
Abstract
Biological networks are known to be robust despite signal disruptions such as gene failures and perturbations. Extensive research is currently under way to explore biological networks and identify the underlying principles of their robustness. Structural properties such as power-law degree distribution and motif abundance have been attributed for robust performance of biological networks. Yet, little has been done so far to quantify such biological robustness. We propose a platform to quantify biological robustness using network simulator (NS-2) by careful mapping of biological properties at the gene level to that of wireless sensor networks derived using the topology of gene regulatory networks found in different organisms. A Support Vector Machine (SVM) learning model is used to measure the correlation of packet transmission rates in such sensor networks. These sensor networks contain important topological features of the underlying biological network, such as motif abundance, node/gene coverage, and transcription-factor network density, which we use to map the SVM features. Finally, a case study is presented to evaluate the NS-2 performance of two gene regulatory networks, obtained from the bacterium Escherichia coli and the baker’s yeast Sachharomyces cerevisiae.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Studies have shown that the current Homo Sapiens have an estimated 250,000 years of evolution. However, the exact functioning of a Human body eludes us till date. Human body is an intricate system of complex mechanisms that continues to interest scientists including biologists, computational and medical researchers. To address this, an ambitious project called The Human Genome Project (HGP) was announced by the US Department of Energy in 1985. Its mission was to identify all the genes in the human genome. HGP was completed in the year 2003 [5]. However, the genome of every human being is unique and the data is still being refined to date [16]. Intensive research in structural genomics and their functional significance followed the completion of HGP creating the new field of Systems Biology, wherein the goal is to study the behavior and dynamics of complex biological systems.
Human body is made up of trillions of cells [23]. Each cell is comprised of genes in which information is encoded. The function of a cell varies depending on the level of gene expression that is regulated by a set of transcription factors. Such interacting genes and transcription factors can be represented as a Gene Regulatory Network (GRN). GRNs have been extensively explored by researchers as it is believed that they hold the key to unravel the mystery behind the working of a human body. Although a major portion of gene-gene interactions is still unknown for higher order organisms, the scientific community has recently focused on simulating the dynamics of GRNs from lower order organisms. In such simulations, it is essential to consider the topological characteristics of GRNs that contribute to their robustness in information transport.
Our contribution lies at this realm of GRNs and in-silico experiments. We propose a framework to quantify biological robustness using NS-2, a network simulator. NS-2 has been primarily used to simulate different computer networks including Wireless Sensor Networks (WSNs). Information in this chapter is categorized as follows. Section 1 presents a discussion on the state of computational modelling of biological systems. Section 2 presents similarities and differences between GRNs and WSNs thereby enabling a way to map a GRN to a WSN for simulation. Section 3 details the simulation setup including the parameters used and the assumptions. Section 3 also explains the network generation procedure and the sink selection strategies necessary for a network to be simulated. A case study is presented in Sect. 4 to identify the suitable model organism, between the bacterium Escherichia coli (abbreviated E. coli) and the baker’s yeast Saccharomyces cerevisiae, for mapping purposes. Finally, future research directions are presented in Sect. 5.
1 Computational Modelling
Ordinary differential equations (ODE) based computational models of biological systems, termed reaction rate equations or mass action kinetics, has received much attention [26]. Here, a homogeneous biological system is represented as a group of biochemical reactions and its dynamics are explored in the continuous-deterministic realm. However, ODE-based models are limited to study the underlying stochastic present in many biological processes such as gene expression and protein synthesis [6]. The limitations of ODE-based models for biological systems are detailed in [26].
Ghosh et al. [7] describes the advantages of using discrete event simulators for modeling biological systems. A fundamental challenge in computational systems biology [12] is the simplification of the biological system complexity without losing the ensemble dynamic behavior. In the system engineering view of complex processes [29], the key notion is to abstract the complexity of the system as a set of discrete time and space variables (random variables), which capture the behavior of the system in time. The entire system is a collection of functional blocks or modules, which are driven by a set of events, where an event defines a large number of micro level state transitions between a set of state variables accomplished within the event execution time. The underlying assumption driving this abstraction is the segregation of the complete state space into such disjoint sets of independent events which can be executed simultaneously without any interaction. The application of this technique in large complex communication networks has demonstrated the accuracy of the approach for the first and higher order dynamics of the system within the limits of input data and state partitioning algorithms [30]. For example, discrete event based system modeling has been effectively applied for designing routers, the key components responsible for routing traffic through the Internet. Discrete event based simulation techniques have also been used in a wide variety of manufacturing processes and studying the system dynamics of complex industrial processes.
Researchers have also tried to adapt existing simulation platforms to model molecular communication. NanoNS is one such [8] simulation framework to model molecular communications. The framework is built over NS-2 software and uses a diffusive molecular communication channel. Researchers in [8] present an extensive review of communication models in nanoscale networks and out of three possible molecular communications, namely diffusive, motor-based and gap junction-based, their work is focused on diffusive-based molecular communication. As an extension of this work, researchers presented a case to build models for a variety of molecular communication channels, intra-body molecular nanonetworks and the network of such intra-body nanonetworks in [17]. This work comprehensively showcases the significance of modelling nanonetworks. Efforts are currently underway to simulate wireless nano sensor networks using NS-3 software (next version of NS-2) [24]. In this work, wireless nano sensor networks are modelled using electromagnetic communication instead of molecular communication as mentioned above. As it is evident by now, the challenges in achieving a simulation framework for communications in molecular networks are multifold [21]. Our core goal here is to identify ground rules for GRN-based robustness—the ability of a biological state to persist despite component errors—by setting up a generic NS-2 simulation platform, rather than developing more detailed molecular communication channels.
Network simulator, NS-2 (NS-2), is a discrete event simulator widely used for studying wireless networks. NS-2 has been used by researchers to model communication in wireless networks and embedded devices. This simulator continues to evolve with the active support of the research community. Taking a step forward, we have used NS-2 as an in-silico platform for quantifying the robustness of biological networks. Specifically, since the primary objective of a wireless sensor network is information transport to specific sink nodes, and because they operate under similar noisy and error prone conditions as biological networks, we define robustness of biological networks as the ability for each node in the network to deliver packets with minimal packet loss. Before envisioning a model for any time-varying functional biological system, it is important to illustrate the preliminary model for the biological system in NS-2. While exclusive simulators to model a molecular network are not present currently, existing simulators can be adjusted to model the desired network. It should be noted that this might not be the perfect approach, but the opportunity to explore the qualitative and quantitative dynamics of molecular networks is not lost. Scenarios are presented below whenever applicable to demonstrate the use of NS-2 to quantify biological robustness.
2 Mapping GRNs to WSNs
Transmission inconsistencies frequently plague WSNs where they suffer from signal disruptions due to sensor failure or from the absence of routing protocols that are sufficiently insensitive to local as well as global network conditions. In a WSN, nodes sense, process and communicate information with each other. Structurally, a GRN can be related to a WSN where every gene or transcription factor is a sensor. Signal transmission within a GRN can be considered as packet transmission in a WSN. The fundamental assumptions in modelling a bio-inspired WSN are [10]:
-
1.
GRN node structure is preserved in WSN.
-
2.
Interactions among nodes in WSN are based on the existing connections in the GRN.
The physical signaling structure of sensors within the WSN must be adapted to reflect the communication between genes in the GRN. If gene G1 up-regulates G2, then the equivalent interaction in the WSN is that sensor S1 sends a packet to S2 according to specific probability distribution defined by gene-gene interactions. For homogeneous sensor nodes, each up-regulation edge in a GRN is replaced by a bi-directional edge; if we allow sensor S1 to send a packet to S2, then S2 should also be able to send a packet to S1. For heterogeneous sensor nodes, however, it is not necessary that both S1 and S2 possess the same transmission radii, giving a directed edge from S1 to S2 and not vice versa.
We recognize that WSNs conceptually operate under noisy and/or adverse conditions similar to the stochastic cellular environment encountered by GRNs. We hypothesize that if it is possible to exploit the simulation platform used for WSNs, namely NS-2, to assess the signal transmission robustness in GRNs, then any observed robust qualities can be explained by fundamental biological processes, such as transcription. The process where signals from nearby neighbors in the form of transcription factors stimulate/inhibit other genes by generating mRNA molecules is transcription. Thus, GRN nodes communicate with one another by sending signals (transcription factors), which are in return processed into output signals (mRNAs). This process is similar to WSNs where sensors receive packets from its neighbors with packet forwarding instructions to other destination nodes. As a result, any node in a network (GRN or WSN) can affect the decision of other nodes and hence the overall network performance.
Here, we considered the transcriptional regulatory network (TRN) of the bacterium E. coli to generate the sample GRN graphs. Such TRNs bear the actual topology of the GRNs with any gene-gene and gene to transcription factor edges deleted. Thus, in such TRNs, a single transcription factor can regulate other transcription factors and genes, while genes do not directly regulate other nodes. Note that our earlier work on WSNs derived from GRN topologies actually considered the TRNs from E. coli which were shown to achieve high packet transmission efficiency [10]; hence such TRNs exhibit the desired biological robustness measures that we seek to model here. The transcription factor molecules having half-lives \(T_{1/2} = \ln 2/k\) [2], where k represents the decay rate constant, are subject to degradation if held at the transcriptional regulation queue. Similarly in the case of WSNs, packets are forwarded from source nodes to sink nodes using multiple hops and can be dropped at intermediate nodes if they exceed the queue length. Hence, genes can be considered as sink nodes and transcription factors as the source nodes. On that account, we describe our measure of robustness in WSNs that adopt the GRN topologies as the ability for each node in the network to deliver information to their local sinks with minimal packet loss.
3 NS-2 Simulation Setup
Consider a biological network topology derived from a well studied organism, E. coli. Sub-networks that are extracted from E. coli comprise of interactions among genes. Let us call this extracted network a Gene Regulatory Network (GRN). Such GRNs comprise two classes of nodes: transcription factors and genes. A transcription factor either up-regulates or down-regulates one (or more) gene. The packet transmission rates are assumed to be identical in NS-2, for all the non-sink nodes; however, in a real biological setting, such rates are directly proportional to the rate constants associated with every edge in the network along with the concentration of the molecules associated with a node. This however creates a roadblock for existing biological network simulators as each of these rate constants need to be experimentally validated which is not currently feasible for the different sample networks generated in this work. The simulation also assumes all packets transmitted to be identical in type and size which correspond to similar signaling molecules affecting the different nodes in the GRN in the context of biological robustness.
Queue limit in NS-2 is useful to limit the number of packets that can be queued at a node. Queue limit in the corresponding GRN represents the half-life of each signal sent from one node to another node. Although this is another approximation in the simulation set-up, it is impossible to characterize all such signaling molecules accurately in the different extracted GRNs. In summary, our proposed NS-2 set-up makes broad assumptions for the pertinent details of biological network signaling but we feel that this is indeed necessary for studying the qualitative dynamics of many sample GRNs wherein such details are not known at length.
Traditionally, robustness of biological networks has been measured by its static graph theoretic characteristics such as network diameter, average shortest path [22], network efficiency [15] amongst others. A network with negligible change in its diameter is considered to be robust when it loses node(s) after an attack. Similarly, negligible change in average shortest path and network efficiency under network perturbations related to temporal fluctuations in the node and/or link availability is attributed to robust networks. Packet receival rate is the ratio of the number of packets received in the network to the number of packets sent. Higher the packet receival rate of a GRN, higher its robustness. Randomly generated WSNs and GRN-derived networks are compared with respect to the packet receival rate. This section discusses the methods used for random network generation to be used as a wireless sensor network. In addition, approaches used to identify the sink node in the network are detailed. A new algorithm for biological network generation is presented in Sect. 3.2.1.
3.1 Network Generation
A script written in the Python programming language [25] is used to generate networks modelled as WSNs. Here, two different nodes within the network are chosen at random, and a link is established between them with probability p.Footnote 1 Networks with 100, 150, 200, 250 and 300 nodes were generated for demonstration purposes as representing “medium” sized sensor networks. 25 networks of each size (100, 150, 200, 250 and 300 nodes) are considered to illustrate the sink node selection approach. Networks of a certain size are spread over an area with specific node transmission range. For example, 25 different networks of size 150 nodes are spread over \(3{\cdot }6\times 10^5\)m\(^2\) (with x = 600 m and y = 600 m) with a node transmission range of 85 m. Node range for a network has been assigned based on the work by [9]. Similarly, networks of size 200 are spread over area of \(4.9\times 10^5\) m\(^2\) (with x = 700 m and y = 700 m) with a node transmission range of 90 m and networks of size 250 are spread over \(8.1\times 10^5\) m\(^2\) (with x = 900 m and y = 900 m) with a node transmission range of 90 m. Networks of size 300 are spread over an area of 10\(^6\) m\(^2\) (with x = 1000 m and y = 1000 m) with a node transmission range of 110 m. Few assumptions are made for simplicity. The directionality of the links between the nodes is ignored. Self-edges,Footnote 2 edges with same source and destination nodes, are removed from the network. Nodes in model organisms such as E. coli and S. cerevisiae self, up- or down-, regulate themselves. However, we ignore self-edges in this case of WSN simulation. In order to compare similar entities, only networks with same number of nodes and edges are considered for comparison. All 25 networks of the same size have exact number of edges. Each network generated using this approach is considered to be a Random Wireless Sensor Network (RWSN).

Sink node selection and respective packet receival rates for different loss models—GRN of 20 nodes [10]
3.2 Sink Selection Strategy
Sink node selection strategy is critical for optimal GRN performance. In [11], we listed three sink selection strategies: (a) Highest Degree (HD), (b) Highest Coverage (HC) and (c) Motif-based (MB) and identified HD strategy as the best approach to provide higher robustness for NS-2 based simulation of GRNs. Nodes with highest degree are selected as a sink node in the HD strategy. Node involved in any three-node motif is selected as a sink node in the HC strategy. Figure 1 shows the comparison of sink selection strategies for a GRN-derived of twenty nodes [10]. In this figure, FHD stands for the node with First Highest Degree, SHD stands for the node with Second Highest Degree, THD stands for the node with Third Highest Degree and PBN stands for node identified with Probabilistic Boolean Network. A PBN is a formalism where set of functions define the expression value of genes in the network. The node with the highest expression is selected as sink node. For detailed information on sink selection strategies including PBN, refer to [10]. Intuitively, using FHD node as a sink makes sense since the node is regulated, in a biological context, by several other regulators and are critical for important biological functionalities. Such nodes also act as hubs in a network.
Three-node motifs have been earlier identified as the building blocks of robust GRNs [20] from a purely topological perspective, and the feed-forward loops, wherein two genes regulate each other and they both regulate a third, were reported to have the most significant impact on GRN robustness. Hence, we also considered nodes involved most in a feed-forward loop (FFL) motif as a sink node in the MB strategy. We considered FFL motifs as they have been identified to play an important role in establishing robustness [13] apart from ensuring important biological functions such as generating signal pulses, and speeding up or delaying response times in target genes [18].

Comparison of best, mean and worst (out of 25 networks) performing RWSNs to GRN—network size 100 [10]

Comparison of best, mean and worst (out of 25 networks) performing RWSNs to GRN—network size 300 [10]

Comparison of average of ten networks between new algorithm and RWSN—network size 1500

Comparison of average of ten networks between new algorithm and RWSN—network size 2500
In [10], we compared several GRN-derived networks with randomly generated networks (network sizes 100, 150, 200, 250 and 300) and showed that GRN-derived networks improve the transmission reliability in our NS-2 based simulation setting. The procedure for generating random networks is described in Sect. 3.1. Figures 2 and 3 present a comparison for best, mean and worst performing RWSNs and GRN of network sizes 100 and 300 respectively. For this experiment, a total of 25 RWSNs are considered and three cases are presented. Comparisons are also made for large-scale predicted GRNs (network size 1500, 1750, 2000, 2250 and 2500). The performance of GRN versus RWSNs in large scale networks (network size 1500 and 2500) is presented in Figs. 4 and 5. The graphs for network sizes 1750, 2000 and 2250 are not reported since they follow similar trend as networks of size 1500 and 2500. This might be possible due to the presence of higher number of FFLs in GRN-derived networks as compared to randomly generated networks. The abundance of FFL motifs in random networks and networks derived from new algorithmFootnote 3 is presented in Table 1. The counts reported in the table are averaged, and approximated to nearest decimal, across ten different networks of a particular type.
3.2.1 New Network Generation Algorithm
Here we discuss the network generation from our work in [19]. For brevity, the Scale-free Directed Network Generator is referred to as SDNG. The algorithm can be utilized to expand existing networks as well as generating directed networks emulating the different distributions of E. coli, namely in-degree, out-degree, cumulative degree and the participation of genes in feed-forward loops. The algorithm is similar to the Barab\(\acute{a}\)si-Albert (BA) model which uses the preferential attachment mechanism [1] for growing scale-free (SF) networks. Networks are grown resembling the phenomena known as the ‘rich get richer and the poor get poorer’, however the BA model was originally employed for undirected networks. The duplication-divergence (DD) model suggested by [28] considers the growth of directed biological networks. The suggested model which was later extended in [4] was predicated by the fact that proteins/genes evolve through copying themselves followed by their subsequent infrequent mutation. In addition to using the cumulative distribution as the sole measure for resembling the original networks, few of the DD grown networks retained a power-law distribution.
To illustrate the dynamics of SDNG, we consider denoting candidate nodes for preferential attachments in an existing network of size n with subscript i, wherein \(K_{i}\) and \(R_{i}\) label the out- and in-degrees respectively. The probability for a candidate node to be connected to a node foreign to the existing network with an edge directed from the candidate node to the foreign node is given by \(A(K_{i}, R_{i})\). The probability that a link is drawn from the foreign node to the candidate node is given by \(B(K_{i}, R_{i})\). Each probability is normalized against all nodes of the existing network to form attachment kernels [14], and their formulas are listed in Table 2.
For this particular work, we considered the power-law attachment kernel for calculating the edge probabilities. Starting with a fully connected eight node network, a candidate node is picked at random with equi-probability. Next, a random number d is selected with equi-probablity from the interval \(d\in (0,1)\). An edge is drawn from the candidate node to the foreign node if \(d\le A(K_{i}, R_{i})\). This process is then repeat for an edge drawn out of the foreign node to the candidate node, provided the probability satisfies \(d\le B(K_{i}, R_{i})\). The above steps are then reiterated \(m_{i}-1\) times, wherein \(m_{i}\) is an another number selected at random from an exponential probability distribution \(\rho (m_{i})=(f^{\frac{1}{1-m_{0}}}-1)f^{-m_{i}/(1-m_{0})}\). The decay of this distribution resembles the degree distribution of E. coli. Here, we considered values of \(f=\frac{1}{4}\) and \(m_{0}=2\).
3.3 SVM Validation
While the network evaluations presented in Figs. 2 and 3 establish the significance of GRN-derived networks, only one sink operates in those networks which is not the case in functional GRNs. To address this, we used multiple sink nodes to model GRN communication. An Support Vector Machine (SVM) model, built using LibSVM [3], is then used to investigate the relative efficiency of packet receival rates based on topological metrics such as network density, genes coverage, transcription factor network density, motif abundance and genes percentage, defined below.
For this, GRNs of varying sizes, \(100<n <500\) were used, where n is the number of nodes in the GRN. Transmission is considered from source nodes (similar to transcription factors) to sink nodes (similar to gene nodes). 410 out of the 490 networks are used to train the learning model and remaining networks are used to test the model. The directionality of the links between the nodes is considered. The topological metrics used in the learning model are briefly described below.
3.3.1 Network Density (ND)
ND is a ratio of the number of edges present in the network to the total number of edges possible in the network.
3.3.2 Genes Coverage (GC)
GC is the summation of the ratios of in-degree of each sink node to the ratio of source nodes having a path to that particular sink node.
3.3.3 Transcription Factor Network Density (TND)
TND is the ratio of the number of edges that transcription factor nodes participate to the total number of edges in the network.
3.3.4 Motif Abundance
Motif abundance is the ratio of abundances of FFL (R\(^{FFL}\)) and bifan (R\(^{BF}\)) motifs that relate to the number of nodes.
3.3.5 Genes Percentage (GP)
GP is the ratio of number of gene nodes to the total number of nodes in the network.
3.4 Contributions of Topological Metrics to GRN Robustness
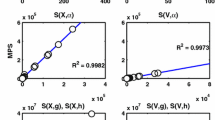
These topological metrics are then used to construct the SVM learning model. Cross validation is used in the training stage; test data is then used to predict the robustness of the networks. The relative importance of the features used in the model in the decreasing order is as follows: ND, R\(^{BF}\), GP, TND, GC and R\(^{FFL}\). Figure 6a shows the weight \(w_{i}\) of features divided by the maximum weight (\(w_{ND}\)): \(|{w{i}}\slash {w_{ND}}|\). Figure 6b shows same ratio but the directions of the weights are considered. It should be noted that a GRN is more communicative when it is sparse implying low ND and high R\(^{BF}\) as shown in Fig. 6b.

a Relative importance of the feature weights, b Relative importance of feature directions

Comparison of best performing networks derived from E. coli and Yeast—20, 35 and 50% loss
4 Case Study: Comparison of Derived Networks from E. Coli and Yeast
We have demonstrated the performance of NS-2 as a platform to quantify robustness in biological networks. In order to exploit the principles of a biological network, it is crucial to evaluate the model organisms. For this purpose, we compare networks derived from two well studied model organisms, E. coli and S. cerevisiae, of sizes consisting 100, 200, 300, 400 and 500 nodes using GeneNetWeaver software [27]. One hundred networks of each size are generated and NS-2 simulations are performed on each of these networks. As comparing the average performance of all networks may not distinguish the performance of the derived networks properly, we compared the best perfoming, average performing and least performing networks. The directionality of the links between the nodes is ignored. The simulation parameters are as follows:
-
1.
Bandwidth = 1Mb
-
2.
Delay = 1.0 ms
-
3.
Queue limit = 5
-
4.
Packet size = 900 bytes
Figure 7 shows the best performing derived networks from E. coli and S. cerevisiae for network sizes: 100, 200, 300, 400, and 500 (nodes) w.r.t. 20, 35 and 50% loss. While the performance of S. cerevisiae derived networks is consistently higher for 500 node network under 20 and 35 and 50% loss, E. coli derived networks perform better, in almost all cases except for 200 network size at 20% loss, for networks of size 100, 200, 300 and 400.

Comparison of mean performing networks derived from E. coli and Yeast–20, 35 and 50% loss
Figure 8 shows the mean performing derived networks from E. coli and S. cerevisiae for network sizes: 100, 200, 300, 400, and 500 (nodes) w.r.t 20, 35 and 50% loss. It can be clearly observed from the figure that the performance of E. coli derived network is better at 20 and 35% loss and S. cerevisiae derived network performs better for higher loss percentage (50%). The difference in performance is \(\sim 0.51\) at 20 % loss (E, coli), \(\sim 0.38\) at 35 % loss (E. coli), \(\sim 0.359\) for 50% loss (Yeast). It appears that S. cerevisiae derived network performs better than E. coli derived network at higher loss percentage. Similarly, Fig. 9 shows the worst performing E. coli and S. cerevisiae derived networks for network sizes: 100, 200, 300, 400, and 500 (nodes) w.r.t 20, 35 and 50% loss. It can also be noticed here that S. cerevisiae derived network performs better than E. coli derived network only for higher network size (500 nodes) and the latter performs better than the former for other network sizes (100, 200, 300 and 400 nodes).

Comparison of worst performing networks derived from E. coli and Yeast—20, 35 and 50% loss

Comparison of 100 node networks derived from E.coli and Yeast respectively—20%, 35%, 50% loss

Comparison of 500 node networks derived from E.coli and Yeast respectively—20%, 35%, 50% loss
Figure 10 shows the comparison of packet receival rates for networks of size 100 (nodes). The difference in the packet receival rates of the best performing E. coli and S. cerevisiae derived networks suggests that E. coli-derived network performs better than yeast-derived network. Figure 11 shows the comparison of packet receival rates for networks of size 500 (nodes).
To arrive at any decisive conclusion on a better model organism for WSN mapping, extensive simulations need to be performed to check if this trend holds for higher network sizes (1000 or 1500 or 2000 node network etc.). Since S. cerevisiae performs marginally better at a high loss rate, sparse WSNs in real-world applications—where communication is essential even at high loss, for instance, during rescue operations after natural disasters—can be modelled using the structural principles of yeast-derived GRN.
Our simulation setup using NS-2 is generic and can be applied to any GRN (e.g.: E. coli, S. cerevisiae), and thus provides a common platform to assess dynamic robustness of biological networks. This also allows to sample several extracted and predicted GRN topologies and measure their signal transmission dynamics thereby identifying specific topological and control properties in these networks that impact their robustness. Such a platform will hence allow one to compare the robustness of the GRN topologies of different organisms, design, validate, test and explore different GRN prediction algorithms besides also serving the greater complex networks community by applying such design rules of robust biological networks to create fault-tolerant and efficient engineered systems.
5 Challenges and Future Directions
NS-2 is a discrete event simulator built for exploring wired networks and then extended to study wireless networks. It is not exclusively built for communication in molecular networks. Creating an environment for simulating molecular networks is extremely challenging. In a biological network, transmission of signals from one node (transcription factor) to another (gene/transcription factor) occurs at a rate that has not been determined yet. Active effort by researchers is focused on estimating such rate constants. Determining the rate constants is critical for modelling the dynamic behavior of a biological system. While our work is preliminary, it allows us to qualitatively and quantitatively simulate biological networks (specifically GRNs) without any knowledge of the underlying rate constants. This will help in establishing the reasons behind the inherent robustness of GRNs as well as motivate the design of efficient WSNs, wherein routing algorithms that intuitively embed biological structural properties in WSNs need to be developed. This can be realized using repeating structural patterns in biological networks termed as motifs.
A WSN can be categorized into several pockets of such patterns and routing can be introduced from different nodes to the sink to achieve higher packet transmission efficiency. Adaptive routing mechanisms can be imagined to improve WSN efficiency. Bandwidth limitations on edges and nodes in a regulatory network need to be studied before bandwidth based studies can be carried out in WSNs. Much needs to be realized in this field before a true bio-inspired WSN is modelled that adheres to structural and dynamic behavior of a biological system.
References
Barabáasi, A-L, Albert (1999) Emergence of scaling in random networks. In: Science 286.5439, pp 509–512
Belle A, Tanay A, Bitincka L, Shamir R, OShea EK (2006) Quantification of protein half-lives in the budding yeast proteome. In: Proceedings of the National Academy of Sciences 103.35, pp 13004–13009. doi:10.1073/pnas.0605420103. eprint: http://www.pnas.org/content/103/35/13004.full.pdf+html. url: http://www.pnas.org/content/103/35/13004.abstract
Chang Chih-Chung, Lin Chih-Jen (2011) LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol (TIST) 2(3):27
Chung, F, Lu L, Dewey TG, Galas DJ (2003) Duplication models for biological networks. J Comput Biol 10(5):677–687
Collins, FS, Morgan M, Patrinos A (2003) The human genome project: lessons from large-scale biology. Science 300(5617):286–290
Ghosh Preetam, Ghosh Samik, Basu Kalyan, Das Sajal K, Zhang Chaoyang (2010) Discrete di usion models to study the e ects of Mg2+ con- centration on the PhoPQ signal transduction system. BMC Genom 11(Suppl 3):S3
Ghosh S, Ghosh P, Basu K, Das SK, S Daefler S (2011) A discrete event based stochastic simulation platform for in silico study of molecular-level cellular dynamics. J Biotechnol Biomater 6:2
Gul E, Atakan B, Akan OB (2010) NanoNS: a nanoscale network simulator framework for molecular communications. Nano Commun Netw 1(2):138–156
Han B, Leblet J, Simon G (2009) Query range problem in wireless sensor networks. Commun Lett IEEE 13(1):55–57. doi:10.1109/LCOMM.2009.081546. Institute, Information-Sciences. NS-2. http://isi.edu.nsnam/ns
Kamapantula BK, Abdelzaher A, Ghosh P, Mayo M, Perkins EJ, Das SK (2012a) Leveraging the robustness of genetic networks: a case study on bio-inspired wireless sensor network topologies. J Amb Intell Hum Comput 1–17
Kamapantula BK, Abdelzaher A, Ghosh P, Mayo M, Perkins E, Das SK (2012b) Performance of wireless sensor topologies inspired by E. coli genetic networks. In: 2012 IEEE International conference on Pervasive Computing and Communications Workshops (PERCOM Workshops). IEEE, pp 302–307
Kitano H (2002) Computational systems biology. Nature 420(6912):206–210
Kitano H (2007) Towards a theory of biological robustness. Mol Syst Biol 3(1)
Krapivsky Paul L, Redner Sidney, Leyvraz Francois (2000) Connectivity of growing random networks. Phys Rev Lett 85(21):4629
Latora V, Marchiori M (2004) The architecture of complex systems. Oxford UP
Lunshof Jeantine E, Bobe Jason, Aach John, Angrist Misha, Thakuria Joseph V, Vorhaus Daniel B, Hoehe Margret R, Church George M (2010) Personal genomes in progress: from the human genome project to the personal genome project. Dialog Clin Neurosci 12(1):47
Malak D, Ozgur BA (2012) Molecular communication nanonetworks inside human body. Nano Commun Netw 3(1):19–35
Mangan S, Uri A (2003) Structure and function of the feed-forward loop network motif. In: Proceedings of the National Academy of Sciences, vol 100, no. 21, pp 11980–11985
Mayo M, Abdelzaher A, Perkins EJ, Ghosh P (2012) Motif participation by genes in E. coli transcriptional networks. Front Physiol 3(357). ISSN: 1664-042X. doi:10.3389/fphys.2012.00357. http://www.frontiersin.org/fractal_physiology/10.3389/fphys.2012.00357/abstract
Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002) Network motifs: simple building blocks of complex networks. Science 298(5594):824–827
Nakano T, Moore MJ, Wei F, Vasilakos AV, Shuai J (2012) Molecular communication and networking: Opportunities and challenges. NanoBiosci IEEE Trans 11(2):135–148
Ng Alex KS, Efstathiou Janet (2006) Structural robustness of complex networks. Phys Rev 3:175–188
NIH (2013) Cells and DNA—Genetics Home Reference. http://ghr.nlm.nih.gov/handbook/basics?show=all
Piro G, Grieco LA, Boggia G, Camarda P, DEE-Dip di Elettrotecnica (2013) Simulating wireless nano sensor networks in the NS-3 platform. In: Proceedings of Workshop on Performance Analysis and Enhancement of Wireless Networks, PAEWN, Barcelona, Spain
Python, Software Foundation (1991) Core Python Programming. http://www.python.org
Samoilov MS, Arkin AP (2006) Deviant effects in molecular reaction pathways. Nat Biotech 24(10):1235–1240
Schaffter T, Marbach D, Floreano D (2011) GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods. Bioinformatics 27(16):2263–2270
Vázquez A, Flammini A, Maritan A, Vespignani A (2002) Modeling of protein interaction networks. Complexus 1(1):38–44
Zeigler BP, Praehofer H, Kim TG et al (1976) Theory of modeling and simulation, vol 19. John Wiley, New York
Zeng X, Bagrodia R, Gerla M (1998) GloMoSim: a library for parallel simulation of large-scale wireless networks. In: Twelfth Workshop on Parallel and Distributed Simulation, 1998. PADS 98. Proceedings. IEEE, pp. 154–161
Acknowledgements
This work is supported by NSF and the US Army’s Environmental Quality and Installations 6.1 basic research program. The Chief of Engineers approved this material for publication.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Kamapantula, B.K., Abdelzaher, A.F., Mayo, M., Perkins, E.J., Das, S.K., Ghosh, P. (2017). Quantifying Robustness in Biological Networks Using NS-2. In: Suzuki, J., Nakano, T., Moore, M. (eds) Modeling, Methodologies and Tools for Molecular and Nano-scale Communications. Modeling and Optimization in Science and Technologies, vol 9. Springer, Cham. https://doi.org/10.1007/978-3-319-50688-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-50688-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50686-9
Online ISBN: 978-3-319-50688-3
eBook Packages: EngineeringEngineering (R0)