
Abstract
Amyotrophic Lateral Sclerosis (ALS ) is a neurodegenerative disease affecting spinal cord and brain motoneurons , leading to paralysis and early death. Multiple etiopathogenic mechanisms appear to contribute in the development of ALS , including glutamate excitotoxicity, oxidative stress , protein misfolding, mitochondrial defects, impaired axonal transport, inflammation and glial cell alterations. The Sigma-1 receptor is highly expressed in motoneurons of the spinal cord, particularly enriched in the endoplasmic reticulum (ER) at postsynaptic cisternae of cholinergic C-terminals. Several evidences point to participation of Sigma-1R alterations in motoneuron degeneration. Thus, mutations of the transmembrane domain of the Sigma-1R have been described in familial ALS cases. Interestingly, Sigma-1R KO mice display muscle weakness and motoneuron loss. On the other hand, Sigma-1R agonists promote neuroprotection and neurite elongation through activation of protein kinase C on motoneurons in vitro and in vivo after ventral root avulsion. Remarkably, treatment of SOD1 mice, the most usual animal model of ALS , with Sigma-1R agonists resulted in significantly enhanced motoneuron function and preservation, and increased animal survival. Sigma-1R activation also reduced microglial reactivity and increased the glial expression of neurotrophic factors. Two main interconnected mechanisms seem to underlie the effects of Sigma-1R manipulation on motoneurons: modulation of neuronal excitability and regulation of calcium homeostasis. In addition, Sigma-1R also contributes to regulating protein degradation, and reducing oxidative stress. Therefore, the multi-functional nature of the Sigma-1R represents an attractive target for treating aspects of ALS and other motoneuron diseases .
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction: Motoneuron Diseases
Motoneuron diseases (MND) are progressive neurodegenerative disorders of wide etiology and clinical spectra, but with a common feature: the loss of lower and/or upper motoneurons . Amyotrophic lateral sclerosis (ALS ) and spinal muscular atrophy (SMA) are the most frequent forms of MND and therefore the most studied. ALS was first described by Charcot in 1869 and is the most common type of MND in adults, with an incidence of 1–5 per 100.000. Concomitant degeneration of both upper (corticospinal/corticobulbar) and lower (spinal/bulbar) motoneurons distinguishes ALS from other forms of MND [1–3]. The main neuropathological features of ALS include extensive loss of motoneurons in the anterior horns of the spinal cord and motor nuclei of the brainstem, degeneration of the corticospinal tract, and degeneration and loss of large pyramidal neurons in the primary motor cortex, also accompanied by reactive gliosis around the areas of degeneration [3]. Cytoplasmic protein inclusions are common in the degenerating neurons, which predominantly comprise a nuclear RNA processing protein, TDP-43 (TAR-DNA binding protein 43) [4] It has been classically considered that despite most ALS cases are sporadic (sALS), 5–10 % are familiar (fALS), related with several genetic mutations [1, 5]. No matter if they are sporadic or familiar, patients develop progressive weakness and muscle atrophy, with spasticity and contractures. Progressive weakness may start distally or proximally in the upper or lower limbs and finally affect all muscles, including those related with breathing, speaking and swallowing. Patients die, mostly due to respiratory failure, by 2–5 years after diagnosis [2, 6].
No effective treatment is presently available for ALS [1]. Patient care focuses on symptomatic treatments and physical therapy. Assisted ventilation and nutrition can transiently overcome the loss of upper airway and respiratory muscular control [2]. A large number of therapeutic trials have been attempted, but it was not until the early 1990s that the first drug approved by the FDA for the treatment of patients with ALS reached the market: riluzole, an antiglutamatergic agent that blocks the presynaptic release of glutamate. However, the efficacy of riluzole is questionable, with minimal therapeutic benefits of about 3–4 months of survival increase [7]. One of the main concerns for developing new therapies is the lack of direct translation from promising preclinical findings to successful clinical results. Although the heterogeneous and complex nature of ALS has been studied extensively, the absence of early detection markers and proper biomarkers for the disease evaluation of patients does not allow identifying whether patients are at different stages or even developing the disease because of different underlying causes. These drawbacks often lead to a difficult interpretation of the results from clinical studies. In this sense, patients who develop the disease mainly because of defects in a particular pathway would display greatest benefit from the compounds that selectively target that pathway. Interestingly and supporting this idea, in most clinical trials, a subset of subjects showed improvement, but none of the compounds displayed an overarching effect on most patients. Therefore it seems that each clinical trial has been successful only in a select subset of individuals. Since ALS is a multifactorial disease, future strategies should be focused on multi-target drugs or on combinatorial treatments that might maximize the translational effects [1].
Frontotemporal lobal degeneration (FTLD or FTD) is the second most common type of dementia after Alzheimer’s disease . It is caused by progressive neuronal atrophy and loss in the frontotemporal cortex, and is characterized by personality and behavioral changes, as well as gradual impairment of language skills [8]. Traditionally, ALS and FTLD were considered as two distinct identities. However, novel evidence suggests that both pathologies belong to a clinical continuum, with pure forms linked by overlapping syndromes. The first link established between FTLD and ALS was the identification of TDP-43 positive ubiquitinated cytoplasmic inclusions in almost all ALS and more than a half FTLD patients [8, 9]. Although neuropsychological testing shows normal cognition in the majority of ALS patients, up to 50 % of them may present some degree of cognitive impairment, while 15–18 % meet the criteria for FTLD [10]. On the contrary, few patients with FLTD develop ALS [11]. Indeed, FTLD-only, ALS -only and coincident FTD-ALS cases were reported to occur inside the same family, supporting the hypothesis of a link between both pathologies. The recent finding of an hexanucleotid expansion in C9ORF72 constitutes a strong link between ALS and FTLD [12–15].
16.2 Pathophysiological Mechanisms Underlying Motoneuron Death
The exact molecular pathway causing motoneuron degeneration in ALS is unknown, but as with other neurodegenerative diseases, it is likely to be a complex interplay between multiple pathogenic mechanisms that may not be mutually exclusive and in which is still unknown the causative relation between them or whether they are the consequence of an upstream disturbance [1, 5, 16].
The identification of underlying genetic defects of familial cases of ALS has allowed the development of relevant animal models of the disease in mice, rats, zebra fish and drosophila [1, 4, 17–19], which have been essential for uncovering morphological and molecular pathogenic events in vivo that are not possible to investigate in humans. The most widely used ALS models are transgenic mice over-expressing human mutated forms of the SOD1 gene, which recapitulate the most relevant clinical and histopathological features of both familial and sporadic ALS .
Among the proposed pathophysiological mechanisms, excitotoxicity has been deeply explored. Neuronal injury caused by excitatory mediators may be due to failure in the neurotransmitter clearance from the synaptic cleft or increased postsynaptic sensitivity to glutamate. This enhanced excitatory input induces a massive calcium influx into the cytoplasm that damages the cells through the activation of calcium-dependent proteases, lipases and nucleases. A large body of evidence implicates excitotoxicity as a mechanism contributing to motoneuron death in ALS , such as threefold increased levels of glutamate in CSF from ALS patients [20, 21]. Furthermore, overactivation of NMDA receptors and increased calcium permeability of AMPA receptors have been described in ALS mouse models [1, 22–25]. Loss of the glial excitatory amino acid transporter 2 (EAAT2) was also reported in ALS mouse models [26, 27].
Oxidative stress results from the imbalance between the production of reactive oxygen species (ROS) and the biological capacity to remove ROS or repair ROS-induced damage. The analysis of CSF and serum from ALS patients showed increased concentration of ROS compared to healthy subjects [28–31]. Evidence of oxidative stress damage to proteins [32], lipids [30] and DNA [33] was also reported to occur in ALS patients. Oxidative stress has been also documented in ALS mouse models [34, 35].
Mitochondria are the cellular organelle in charge of ATP production, calcium homeostasis maintenance and intrinsic apoptosis regulation. An important core of evidences implicates mitochondria as key players in ALS physiopathology [36]. Reduced mitochondrial DNA content associated with increased mutations of mitochondrial DNA, and respiratory chain complexes dysfunction have been described in the spinal cord of ALS patients [37]. Mitochondrial function impairments affect also the skeletal muscle of ALS patients [38]. In vitro studies showed mitochondrial morphological and functional alterations in NSC-34 cells expressing mutant SOD1 [39]. Experiments performed in mSOD1 mice also revealed early mitochondrial morphological abnormalities prior to onset of symptoms [40].
Neurons are polarized cells that require efficient mechanisms to direct axonal vs. dendritic transport. Since neurons transmit signals along long distances, proteins and organelles have to travel more than in other cell types (axons of human motoneurons can reach 1 m long). Even within the axon, cargos must be delivered to specific compartments, thereby increasing the importance of axonal transport in motoneurons. Several works demonstrated the accumulation of neurofilaments in motoneuron cell bodies in human patients, suggesting that axonal transport is impaired in these cells [41–44]. Additionally, abnormalities of organelle axonal trafficking occur in ALS patients [45]. Axonal transport has been widely studied in animal models mimicking ALS . It has been demonstrated that transgenic mice overexpressing SOD1 transgene develop neuronal cytoskeletal pathology resembling human ALS [46]. Controversially, recent evidence suggests that axonal transport deficits may evolve independently from motoneuron degeneration in mutant SOD1 mice [47]. Marinkovic et al. [47] demonstrated that mutant SOD1 axons are able to survive despite long-lasting transport deficits since these are present soon after birth, months before the first signs of muscle denervation [48–50].
Protein aggregates or inclusions have long been recognized as a pathological hallmark of several neurodegenerative disorders, including ALS , in which protein aggregates are common in spinal motoneurons [1]. Ubiquitin-positive inclusions are characteristic of ALS histopathology. Nevertheless, it remains unclear whether inclusion formation is responsible for cellular toxicity and ALS pathogenesis, if aggregates may be innocuous neurodegeneration -derived products, or if they may represent a protective reaction of the cell to reduce intracellular concentrations of toxic proteins. Several proteins are found forming the intracellular inclusions in ALS , including neurofilaments [42–44], SOD1 [51–53], TDP-43 [9, 54], FUS [55, 56], ubiquilin 2 [57] and C9OFF72 [12, 13].
Physiologically, accumulation of misfolded proteins elicits the endoplasmic reticulum (ER) stress response. ER-resident chaperones sense the accumulation of misfolded proteins and activate the Unfolded-Protein Response (UPR), which leads to the suppression of general translation and ER-associated protein degradation. However, prolonged UPR activation may trigger apoptotic signaling [58]. The addition of CSF from ALS patients induces ER stress on cultured NSC-34 cells and primary rat spinal motoneuron cells [59]. Considerable evidence implicates ER stress as an important feature of motoneuron degeneration in ALS . UPR markers are up-regulated in sALS patients [60] as well as in mutant SOD1 rodent models [61, 62]. Interestingly, a longitudinal gene expression profile in mutant SOD1 mice revealed early up-regulation of several UPR markers prior to muscle denervation in vulnerable motoneurons (innervating fast fatigable muscles, e.g. extensor digitorum longus) compared to resistant motoneurons (innervating slow muscle fibers, e.g. soleus). Similar changes eventually occurred in disease-resistant motoneurons but 25–30 days later [62], suggesting a role for ER stress in determining the susceptibility of motoneurons in ALS .
Neighboring glial cells also play a crucial role in the motoneuron degeneration occurring in ALS [1, 63, 64]. Clement et al. [65] generated chimeric mice expressing mSOD1 in specific cell lines and demonstrated that normal motoneurons developed ALS signs when surrounded by mutant SOD1-expressing glia. To further explore the contribution of microglia in ALS , double transgenic mice were generated expressing the Cre–Lox recombination system to selectively suppress the mutant SOD1 expression in motoneurons or microglia. Mutant SOD1 deletion in motoneurons lead to delayed disease onset but no modifications of disease progression once initiated. On the other hand, mutant SOD1 suppression in microglia and macrophages did not alter disease onset but significantly prolonged mice survival. These findings suggest that disease onset and progression might be related to different mechanisms [66, 67]. It is also accepted that astrocytes play a role in ALS . Astrocytes derived from postmortem tissue of familial and sporadic ALS patients are toxic to motoneurons but not to GABAergic neurons. Blocking mSOD1 expression produced significant neuroprotective effects on ALS -derived astrocytes [68].
Neuroinflammation is a common pathological event of neurodegenerative disorders [69] and its modulation has been proposed as an important potential therapeutic target [70]. In ALS , motoneuron damage leads to the activation of microglia, astrocytes and the complement system, further contributing to neurodegeneration [71, 72]. Spinal cord tissue and CSF from sporadic and familial ALS cases present increased microglial activation and T cells infiltration [73, 74] as well as higher concentration of some proinflammatory mediators, including monocyte chemoattractant protein 1 (MCP-1) and IL-8 [75]. Gene array analysis of mutant SOD1 mice revealed an enhanced expression of inflammatory-related molecules especially at late stages of the disease [76, 77].
RNA processing abnormalities were first related to MND by the description of Spinal Motor Neuron protein 1 (SMN1) mutations as a cause of SMA [78]. The SMN proteins play a role in the assembly of small ribonucleoproteins, which participate in pre-mRNA splicing [79]. Later identification of TDP-43, a RNA-DNA binding protein, as a major component of the ubiquitinated protein inclusions in ALS patients [9] focused the attention to RNA processing alteration as an important pathophysiological mechanism of the disease. TDP-43 is predominantly localized in the nucleus where it is implicated in several events for RNA processing, including transcriptional regulation, alternative splicing and microRNA processing. ALS -related TDP-43 positive cytoplasmic inclusions are present in neuronal and non-neuronal cells, excluding those based on mSOD1 and FUS mutations [54, 80]. Recent studies have evaluated the RNA-binding targets of TDP-43 [81–83] and revealed that TDP-43 binds to several RNA target molecules (about 30 % of the mouse transcriptome). Such high level of intronic binding suggests a nuclear function for TDP-43. In fact, blocking tardbp43 expression using antisense oligonucleotides in adult mouse striatum altered the expression levels of 601 mRNA and changed the splicing pattern of 965 mRNA transcripts, including some relevant to neurodegeneration , such as progranulin, choline acetyltransferase or FUS [81]. TDP-43 alteration might potentially alter the transcriptional process of crucial genes for motoneuron homeostasis. Additional evidence about dysregulated RNA processing as motoneuron injury contributor in ALS arises from the detection of RNA oxidation biomarkers in human ALS and mSOD1 mice [84]. Since the discovery of C9ORF72 hexanucleotide (G4C2) repeat expansions as a frequent cause of ALS and FTD, efforts have been conducted for investigating linked pathophysiological abnormalities. Repeat containing RNA foci in these patients suggested a deleterious gain of function. Repeats are able to form G-quadruplexes, which may be able to facilitate the binding and sequestration of different RNA binding proteins to the repeat [85]. Subsequently, these proteins are not able to execute their normal functions. Another mechanism is the possible occurrence of repeat-associated non-ATG (RAN) translation along the hairpin-forming repeat. This results in aggregates containing different dipeptide repeat proteins in patients with the C9ORF72 repeat expansion [15, 86]. Recently, two independent studies used engineered drosophila to express high repeat expansions of G4C2 [87, 88], and established a strong connection between defective nuclear trafficking and neurodegeneration in these flies.
16.3 Structure and Functions of Sigma-1 Receptor
The Sigma-1R is a transmembrane protein found in the ER [89, 90], which is highly expressed in motoneurons and other cells in the spinal cord [89, 91–94]. Although it was initially classified as an opioid receptor, further experiments showed that its properties were distinct from known opioid receptors [95]. A 223 amino acids Sigma-1R protein has been cloned from several mammals, and contains 90 % identical and 95 % similar amino acid sequences across species, with both the N- and C-termini on the same side of the membrane facing the cytoplasm [90, 96], and can be present in monomeric or oligomeric forms even in the absence of ligand [97, 98]. The N-terminus, of approximately 110 amino acids, determines the diversity of intracellular interactions of Sigma-1R with a variety of proteins [99–102]. Many synthetic compounds have been characterized as selective modulators of the Sigma-1R [103], and several endogenous molecules have been proposed to be Sigma-1R ligands as well, including lipid steroids (DHEA, progesterone and pregnenolone sulfate) [104], lipid sphingosine derivatives [105] and N,N-dimethyl tryptamine (DMT) [106]. It is plausible that these compounds regulate Sigma-1R function in different tissues according to their availability.
This receptor has the ability to translocate from the ER to the plasma membrane and mitochondria-associated membranes [90, 107, 108]. In the nervous system, Sigma-1R mediates regulation of a wide range of processes, such as neuritogenesis [109], modulation of K+ channels [110] and N-methyl-D-aspartate (NMDA) receptors activity [111], ER-mitochondria communication [90], modulation of G-protein couples receptors (GPCRs), Ca2+ homeostasis [90], and microglial activity [112]). The Sigma-1R appears as a pluri-functional target involved in a broad range of cellular processes and, thereby, its modulation might provide better translational outcomes than drugs acting selectively on one of these multiple aspects.
Langa et al. [113] developed homozygous Sigma-1R knock out mice, which showed to be fully fertile and with no obvious behavioral alterations. However, further careful analyses revealed alterations of hippocampal neurogenesis [114, 115], ethanol consumption [116], retinal function [117, 118], anxiety, memory impairments [119] and, most relevant, motor dysfunction and loss of neuromuscular junctions [120, 121].
16.4 Sigma-1 Receptor and Motoneurons
To fully understand the mechanisms underlying Sigma-1R role in motoneurons , it is important to know its subcellular localization in the cells. It has been shown that Sigma-1R is enriched in the subsurface cisternae in postsynaptic C-terminals of motoneurons [120]. Synaptic innervation onto motoneurons is complex, with synapses involving all the major neurotransmitters, that have been classified as S, M, T F, P and C-boutons/terminals (referring to the pre- or postsynaptic structure, respectively) [122]. C-terminals are large cholinergic postsynaptic sites with a unique ultrastructure seen at the electron microscopy level. They are referred to as “C” because of the subsurface cisternae of smooth endoplasmic reticulum adjacent to the plasma membrane, and are large synapses found only on soma and proximal dendrites of motoneurons [123]. Presynaptic C-boutons originate from a group of cholinergic interneurons located near the spinal cord central canal, which have been shown to increase motoneuron excitability and, thus, potentiate muscle contraction [124]. Interestingly, Sigma-1R is specially enriched in the subsurface cisternae underlying the postsynaptic membrane of C-boutons in motoneurons. Diverse alterations of C-boutons have been reported in animal models of ALS and spinal cord injury [125–128]. The postsynaptic membrane of C-boutons is rich in numerous proteins, including m2-type muscarinic receptors (m2AChR) [129–131], voltage-gated Kv2.1 [132], Kv1.4, Kv1.5 [110] and Ca2+-activated K+ (SK) channels [133], connexin 32 [134], VAMP-2 [129], Sigma-1R [135] and neuregulin-1 [136]; whereas the presynaptic element contains, at least, neuregulin-1 receptors ErbB2 and ErbB4 [136]. In contrast, the role of subsurface cisternae in postsynaptic densities where Sigma-1 receptors are located is still unknown, but believed to couple the electrical activity of the plasma membrane with intracellular signaling involving the ER [137, 138].
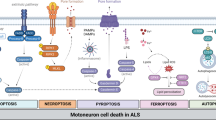
Cholinergic innervation onto motoneurons plays a role in modulating the excitability of the cells during locomotion [124, 139]. Interestingly, Sigma-1R, m2AChR and SK channels have special relevance regarding motoneurons excitability. Indeed, it has been proposed that differential expression of SK2.2 and SK2.3 channels in neurons is a marker for α-motoneurons innervating fast or slow muscle fibers modifying the hyperpolarization properties of the plasma membrane [133]. Miles et al. [139] described how cholinergic innervation on motoneurons increases excitability during fictive locomotion by acting on m2AChR, whereas motoneurons lacking Sigma-1R have increased excitability [140]. Sigma-1R has been also shown to interact with diverse potassium channels, thereby shaping neuronal excitability [99, 110, 141, 142] (Fig. 16.1).

Sigma-1 receptor localization at the C-boutons and its pleiotropic role in the motoneuron. Sigma-1 receptor is located at the endoplasmic reticulum subsynaptic cisterna of the cholinergic synapses, from where it may interact both with elements of the plasma membrane (e.g. ion channels ) or the cytoplasm (e.g. mitochondria). Sigma-1 receptor modulates the activity of several ionotropic and metabotropic receptors, including M2AchR, NMDA, dopaminergic D1 and opioid receptors. Further studies are needed to elucidate how Sigma-1 receptor interacts with ion channels (Kv or SK) and other elements present at the C-boutons, such as Connexin32, VAMP-2 and Neuregulin1. The Sigma-1 receptor is also able to interact with BiP, a chaperone of the endoplasmic reticulum, and to participate in the interactions between the endoplasmic reticulum and the mitochondria. For further details, see the Sect. 16.4 text
Sigma-1R co-localizes with neuregulin-1 expressed at the motoneuron C-boutons postsynaptic membrane [136]. Neuregulin-1 is a neurotrophic factor essential for the normal development and function of the nervous system [143]. Neuregulin-1 ErbB receptors are also located in the presynaptic terminals of C-boutons. Neuregulin-1/ErbB system alterations have been related to ALS , with reduced neuregulin-1 type III expression in the spinal cord of ALS patients and mouse models [144]. Loss-of-function mutations on the gene encoding for ErbB4 receptor produce late-onset ALS in patients [145]. Although no link between Sigma-1R and neuregulin-1 has been established yet, it is likely that Sigma-1R serves as a chaperone for neuregulin-1 at subsurface cisternae of motoneurons , as it has been shown to participate in the post-translational processing of other neurotrophic factors [146] (Fig. 16.1).
Little is known about endogenous ligands for Sigma-1R. It has been shown that N,N-dimethyltryptamine (DMT) is an endogenous agonist for the Sigma-1R [147] and that Indole(ethyl)amine N-methyltransferase (INMT ) , the enzyme that converts the amino acid tryptophan into DMT, co-localizes with Sigma-1R at C-terminals of motoneurons [135]. Endogenous steroids have been shown to act as Sigma-1R agonists , including dehydroepiandrosterone (DHEA) sulfate and pregnenolone sulfate [148]. Nevertheless, further studies are needed to elucidate the mechanisms by which Sigma-1R function is endogenously modulated and how this affects motoneuron physiology (Fig. 16.1) .
16.5 Evidences of Sigma-1 Receptor Contribution in Motoneuron Disease
There is a body of evidence suggesting that Sigma-1R alterations lead to motoneuron dysfunction and degeneration [121, 140]. Mutations in a highly conserved region of the transmembrane domain of the Sigma-1R were described in ALS patients. The mutation produces an aberrant subcellular distribution of the receptor in NSC34 cells, and cells expressing the mutant protein are more prone to undergo apoptosis induced by ER stress [149]. Sigma-1R was found to abnormally redistribute in alpha-motoneurons of ALS patients and form ubiquitinated aggregates that lead to UPR. Additionally, Sigma-1R levels were found reduced in samples of ALS patients [150]. Other mutations in the 3′-untranslated region (UTR) of the Sigma-1R gene were described in affected individuals with the FTD-ALS pedigree [151].
Interestingly, Sigma-1R KO mice display locomotor deficits associated with muscle weakness, axonal degeneration and motoneuron loss [121, 140]. Altered Sigma-1R function in motoneurons has been also shown to disrupt ER-mitochondria contacts and affect intracellular calcium signaling, leading to activation of ER stress and to defects in mitochondrial dynamics and transport [121]. Crossing Sigma-1R KO mice with mutant SOD1 mice (SOD1G93A) exacerbated the motor phenotype and accelerated the end stage of the disease [140]. Conversely, stimulating Sigma-1R function using the agonists PRE-084 or SA4503 has been shown protective in both in vitro and in vivo models of mutant SOD1 ALS [94], as well as in non-SOD1 linked MND [152].
A note of caution must be taken since there is controversy about the expression profile of Sigma-1R in mutant SOD1 ALS models. Analysis of Sigma-1R in protein extracts from lumbar anterior spinal cord showed no changes in the amount of Sigma-1R expressed [94], whereas immunohistochemical analysis revealed decreased labeling of Sigma-1R at the C-boutons of SOD1 lumbar motoneurons at early pre-symptomatic stages of the disease [127].
16.6 Potential Mechanisms on Sigma-1 Receptor-Mediated Therapeutic Actions
The Sigma-1R has been shown to be a target for the treatment of a variety of chronic neurological diseases, including pain [153–155], depression [148], Alzheimer’s [156–158], Parkinson’s [159], and Huntington [160] diseases, schizophrenia [161], stroke [162, 163], ischemia [164], degeneration of retinal neurons [117, 118], and selective cholinergic lesions [156]. The administration of Sigma-1R ligands has promoted neuroprotection after several types of insults, including excitotoxic damage [165], hypoxia-mediated neurotoxicity [166], oxidative stress -induced cell death [167] and glucose deprivation [164].
Regarding motoneurons , the selective Sigma-1R agonist PRE-084 has been reported to exert positive effects on motoneuron death. PRE-084 administration promotes neuroprotection and neurite elongation through activation of protein kinase C (PKC) on motoneurons in an in vitro organotypic model of excitotoxic lesion [168]. Moreover, administration of PRE-084 significantly prevented the marked death of spinal motoneurons after spinal root avulsion in adult rats, an effect that was associated with attenuating ER stress within motoneurons and promoting the expression of GDNF by surrounding glial cells [93]. Remarkably, treatment of SOD1 mice with Sigma-1R agonists resulted in significantly improved motoneuron function and preservation, and increased animal survival [94, 152, 169, 170]. Several mechanisms have been hypothesized to underlie motoneuron protection in ALS models (Fig. 16.2). Sigma-1R agonists administration resulted in increased PKC-specific phosphorylation of NR1 subunits present in spinal motoneurons, likely reducing the calcium permeability of NMDA receptors and its influx into motoneurons, thereby attenuating excitotoxicity [94, 111]. Sigma-1R agonists , such as SKF10097 and PRE-084, have been reported to also suppress NMDA currents in rat retinal ganglion cells and cortical neurons through a PKC-dependent mechanism, leading to reduction of calcium influx into the cytoplasm [111, 166]. Sigma-1R agonists administration also reduced microglial and astroglial reactivity in the mutant SOD1 and in the wobbler ALS mouse models, and enhanced glial expression of neurotrophic factors, such as BDNF [94, 152]. In this sense, Sigma-1R activation has been linked to modulation of multiple aspects of microglial activation in vitro [171, 172], as well as to increase the glial expression of neurotrophic factors after spinal root avulsion [93].

Schematic representation of the effect of agonizing the Sigma-1 receptor in ALS mouse models. (a) Wild type spinal cord motoneurons project their axons from the anterior horn of the spinal cord through the anterior root to reach the skeletal muscles. (b) ALS spinal cord suffers a dramatic death of motoneurons, accompanied by loss of neuromuscular connections and ventral root motor axons. In addition, non-neuronal cells proliferate and become activated across the spinal cord, contributing to the disease progression. (c) Sigma-1 receptor agonists are able to prevent the loss of neuromuscular connections and motor axons, as well as the death of motoneuron cell bodies in the spinal cord. Furthermore, Sigma-1R agonists reduce microglial reactivity, despite no changes are observed in astrocytosis
Overall, two main interconnected mechanisms are likely to underlie the direct effect of Sigma-1R manipulation on motoneurons : the modulation of the neuronal excitability and the calcium homeostasis. The Sigma-1R is located in C-terminals in close proximity to Kv2.1 and SK channels, which appear as two suitable candidates for the Sigma-1R modulation of postsynaptic excitability of motoneurons. A body of evidence indicates that inhibition of m2AChR and/or activation of Kv2.1 and/or SK channels in C-terminals contribute toward reduction of motoneuron excitability [124, 133, 139]. Although the mechanisms by which Sigma-1R activates Kv2.1 and/or SK channels and thus decreases motoneuron excitability are still unclear, it has been shown within other systems that Sigma-1R can modulate activities of SK channels and a variety of Kv type channels [99, 142, 173]. Sigma-1R can form complexes with a variety of G-protein coupled receptors (GPCRs) that can subsequently alter ionotropic receptors including opioid and dopaminergic D1 receptors [174, 175] (Fig. 16.1).
As previously mentioned, Sigma-1R is located in the subsurface cisternae of C-terminals underlying the plasma membrane of motoneurons [89, 120]. Such physical proximity between the plasma membrane and the subsynaptic cisternae in C- terminals (less than 10 nM) makes direct molecular interaction possible for proteins located in adjacent membranes. Indeed, the Sigma-1R is characterized by a unique mode of action in regulating both the calcium entry at the plasma membrane level (e.g. via potassium channels, voltage-sensitive Ca2+ channels, etc.) and calcium mobilization from the endoplasmic stores (e.g. via IP3 receptors). The ER supplies calcium directly to mitochondria via inositol 1,4,5-triphosphate receptors (IP3 receptors) at close contacts between the two organelles referred to as mitochondrial-associated ER membranes (MAM). Sigma-1R is a calcium-sensitive and ligand operated chaperone at MAM, normally forming a complex with another chaperone, binding immunoglobulin protein (BiP), which normally prevents the Sigma-1R from translocation. Upon ER calcium depletion or via ligand stimulation, Sigma-1R dissociates from BiP, leading to prolonged calcium signaling into mitochondria via IP3 receptors. Sigma-1R translocation has been shown to occur under chronic ER stress conditions. Indeed, increasing Sigma-1R in cells counteracts ER stress response, whereas decreasing its expression enhances apoptosis [90]. Subsequently, activity of both Kv2.1 and SK channels has been shown to be modulated by calcium, either directly or indirectly through Ca/calmodulin/calcineurin dependent mechanisms [176, 177] (Fig. 16.1).
In addition, Sigma-1R also contributes to maintenance of protein quality by regulating protein degradation and stability. Indeed, abnormal Sigma-1R accumulation is found in neuronal nuclear inclusions in neurodegenerative diseases [151, 178]. Sigma-1R participation in the degradation of misfolded protein via the ER machinery linked to the ubiquitin-mediated UPR suggests that Sigma-1R may function to counteract this pathological mechanism and promote survival in affected motoneurons . Ligand activation may promote and stabilize Sigma-1R oligomers, thus conferring improved chaperone functionality to the receptor [90].
Finally, modulation of Sigma-1R may also contribute to neuroprotection by reducing oxidative stress . It was shown that depletion of Sigma-1R leads to increased oxidative stress and abnormal mitochondrial membrane potential, thus triggering cytochrome C release and elevated caspase-3 cleavages [179].
16.7 Bases of Motoneuron Vulnerability
Understanding the bases of motoneuron vulnerability is crucial for developing novel strategies to cope with MND. In this section we focus on those aspects of motoneuron vulnerability that are related to mechanisms in which Sigma-1R plays a relevant role: the alteration of excitability properties of motoneurons and calcium homeostasis. As previously mentioned, ALS is a degenerative disease in which lower and upper motoneurons are selectively vulnerable, but interestingly some groups of motoneurons are relatively resistant to the disease process. It has been hypothesized that the differential susceptibility of motoneuron populations might be related to their excitability properties. Indeed, a consistent clinical feature of ALS is the preservation of eye movements and the external sphincters function. Pathological studies confirmed that there is relative sparing of the cranial motor nuclei of the oculomotor, trochlear and abducens nerves, and of the Onuf’s nucleus in the sacral spinal cord, which innervates the external sphincter of the pelvic floor [180]. Although neuronal numbers are relatively well-preserved in these resistant motor nuclei, some pathological changes resembling those observed in ventral spinal cord motoneurons are present, but to a lesser degree [181, 182]. Oculomotor nuclei are also relatively spared in mutant SOD1 mouse models [183]. The pattern of innervation of extraocular muscles is different from other skeletal muscles. Neuromuscular junctions are distributed throughout the fiber length at a high density [184], and show some structural peculiarities [185]. About 20 % of the extraocular muscles fibers are innervated by multiple neuromuscular junctions [186]. Oculomotor motor units are amongst the smallest seen in any skeletal muscle [187], with high firing discharge rates. Even in the primary position of gaze, 70 % oculomotor neurons are active, commonly discharging at 100 Hz [188]. In contrast, there is strong experimental evidence of a special susceptibility of large, phasic motoneurons in the degenerative process of ALS . Electromyographic analysis performed in ALS patients revealed that the larger and stronger motor units are clearly more affected by the disease [189], and histopathological studies have described a preferential degeneration of large motoneurons in ALS [190]. In mutant SOD1 models, selective vulnerability of large fast-fatigable hindlimb motor units before the onset of clinical symptoms was reported, followed by affectation of fast fatigue-resistant motor units at symptoms onset, but with sparing of slow motor units [191]. This is consistent with the rapid denervation of extensor digitorum longus muscle (rich in fast fatigable motor units) and the resistance of soleus muscle (with mainly slow motor units) described along disease progression in SOD1G93A mice [192, 193].
Understanding the differences in properties of vulnerable vs. resistant motoneurons may provide insights into the mechanisms of neuronal degeneration, and identify targets for therapeutic manipulation. In an interesting study Brockington et al. [194] performed a microarray analysis to compare the gene expression profile of isolated motoneurons from the ALS -resistant oculomotor nuclei and ALS -vulnerable spinal cord motoneurons from post-mortem ALS patients tissue. They found nearly 2000 genes differentially expressed by the two motoneurons subtypes, participating in synaptic transmission, ubiquitin-dependent proteolysis, mitochondrial function, transcriptional regulation, immune system functions and the extracellular matrix. They further focused on glutamate and GABA neurotransmission. The AMPA glutamate receptor consists of four subunits, GluR1–GluR4, and the presence of the GluR2 subunit determines the calcium permeability of the receptor. In the absence of GluR2, the AMPA receptor–ion channel complex becomes permeable to calcium. Gene array results showed up-regulation of the GluR2 subunit in resistant oculomotor motoneurons relative to the vulnerable lumbar motoneurons, thus reducing calcium influx into the cells. On the other hand, GABA is the most widely distributed inhibitory neurotransmitter in the CNS and acts through the interaction with GABA-A (ligand-gated chloride channel) and GABA-B (metabotropic) receptors. In oculomotor motoneurons, there is up-regulation of six GABA-A receptor subunits and of GABA-B receptor subunit 2 relative to spinal motoneurons, leading to an increased inhibition. Other studies performed in mSOD1 models confirmed these findings, revealing an excitatory/inhibitory imbalance affecting synaptic inputs into spinal motoneurons [23]. To test the hypothesis that inhibitory interneuron innervation of motoneurons was abnormal in ALS , Chang and Martin [195, 196] measured GABAergic, glycinergic and cholinergic immunoreactive terminals on spinal motoneurons of SOD1G93A mice. They found reduction of glycinergic innervation from pre-symptomatic age (8 weeks), before loss of choline acetyltransferase-positive boutons, whereas no significant differences in GABAergic boutons density were found along age.
Interestingly, the increased excitation and reduced inhibition onto motoneurons has been hypothesized as a protective compensatory reaction rather a detrimental phenomenon [197]. As above mentioned, oculomotor nucleus motoneurons are strongly resistant to degeneration, but have particular physiological characteristics, including high discharge rates [188]. In turn, vulnerable fast-fatigable spinal motoneurons are those with larger cell bodies and more phasic activity pattern. Surprisingly, early administration of an AMPA receptor agonist protected spinal motoneurons whereas an AMPA receptor antagonist enhanced motoneurons pathology in SOD1G93A mice [197]. Furthermore, the authors proposed that reduction of gephyrin (an inhibitory synapse marker), increase of serotonin labeled area in the ventral spinal cord and increased C-boutons size and number are protective compensatory reactions that promote motoneuron survival. In agreement with these findings an abnormal response of the potassium-chloride co-transporter 2 (KCC2) in mutant SOD1 motoneurons in response to axonal damage and deafferentation [198] was recently described. KCC2 is a transmembrane chloride extruder that maintains low intracellular chloride levels, thereby allowing GABA and glycine to exert inhibitory transmission during adulthood [199–201]. Under normal conditions, KCC2 is down-regulated after motoneuron insults thus promoting increased excitability needed for axonal regeneration [202, 203]. In contrast, mutant SOD1 motoneurons were unable to down-regulate their KCC2 and thus did not become hyperexcitable even when already disconnected from their muscles [198]. Further studies revealed that functional overload is able to rescue motor units in mutant SOD1G93A mice [204], supporting the hypothesis of hypoexcitability as one potential factor underlying selective motoneuron damage.
In vitro studies of motoneuron excitability also show discrepancies regarding whether hypo- or hyperexcitability is a susceptibility factor for motoneurons in ALS . Changes in excitability have been reported to occur very early in mutant SOD1 mice [205]. Motoneurons from mutant SOD1 embryos recorded in culture show signs of hyperexcitability [206, 207], as well as motoneurons in in vitro preparation of mutant SOD1 embryonic spinal cords [208] or from the hypoglossal nucleus in the brainstem [209]. Contrarily, Pambo–Pambo et al. [210] did not observe any change in spinal motoneurons excitability properties, whereas Bories et al. [211] and Leroy et al. [212] reported spinal motoneurons to be hypoexcitable. A note of caution must be taken within this context since most of these studies were performed at developmental stages, when the maturation of the spinal circuitry is not yet completed.
Motoneurons express low levels of cytosol calcium-binding proteins compared to other neuronal populations, with motoneuron populations that are typically lost earlier during the disease course showing the lowest expression levels, suggesting that reduced cytosol calcium buffering contributes to the selective vulnerability of motoneurons [213, 214]. In fact, ALS -vulnerable spinal and brainstem motoneurons display low endogenous Ca2+ buffering capacity, 5–6 times lower than that of ALS -resistant motoneurons (i.e. oculomotor motoneurons), making them more susceptible to excitotoxic insults [215]. However, this view may not agree with the above mentioned oculomotor motor units properties since, although this motoneuron population is highly active, it is not vulnerable to ALS .
Interestingly, novel evidence has recently pointed out the potential contribution of C-boutons as participating in ALS pathophysiology [127, 140]. As described in Sect. 16.4, the postsynaptic membrane of C-boutons is rich in numerous proteins, including Sigma-1R [135], M2 muscarinic receptors [129–131], voltage-gated Kv2.1 [132] and Ca2+−activated K (SK) channels [133], connexin 32 [134], VAMP-2 [129], and neuregulin-1 [136]; whereas the presynaptic element contains, at least, neuregulin-1 receptors ErbB2 and ErbB4 [136]. Several alterations that may be related to C-bouton have been reported in ALS . It has been shown that mutations in Sigma-1R cause juvenile ALS [149, 150]. In agreement with this observation, knocking down Sigma-1R in mutant SOD1 mice leads to reduced lifespan [140], whereas treatment with a Sigma-1R agonist is neuroprotective [94]. Other morphological alterations appear to be present in ALS -linked mutations of VAMP-associated protein B, which is abnormally aggregated in C-boutons altering their function (VAPB, ALS8) [216]. The neuregulin1/ErbB system is also involved in ALS pathogenesis since ErbB4 mutations leading to a reduced autophosphorylation of ErbB4 receptors are associated with a hereditary late onset form of ALS [145], and neuregulin1/ErbB signaling alterations have been also observed in SOD1G93A mice [144].
16.8 Conclusions
Overall, mutations of Sigma1-R have been reported in ALS in human patients, and sigma-1R modulation has proven to protect motoneurons in vitro and in in vivo models of traumatic injury to motoneurons and neurodegeneration . Although the exact molecular mechanisms underlying such effect have not been elucidated yet, Sigma-1R is a pleiotropic target, involved in several functions, many of them related to the pathophysiology of MND, including modulation of neuronal excitability, calcium homeostasis, and ER and mitochondrial activity. Thus, the multi-functional nature of the Sigma-1R provides an attractive target for treating ALS . Further human trials will be needed to assess whether pharmacologically targeting Sigma-1R is a suitable tool to protect motoneurons in MND.
References
Mancuso R, Navarro X (2015) Amyotrophic lateral sclerosis: current perspectives from basic research to the clinic. Prog Neurobiol 133:1–26
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3
Kiernan MC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955
Ince PG et al (2011) Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122:657–671
Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 76:1046–1057
Worms PM (2001) The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci 191:3–9
Ludolph AC, Jesse S (2009) Evidence-based drug treatment in amyotrophic lateral sclerosis and upcoming clinical trials. Ther Adv Neurol Disord 2:319–326
Van Langenhove T, van der Zee J, Van Broeckhoven C (2012) The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med 44:817–828
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Ringholz GM et al (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65:586–590
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
DeJesus-Hernandez M et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 72:245–256
Renton AE et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Ling S-C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438
Mori K et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723
Turner B, Talbot K (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol 85:94–134
McGoldrick P, Joyce PI, Fisher EMC, Greensmith L (2013) Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta 1832:1421–1436
Babin PJ, Goizet C, Raldúa D (2014) Zebrafish models of human motor neuron diseases: advantages and limitations. Prog Neurobiol 118:36–58
Perry TL, Krieger C, Hansen S, Eisen A (1990) Amyotrophic lateral sclerosis: amino acid levels in plasma and cerebrospinal fluid. Ann Neurol 28:12–17
Shaw PJ, Forrest V, Ince PG, Richardson JP, Wastell HJ (1995) CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration 4:209–216
Texidó L et al (2011) Sera from amyotrophic lateral sclerosis patients induce the non-canonical activation of NMDA receptors “in vitro”. Neurochem Int 59:954–964.
Sunico CR et al (2011) Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol 21:1–15
Kawahara Y et al (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801
Kawahara Y et al (2006) Underediting of GluR2 mRNA, a neuronal death inducing molecular change in sporadic ALS, does not occur in motor neurons in ALS1 or SBMA. Neurosci Res 54:11–14
Bristol LA, Rothstein JD (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 39:676–679
Barbeito LH et al (2004) A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Rev 47:263–274
Lyras L, Evans PJ, Shaw PJ, Ince PG, Halliwell B (1996) Oxidative damage and motor neurone disease difficulties in the measurement of protein carbonyls in human brain tissue. Free Radic Res 24:397–406
Mitsumoto H et al (2008) Oxidative stress biomarkers in sporadic ALS. Amyotroph Lateral Scler 9:177–183
Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH (2004) Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology 62:1758–1765
Smith RG, Henry YK, Mattson MP, Appel SH (1998) Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 44:696–699
Shaw PJ, Ince PG, Falkous G, Mantle D (1995) Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol 38:691–695
Bogdanov M et al (2000) Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med 29:652–658
Barber SC, Shaw PJ (2010) Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 48:629–641
Parakh S, Spencer DM, Halloran MA, Soo KY, Atkin JD (2013) Redox regulation in amyotrophic lateral sclerosis. Oxidative Med Cell Longev 2013:1–12
Shi P, Gal J, Kwinter DM, Liu X, Zhu H (2010) Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta 1802:45–51
Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA (2002) Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem 80:616–625
Wiedemann FR et al (1998) Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci 156:65–72
Menzies FM et al (2002) Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125:1522–1533
Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18:3241–3250
Hirano A, Donnenfeld H, Sasaki S, Nakano I (1984) Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43:461–470
Julien JP (1997) Neurofilaments and motor neuron disease. Trends Cell Biol 7:243–249
Julien JP, Couillard-Després S, Meier J (1998) Transgenic mice in the study of ALS: the role of neurofilaments. Brain Pathol 8:759–769
Schmidt ML, Carden MJ, Lee VM, Trojanowski JQ (1987) Phosphate dependent and independent neurofilament epitopes in the axonal swellings of patients with motor neuron disease and controls. Lab Investig 56:282–294
Breuer AC et al (1987) Fast axonal transport in amyotrophic lateral sclerosis: an intra-axonal organelle traffic analysis. Neurology 37:738–748
Tu PH et al (1996) Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci U S A 93:3155–3160
Marinkovic P et al (2012) Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 109:4296–4301
Fischer LR et al (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240
Mancuso R, Santos-Nogueira E, Osta R, Navarro X (2011) Electrophysiological analysis of a murine model of motoneuron disease. Clin Neurophysiol 122:1660–1670
Azzouz M et al (1997) Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve 20:45–51
Shibata N et al (1994) Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett 179:149–152
Shibata N et al (1994) superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett 179:149–152
Bruijn LI et al (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Mackenzie IRA et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Groen EJN et al (2010) FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol 67:224–230
Hewitt C et al (2010) Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 67:455–461
Deng H-X et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215
Kaufmann P, Mitsumoto H (2002) Amyotrophic lateral sclerosis: objective upper motor neuron markers. Curr Neurol Neurosci Rep 2:55–60
Vijayalakshmi K et al (2011) Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol Dis 41:695–705
Atkin JD et al (2008) Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis 30:400–407
Atkin JD et al (2006) Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem 281:30152–30165
Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12:627–636
Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772
Valori CF, Brambilla L, Martorana F, Rossi D (2014) The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell Mol Life Sci 71:287–297
Clement AM et al (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Boillee S, Vandervelde C, Cleveland D (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59
Boillee S et al (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Haidet-Phillips AM et al (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828
Khandelwal PJ, Herman AM, Moussa CEH (2011) Inflammation in the early stages of neurodegenerative pathology. J Neuroimmunol 238:1–11
Yong VW, Rivest S (2009) Taking advantage of the systemic immune system to cure brain diseases. Neuron 64:55–60
Troost D, Van den Oord JJ, Vianney de Jong JM (1990) Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 16:401–410
Zhao W, Beers DR, Appel SH (2013) Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J NeuroImmune Pharmacol 8:888–899
Henkel JS et al (2004) Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 55:221–235
Sta M et al (2011) Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis 42:211–220
Kuhle J et al (2009) Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol 16:771–774
Ferraiuolo L et al (2007) Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci 27:9201–9219
Lincecum JM et al (2010) From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet 42:392–399
Lefebvre S et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–165
Burghes AHM, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10:597–609
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007
Polymenidou M et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468
Tollervey JR et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14:452–458
Xiao S et al (2011) RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci 47:167–180
Chang Y et al (2008) Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 3:e2849
Lee Y-B et al (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186
Ash PEA et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646
Zhang K et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61
Freibaum BD et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133
Alonso G et al (2000) Immunocytochemical localization of the sigma1 receptor in the adult rat central nervous system. Neuroscience 97:155–170
Hayashi T, Su T-P (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610
Gekker G et al (2006) Cocaine-induced HIV-1 expression in microglia involves sigma-1 receptors and transforming growth factor-beta1. Int Immunopharmacol 6:1029–1033
Palacios G et al (2003) Immunohistochemical localization of the sigma1-receptor in oligodendrocytes in the rat central nervous system. Brain Res 961:92–99
Penas C et al (2011) Sigma receptor agonist 2-(4-morpholinethyl)1 phenylcyclohexanecarboxylate (Pre084) increases GDNF and BiP expression and promotes neuroprotection after root avulsion injury. J Neurotrauma 28:831–840
Mancuso R et al (2012) Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9:814–826
Quirion R et al (1992) A proposal for the classification of sigma binding sites. Trends Pharmacol Sci 13:85–86
Su T-P, Hayashi T, Maurice T, Buch S, Ruoho AE (2010) The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci 31:557–566
Gromek KA et al (2014) The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem 289:20333–20344
Mishra AK et al (2015) The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J 466:263–271
Balasuriya D et al (2014) A direct interaction between the sigma-1 receptor and the hERG voltage-gated K+ channel revealed by atomic force microscopy and homogeneous time-resolved fluorescence (HTRF®). J Biol Chem 289:32353–32363
Balasuriya D, Stewart AP, Edwardson JM (2013) The σ-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. J Neurosci 33:18219–18224
Carnally SM, Johannessen M, Henderson RM, Jackson MB, Edwardson JM (2010) Demonstration of a direct interaction between sigma-1 receptors and acid-sensing ion channels. Biophys J 98:1182–1191
Wu Z, Bowen WD (2008) Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem 283:28198–28215
Maurice T, Grégoire C, Espallergues J (2006) Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol Biochem Behav 84:581–597
Su TP, London ED, Jaffe JH (1988) Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science 240:219–221
Ramachandran S et al (2009) The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur J Pharmacol 609:19–26
Fontanilla CV et al (2012) Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 205:185–193
Morin-Surun M, Collin T, Denavit-Saubié M, Baulieu EE, Monnet F (1999) Intracellular σ1 receptor modulates phospholipase C and protein kinase C activities in the brainstem. Proc Natl Acad Sci U S A 96:8196
Mavlyutov TA, Ruoho AE (2007) Ligand-dependent localization and intracellular stability of sigma-1 receptors in CHO-K1 cells. J Mol Signal 2:8
Takebayashi M, Hayashi T, Su T-P (2004) Sigma-1 receptors potentiate epidermal growth factor signaling towards neuritogenesis in PC12 cells: potential relation to lipid raft reconstitution. Synapse 53:90–103
Aydar E, Palmer CP, Klyachko VA, Jackson MB (2002) The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34:399–410
Zhang X-J, Liu L-L, Jiang S-X, Zhong Y-M, Yang X-L (2011) Activation of the ζ receptor 1 suppresses NMDA responses in rat retinal ganglion cells. Neuroscience 177:12–22
Hall AA, Herrera Y, Ajmo CT Jr, Cuevas J, Pennypacker KR (2009) Sigma receptors suppress multiple aspects of microglial activation. Glia 57:744–754
Langa F et al (2003) Generation and phenotypic analysis of sigma receptor type I (sigma1) knockout mice. Eur J Neurosci 18:2188–2196
Sha S et al (2013) Sigma-1 receptor knockout impairs neurogenesis in dentate gyrus of adult hippocampus via down-regulation of NMDA receptors. CNS Neurosci Ther 19:705–713
Sha S et al (2015) Sex-related neurogenesis decrease in hippocampal dentate gyrus with depressive-like behaviors in sigma-1 receptor knockout mice. Eur Neuropsychopharmacol 25:1275–1286
Valenza M, DiLeo A, Steardo L, Cottone P, Sabino V (2015) Ethanol-related behaviors in mice lacking the sigma-1 receptor. Behav Brain Res 297:196–203
Ha Y et al (2011) Late-onset inner retinal dysfunction in mice lacking sigma receptor 1 (σR1). Invest Ophthalmol Vis Sci 52:7749–7760
Mavlyutov TA, Nickells RW, Guo L-W (2011) Accelerated retinal ganglion cell death in mice deficient in the Sigma-1 receptor. Mol Vis 17:1034–1043
Chevallier N, Keller E, Maurice T (2011) Behavioural phenotyping of knockout mice for the sigma-1 ( 1) chaperone protein revealed gender-related anxiety, depressive-like and memory alterations. J Psychopharmacol 25:960–975
Mavlyutov TA, Epstein ML, Andersen KA, Ziskind-Conhaim L, Ruoho AE (2010) The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 167:247–255
Bernard-Marissal N, Médard J-J, Azzedine H, Chrast R (2015) Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138:875–890
Conradi S, Skoglund S (1969) Observations on the ultrastruture and distribution of neuronal and glial elements on the motoneuron surface in the lumbosacral spinal cord of the cat during postnatal development. Acta Physiol Scand Suppl 333:5–52
Deardorff AS, Romer SH, Sonner PM, Fyffe REW (2014) Swimming against the tide: investigations of the C-bouton synapse. Front Neural Circuits 8:106
Zagoraiou L et al (2009) A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64:645–662
Pullen AH, Athanasiou D (2009) Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur J Neurosci 29:551–561
Herron LR, Miles GB (2012) Gender-specific perturbations in modulatory inputs to motoneurons in a mouse model of amyotrophic lateral sclerosis. NSC 226:313–323
Casas C et al (2013) Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav 3:145–158
Ferrucci M et al (2010) A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis 37:370–383
Hellström J, Arvidsson U, Elde R, Cullheim S, Meister B (1999) Differential expression of nerve terminal protein isoforms in VAChT-containing varicosities of the spinal cord ventral horn. J Comp Neurol 411:578–590
Hellström J, Oliveira ALR, Meister B, Cullheim S (2003) Large cholinergic nerve terminals on subsets of motoneurons and their relation to muscarinic receptor type 2. J Comp Neurol 460:476–486
Li W, Ochalski PA, Brimijoin S, Jordan LM, Nagy JI (1995) C-terminals on motoneurons: electron microscope localization of cholinergic markers in adult rats and antibody-induced depletion in neonates. NSC 65:879–891
Muennich EAL, Fyffe REW (2004) Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J Physiol Lond 554:673–685
Deardorff AS et al (2013) Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar -motoneurons. J Physiol Lond 591:875–897
Yamamoto T, Hertzberg EL, Nagy JI (1991) Subsurface cisterns in alpha-motoneurons of the rat and cat: immunohistochemical detection with antibodies against connexin32. Synapse 8:119–136
Mavlyutov TA et al (2012) Development of the sigma-1 receptor in C-terminals of motoneurons and colocalization with the N,N′-dimethyltryptamine forming enzyme, indole-N-methyl transferase. Neuroscience 206:60–68
Gallart-Palau X et al (2014) Neuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to a-motoneurons and altered during motoneuron diseases. FASEB J 28:3618–3632
Henkart M, Landis DM, Reese TS (1976) Similarity of junctions between plasma membranes and endoplasmic reticulum in muscle and neurons. J Cell Biol 70:338–347
Henkart M (1980) Identification and function of intracellular calcium stores in axons and cell bodies of neurons. Fed Proc 39:2783–2789
Miles GB, Hartley R, Todd AJ, Brownstone RM (2007) Spinal cholinergic interneurons regulate the excitability of motoneurons during locomotion. Proc Natl Acad Sci U S A 104:2448–2453
Mavlyutov TA et al (2013) Lack of sigma-1 receptor exacerbates ALS progression in mice. Neuroscience 240:129–134
Kourrich S et al (2013) Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 152:236–247
Kinoshita M, Matsuoka Y, Suzuki T, Mirrielees J, Yang J (2012) Sigma-1 receptor alters the kinetics of Kv1.3 voltage gated potassium channels but not the sensitivity to receptor ligands. Brain Res 1452:1–9
Esper RM, Pankonin MS, Loeb JA (2006) Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev 51:161–175
Song F, Chiang P, Wang J, Ravits J, Loeb JA (2012) Aberrant neuregulin 1 signaling in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 71:104
Takahashi Y et al (2013) ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet 93:900–905
Fujimoto M, Hayashi T, Urfer R, Mita S, Su T-P (2012) Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse 66:630–639
Fontanilla D et al (2009) The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323:934–937
Maurice T, Su T-P (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther 124:195–206
Al-Saif A, Al-Mohanna F, Bohlega S (2011) A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70:913–919
Prause J et al (2013) Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet 22:1581–1600
Luty AA et al (2010) Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol 68:639–649
Peviani M et al (2014) Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis 62:218–232
Mei J, Pasternak GW (2002) Sigma1 receptor modulation of opioid analgesia in the mouse. J Pharmacol Exp Ther 300:1070–1074
Mei J, Pasternak GW (2007) Modulation of brainstem opiate analgesia in the rat by 1 receptors: a microinjection study. J Pharmacol Exp Ther 322:1278–1285
Zamanillo D, Romero L, Merlos M, Vela JM (2013) Sigma 1 receptor: a new therapeutic target for pain. Eur J Pharmacol 716:78–93
Antonini V et al (2011) Anti-amnesic and neuroprotective actions of the sigma-1 receptor agonist (-)-MR22 in rats with selective cholinergic lesion and amyloid infusion. J Alzheimers Dis 24:569–586
Yin J et al. (2015) Sigma-1 (σ1) receptor deficiency reduces β-amyloid (25–35) -induced hippocampal neuronal cell death and cognitive deficits through suppressing phosphorylation of the NMDA receptor NR2B. Neuropharmacology 89:215–224.
Villard V, Espallergues J, Keller E, Vamvakides A, Maurice T (2011) Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 ( 1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J Psychopharmacol 25:1101–1117
Francardo V et al (2014) Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 137:1998–2014
Hyrskyluoto A et al (2013) Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-κB pathway. Cell Death Dis 4:e646
Ishiguro H et al (1998) Association between polymorphisms in the type 1 sigma receptor gene and schizophrenia. Neurosci Lett 257:45–48
Allahtavakoli M, Jarrott B (2011) Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res Bull 85:219–224
Ajmo CT Jr, Vernon DOL, Collier L, Pennypacker KR, Cuevas J (2006) Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats. Curr Neurovasc Res 3:89–98
Katnik C, Guerrero WR, Pennypacker KR, Herrera Y, Cuevas J (2006) Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J Pharmacol Exp Ther 319:1355–1365
Griesmaier E et al (2012) Neuroprotective effects of the sigma-1 receptor ligand PRE-084 against excitotoxic perinatal brain injury in newborn mice. Exp Neurol 237:388–395
Lockhart BP, Soulard P, Benicourt C, Privat A, Junien JL (1995) Distinct neuroprotective profiles for sigma ligands against N-methyl-D-aspartate (NMDA), and hypoxia-mediated neurotoxicity in neuronal culture toxicity studies. Brain Res 675:110–120
Tuerxun T et al (2010) SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neurosci Lett 469:303–308
Guzmán-Lenis M-S, Navarro X, Casas C (2009) Drug screening of neuroprotective agents on an organotypic-based model of spinal cord excitotoxic damage. Restor Neurol Neurosci 27:335–349
Mancuso R et al (2014) Lack of synergistic effect of resveratrol and sigma-1 receptor agonist (PRE-084) in SOD1G93A ALS mice: overlapping effects or limited therapeutic opportunity? Orphanet J Rare Dis 9:1–11
Ono Y et al (2014) SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci Lett 559:174–178
Zhao J et al (2014) Sigma receptor ligand, (+)-pentazocine, suppresses inflammatory responses of retinal microglia. Invest Ophthalmol Vis Sci 55:3375–3384
Wegleiter K et al (2014) The sigma-1 receptor agonist 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) protects against newborn excitotoxic brain injury by stabilizing the mitochondrial membrane potential in vitro and inhibiting microglial activation in vivo. Exp Neurol 261:501–509
Martina M, Turcotte M-EB, Halman S, Bergeron R (2007) The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J Physiol Lond 578:143–157
Kim FJ et al (2010) Sigma(1) receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol 77:695–703
Navarro G et al (2010) Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc Natl Acad Sci U S A 107:18676–18681
Misonou H, Mohapatra DP, Trimmer JS (2005) Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26:743–752
Schumacher MA, Rivard AF, Bachinger HP, Adelman JP (2001) Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410:1120–1124
Miki Y et al (2014) Accumulation of the sigma-1 receptor is common to neuronal nuclear inclusions in various neurodegenerative diseases. Neuropathology 34:148–158
Tsai S-Y, Hayashi T, Mori T, Su T-P (2009) Sigma-1 receptor chaperones and diseases. Cent Nerv Syst Agents Med Chem 9:184–189
Mannen T, Iwata M, Toyokura Y, Nagashima K (1977) Preservation of a certain motoneurone group of the sacral cord in amyotrophic lateral sclerosis: its clinical significance. J Neurol Neurosurg Psychiatry 40:464–469
Okamoto K, Hirai S, Ishiguro K, Kawarabayashi T, Takatama M (1991) Light and electron microscopic and immunohistochemical observations of the Onuf’s nucleus of amyotrophic lateral sclerosis. Acta Neuropathol 81:610–614
Okamoto K et al (1993) Oculomotor nuclear pathology in amyotrophic lateral sclerosis. Acta Neuropathol 85:458–462
Nimchinsky EA et al (2000) Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J Comp Neurol 416:112–125
Harrison AR, Anderson BC, Thompson LV, McLoon LK (2007) Myofiber length and three-dimensional localization of NMJs in normal and botulinum toxin treated adult extraocular muscles. Invest Ophthalmol Vis Sci 48:3594–3601
Khanna S, Richmonds CR, Kaminski HJ, Porter JD (2003) Molecular organization of the extraocular muscle neuromuscular junction: partial conservation of and divergence from the skeletal muscle prototype. Invest Ophthalmol Vis Sci 44:1918–1926
Pachter BR (1983) Rat extraocular muscle. 1. Three dimensional cytoarchitecture, component fibre populations and innervation. J Anat 137(Pt 1):143–159
Porter JD et al (2001) Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc Natl Acad Sci U S A 98:12062–12067
Robinson DA (1970) Oculomotor unit behavior in the monkey. J Neurophysiol 33:393–403
Dengler R et al (1990) Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve 13:545–550
Sobue G et al (1983) Degenerating compartment and functioning compartment of motor neurons in ALS: possible process of motor neuron loss. Neurology 33:654–657
Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419
Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28:154–164
Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol Lond 586:3337–3351
Brockington A et al (2012) Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol 125:95–109
Chang Q, Martin LJ (2009) Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol 174:574–585
Chang Q, Martin LJ (2011) Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 31:2815–2827
Saxena S et al (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96
Mòdol L, Mancuso R, Alé A, Francos-Quijorna I, Navarro X (2014) Differential effects on KCC2 expression and spasticity of ALS and traumatic injuries to motoneurons. Front Cell Neurosci 8:7
Ben-Ari Y, Gaiarsa J-L, Tyzio R, Khazipov R (2007) GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87:1215–1284
Bos R et al (2013) Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci U S A 110:348–353
Boulenguez P et al (2010) Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16:302–307
Coull JAM et al (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–1021
Cramer SW et al (2008) The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain 4:36
Gordon T, Tyreman N, Li S, Putman CT, Hegedus J (2010) Functional over-load saves motor units in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 37:412–422
ElBasiouny SM, Schuster JE (2011) The effect of training on motoneuron survival in amyotrophic lateral sclerosis: which motoneuron type is saved? Front Physiol 2:18
Pieri M et al (2003) Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 351:153–156
Kuo JJ et al (2004) Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91:571–575
Martin E, Cazenave W, Cattaert D, Branchereau P (2013) Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54:116–126
van Zundert B et al (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28:10864–10874
Pambo-Pambo A, Durand J, Gueritaud J-P (2009) Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J Neurophysiol 102:3627–3642
Bories C, Amendola J, Lamotte d’Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:451–459
Leroy F, Lamotte d’Incamps B, Imhoff-Manuel RD, Zytnicki D (2014) Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. eLife 3
Appel SH, Beers DR, Siklos L, Engelhardt JI, Mosier DR (2001) Calcium: the darth vader of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2(Suppl 1):S47–S54
Reiner A, Medina L, Figueredo-Cardenas G, Anfinson S (1995) Brainstem motoneuron pools that are selectively resistant in amyotrophic lateral sclerosis are preferentially enriched in parvalbumin: evidence from monkey brainstem for a calcium-mediated mechanism in sporadic ALS. Exp Neurol 131:239–250
Alexianu ME et al (1994) The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol 36:846–858
Aliaga L et al (2013) Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum Mol Genet 22:4293–4305
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG (outside the USA)
About this chapter
Cite this chapter
Mancuso, R., Navarro, X. (2017). Sigma-1 Receptor in Motoneuron Disease. In: Smith, S., Su, TP. (eds) Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology, vol 964. Springer, Cham. https://doi.org/10.1007/978-3-319-50174-1_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-50174-1_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50172-7
Online ISBN: 978-3-319-50174-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)