often uses microhomology: implications for alternative end joining. DNA Repair 17:74–80
Park J, Feng J, Li Y, Hammarsten O, Brazil DP, Hemmings BA (2009) DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J Biol Chem 284:6169–6174
Raaphorst GPGP, Leblanc J-M, Li LF (2005) A comparison of response to cisplatin, radiation and combined treatment for cells deficient in recombination repair pathways. Anticancer Res 25:3–58
Raleigh DR, Haas-Kogan DA (2013) Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol 9:219–233
Sanjiv K, Hagenkort A, Calderon-Montano JM, Koolmeister T, Reaper PM et al (2016) Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Reports 14:298–309
Sears CR, Turchi JJ (2012) Complex cisplatin-double strand break (DSB) lesions directly impair cellular non-homologous end joining (NHEJ) independent of downstream damage response (DDR) pathways. J Biol Chem 287:24263–24272
Sears CR, Cooney SA, Chin-Sinex H, Mendoca MS, Turchi JJ (2016) DNA damage response (DDR) pathway engagement in cisplatin radiosensitization of non-small cell lung cancer. DNA Repair 40:35–46
Sekhar KR, Reddy YT, Reddy PN, Crooks PA, Venkateswaran A, McDonald WH, et al (2011) The novel chemical entity YTR107 inhibits recruitment of nucleophosmin to sites of DNA damage, suppressing repair of DNA double-strand breaks and enhancing radiosensitization. Clin Cancer Res 17:6490–6499
Seno JD, Dynlacht JR (2004) Intracellular redistribution and phosphorylation of proteins of the Mre11/Rad50/Nbs1 repair complex following irradiation and heat shock. J Cell Physiol 199:157–170
Seshacharyulu P, Ponnusamy MP, Harida D, Jain M, Ganti AK, Batra SK (2012) Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 16:15–31
Shewach DS, Lawrence TS (1995) Radiosensitization of human tumor cells by gemcitabine in vitro. Semin Oncol 22:68–71
Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois M-M, Maity R et al (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53:7–18
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R et al (2006) Acetylation of the p53 DNA binding domain regulates apoptosis induction. Mol Cell 24:841–851
Tanno S, Yanagawa N, Habiro A, Koizumi K, Nakano Y, Osanai M et al (2004) Serine/threonine kinase AKT is frequently activated in human bile duct cancer and is associated with increased radioresistance. Cancer Res 64:3486–3490
Tarish FL, Schultz N, Tanoglidi A, Hamberg H, Letocha H et al (2015) Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Science Trans Med 7:1–6
Thijssen R, Ter Burg J, Garrick B, van Bochove GG, Brown JR et al (2016) Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 128:574–583
Toulany M, Roderman HP (2015) Phosphatidylinositol 3-kinase/Akt signaling as a key mediation of tumor cell responsiveness to radiation. Semin Can Biol 35:180–190
Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J et al (2008) Targeting ofAKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther 7:1772–1781
Toulany M, Lee KJ, Fattah KR, Lin YF, Fehrenbacher B, Schaller M et al (2012) Akt1promotes post-irradiation survival of human tumor cells through initiation, progression and termination of DNA-PKcs-dependent DNA-double strand break repair. Mol Cancer Res 10:945–957
Van Putten JW, Groen HJ, Smid K et al (2001) End-joining deficiency and radiosensitization by gemcitabine. Cancer Ras 61:1585–1591
Wachters FM, Van Putten JW, Maring JG, Zdzienicka MZ, Grown HJ, Kampinga HH (2001) Selective targeting of homologous DNA recombination repair by gemcitabine. Int J Rad Oncol Biol Phys 57:553–562
Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA (2014) Nonhomologous end joining: a good solution for bad ends. DNA Repair 17:39–51
Weinberg R, Hanahan D (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
West AC, Johnstone RW (2014) New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124:30–39
Zheng M-h, Sun H-t, Xu J-g, Gang Y, Lei-ming H et al (2016) Combining whole brain radiotherapy with Gefitinib/Erlotinib for brain metastases from non-small-cell lung cancer: a meta analysis. RioMed Res Int 2016:5807346
Zhu J, Zhou L, Wu G, Konig H, Lin S et al (2013) A novel small molecule RAD51 inactivator overcomes imatinib resistance in chronic myeloid leukaemia. EMBO Mol Med 5:353–365
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Radany, E.H. (2017). The Mammalian DNA Damage Response as a Target for Therapeutic Gain in Radiation Oncology. In: Wong, J., Schultheiss, T., Radany, E. (eds) Advances in Radiation Oncology. Cancer Treatment and Research. Springer, Cham. https://doi.org/10.1007/978-3-319-53235-6_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-53235-6_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53233-2
Online ISBN: 978-3-319-53235-6
eBook Packages: MedicineMedicine (R0)