
Abstract
At first the role of σ1 receptors in various neurological, psychiatric and neurodegenerative disorders is discussed. In the second part, the principle of positron emission tomography (PET ) is described and the known fluorinated PET tracers for labeling of σ1 receptors are presented. The third part focuses on fluoroalkyl substituted spirocyclic PET tracers, which represent the most promising class of fluorinated PET tracers reported so far. The homologous fluoroalkyl derivatives 12–15 show high σ1 affinity (K i = 0.59–1.4 nM) and high selectivity over the σ2 subtype (408–1331-fold). The enantiomers of the fluoroethyl derivative fluspidine 13 were prepared and pharmacologically characterized. Whereas the (S)-configured enantiomer (S)-13 (K i = 2.3 nM) is 4-fold less active than the (R)-enantiomer (R)-13 (K i = 0.57 nM), (S)-13 is metabolically more stable. The interactions of (S)-13 and (R)-13 with the σ1 receptor were analyzed at the molecular level using the 3D homology model. In an automated radiosynthesis [18F](S)-13 and [18F](R)-13 were prepared by nucleophilic substitution of the tosylates (S)-17 and (R)-17 with K[18F]F in high radiochemical yield, high radiochemical purity and short reaction time. Application of both enantiomers [18F](S)-13 and [18F](R)-13 to mice and piglets led to fast uptake into the brain, but [18F](R)-13 did not show washout from the brain indicating a quasi-irreversible binding. Both radiotracers [18F](S)-13 and [18F](R)-13 were able to label regions in the mouse and piglet brain with high σ1 receptor density. The specific binding of the enantiomeric tracers [18F](S)-13 and [18F](R)-13 could be replaced by the selective σ1 ligand SA4503.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
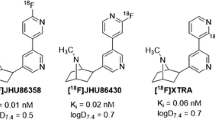

Keywords
- Neuroimaging
- PET , synthesis
- Radiosynthesis
- σ1 receptors
- Ligand receptor interaction
- Enantioselective kinetics
- Non-covalent quasi irreversible binding
4.1 Introduction: σ1 Receptors in Brain Diseases
σ1 receptors play a major role in various pathological conditions in the periphery (e.g. vascular diseases, cancer ) and in the central nervous system (CNS), where they are involved in various neurological, psychiatric and neurodegenerative disorders. Non-invasive imaging of σ1 receptors by positron emission tomography (PET ) can be useful for studying the pathophysiology of these CNS diseases. Moreover, a PET tracer for labeling of σ1 receptors can be used for target validation, visualization and quantification of metabolic and biochemical processes as well as diagnosis and prognosis of a particular disease [1, 2].
4.1.1 Pain
It has been shown that the σ receptor system functions as endogenous anti-opioid system. Whereas σ1 agonists , such as (+)-pentazocine (Fig. 4.1) lead to reduced opioid receptor-mediated analgesia , the opposite effect is produced by σ1 antagonists , e.g. haloperidol [3]. The combination of opioid analgesics with σ1 antagonists allows the reduction of opioid dose, while maintaining strong analgesia but reducing opioid- mediated side effects. Furthermore, opioid receptor mediated analgesia is potentiated by downregulation of σ1 receptors [4]. In addition to the modulation of opioid mediated analgesia , σ1 receptor antagonists show promising analgesic activity in various neurogenic pain models. The role of σ1 receptors in neuropathic pain conditions was confirmed by σ1 receptor knock-out mice: capsaicin could not induce mechanical allodynia in σ1 receptor knock-out mice, but it was able to induce mechanical allodynia in wild-type mice. This mechanical allodynia was inhibited dose-dependently by σ1 antagonists . Moreover, selective σ1 agonists were able to reverse this analgesic effect [5]. The most developed drug in this field is the σ1 antagonist S1RA (Fig. 4.1), which showed high analgesic activity in various models of neurogenic pain. After successful completion of the phase I clinical trial, a phase II clinical trial with S1RA for the treatment of neuropathic pain caused by various conditions is currently ongoing. Moreover, S1RA is investigated as add-on therapy to analgesic opioids with the aim to enhance the analgesic effect and reduce dose and adverse side effects pf the opioid [6, 7].

Important σ1 receptor ligands: (+)-pentazocine , the prototypical benzomorphan σ1 receptor agonist ; S1RA in phase II clinical trials for pain management /neuropathic pain , add-on to opioids); antidepressants imipramine and fluoxetine; antipsychotics haloperidol and panamesine; cocaine as example for an abuse compound; anti-Alzheimer drug (acetylcholinesterase inhibitor) donepezil
4.1.2 Depression
There is strong evidence that σ1 receptors are involved in the pathophysiology of depression . It was observed that σ1 receptor knock-out mice develop a depressive-like behavior. [8] In animal models of depression (e.g. forced swim test) some σ1 agonists showed antidepressive properties [9, 10]. Antidepressant activity of σ1 agonists was also observed in animal models of Alzheimer’s disease (AD ) [11, 12]. In addition to their main pharmacological mechanism several clinically used antidepressants reveal high to moderate σ1 affinity (Ki(σ1) = 20–200 nM). In Fig. 4.1 the tricyclic antidepressant imipramine and the selective serotonin reuptake inhibitor fluoxetine are shown exemplarily. It is assumed that the interaction of these drugs with the σ1 receptor contributes to their overall antidepressive effects. Moreover, a downregulation of σ1 receptors in the striatum, hippocampus and cerebral cortex was reported after repeated treatment of rats with imipramine and fluoxetine [13]. The same effect was observed after repeated treatment with the σ1 agonist (±)-pentazocine [14].
4.1.3 Psychosis
In addition to its dopamine D2 receptor antagonistic activity the clinically used prototypical antipsychotic haloperidol (Fig. 4.1) reacts as a potent antagonist at σ1 receptors (K i(σ1) = 3.9 nM). Moderate to high σ1 affinity has also been reported for other clinically used antipsychotics. It is supposed that σ1 antagonistic activity contributes significantly to the observed antipsychotic activity of these antipsychotics. Recently, a correlation between a polymorphism within the σ1 receptor gene and increased risk of schizophrenia was reported [15]. During the past 10 years five σ1 antagonists (panamesine (EMD57445), eliprodil (SL82.0715), rimcazole (BW234U), BMY14802 (BMS181100), DuP734) entered clinical trials for the treatment of schizophrenia [16]. In Fig. 4.1 panamesine is depicted exemplarily for this class of ligands.
4.1.4 Addiction
Activation of σ1 receptors contributes considerably to plasticity processes underlying reinforcement and addiction . The high density of σ1 receptors in addicted rats may play a crucial role in the reinforcing effects of addictive drugs, such as methamphetamine , cocaine and ethanol [17]. After chronic self-administration of methamphetamine to rats, σ1 receptor upregulation has been found [18–20]. It was postulated that methamphetamine-induced dopamine D2 autoreceptor downregulation leads to increased protein kinase A activity resulting in increased production of σ1 receptors [21]. The existence of heteromeric receptors consisting of both σ1 and dopamine D1 receptors supports the hypothesis of the involvement of σ1 receptors in addictive processes [22]. The behavioral effects caused by methamphetamine could be inhibited by the σ1 antagonist MS-377. Cocaine (Fig. 4.1) binds with high affinity at the σ1 receptor and behaves as σ1 receptor agonist [23]. Cocaine responses of addicted rats could be blocked by σ1 antagonists . Whereas the σ1 antagonist BD1047 diminishes ethanol induced behavioral effects, the σ1 agonist PRE-84 reinforced these addictive responses [24].
4.1.5 Alzheimer’s Disease
Although σ1 receptors are not involved in learning and memory processes, since these processes cannot be modulated by σ1 agonists or σ1 antagonists , they are involved in diseases associated with memory deficit. The role of σ1 receptors was investigated in a mouse model of Alzheimer’s disease , which was generated by central application of amyloid β25–35. It was shown that the selective σ1 agonist (+)-pentazocine could attenuate dose-dependently the memory deficits occurring seven days after amyloid β25–35 injection. Acetylcholinesterase inhibitors, which are clinically used for the treatment of Alzheimer’s disease , represent the first-line therapy. It has been reported that donepezil (Fig. 4.1) not only inhibits the acetylcholinesterase but also activates σ1 receptors . This interaction with σ1 receptors was postulated to contribute to the overall neuroprotective and anti-amnesic effects of donepezil [25]. In a PET study with early Alzheimer’s patients a low density of σ1 receptors was observed. It may be concluded that activation of σ1 receptors might be a useful strategy for the treatment of Alzheimer’s disease .
4.2 Fluorinated PET Tracers for σ1 Receptors
4.2.1 Principle of Positron Emission Tomography (PET )
Positron emission tomography (PET ) represents a promising modality for studying biological processes in a non-invasive manner. For PET a positron emitter is required, i.e. a radioactive nuclide with increased number of protons in the nucleus. This type of isotope can release a positron and a neutrino (ν) upon conversion of a proton into a neutron. The emitted positron travels in matter until it meets its antiparticle, an electron. The positron and the electron react with each other in an annihilation process, i.e. transformation of the complete mass of the particles into irradiation energy. The annihilation process generates two gamma quants with an energy of 511 keV, which move in opposite directions (angle = 180°). A signal is only accepted as an annihilation event, when two gamma quants are registered simultaneously in opposite directions. The registration of two signals allows the identification of the origin of the irradiation and thus the original position of the PET tracer (Fig. 4.2).

Principle of PET
The most commonly used non-metallic positron emitters are 11C, 13N, 15O, and 18F. The corresponding decay reactions and half-lives are depicted in Table 4.1. Since the radionuclides 11C, 13N and 15O decay with very short half-lives, the isotope 18F with a half-life of 110 min represents the most interesting radionuclide for the development of PET tracers. In particular labeling with 18F does not require a cyclotron on bedside. However, C, O, and N atoms are present in almost all pharmacologically active compounds. The corresponding radionuclides can be introduced without changing the structure of the compound. In contrast, F atoms are not present in all drugs, and therefore a potent fluorine containing drug has to be developed first before the corresponding 18F–labeled ]PET tracer can be developed. This report is focusing on fluorinated PET tracers for imaging of σ1 receptors .
4.2.2 Fluorinated PET Tracers for Imaging of σ1 Receptors
In the literature a great variety of fluorinated PET tracers for imaging of σ1 receptors is reported [26]. In the following part the fluorinated PET tracers are classified into compounds bearing 18F at the aromatic ring (Fig. 4.3) and compounds with an aliphatic 18F atom (Fig. 4.4).

Structure, σ1 and σ2 affinity of PET tracers with aromatic 18F

Structure, σ1 and σ2 affinity of PET tracers with aliphatic 18F
Although methods for the introduction of [18F] fluoride into the aromatic ring have been reported, very often several further reaction steps are required to obtain the final PET tracer. The additional reaction steps lead to a longer production time and thus reduced radiochemical yields.
Cyclopropyl-(4-nitrophenyl) methanone served as starting material for the synthesis of [18F]haloperidol and [18F]BMY14802. (Fig. 4.3) At first [18F]fluoride was introduced by nucleophilic aromatic substitution of the nitro moiety with Cs[18F]F. Subsequent cleavage of the cyclopropyl ring with HCl and nucleophilic substitution of the resulting chlorobutyrophenone with the corresponding N-heterocycle provided the PET tracers [18F]haloperidol and [18F]BMY14802 [27–29], whereby the synthesis of [18F]BMY14802 required additional reduction of the ketone [30]. The selectivity of haloperidol and BMY-14802 for the σ1 receptor is rather low, since both compounds show strong interactions with dopamine and σ2 receptors as well. Therefore, both PET tracers can be used for the determination of uptake, distribution, penetration of the blood brain barrier, metabolism, and further pharmacokinetic parameters. The selective labeling of σ1 or dopamine receptors with these tracers is however not possible.
The [18F]fluorobenzylamines [18F]1, [18F]2a, and [18F]2b were obtained in a four-step process. Nucleophilic substitution of 2- or 4-nitrobenzaldehyde with Cs[18F]F or K[18F]F led to the radioactively labeled fluorinated benzaldehyde. Reduction of the aldehyde and nucleophilic substitution afforded the fluorinated benzyl iodide, which was coupled with the corresponding piperidine to provide the PET tracers [18F]1, [18F]2a, and [18F]2b in 3–10 % radiochemical yield. In rats and monkeys the PET tracer [18F]1 showed rapid brain uptake and fast washout. Replacement studies with haloperidol confirmed selective labeling of σ1 receptors without addressing the σ2 subtype [31]. In rat distribution experiments high penetration of [18F]2a into the brain was detected. However, the high liver uptake could not be blocked by haloperidol. Furthermore, the high σ2 affinity of [18F]2a and [18F]2b has to be considered [32].
In contrast to the radiosynthesis of the previous compounds, the radiosynthesis of [18F]3 was performed in a single step at the end of the synthesis by nucleophilic aromatic substitution of the corresponding 2-NO2 derivative with K[18F]F/Kryptofix 2.2.2/K2CO3 complex. The radiochemical yield was 5–10 % (decay corrected). In mice, high uptake of the PET tracer [18F]3 in the brain and peripheral organs was observed. The specific binding could be displaced by administration of haloperidol [33].
Fluorinated PET tracers with [18F]fluoride bound at a sp3-hybridized C-atom (aliphatic C-atom) are prepared by nucleophilic substitution (SN2 reaction) of mesylate ([18F]4, [18F]5) or tosylate precursors ([18F]8, [18F]9, [18F]10, [18F]fluspidine). This transformation is usually performed as the last step of the synthesis , i.e. the labeled compound is produced and purified without further transformations.
In an alternative strategy, precursor molecules with a phenol or other nucleophilic functional group are coupled with [18F]fluoroethyl ([18F]FE-SA4503, [18F]6) or [18F]fluoromethyl ([18F]FM-SA4503, [18F]7) moieties at the end of the synthesis . In these cases the fluorinated reagents [18F]FCH2CH2OTos and [18F]H2CBrF have to be prepared first by nucleophilic substitution of TosOCH2CH2OTos and H2CBr2 with K[18F]F/Kryptofix system, respectively. Whereas the production of [18F]FCH2CH2OTos represents a standard procedure in radiochemistry [34], the synthesis of the [18F]H2CBrF is more challenging and not available everywhere.
In a first human study, the fluoropropyl derivative [18F]4 did not reach a transient equilibrium in the brain within 3 h after injection of the radiotracer. A significant washout of the radiotracer from the brain was not observed. Moreover, instability of the radiotracer [18F]4 was reported [35, 36]. In rodents, the fluoroethyl derivative [18F]5 showed a much faster clearance from the brain than [18F]4. Human studies with [18F]5 have not been reported thus far [37–39].
Originally a low σ1: σ2 selectivity of FE-SA4503 (6) was reported [40, 41], which was later corrected to be in the range of 14 [42]. Therefore the PET tracer [18F]FE-SA4503 ([18F]6) was investigated in various animal models. A PET study with rhesus monkeys resulted in a fast uptake of [18F]6 in the brain and enrichment of [18F]6 in σ1 receptor rich regions, but the receptor ligand binding did not reach an equilibrium within 90 min [40]. The fluoromethoxy derivative [18F]FM-SA4503 ([18F]7) represents an uncommon PET tracer, since fluoromethoxy derivatives usually undergo fast metabolic degradation and extensive defluorination due to the acetalic nature of this group. However, these reactions were not observed for [18F]7. Moreover, replacement studies with haloperidol in monkeys revealed a higher specific binding for [18F]7 than for [11C]SA4503 rendering [18F]7 a more potent PET tracer for labeling of σ1 receptors in the brain [41].
The synthesis and biological evaluation of the [18F]fluoroethoxy and [18F]fluoroalkyl labeled PET tracers [18F]8, [18F]9, [18F]10, and [18F]fluspidine ([18F]13) were reported very recently. In animal studies with monkeys and mice, the biodistribution of the PET tracers was analyzed. They show accumulation in regions of the CNS and the periphery with high σ1 receptor density. The specificity of σ1 receptor binding was proven by displacement experiments with haloperidol [43–45]. In σ1 receptor knock-out mice the extraordinarily potent tracer [18F]10 (K i = 2.5 pM) [44] showed rapid brain uptake and rapid clearance without specific interaction with any other brain target. In animal studies signs of toxicity could not be detected. Due to its high σ1 receptor affinity and high specificity, [18F]10 is currently evaluated for imaging of σ1 receptors in various neurological disorders, such as chronic pain and Alzheimer’s disease [46].
The spirocyclic PET tracer [18F]fluspidine ([18F]13) [47], which is the only fluorinated PET tracer with a center of chirality, belongs to the most promising PET tracers reported so far and will be discussed in more detail in part 3 “Spirocyclic PET tracers” of this report.
4.3 Spirocyclic PET Tracers
4.3.1 Homologous Fluoroalkyl Derivatives 12–15
The development of spirocyclic PET tracers started with the 2-benzofuran 11 [48, 49]. It was found that 11 interacts with very high affinity with σ1 receptors (K i = 1.1 nM). Since the σ2 affinity (K i = 1280 nM) is very low, 11 shows an excellent 1100-fold selectivity for σ1 receptors over the σ2 subtype. Cross reaction with other targets could not be detected during a screening against more than 60 other receptors, ion channels , transporters and enzymes. 11 did not interact with the hERG channel [50]. The hERG channel is a voltage gated K+-channel in the heart, whose blockade can lead to life threatening arrhythmia caused by QT time prolongation. During drug development, hERG channel interactions are determined very early to avoid heart problems. In the field of σ1 receptors the hERG channel is of particular importance, since the pharmacophores of σ1 receptor ligands and hERG channel blockers are very similar [51]. In the mouse capsaicin assay, 11 showed high analgesic activity, which is in the same range as the analgesic activity of S1RA (Fig. 4.1). Therefore, 11 is regarded as σ1 receptor antagonist. Incubation with rat liver microsomes led to the identification of seven metabolites. The acetalic functional group represents a major position for metabolic transformations [50].
In order to remove the chemically and metabolically labile acetalic functionality and to install a structural element, which allows the introduction of a fluorine atom at the very end of the synthesis , the methoxy group of 11 was replaced by homologous fluoroalkyl residues (compounds 12–15 in Fig. 4.5). The homologous fluoroalkyl derivatives 12–15 show low nanomolar up to subnanomolar σ1 affinity (see Fig. 4.5). Moreover, all four homologs display very high subtype selectivity [52–57]. The fluoroethyl derivative 13, which was termed fluspidine, showed the highest σ1 affinity (K i = 0.59 nM) and the highest subtype selectivity (1331-fold) of this series of compounds.

Design of fluoroalkyl substituted spirocyclic 2-benzofurans 12–15 starting from the methoxy derivative 11
4.3.2 Radiosynthesis of [18F]12-[18F]15
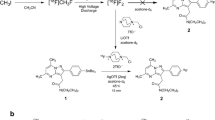
Due to the high σ1 affinity all four fluoroalkyl derivatives 12–15 were synthesized in radioactive form (Scheme 4.1). For this purpose the tosylates 16–19 were reacted with K[18F]F complexed with the cryptand K2.2.2. (Kryptofix®). The higher homologs [18F]13-[18F]15 were obtained in high radiochemical yield and purity within 20–30 min in refluxing acetonitrile [47, 55, 57]. However, the fluorination of the tosyloxymethyl derivative 16 required 20 min heating in DMSO at 150 °C to yield [18F]12 [52].

Radiosynthesis of [18F]12-[18F]15
Reagents and reaction conditions: (a) K[18F]F, K2.2.2., DMSO, K2CO3, 150 °C, 20 min, for [18F ]12; (b) K[18F]F, K2.2.2., acetonitrile, K2CO3, 85 °C, 20–30 min for [18F]13-[18F]15
a procedure time: time for the overall procedure including synthesis , purification and formulation
b rad. yield: radiochemical yield, decay corrected
c spec. activ.: specific activity of the final PET tracer
The complete procedure for the radiosynthesis , purification and formulation of the PET tracers [18F]12-[18F]15 usually took less than 120 min (approx. 1 half-life of 18-fluorine). The radiochemical yield was in the range of 40–50 % and the radiochemical purity of the final PET tracers was >98.6 %.
Due to the high σ1 affinity and convenient radiosynthesis the four PET tracers [18F]12-[18F]15 were evaluated in vivo. Although all four PET tracers [18F]12-[18F]15 were suitable for imaging of σ1 receptors in the brain, the fluoroethyl derivative fluspidine (13) appeared to be the most promising candidate in terms of radiochemical availability, brain uptake (4.7 % ID/g, 30 min post injection (p.i.)), brain-to-plasma ratio (13, 60 min p.i.), specific binding (specific displacement by haloperidol), formation of radiometabolites (94 % of parent compound in plasma 30 min p.i., only one major radiometabolite was detected) and imaging contrast [54]. Therefore, the enantiomers of fluspidine (13) were separated before further studies were performed in vivo .
4.3.3 Synthesis of Enantiomerically Pure [18F]-(R)-and -(S)-Fluspidine
For the synthesis of racemic fluspidine two independent routes are reported (Scheme 4.2). According to the first route, 2-bromobenzaldehyde was reacted with the Wittig reagent [(1,3-dioxolan-2-yl)methyltriphenylphosphonium bromide and K2CO3 to afford an α, β-unsaturated acetal [54]. Halogen metal exchange with n-BuLi, subsequent addition of the aryllithium intermediate at 1-benzylpiperidin-4-one and treatment of the product with HCl gave the aldehyde 21. Reduction of the aldehyde 21 with NaBH4 led to an alcohol, which was converted directly into the fluoroethyl derivative 13 upon treatment with DAST (diethylaminosulfur trifluoride). For the radiosynthesis of enantiomerically pure fluspidine enantiomers the tosylate 17 represented the central intermediate.

Synthesis of enantiomerically pure (R)- and (S)-fluspidine (R)-13 and (S)-13
In an alternative route, the tosylate 17 was obtained starting with the hemi acetal 23. The P-ylide Ph3P=CHCO2Et reacted with 23 in a Domino reaction, consisting of ring opening of the hemi acetal 23 to give an hydroxyaldehyde, Wittig reaction of the aldehyde with the P-ylide and subsequent intramolecular conjugate addition to the α,β-unsaturated ester [49]. Reduction of the ester 22 with LiAlH4 provided the alcohol, which was transformed into the tosylate 17.
Reagents and conditions: (a) [(1,3-dioxolan-2-yl)methyltriphenylphosphonium bromide, [(CH2O)2CHCH2PPh3 Br], K2CO3, tris[methoxyethoxyethyl)amine. (b) n-BuLi, −78 °C, 1-benzylpiperidin-4-one. (c) HCl, THF. (d) NaBH4, CH3OH. (e) TosCl, NEt3, DMAP. (f) Ph3P=CHCO2Et, Cs2CO3, toluene, reflux. (g) LiAlH4, Et2O, −15 °C. (h) chiral preparative HPLC, Daicel Chiralpak IB®. (i) K[18F]F/K2.2.2, K2CO3, CH3CN, 85 °C.
The enantiomeric tosylates (R)-17 and (S)-17 were separated by a chiral preparative HPLC using a Daicel Chiralpak IB® column [58]. The enantiomeric tosylates (R)-17 and (S)-17 were isolated in 98.2 % ee and 97.8 % ee, respectively. Reaction of the enantiomeric tosylates (R)-17 and (S)-17 with tetrabutylammonium fluoride (TBAF) in THF resulted in the fluspidine enantiomers (R)-13 (99.6 % ee) and (S)-13 (96.4 % ee). The absolute configuration was determined by circular dichroism. The σ1 affinity of the fluspidine enantiomers (R)-13 and (S)-13 was 0.57 nM and 2.3 nM indicating the (R)-enantiomer being the eutomer with an eudismic ratio of 4.
Due to the high σ1 affinity of both fluspidine enantiomers (R)-13 and (S)-13, the radiosynthesis of both [18F](R)-13 and [18F](S)-13 was performed by nucleophilic substitution of the tosylates (R)-17 and (S)-17 with K[18F]F complexed with the cryptand Kryptofix(R). For the careful preclinical evaluation of the PET tracer fluspidine [18F]13 and its enantiomers [18F](R)-13 and [18F](S)-13 an automated radiosynthesis was developed [59]. The key features of the automated radiosynthesis are a reaction time of 15 min in boiling acetonitrile (85 °C), 59 ± 4 min time for the complete process, 37 % radiochemical yield (decay corrected) and >98.8 % radiochemical purity.
4.3.4 Interaction of (R)- and (S)-Fluspidine with the σ1 Receptor
For a molecular-level description of the binding of fluspidine enantiomers to the σ1 receptor , the putative binding modes of (R)-13 and (S)-13 on our σ1 receptor 3D homology model were retrieved [60–62]. The two enantiomers were then docked into the putative binding site of the σ1 receptor, and their affinity toward the receptor was scored after long runs of Molecular Dynamics (MD) simulation by MM/PBSA (Molecular Mechanics/Poisson Boltzmann Surface Area) analysis [63].
In a typical structure of the MD-simulated σ1–ligand complexes, both (R)-13 and (S)-13 are oriented horizontally inside the receptor binding pocket and adopt similar binding poses, as illustrated in Fig. 4.6. For both enantiomers, the -NH+ moiety of the ligand piperidine ring is anchored around the negatively charged side chain of D126 of the σ1 protein, interacting with each other through a permanent salt bridge. As tracked by MD simulations, the average distance for the salt bridge through the proton at the cationic moiety of 13 and the COO− group of σ1 D126 is 3.1 ± 0.1 Å for the (R)-enantiomer and 3.3 ± 0.1 Å for the (S)-enantiomer (Fig. 4.6, upper panels). A stable hydrogen bond between the donor hydroxyl group of T151 and the acceptor counterpart in the benzofuran moiety of fluspidine is also detected during the entire course of the MD simulation. Of note, the hydrophobic pocket lined by the side chains of the receptor residues I128, F133, Y173 and L186 with the further stabilizing contribution of E172 perfectly encases the 2-benzofuran portion of both enantiomers. Finally, the equilibrated MD trajectories revealed the presence of stabilizing π/cation and π/π interactions between the N-benzyl ring of (R)-13 and (S)-13 and the side chains of R119 and W121.

Details of the key interactions detected in the equilibrated MD snapshots of (R)-13 (upper left panel) and (S)-13 (upper right panel) in complex with the σ1 receptor . The main protein residues involved in ligand/receptor interactions are R119, W121 (π-interaction; cyan), D126 (salt bridge; red), I128, F133, E172, Y173, L186 (hydrophobic cavity; steel blue) and T151 (hydrogen bond; green). Compounds (R)-13 and (S)-13 are shown in atom-colored sticks and-balls: C, gray; O, red; and N, blue. H atoms are not shown but the salt bridges and the hydrogen bonds are indicated as black broken lines. In both panels, water molecules, ions and counterions are not shown for clarity. (bottom left panel) Binding free energy (ΔGbind) and its components for (R)-13 and (S)-13 in complex with the σ1 receptor. All energy values are in kcal/mol. The calculated Ki(σ1)calcd. Values (nM), obtained using the relationship ΔG = −RTln(1/Ki) are also reported. (bottom right panel) Per residue enthalpic contribution (ΔHbind,res) to binding for the σ1 receptor in complex with (R)-13 and (S)-13. Only σ1 residues critical for receptor binding are shown
All of the interactions described above are quantified by a calculated free energy of binding ΔGbind (Fig. 4.6, bottom left panel) for σ1 equal to −11.98 ± 0.31 kcal/mol for (R)-13 and −11.72 ± 0.32 for (S)-13 corresponding to an estimated affinity Ki(σ1)calcd. Value of 1.66 nM and 2.58 nM, respectively, in stringent agreement with the experimentally determined Ki values.
Analyzing the single energetic component of the binding free energy, we can see that for both compounds the overall polar component disfavors binding (i.e., ΔEELE + ΔGPB = +11.88 kcal/mol for (R)-13 and +11.67 kcal/mol for (S)-13, respectively). However, the decomposition of the polar interactions into its Coulombic (ΔEELE) and solvation contributions (ΔGPB) shows that indeed the direct intermolecular electrostatic interactions (ΔEele) are always favorable to binding but their contribution cannot compensate the large, unfavorable term (ΔGPB) stemming from desolvation penalties associated with the binding event, thereby ultimately leading to an unfavorable contribution. In contrast, the intermolecular van der Waals interactions (ΔEVDW) and the nonpolar solvation term (ΔGNP) provide the driving force for binding. The highly favorable total nonpolar binding free energy reproduces the considerable contribution afforded by the stabilizing interactions from the various hydrophobic residues that line the surface of the binding cavity between the receptor and the ligands (i.e., ΔEVDW + ΔGNP = −52.01 kcal/mol for (R)-13 and −51.47 kcal/mol for (S)-13, respectively). Finally, the entropic terms for both enantiomers are almost equal (i.e., TΔSbind = +28.15 kcal/mol for (R)-13 and +28.08 kcal/mol for (S)-13, respectively), as could be expected since the two molecules are identical both from the standpoint of the molecular structure and the loss in degrees of freedom they undergo upon binding to the σ1 receptor . As illustrated in the bottom right panel of Fig. 4.6, a deconvolution of the enthalpic component (ΔHbind,res) of the binding free energy into contributions from each protein residue was carried out to investigate in detail the binding mode of both enantiomers (R)-13 and (S)-13 to the σ1 receptor. Specifically, the stable hydrogen bond involving T151 (Average Dynamic Length (ADL) = 2.00 ± 0.02 Å for (R)-13 and 2.06 ± 0.02 Å for (S)-13) and the stable salt bridge featured by D126 are responsible for stabilizing contributions of −1.53 kcal/mol for (R)-13 and −1.46 kcal/mol for (S)-13 and −2.70 kcal/mol for (R)-13 and −2.68 kcal/mol for (S)-13, respectively. Moreover, substantial van der Waals and electrostatic interactions are contributed by residues R119 (−0.89 kcal/mol for (R)-13 and −0.90 kcal/mol for (S)-13) and W121 (−1.95 kcal/mol for (R)-13 and −1.89 kcal/mol for (S)-13), through the aforementioned π-cation and T-stacking π−π interaction, respectively, and by the residues belonging to the hydrophobic pocket I128, F133, E172, Y173, L186 (∑ΔHbind,res = −6.42 kcal/mol for (R)-13 and ∑ΔHbind,res = −6.19 kcal/mol for (S)-13). All other receptor residues were characterized by negligible interaction enthalpy values (|ΔHbind,res| < 0.30 kcal/mol).
4.3.5 In Vitro Biotransformation of (R)- and -(S)-Fluspidine
The biotransformation was investigated in vitro upon incubation of both non-labeled fluspidine enantiomers (R)-13 and (S)-13 with rat liver microsomes and NADPH/H+. At first the rate of degradation was determined in kinetic experiments over a period of 90 min[57]. After an incubation period of 30 min approx. 72 % of both parent fluspidine enantiomers were detected. However, after a period of 90 min, only 33 % of (R)-13 was left, whereas 58 % of unchanged (S)-13 was found. These experiments indicate a higher metabolic stability of (S)-fluspidine (S)-13 compared to its (R)-configured enantiomer (R)-13.
In the next step, the number and structure of the formed metabolites was analyzed [58]. For (S)-fluspidine (S)-13 eight metabolites were identified by LC-MS experiments: the N-debenzylated metabolite (S)-13A, four monooxygenated metabolites (S)-13B–E and three metabolites containing two additional O-atoms. In Fig. 4.7 the primary metabolites of (S)-13 are depicted showing the metabolic labile positions, which are attacked by CYP enzymes. Regarding the biotransformation the N-benzyl moiety represents a privileged structural element for degradation, since the N-debenzylated metabolite (S)-13A, the 4-hydroxybenzyl metabolite (S)-13B and the N-oxide (S)-13C were identified. Additionally, metabolites with an OH moiety at the piperidine ring ((S)-13D) and at the fluoroethyl side chain ((S)-13E) were identified. The dioxygenated metabolites were formed by further hydroxylation of the 4-hydroxybenzyl metabolite (S)-13B. at the piperidine ring, the N-atom, and the hydroxybenzyl moiety. Although (R)-13 was transformed faster than (S)-13, only seven metabolites were formed upon incubation of (R)-13 with rat liver microsomes and NADPH/H+. The metabolite (R)-13E bearing the OH moiety in the fluoroethyl side chain could not be detected for the enantiomer (R)-13.

Metabolically labile positions of (S)-fluspidine (S)-13
4.3.6 In Vivo PET Studies with [18F](R)- and [18F](S)-Fluspidine
The in vivo kinetics was carefully investigated after application of the radiotracers [18F](S)-13 and [18F](R)-13 to mice and piglets [64]. In the piglet study the brain uptake and wash-out kinetics of the enantiomers [18F](S)-13 and [18F](R)-13 showed significant differences. The initial uptake of the enantiomers was very similar, but the wash-out of [18F](R)-13 was very slow. After 120 min the concentration of [18F](R)-13 in the brain was almost the same as after 5 min indicating a quasi-irreversible but non-covalent binding of the more affine enantiomer [18F](R)-13. The specific interactions of [18F](S)-13 and [18F](R)-13 with σ1 receptors was reduced by administration of the selective σ1 ligand SA4503 confirming the selective labeling of σ1 receptors (Fig. 4.8, part c). The recorded data were used to establish a tracer kinetic model for both enantiomers, the influx rate constant k 1, the clearance rate constant k2’, and the binding potential (k 3’/k 4) for various brain regions were estimated and the distribution volume in the whole brain was calculated.

Studies with the PET tracer [18F](S)-fluspidine ([18F](S)-13): (a): ex vivo autoradiography of mouse brain; (b): PET image of whole piglet brain; (c): inhibition study with the σ1 ligand SA4503; the region of the thalamus is shown exemplarily (Modified according to Ref. [2])
Determination of the plasma concentration of the parent compounds showed that the metabolic degradation of [18F](R)-13 is significantly faster than that of [18F](S)-13, which confirms the in vitro experiment with rat liver microsomes. For both enantiomers lipophilic radiometabolites, which would be able to penetrate into the brain, were not detected.
In Fig. 4.8 brain images obtained after treatment of the animals with the fluorinated PET tracer [18F](S)-fluspidine ([18F](S)-13) are shown. The ex vivo autoradiography of a mouse brain (Fig. 4.8 part a) reveals high concentration of the radioligand in those regions, which are reported to be rich in σ1 receptors . The resolution of the PET image of the whole piglet brain (Fig. 4.8 part b) is reduced compared to the mouse autoradiography, which directly detects β+ (e+) particles, whereas PET detects gamma ray coincidences. The specificity of [18F](S)-fluspidine binding to σ1 receptors was investigated with a blocking experiment. In which a large amount of SA4503 was administered. In Fig. 4.8, part c, the thalamus of piglet brain is displayed exemplarily indicating that labeling with [18F](S)-fluspidine can be inhibited by the σ1 receptor ligand SA4503
4.4 Conclusion
Preclinical evaluation performed with [18F](S)- and [18F](R)-fluspidine ([18F](S)-13) and ([18F](R)-13) indicates that both tracers are valuable tools for selective non-invasive visualization and quantification of σ1 receptors in the brain under healthy and diseased conditions. To provide a molecular rationale of the interactions between fluspidine enantiomers and the σ1 receptor , the two enantiomers were docked into the putative binding site of our σ1 3D receptor model, and their affinity toward the receptor was scored by MM/PBSA analysis. The results of our modeling investigations confirm that both enantiomers of 13 can be accommodated within the σ1 binding site and establish the same network of stabilizing interactions with the target receptor. Although (S)-fluspidine shows lower σ1 affinity than (R)-fluspidine, the higher metabolic stability of (S)-fluspidine and its common reversible binding kinetics favors slightly this enantiomer for further development. However, the potential of the non-covalent quasi-irreversible σ1 binding of [18F](R)-fluspidine remains to be further elucidated in clinical studies.
References
Toyohara J, Sakata M, Ishiwata K (2009) Imaging of σ1 receptors in the human brain using PET and [11C]SA4503. Cent Nerv Syst Agents Med Chem 9(3):190–196
Brust P, van den Hoff J, Steinbach J (2014) Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci Bull 30(5):777–811
Chien CC, Pasternak GW (1994) Selective antagonism of opioid analgesia by a sigma system. J Pharmacol Exp Ther 271:1583–1590
Maurice T, Su TP (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther 124:195–206
Entrena JM, Cobos EJ, Nieto FR, Cendan CM, Gris G, Del Pozo E, Zamanillo D, Baeyens JM (2009) σ1 receptors are essential for capsaicin-induced mechanical hypersensitivity: studies with selective σ1 ligands and σ1 knockout mice. Pain 143(3):252–261
Diaz JL, Cuberes R, Berrocal J, Contijoch M, Christmann U, Fernandez A, Port A, Holenz J, Buschmann H, Laggner C, Serafini MT, Burgueno J, Zamanillo D, Merlos M, Vela JM, Almansa C (2012) Synthesis and biological evaluation of the 1-arylpyrazole class of σ1 receptor antagonists: identification of 4-{2-[5-methyl-1-(naphthalen-2-yl)-1H-pyrazol-3-yloxy]ethyl}morpholine (S1RA, E-52862). J Med Chem 55(19):8211–8224
Wünsch B (2012) The σ1 receptor antagonist S1RA is a promising candidate for the treatment of neurogenic pain. J Med Chem 55(19):8209–8210
Sabino V, Cottone P, Parylak SL, Steardo L, Zorrilla EP (2009) σ1 receptor knockout mice display a depressive-like phenotype. Behav Brain Res 198(2):472–476
Hayashi T, Su TP (2008) An update on the development of drugs for neuropsychiatric disorders: focusing on the σ1 receptor ligand. Expert Opin Ther Targets 12(1):45–58
Lucas G, Rymar VV, Sadikot AF, Debonnel G (2008) Further evidence for an antidepressant potential of the selective σ1 agonist SA 4503: electrophysiological, morphological and behavioural studies. Int J Neuropsychopharmacol 11(4):485–495
Urani A, Romieu P, Roman FJ, Yamada K, Noda Y, Kamei H, Manh Tran H, Nagai T, Nabeshima T, Maurice T (2004) Enhanced antidepressant efficacy of σ1 receptor agonists in rats after chronic intracerebroventricular infusion of β-amyloid-(1-40) protein. Eur J Pharmacol 486(2):151–161
Urani A, Romieu P, Roman FJ, Maurice T (2002) Enhanced antidepressant effect of sigma1 (σ1) receptor agonists in β25-35-amyloid peptide-treated mice. Behav Brain Res 134(1–2):239–247
Shirayama Y, Nishikawa T, Umino A, Takahashi K (1993) p-chlorophenylalanine-reversible reduction of binding sites by chronic imipramine treatment in rat brain. Eur J Pharmacol 237(1):117–126
Shirayama Y, Nishikawa T, Takahashi K (1994) Differential effects of repeated dl-pentazocine treatment on σ binding sites in discrete brain areas of the rat. Neurosci Lett 165(1–2):219–222
Ohi K, Hashimoto R, Yasuda Y, Fukumoto M, Yamamori H, Umeda-Yano S, Kamino K, Ikezawa K, Azechi M, Iwase M, Kazui H, Kasai K, Takeda M (2011) The SIGMAR1 gene is associated with a risk of schizophrenia and activation of the prefrontal cortex. Prog Neuro-Psychopharmacol 35(5):1309–1315
Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP (2011) Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets 15(5):557–577
Sabino V, Cottone P, Blasio A, Iyer MR, Steardo L, Rice KC, Conti B, Koob GF, Zorrilla EP (2011) Activation of σ-receptors induces binge-like drinking in sardinian alcohol-preferring rats. Neuropsychopharmacology 36(6):1207–1218
Guitart X, Codony X, Monroy X (2004) Sigma receptors: biology and therapeutic potential. Psychopharmacology 174(3):301–319
Robson MJ, Noorbakhsh B, Seminerio MJ, Matsumoto RR (2012) σ1 receptors: potential targets for the treatment of substance abuse. Curr Pharm Des 18(7):902–919
Katz JL, Su TP, Hiranita T, Hayashi T, Tanda G, Kopajtic T, Tsai SY (2011) A role for σ receptors in stimulant self administration and addiction. Pharmaceuticals 4(6):880–914
Stefanski R, Justinova Z, Hayashi T, Takebayashi M, Goldberg SR, Su TP (2004) Sigma1 receptor upregulation after chronic methamphetamine self-administration in rats: a study with yoked controls. Psychopharmacology 175(1):68–75
Navarro G, Moreno E, Aymerich M, Marcellino D, McCormick PJ, Mallol J, Cortes A, Casado V, Canela EI, Ortiz J, Fuxe K, Lluis C, Ferre S, Franco R (2010) Direct involvement of σ1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc Natl Acad Sci U S A 107(43):18676–18681
Sharkey J, Glen KA, Wolfe S, Kuhar MJ (1988) Cocaine binding at sigma receptors. Eur J Pharmacol 149:171–174
Maurice T, Su TP (2009) The pharmacology of sigma-1 receptors. Pharm Ther 124:195–206
Meunier J, Ieni J, Maurice T (2006) The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol 149(8):998–1012
Brust P, Deuther-Conrad W, Lehmkuhl K, Jia H, Wünsch B (2014) Molecular imaging of σ1 receptors in vivo: current status and perspectives. Curr Med Chem 21:35–69
Yousef KA, Fowler JS, Volkow ND, Dewey SL, Shea C, Schlyer DJ, Gatley SJ, Logan J, Wolf AP (1996) [18 F]haloperidol binding in baboon brain in vivo. Nucl Med Biol 23(1):47–52
Schlyer DJ, Volkow ND, Fowler JS, Wolf AP, Shiue CY, Dewey SL, Bendriem B, Logan J, Raulli R, Hitzemann R, Brodie J, Alavi AA, MacGregor RR (1992) Regional distribution and kinetics of haloperidol binding in human brain: a PET study with [18F] haloperidol. Synapse 11:10–19
Shiue CY, Fowler JS, Wolf AP, McPherson DW, Arnett CD, Zecca L (1986) No-carrier-added fluorine-18-labeled N-methylspiroperidol: synthesis and biodistribution in mice. J Nucl Med 27:226–234
Ding YS, Fowler JS, Dewey SL, Wolf AP, Logan J, Gatley SJ, Volkow ND, Shea C, Taylor DP (1993) Synthesis and PET studies of fluorine-18-BMY 14802: a potential antipsychotic drug. J Nucl Med 34(2):246–254
Mach RH, Gage HD, Buchheimer N, Huang Y, Kuhner R, Wu L, Morton TE, Ehrenkaufer RL (2005) N-[18F]4′-fluorobenzylpiperidin-4yl-(2-fluorophenyl) acetamide ([18F]FBFPA): a potential fluorine-18 labeled PET radiotracer for imaging σ1 receptors in the CNS. Synapse 58(4):267–274
Dence CS, John CS, Bowen WD, Welch MJ (1997) Synthesis and evaluation of [18F] labeled benzamides: high affinity s receptor ligands for PET imaging. Nucl Med Biol 24(4):333–340
Shiue CY, Shiue GG, Zhang SX, Wilder S, Greenberg JH, Benard F, Wortman JA, Alavi AA (1997) N-(N-benzylpiperidin-4-yl)-2-[18F]fluorobenzamide: a potential ligand for PET imaging of s receptors. Nucl Med Biol 24(7):671–676
Kniess T, Laube M, Brust P, Steinbach J (2015) 2-[18F]Fluoroethyl tosylate – a versatile tool for building 18F-based radiotracers for positron emission tomography. Med Chem Commun 6:1714–1754
Waterhouse RN, Lombardo I, Simpson N, Kegeles LS, Laruelle M (2000) Evaluation of the novel σ1 receptor radioligand 1-(3-[F-18]Fluoropropyl)-4-[(4-cyanophenoxy)methyl]piperidine, [F-18]FPS: PET imaging studies in baboons. Neuroimage 11(Suppl):S16
Waterhouse RN, Nobler MS, Zhou Y, Chang RC, Morales O, Kuwabara H, Kumar A, VanHeertum RL, Wong DF, Sackeim HA (2004) First evaluation of the sigma-1 receptor radioligand [18F]1-3-fluoropropyl-4-((4-cyanophenoxy)-methyl)piperidine ([18F]FPS) in healthy humans. NeuroImage 22(S2):T29
Waterhouse RN, Mardon K, Giles KM, Collier TL, O’Brien JC (1997) Halogenated 4-(phenoxymethyl)piperidines as potential radiolabeled probes for σ1 receptors: in vivo evaluation of [123I]-1-(iodopropen-2-yl)-4-[(4-cyanophenoxy)methyl]piperidine. J Med Chem 40(11):1657–1667
Waterhouse RN, Chang RC, Zhao J, Carambot PE (2006) In vivo evaluation in rats of [18F]1-(2-fluoroethyl)-4-[(4-cyanophenoxy)methyl]piperidine as a potential radiotracer for PET assessment of CNS σ1 receptors. Nucl Med Biol 33(2):211–215
Waterhouse RN, Zhao J, Stabin MG, Ng H, Schindler-Horvat J, Chang RC, Mirsalis JC (2006) Preclinical acute toxicity studies and dosimetry estimates of the novel sigma-1 receptor radiotracer, [18F]SFE. Mol Imaging Biol 8(5):284–291
Elsinga PH, Kawamura K, Kobayashi T, Tsukada H, Senda M, Vaalburg W, Ishiwata K (2002) Synthesis and evaluation of [18F]fluoroethyl SA4503 as a PET ligand for the σ receptor. Synapse 43(4):259–267
Kawamura K, Tsukada H, Shiba K, Tsuji C, Harada N, Kimura Y, Ishiwata K (2007) Synthesis and evaluation of fluorine-18-labeled SA4503 as a selective σ1 receptor ligand for positron emission tomography. Nucl Med Biol 34(5):571–577
Lever JR, Gustafson JL, Xu R, Allmon RL, Lever SZ (2006) σ1 and σ2 receptor binding affinity and selectivity of SA4503 and fluoroethyl SA4503. Synapse 59(6):350–358
Moussa IA, Banister SD, Giboureau N, Meikle SR, Kassiou M (2011) Synthesis and in vivo evaluation of [18F]N-(2-benzofuranylmethyl)-N′-[4-(2-fluoroethoxy)benzyl]piperazine, a novel σ1 receptor PET imaging agent. Bioorg Med Chem Lett 21(22):6820–6823
Wang X, Li Y, Deuther-Conrad W, Xie F, Chen X, Cui MC, Zhang XJ, Zhang JM, Steinbach J, Brust P, Liu BL, Jia HM (2013) Synthesis and biological evaluation of 18F labeled fluoro-oligo-ethoxylated 4-benzylpiperazine derivatives for sigma-1 receptor imaging. Bioorg Med Chem 21(1):215–222
James ML, Shen B, Zavaleta CL, Nielsen CH, Mesangeau C, Vuppala PK, Chan C, Avery BA, Fishback JA, Matsumoto RR, Gambhir SS, McCurdy CR, Chin FT (2012) New positron emission tomography (PET) radioligand for imaging σ-1 receptors in living subjects. J Med Chem 55(19):8272–8282
Shen B, James MJ, Andrews L, Lau C, Chen S, Palner M, Miao Z, Arksey NC, Shuhendler AJ, Scatliffe S, Kaneshige K, Parsons SM, McCurdy CR, Salehi A, Gambhir SS, Chin FT (2015) Further validation to support clinical translation of [18F]FTC-146 for imaging sigma-1 receptors. EJNMMI Res 5:49
Fischer S, Wiese C, Grosse Maestrup E, Hiller A, Deuther-Conrad W, Scheunemann M, Schepmann D, Steinbach J, Wünsch B, Brust P (2011) Molecular imaging of σ receptors: synthesis and evaluation of the potent σ1 selective radioligand [18F]fluspidine. Eur J Nucl Med Mol Imaging 38(3):540–551
Maier CA, Wünsch B (2002) Novel spiropiperidines as highly potent and subtype selective σ-receptor ligands. Part 1. J Med Chem 45:438–448
Maier CA, Wünsch B (2002) Novel σ receptor ligands. Part 2. SAR of spiro[[2]benzopyran-1,4′-piperidines] and spiro[[2]benzofuran-1,4′-piperidines] with carbon substituents in position 3. J Med Chem 45:4923–4930
Wiese C, Große Maestrup E, Schepmann D, Vela JM, Holenz J, Buschmann H, Wünsch B (2009) Pharmacological and metabolic characterisation of the potent σ1 receptor ligand 1′-benzyl-3-methoxy-3H-spiro[[2]benzofuran-1,4′-piperidine]. J Pharm Pharmacol 61:631–640
Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F (2005) QT Prolongation Through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev 25(2):133–166
Maisonal A, Große Maestrup E, Wiese C, Hiller A, Schepmann D, Fischer S, Deuther-Conrad W, Steinbach J, Brust P, Wünsch B (2012) Synthesis, radiofluorination and pharmacological evaluation of a fluoromethyl spirocyclic PET tracer for central σ1 receptors and comparison with fluoroalkyl homologs. Bioorg Med Chem 20:257–269
Holl K, Schepmann D, Daniliuc CG, Wünsch B (2014) Sharpless Asymmetric Dihydroxylation as key step in the enantioselective synthesis of spirocyclic σ1 receptor ligands. Tetrahedron Asymmetry 25:268–277
Große Maestrup E, Wiese C, Schepmann D, Brust P, Wünsch B (2011) Synthesis, pharmacological activity and structure affinity relationships of spirocyclic σ1 receptor ligands with a (2-fluoroethyl) residue in 3-position. Bioorg Med Chem 19:393–405
Große Maestrup E, Fischer S, Wiese C, Schepmann D, Hiller A, Deuther-Conrad W, Steinbach J, Wünsch B, Brust P (2009) Evaluation of spirocyclic 3-(3-fluoropropyl)-2-benzofurans as σ1 receptor ligands for neuroimaging with positron emission tomography. J Med Chem 52:6062–6072
Wiese C, Große Maestrup E, Schepmann D, Grimme S, Humpf H-U, Brust P, Wünsch B (2011) Enantioselective σ1 receptor binding and biotransformation of the spirocyclic PET tracer 1-benzyl-3-(3-fluoropropyl)-3H-spiro[[2]benzofuran-1,4′-piperidine]. Chirality 23:148–154
Maisonial A, Große Maestrup E, Fischer S, Hiller A, Scheunemann M, Wiese C, Schepmann D, Steinbach J, Deuther-Conrad W, Wünsch B, Brust P (2011) A 18F-labeled fluorobutyl-substituted spirocyclic piperidine derivative as a selective radioligand for PET imaging of sigma1 receptors. Chem Med Chem 6:1401–1410
Holl K, Falck E, Köhler J, Schepmann D, Humpf H-U, Brust P, Wünsch B (2013) Synthesis, characterization, and metabolism studies of fluspidine enantiomers. Chem Med Chem 12:2047–2056
Maisonal-Besset A, Funke U, Wenzel B, Fischer S, Holl K, Steinbach J, Brust P, Wünsch B (2014) Automation of the radiosynthesis and purification procedures for [18F]Fluspidine preparation, a new radiotracer for clinical investigations in PET imaging of σ1 receptors in brain. Appl Radiat Isot 84:1–7
Laurini E, Dal Col V, Mamolo MG, Zampieri D, Posocco P, Fermeglia M, Vio L, Pricl S (2011) Homology model and docking-based virtual screening for ligands of the sigma1 receptor. ACS Med Chem Lett 2:834–839
Meyer C, Schepmann D, Yanagisawa S, Yamaguchi J, Dal Col V, Laurini E, Itami K, Pricl S, Wunsch B (2012) Pd-catalyzed direct C-H bond functionalization of spirocyclic sigma1 ligands: generation of a pharmacophore model and analysis of the reverse binding mode by docking into a 3D homology model of the sigma1 receptor. J Med Chem 55:8047–8065
Brune S, Schepmann D, Klempnauer KH, Marson D, Dal Col V, Laurini E, Fermeglia M, Wunsch B, Pricl S (2014) The sigma enigma: in vitro/in silico site-directed mutagenesis studies unveil sigma1 receptor ligand binding. Biochemistry 53:2993–3003
Massova I, Kollman PA (2000) Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discovery Des 18:113–135
Brust P, Deuther-Conrad W, Becker G, Patt M, Donat CK, Stittsworth S, Habermann B, Fischer S, Hiller A, Wenzel B, Dukic-Stefanovic S, Mishchenko L, Hesse S, Steinbach J, Lever SS, Sabri O, Wünsch B (2014) Distinctive in vivo kinetics of the new sigma1 receptor ligands (R)-(+)- and (S)-(−)-18F-fluspidine in porcine brain. J Nucl Med 55:1–7
Acknowledgement
This work was supported by the Deutsche Forschungsgemeinschaft, which is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG (outside the USA)
About this chapter
Cite this chapter
Weber, F., Brust, P., Laurini, E., Pricl, S., Wünsch, B. (2017). Fluorinated PET Tracers for Molecular Imaging of σ1 Receptors in the Central Nervous System. In: Smith, S., Su, TP. (eds) Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology, vol 964. Springer, Cham. https://doi.org/10.1007/978-3-319-50174-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-50174-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50172-7
Online ISBN: 978-3-319-50174-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)