
Abstract
In 1990, Woese et al. divided the Tree of Life into three separate domains: Eukarya, Bacteria, and Archaea. Archaea were originally perceived as little more than “odd bacteria” restricted to extreme environmental niches, but later discoveries challenged this assumption. Members of this domain populate a variety of unexpected environments (e.g. soils, seawater, and human bodies), and we currently witness ongoing massive expansions of the archaeal branch of the Tree of Life. Archaea are now recognized as major players in the biosphere and constitute a significant fraction of the earth’s biomass, yet they remain underexplored. An ongoing surge in exploration efforts is leading to an increase in the (a) number of isolated strains, (b) associated knowledge, and (c) utilization of Archaea in biotechnology. They are increasingly employed in fields as diverse as biocatalysis, biocomputing, bioplastic production, bioremediation, bioengineering, food, pharmaceuticals, and nutraceuticals. This chapter provides a general overview on bioprospecting Archaea, with a particular focus on extreme halophiles. We explore aspects such as diversity, ecology, screening techniques and biotechnology. Current and future trends in mining for applications are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 The Archaea
5.1.1 Archaeal Diversity
Pioneer work in the 1970s first recognized the non-monolithic nature of prokaryotes, and the distinctiveness of “archaeabacteria” (Balch et al. 1977; Fox et al. 1977; Woese and Fox 1977). Ribosomal RNA (rRNA)-based analyses further supported these findings and this uniqueness was eventually formalized with the establishment of the three domains of Life: Eukarya, Bacteria and Archaea (Woese et al. 1990).
The first visual representations of the archaeal branch of the Tree of Life were sparsely populated, and sub-divided into Euryarchaeota and Crenarchaeota (Woese et al. 1990). They included only cultured representatives, all of which originating from extreme environments. Advances in cultivation-independent techniques, and particularly the use of metagenomics and single amplified genomes (SAGs), revealed several novel phylogenetic groups, which are quickly reshaping our view of the Archaea (e.g. Eme and Doolittle 2015). Indeed, we currently witness a flurry of ongoing additions of new archaeal phyla, which culminated in the creation of two superphyla, commonly referred to as TACK (Guy and Ettema 2011) and DPANN (Rinke et al. 2013).
The TACK superphylum encompasses the Thaumarchaeota (Brochier-Armanet et al. 2008), Aigarchaeota (Nunoura et al. 2010), Crenaerchaeota (Woese et al. 1990), Korarchaeota (Barns et al. 1996), and the recently suggested Bathyarchaeota (Meng et al. 2014). The DPANN superphylum includes Diapherotrites, Parvarchaeota, Aenigmarchaeota, Nanohaloarchaeota, and Nanoarchaeota (Rinke et al. 2013). However, there has been debate on the taxonomic ranking of the Nanoarchaeota since their discovery (Brochier et al. 2005; Huber et al. 2002; Waters et al. 2003). The rapidly evolving nature of the archaeal branch is further highlighted by the (a) even more recent description of the Pacearchaeota and the Woesearchaeota, two massive novel groups within the DPANN superphylum (Castelle et al. 2015), (b) description of the putative new order “Altiarchaeales”, placed within the Euryarchaeota (Probst and Moissl-Eichinger 2015), and (c) discovery of the “Lokiarchaeota” (Spang et al. 2015). The “Lokiarchaeota” represent a candidate novel phylum within the TACK superphylum and seem to bridge the phylogenetic gap between Archaea and Eukarya (Spang et al. 2015). Some authors believe that this discovery might result in the future fusion of both branches of the Tree of Life into a single domain (Fig. 5.1).

The Tree of Life, as originally envisioned (a), and expanded view of latest additions to the Archaea branch (b). Recent findings point suggest an alternative placement for the Eukarya branch, and possible fusion with Archaea (c). Reprinted from Current Biology, Vol 25 No 6, Eme, L. & Doolittle, F., Microbial Diversity: A Bonanza of Phyla, R228, Copyright (2015), with permission from Elsevier
5.1.2 Archaeal Ecology
From an ecological perspective, members of the domain Archaea were traditionally split into thermophilic, methanogenic, and halophilic, all of them isolated from extreme environments (e.g. Ferrera et al. 2008). The widespread use of molecular-based methodologies brought drastic changes, and revealed members of the Archaea to be much more diverse and ubiquitous than previously expected (see Sect. 5.1.1). Archaea populate and thrive in a variety of cold and moderate environments, including landfills, soils, fresh-water sediments, and deep-sea locations, and are involved in symbiotic relationships, e.g. with sponges (e.g. Antunes et al. 2011a; Ferrera et al. 2008; Karner et al. 2001; Leininger et al. 2006; Preston et al. 1996). They contribute to global energy and element cycles, most noticeably via ammonia oxidation in pelagic environments and soils (Ferrera et al. 2008). Archaea might also play important roles in the human body with methanogenic archaea present in the human gut and oral cavities (de Macario and Macario 2009), and abundant Thaumarchaeota detected on human skin (Probst et al. 2013).
5.2 Archaea in Saline Environments
5.2.1 High Salinity Biotopes
An environment is considered hypersaline when its salt concentration surpasses that of the average seawater (i.e. 3.5 % total dissolved salts). Many of the known hypersaline water bodies derive from simple evaporation of seawater, therefore closely mirroring its ionic composition and proportions. These are known as thalassohaline, as opposed to athalassohaline environments, which are usually derived from inland water bodies, hence the dissolved ions are of non-marine proportions (DasSarma and Arora 2001; Rodríguez-Valera 1988).
Hypersaline biotopes occur in high abundance in arid, coastal and deep-sea locations across the globe. Seawater often penetrates through seepage or narrow inlets near coastal areas creating several small evaporation ponds. Well known examples are (a) the Solar Lake and Gavish Sabkha near the Red Sea coast, (b) Guerrero Negro on the Baja California coast, (c) Lake Sivash near the Black Sea, (d) Sharks Bay in Western Australia, and (e) several locations in Antarctica (e.g. Deep Lake, Organic Lake and Lake Suribati). The number of hypersaline water bodies in coastal areas is further augmented by the numerous artificial solar salterns constructed throughout the ages for the production of sea salt. Natural inland hypersaline lakes have higher salinities than coastal ones and include the Dead Sea (Middle East) and the Great Salt Lake (USA), which are the two largest and best-studied examples. The conjugation of high salinity and alkaline conditions produces unusual alkaline hypersaline soda brines. Some of the better-known examples include the Wadi Natrum lakes, Egypt; Lake Magadi, Kenya; the Great Basin lakes, western United States (Mono Lake, Owens Lake, Searles Lake, and Big Soda Lake), and several others in China, India and throughout the world.
Another type of hypersaline biotopes are the often-overlooked saline soils. These include desolate areas present in, for example, Death Valley (California, USA), Alicante (Spain), dispersed locations across Iraq, and Dry Valleys (Antarctica), among several others (Ventosa et al. 1998). Additional examples of less conspicuous highly saline environments include pickled food, fermented products of oriental cuisine (soy sauce, fish paste), surfaces of salt-excreting desert shrubs, human or animal skin, and other places exposed to periodical drying (Galinsky & Trüper, Galinski and Trüper 1994; Lee 2013).
The least explored hypersaline environments include subterranean brines, evaporite deposits, and brine-filled deep-sea basins. Exploration of such environments has been hampered by the remoteness of such locations, and technical and sampling impediments. The underexplored potential of such locations has attracted considerable attention in the last decades, and resulted in several interesting studies (e.g. Antunes et al. 2008, 2011a, b, 2015; Bougouffa et al. 2013; Daffonchio et al. 2006; Fish et al. 2002; Guan et al. 2015; Joye et al. 2009; Mapelli et al. 2012; McGenity et al. 2000; Siam et al. 2012; van der Wielen et al. 2005; Vreeland et al. 2000; Wang et al. 2011), and some preliminary insights into potential applications of their microbial inhabitants (e.g. Antunes et al. 2011b; Mohamed et al. 2013; Sagar et al. 2013; Sayed et al. 2014).
5.2.2 The Halobacteria: Extremely Halophilic Archaea
Microbes living in hypersaline environments are called halophiles. Based on their preferred salinity, they can be categorized as slight (0.3–0.8 M or 1.7–4.8 % NaCl), moderate (0.8–3.4M or 4.7–20 % NaCl), or extreme halophiles (above 3.4 M or 20 % NaCl) (Ollivier et al. 1994). Extreme halophiles are traditionally associated with the members of the euryarchaeal class Halobacteria. This class contains the single order Halobacteriales and its single family Halobacteriaceae, although a recent proposal argues for splitting Halobacteria into Haloferacales, Natrialbales, and an emended order Halobacteriales (Gupta et al. 2015). The class Halobacteria currently includes 177 species with validly published names, placed in 48 genera (LPSN- List of Prokaryotic Names with Standing in Nomenclature 2015; Table 5.1).
Extremely halophilic behaviour is not, however, an exclusive characteristic of the Halobacteriales, as it is also observed in Methanohalobium and Methanohalophilus, of the family Methanosarcinaceae, within the Euryarchaeota (Oren 2000). No halophiles have, thus far, been described within the kingdom Crenarchaeota (Oren 2002).
5.3 Applications of Halophilic Archaea
Mankind has been using halophiles for at least 5000 years. For example, the characteristic red coloration seen in salterns across the globe is imparted mostly by halophilic archaea, and aids in the process of salt crystallization. Other ancient applications include the production of fish sauce, soy sauce and other traditional fermented foods (e.g. Lee 2013).
However, an exponential increase in the applications for halophiles has been observed in the last few decades, particularly after the discovery of extremophiles and extreme-condition adapted enzymes (extremozymes). The industrial application of extremozymes, is clearly the most prominent direct applications for halophiles, and most other extremophiles. However, further exploration is leading to an increase in the number of isolated strains, general knowledge, and number of applications for halophiles and halophilic archaea (Table 5.1; Fig. 5.2).

Overview of the evolution in number of publications and patents associated with Archaea and Halophilic Archaea (Publications: data collected using PUBMED; Patents: data collected using Google Scholar)
5.3.1 Archaeal Pigments
Environments with high densities of halophilic archaea frequently have a characteristic red coloration. This is mostly due to the production of C-50 carotenoid pigments (α-bacterioruberin and its derivatives mono-anhydrobacterioruberin (MABR) and bis-anhydrobacterioruberin (BABR), along with small fractions of C-40 carotenoids such as lycopene and β-carotene) (Yatsunami et al. 2014), which are found in the membranes of several halophiles that thrive in such environments. The reddening of the brines contributes to the absorption of light energy, thereby increasing water evaporation and speeding up the process of salt crystallization (Oren 2002). Within these pigments, β-carotene is the most widely used, mainly as a natural food colorant and as an antioxidant, but also as an important additive in cosmetics, multivitamin preparations, and health food products (nutraceuticals) (Margesin and Schinner 2001; Oren 2002, 2010).
5.3.2 Bacteriorhodopsin
Some of the most interesting uses of halophilic archaea arise from the different proposed applications of bacteriorhodopsin. This molecule, discovered in the early 1970s, is the key protein of the halobacterial photosynthetic system (Hampp 2000; Oren 2002). It is present in Halobacterium salinarum and a few other representatives of the Halobacteriaceae where it forms a two-dimensional crystal integrated into the cellular membrane in patches (usually referred to as “purple membrane”). Bacteriorhodopsin is involved in the light-driven ejection of protons from the cell, establishing a protonic gradient across the membrane. Cells couple the dissipation of this gradient to the production of energy (i.e. ATP) by a membrane-bound ATPase.
The naturally occurring two-dimensional crystalline structure of bacteriorhodopsin is responsible for its (a) astonishing stability toward chemical and thermal degradation, and (b) photosensitivity and cyclicity to illumination. This favourable combination of properties clearly distinguishes the halophilic protein from synthetic materials and makes it attractive for numerous applications (Hampp 2000). These include holography, spatial light modulators, artificial retina, artificial neural networks, optical computing, and new types of optical memories (Margesin and Schinner 2001). From 2005–2010, over 50 patents were granted associated with different uses of bacteriorhodopsin (Trivedi et al. 2011).
5.3.3 Bioplastics
Polyhydroxyalkanoates (PHAs) are a heterogenous family of polyesters, usually used as intracellular carbon storage compounds (most frequently in the form of poly-β-hydroxybutyrate, PHB). The properties of some PHAs are comparable to those of polyethylene and polypropylene with further advantages such as biodegradability, complete water impermeability, and biocompatibility, making them a viable alternative to oil-derived thermoplastics (Divya et al. 2013; Margesin and Schinner 2001; Ventosa and Nieto 1995).
Some halophilic archaea such as Haloarcula marismortui and Haloferax mediterranei were successfully used to produce high amounts of PHA (Han et al. 2007). H. mediterranei can accumulate up to 6 g (60 % of the total biomass dry weight) of PHB per liter of culture using inexpensive starch (DasSarma et al. 2010) or rice bran as carbon source (Huang et al. 2006). The vulnerability of the haloarchaeal cells to pure water (no salt) facilitates isolation of PHA granules by hypoosmotic shock treatment (Quillaguamán et al. 2010). This cheap, straightforward and high yielding harvest procedure reduces downstream processing costs which can account up to 40 % of the total production costs for bacterial PHA production (Choi and Lee 1999).
5.3.4 Enzymes
The inability of “normal” enzymes to operate under the harsh conditions imposed by many industrial processes has limited their widespread use. The discovery of extremophiles and their extreme-adapted extremozymes, is revolutionizing this field with an apparently unceasing range of novel industrial applications. Furthermore, as extremozyme discovery is coupled with enzyme tailoring by rational engineering or directed evolution, the development of economical bioprocesses will accelerate and be enabled on larger scales (Demirjian et al. 2001; DasSarma et al. 2010; Liszka et al. 2012).
The special characteristics of halophilic enzymes, which allow them to function properly under high salinities (Reed et al. 2013), are also responsible for their frequently very poor solubility and denaturation at lower salinities, which could limit their applicability (Madern et al. 2000; van den Burg 2003). These same specific properties seem, however, to make them particularly advantageous in aqueous/organic and non-aqueous media (DasSarma and Arora 2001; Karan et al. 2012; van den Burg 2003). Furthermore, the combination of reverse micelles with halophilic enzymes is further extending the range of applications for these enzymes (van den Burg 2003; Marhuenda-Egea and Bonete 2002).
Relevant enzymes from halophilic archaea include glycosyl hydrolases, proteases, and lipases (Table 5.2). Such enzymes have great potential for biocatalysis in high-salt environments (used in, e.g. the food and detergent industries; Delgado-García et al. 2012; Liszka et al. 2012).
5.3.5 Food Industry
In general, halotolerant and halophilic microorganims (bacteria and archaea) play an essential role in the production of several traditional fermented foods, giving them their characteristic taste, flavor, and aroma. Their salinities range from low to intermediary as present in Sauerkraut, pickles or olives, to the concentrated brines used for fermentation of several traditional food products found in the Pacific Rim area. Within the halophilic archaea the importance of Halobacterium salinarum and Halococcus strains in the production of nam pla, a Thai fish sauce, is well recognized (Ventosa and Nieto 1995). Also, Natrinema gari and Halococcus thailandensis, which were originally isolated from fish sauce, are implicated as important players in the fermentation process (Tapingkae et al. 2008; Namwong et al. 2007), while a protease secreting Halobacterium strain was reported to enhance the overall sauce fermentation process (Akolkar et al. 2010). More modern applications include the use of halophilic archaea for the production of food additives (e.g. polyunsaturated fatty acids; Ventosa and Nieto 1995) and pigments (see Sect. 5.3.1).
5.3.6 Halocins
Halocins are archaeal bacteriocin-like antimicrobial peptides, produced by many members of the Halobacteriales, which inhibit the growth of closely related microbes (Riley and Wertz 2002). According to Kis-Papo and Oren (2000), they could have a role in interspecies competition, particularly on solid substrates.
To name but a few examples, species within Haloferax, Haloarcula and Halobacterium are reported to secrete specific halocins such as S8, H1, H4, C8, H6/H7, and R1 (Salgaonkar et al. 2012; O’Connor and Shand 2002). Despite the almost universal production of these compounds by haloarchaea (Torreblanca et al. 1994), they have been generally overlooked in the ongoing search for new antibiotics (Litchfield 2011). Possible reasons are that many of these purified halocins are not active against the classic group of tested bacteria, and also that many are only active after proteolytic cleavage (Li et al. 2003; Litchfield 2011).
5.3.7 Metal Bioremediation and Nanoparticles
Natural and anthropogenic activities such as erosion and mining have resulted in deposition of toxic heavy metals and their derivatives in soils, rivers and oceans (Paula et al. 2013). The use of microbial-based bioremediation attracts considerable interest, and research on the use of halophiles for metal bioremediation is flourishing (Bini 2010). Several taxa of halophilic archaea are interesting in that, potentially, their metal(loid)s resistance capabilities can be harnessed. Al-Mailem et al. (2011) reported the capability of Halococcus, Halobacterium and Haloferax to resist and volatilize mercury (Hg). Williams et al. (2013) discussed the tolerance of Natronobacterium gregoryi and Halobacterium saccharovorum to 0.001 and 0.01 mM of cadmium (Cd) and zinc (Zn), respectively. Das et al. (2014) investigated the tolerance and intracellular accumulation of Cd by Haloferax, whereas Salgaonkar et al. (2015) reported the resistance of halophilic archaea to zinc oxide nanoparticles (ZnO NPs) for the first time.
Metal(loid)s resistance in halophilic archaea also make them possible candidates for the environmentally-sound synthesis of metal nanoparticles (NPs) which can be employed in various fields. For example, the selenium nanoparticles (SeNPs) synthesized by Halococcus salifodinae BK18 could be used as a chemotherapeutic agent against cancer as they stopped the proliferation of cancerous HeLa cell lines when studied in vitro (Srivastava et al. 2014). Also, silver nanoparticles (AgNPs) synthesized by Halococcus salifodinae BK3 are reported to have anti-bacterial activity against both Gram-positive (Staphylococcus aureus and Micrococcus luteus) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacteria (Srivastava et al. 2013). Since metal uptake and synthesis of NPs are intracellular, haloarchaea have an added advantage as they can be used for metal(loid)s bioremediation and NPs synthesis.
5.3.8 Other Applications
The use of halophilic archaea for exo-polysaccharide production has also a large potential with current utilisation as stabilisers, thickeners, gelling agents and emulsifiers in the pharmaceutical, paint, paper and textile industries (Litchfield 2011; Ventosa and Nieto 1995). Further examples of the wide range of applications for halophiles include such diverse areas as microbially enhanced oil recovery (MEOR) processes, use of gas vesicles for bioengineering, liposomes with increased resistance for cosmetic industry, and saline soil recovery for agriculture, among several others (Litchfield 2011; Oren 2002; Ventosa and Nieto 1995).
5.4 Screening Methodologies
5.4.1 Archaeal Pigments
When grown on agar medium containing high NaCl concentrations, most extremely halophilic archaea display bright red-orange pigmentation, imparted by carotenoids, and can therefore be easily segregated from their non-archaeal counterparts.
5.4.1.1 Haloarchaeal Pigments Extraction and Characterization
Haloarchaeal pigments can be extracted from cells by using solvents, individually or combined (Salgaonkar et al. 2015). In particular, the ultraviolet (UV)-visible spectra of the haloarchaeal C-50 bacterioruberin pigment show characteristic absorption maxima and peaks, while high-performance liquid chromatography (HPLC) analysis presents multiple elution peaks (Yatsunami et al. 2014; Bodaker et al. 2009).
5.4.2 Polyhydroxyalkanoates
Various methods are employed for the screening of intracellular accumulated PHA. The primary method relies on cell staining or the staining agent being incorporated during growth, with binding to PHA granules which fluorescence when exposed to UV light (Legat et al. 2010; Ostle and Holt 1982; Spiekermann et al. 1999). Quantitative PHA production is estimated by acidic hydrolysis, and characteristic absorption peaks (Slepecky 1961). The presence of intracellular PHA granules can also be detected with transmission electron microscopy, Fourier transform infrared spectroscopy (FTIR), or screening of target strains for the genes encoding PHA synthase (further details in e.g. Han et al. 2010; Salgaonkar and Bragança 2015).
5.4.2.1 Extraction of PHA
PHAs can be recovered by lysing cells, followed by polymer solubilization and purification (Tan et al. 2014). As halophilic archaea thrive under very high salinities, their use is associated with very low risks of contamination. Furthermore, their cells lyse in water or in low osmolarity solutions, greatly facilitating the extraction of intracellular PHA granules, and reducing production costs (Quillaguamán et al. 2010).
5.4.2.2 PHA Characterization
Characterization of PHA is very important for their application, as more than 150 monomeric units are available, which impart different properties to the polymer (Tan et al. 2014). Monomer composition is determined by techniques such as gas chromatography (GC), nuclear magnetic resonance (NMR) and spectroscopy after depolymerization (Tan et al. 2014). Furthermore gel permeation chromatography (GPC) is used to determine the polymer’s average (a) molecular mass (Mw), (b) molecular mass distribution (Mn), and (c) polydispersity index (PDI; Mw/Mn) (Ashby et al. 2002).
PHA thermal properties determine the temperature conditions at which the polymer can be processed and utilized (Tan et al. 2014; Chen 2010). Thermal properties include glass transition temperature, melting temperature, and thermodegradation temperature, which are obtained using differential scanning calorimetry, differential thermal analysis, and thermogravimetric analysis. The absolute crystallinity of produced PHA polymers can be measured by X-ray diffraction (XRD) analysis (see Chanprateep 2010 and Sánchez et al. 2003 for more detailed information).
Note that PHA polymers can either be a soft elastomeric material or a hard rigid material, displaying a wide elongation at break values between 2 % and 1000 % (Chen 2010). PHA mechanical properties that are commonly evaluated include: (a) Young’s modulus which provide a measure of the polymer’s stiffness and ranges from the very ductile mcl-PHA to the stiffer scl-PHA (Rai et al. 2011); (b) elongation at break, which measures the extent that a material will stretch before it breaks and is expressed as a percentage of the material’s original length; and (c) tensile strength, which measures the amount of force required to pull a material until it breaks (Rai et al. 2011). These assays can be performed with tensile tester instrument by standardized test methods such as the ones recommended by the American Society for Testing and Materials (ASTM) standards (Wu and Liao 2014).
5.4.3 Enzymes
Quantitative analysis of hydrolytic enzyme production in halophilic archaea traditionally relies on screening by plate assays wherein the substrate of the enzyme in question is provided as the sole carbon source (Kharroub et al. 2014; Kakhki et al. 2011). Any minimal halophilic medium supplemented with 20–25 % salt and having a proper nitrogen source can be used for enzymatic screening. Examples of preparation and screening methodologies are abundant and include different hydrolytic activities such as e.g. (a) amylase (Amoozegar et al. 2003), (b) cellulose and xylanse (Wejse et al. 2003), (c) pectinase (Soares et al. 1999), (d) extracellular protease (Amoozegar et al. 2008), (e) DNase (Onishi et al. 1983), and (f) chitinase (Park et al. 2000). Examples of purification procedure of enzymes obtained from halophilic archaea can be found in multiple references (e.g. Delgado-García et al. 2012; Moshfegh et al. 2013; Pérez-Pomares et al. 2003; Vidyasagar et al. 2006).
A faster alternative to plate screening is the in silico approach where genomic data is checked for putative enzyme genes. But the fact that the whole genome of the organism has to be known, clearly limits the use of this method.
5.4.4 Halocins
Halocins are commonly found in the cell-free supernatants (CFS) of halophilic archaea. Standard methodologies employ the agar well diffusion assay, in which the indicator organism is surface-spread or seeded into agar and the CFS of the producer strain is placed in wells within the same plate and allowed to diffuse. The minimum inhibitory concentration (MIC) of the halocin is assayed by serial dilution of the CFS and the activity is presented in Arbitrary Units (AU) (Atanasova et al. 2013; Salgaonkar et al. 2012).
5.4.4.1 Characterization and Purification of Halocins
After achieving significant MIC results, additional steps of characterization and purification are employed. Initial characterization plots halocin activity profiles versus growth phase. This provides insights on the phase of growth during which the halocin is produced. To further characterize halocin activity several parameters are tested: pH, temperature, NaCl concentration, and different solvents. It is worth noting that almost all reported halocins are hydrophobic, and reverse-phase HPLC is commonly employed for their complete purification (Meknaci et al. 2014; Price and Shand 2000).
5.4.5 Bioremediation of Metal(loid)s/Metal Nanoparticles
Resistance of haloarchaeal strains to metal(loid)s can be checked by growing strains in media with increasing concentrations of the respective metals. This will also determine the MIC, which is the minimum concentration of metal(loid)s that inhibits archaeal growth. It is worth mentioning that growth of halophilic archaea in the presence of certain metals such as silver/tellurium and selenium changes its pigmentation from red-orange to black and brick-red, respectively.
5.4.5.1 Detection of Metal(loid)s Uptake
Cells grown in the presence of metal(loid)s are hydrolysed using a solution of concentrated nitric acid: sulphuric acid (v/v), followed by complete digestion at 100° C and analysis by absorption spectrophotometry (AAS) (Das et al. 2014).
5.4.5.2 Characterization of the Nanoparticles
The cells grown in the presence of metal(loid)s are harvested, dialyzed, dried and ground using motor and pestle to fine powder (nm range). This powder is analyzed using techniques such as scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX), XRD and TEM. The UV-visible spectra of silver and selenium nanoparticles show absorption maxima at 440 and 270nm, respectively.
5.5 Current and Future Trends in Mining for Applications
Intensive research efforts currently aim to unleash the full biotechnological potential of halophilic archaea. The recent introduction of genetically optimized efficient expression systems for genes from halophilic sources, has removed a major limitation for large-scale applications. The most promising systems are based on fast growing aerobic extreme halophiles, such as Haloferax volcanii (Allers et al. 2010) or Halobacterium sp. NRC-1 (Karan et al. 2013), which can even be used for high-yielding protein expression in bioreactors (Strillinger et al. submitted). Additionally, different strategies were reported to optimize E. coli for archaeal protein expression (e.g. Connaris et al. 1998; Cao et al. 2008). With the appropriate molecular biotechnology tools in place, developments of more efficient and reliable bioprospecting tools are underway to eliminate remaining bottlenecks. Comprehension of the full capacity of halophilic archaea will arise from understanding their biodiversity and a detailed insight into their molecular functions. Hence, since less than 1 % of the viable organisms within a particular niche are cultivable (Amann et al. 1995), accessing and harvesting genomic material of these microorganisms represents the main challenge. To some extent, introducing specialized laboratory equipment to mimic the extreme conditions of the native habitats will facilitate more efficient laboratory cultivation of halophiles from samples. However, major contributions are expected to come from metagenomic approaches as well as SAG libraries.
5.5.1 Next Generation Sequencing Methods
The advent of cheaper and faster second or next-generation sequencing (NGS) platforms, enabled a shift towards novel culture-independent genome and transcriptome analysis methods. These methods are based on direct DNA and/or RNA isolation from environmental samples and fall into the following classes: (i) metagenomics (DNA based), (ii) metatranscriptomics (RNA based) and (iii) single cell genomics (DNA based).
Metagenomics identification of microbial communities commonly relies on sequencing of the 16S rRNA; however, the same concept can be applied directly for the sequencing of metagenomic DNA samples (Von Mering et al. 2007). Introduction of metagenomics lead to the identification of thousands of novel protein families from diverse environments (Yooseph et al. 2007). Metatranscriptomics based on mRNA (Sorek and Cossart 2010) complements the DNA-based metagenomic approach and provides an understanding of the genomically active genes of microbial population at a given time point from a specific environment. This method requires the isolation of mRNA, which is translated into cDNA before sequencing. The resulting short sequences (reads) of typically a few hundred base pairs for NGS are subsequently assembled and annotated.
5.5.2 DNA Assembly, the First Milestone for Successful Data Mining
Assembling the comparatively short reads is a challenging task, since reads are derived from a myriad of organisms, which form the sampled community. Hence the bioinformatic assembly algorithms applied need to accurately resolve the correct position, and the specific biological entity (e.g. DNA from microbial genomes, viruses or plasmids) for each read (Mick and Sorek 2014). Every single DNA fragment is therefore compared, to all others, to identify overlapping sequences as merging points. Bioinformatics challenges include (a) defining the exact length of naturally occurring and quite common repeats (identical sequence repetitions), (b) differentiating between random overlaps and defined overlaps, (c) defining the correct orientation of the DNA sequence, (d) identifying sequencing errors from the real sequence, (e) correctly identifying the organism (from the pool of diverse genetic material in the environmental sample) from which the sequence originates, and (f) accounting for different sequence depths (amount of sequencing). One should note that before sequencing, the DNA is amplified using random primers, which show statistical variations in binding DNA, resulting in regions that are more or less often amplified per amplification cycle. Assembler programs therefore require about 8 copies of each piece of genome (Baker 2012).
Until recently, the assembly of genomes relied on the genetic material from a single organism or on reference genomes. These approaches led to problems when trying to separate complex metagenomic data into specific biological entities. For samples from archaeal and/or extremophilic communities, which include genomic material that commonly extend far beyond what is covered by reference databases, other assembly strategies are required to interpret metagenomic data without relying on reference sequences. Nielsen et al. (2014) established a new method and demonstrated its power on the analysis of the complex human gut microbiome. The protocol facilitates the extraction of single genomes from complex microbial samples and uses the relative abundance of an organism in the community, which fluctuates over time between different samplings of the same environment. By tracking the changes in abundance of genes between different sampling times, genes showing highly correlated abundance are clustered together. It was shown that such a correlation corresponds with a high probability of belonging to the same genome (Mick and Sorek 2014). Strain-level resolution in metagenomics can be used to identify variations in highly flexible genomic parts, which are coexisting with the relatively stable core components (Kashtan et al. 2014) and thus provides insight into genes essential for adaptation to dramatic changes in environment. Those genes may illuminate the microbial mechanisms involved in environmental adaption. Limitations of this approach include the need for access to a fairly large number of independent samplings of one niche, or related niches, which is required for statistical analysis. However, due to the amount of sequences per sample, the sequence depth can be reduced (Mick and Sorek 2014).
The advent of single-cell genomics (Lasken 2007) allowed identification of different species in an environment, while eliminating the challenge of assigning DNA reads (fragments) to different genomes. Single cell genomics is based on multiplication of the DNA of a single cell through multiple displacement amplifications. As a result, a few femtograms of DNA are enough to provide the microgram amounts of DNA necessary for library construction and sequencing (Lasken 2007). Equal and complete amplification of the minimal amount of source DNA must be achieved to obtain unbiased results, which represents the major challenge of this method.
5.5.3 Current Representation of Archaeal Genomes in Largest Databases
Compared to other domains of life, genomic analysis of archaea is still in its infancy, but interest is growing. Correspondingly, current genomic information of archaeal origin represents only 0.4 to 3 % of the data available from major databases listing genomic and oligonucleotide sequences (Table 5.3). The availability of reference sequences is crucial for genome annotation (see below) and therefore continuous publication of fully assembled and annotated archaeal genomes is required to facilitate genomic assembly, improve reliability and accelerate bioinformatic processing of archaeal data.
5.5.4 Genome Annotation, the Second Milestone in Successful Information Mining
Genome annotation connects DNA sequences to biological information. The value of a genome is determined by its annotation (Stein 2001). Inaccurate annotations lead to incorrect in silico identification of enzymes of interest and are particularly problematic when systems biology approaches are used to understand the functions of a cell at the molecular level based on a model of pathways or specific enzymes. Starting from an assembled standard genome, the annotation can be divided into three steps: First, parts of the genome that do not code for proteins are excluded (non-coding RNA); second, the prediction of protein-coding genes (open reading frames) in the genome is undertaken; and third, a biological function is assigned to the proteins. Depending on the goal of the genome annotation a further focus might be the identification of regulatory elements or non-coding RNA (e.g. tRNA, and rRNA).
The standard gene annotation approach relies on gene homology to genes already annotated and available from the common genomic databases. Unfortunately, annotation reliability is indirectly proportional to the variance of the two compared respective proteins’ primary structures. Since novel genomes from uncommon habitats are expected to show a lower homology to any gene described so far, the reliability of genome annotation is, in general, decreased. The situation is complicated by error propagation. Also, experimental validation of the encoded protein’s function exists only for a small and continuously diminishing fraction of gene sequences available from databases. Originally, the functions of novel genes were annotated based on gene sequences with experimentally verified function. Based on these novel determined genes, further genes were annotated and so on. While in this chain, two proteins in a row are always highly similar, a low similarity of the last annotated gene and the experimental verified source may result, depending on how many non experimentally verified genes are in-between. From an experimental and protein engineering point of view, faulty annotations are a fundamental problem.
Analysis of state-of-the-art annotation pipelines reveals a surprisingly high level of uncertainty in gene annotation. Annotations of the same E. coli strain by the leading annotation pipelines yielded about 5.5 % false positives and a significantly higher rate of false positives may be expected for novel genomes (Alam et al. 2013, Grötzinger et al. 2014). Hence, several bioinformatics groups work on strategies to increase annotation reliability, typically by including additional data. For example Alam et al. (2013), combined several strategies, including comparison of predicted 16S rRNA genes with the NCBI prokaryotic 16S rRNA gene database to retrieve taxonomic information and rank the obtained BLAST hits (Altschul et al. 1990). BLAST against several databases resulted in coverage of most known genes. Additionally, the analysis of gene distribution in different pathways helped to evaluate expected and annotated gene presence. Software such as the InterProScan database (Jones et al. 2014; Mitchell et al. 2015) introduced predictions of protein functions based on the number of domains or active sites. Other approaches are focusing on highly reliable annotation of a selected set of single proteins instead of a whole genome annotation, e.g. when mining genomic data for enzymes of interest to biotechnology (Grötzinger et al. 2014). The analysis of annotation metadata is particularly useful for this approach. These metadata contain information on the presence of conserved domains such as active centers or binding pockets, and can be identified during the annotation process. Presence of domains that are relevant for protein activity should increase annotation reliability. Despite the progress made in annotation of proteins with described function, the correct assignment of function and pathway location of proteins that are not described remains a major hurdle.
5.5.5 Potential and Challenges of Upcoming Generations of DNA Sequencing
The advent of the third-generation DNA sequencing (single molecule sequencing) not only brings a further reduction in sequencing costs, but also increases read lengths to several thousand base pairs. This not only reduces the complexity of the genome assembly process, or the assignment of specific genomes from a metagenomic DNA pool, but also increases the overall quality of genomes and therefore may even eliminate the concept of draft genomes completely (Land et al. 2015). At the moment about 10 % of all draft genomes are of too poor quality to be used (Land et al. 2014). Third generation sequencing can theoretically produce a finished genome in a few hours and simultaneously identify specific methylation sites (Land et al. 2015).
Although DNA assembly might be simplified in the future, the challenge of proper genomic annotation remains and new challenges will arise from the management of the constantly increasing stream of data. Experimentalists are in need for tools to help them make sense of their massive amount of data, while currently bioinformatics research is struggling to analyze, compare, interpret and visualize data at the pace at which sequencing throughput increase (Land et al. 2015). Bioinformatics progress heavily relies on the use of supercomputers because the amount and the complexity of genomic data are growing significantly faster than the increase in computing and storage capabilities of current systems. The development of new algorithms will require dividing the entire data processing into more manageable tasks, so that it can be addressed on smaller computer clusters, by cloud computing, or by outsourcing and accession via the web. This will assist the end-user as it does not require direct access to a supercomputer.
The need to minimize the amount of metadata included in every sequenced data (Kottmann et al. 2008) illustrates the problems that arise from handling the increasing data volume and complexity. Such metadata include, e.g. geographic location and habitat from which the sample was taken, and details of the sequencing method used which is necessary for efficient assembly, and assigning specific features such as tolerance to specific extreme environments. However, as described above, insufficient reliability of annotations for genomic material from uncommon environments mandates that annotation pipelines include a significantly enriched body of metadata. Future annotation of halophilic archaea could particularly benefit from metadata-based precise domain architecture prediction (e.g. if functionally associated domains are in close proximity such as active catalytically center and cofactor binding pocket). In detail pathway analysis can be used to evaluate how many of the other enzymes, required to provide the cofactor, or substrate, or use of a product, are represented in the organism. It may therefore provide a reliable measure for the probability of correct annotation.
Static tables or images such as charts or plots cannot illustrate accurately the highly complex information available within genomic datasets. Therefore, new approaches to analyse and visualize data are also necessary, apart from novel algorithms,. Linking integrated databases/warehouses (e.g. INDIGO (Alam et al. 2013), to visualization tools such as Krona (Ondov et al. 2014), can be used to illustrate clusters or correlations of genomic information. The integrated databases provide annotations, and direct access to metadata quickly, which can be visualized on multi-level pie charts using standard web browsers. The unprecedented rate of development of genomic sequencing methods effectively shifted the major costs of biomining from sequencing to the genome assembly and functional annotation, and data analysis and management procedures (Land et al. 2015).
The combination of novel culture independent sequencing techniques with new bioinformatics annotation and data analysis tools now permits the analysis of natural microbial communities in situ. Future results will therefore provide insights into microbial distribution patterns, and their individual (SAG & assigned genomes), or uniform (meta-genomics/transcriptomics) molecular functions. Hence, a powerful set of techniques is at hand to mine archaeal sources, which will harvest an increasing amount of the biotechnological potential of halophilic archaea. The appropriate utilization of these tools in combination with laboratory-based analysis, will not only increase our understanding of symbiotic and other interactions in microbial communities, but will also provide access to whole sets of enzymes from the same environments. This information can be used to establish multi-enzyme reactions in industry and consequently provide more sustainable solutions for the pharmaceutical and biotechnological industry.
5.6 Research Initiatives of Interest for Bioprospecting Archaea
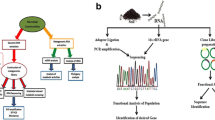
It is a difficult task to list research initiatives on bioprospecting Archaea. First, there is no agreed definition of the term “bioprospecting”, and although there is a general understanding that it involves research for commercial purposes (outlined in Fig. 5.3), it is usually difficult to distinguish, in practice, between basic and applied research (Arico and Salpin 2005). Additionally, large-scale research initiatives usually have a wide-scope and unsurprisingly no such program has specifically targeted Archaea. However, given their importance in extreme environments, and their newly found relevance in marine ecosystems, one can rightfully assume that research initiatives focusing on such locations include Archaea as major targets.

General outline of the different possible steps involved in bioprospecting activities
Research on extremophiles and their applications has boomed recently as evidenced by an increasing number of publications in high-impact journals and patents. The importance of this field is further attested by concerted funding initiatives in the USA (NSF and NASA’s programs Life in Extreme Environments, Exobiology and Astrobiology), the EU (Biotechnology of Extremophiles, Extremophiles as Cell Factories, ILEE- Investigating Life in Extreme Environments, and CAREX- Coordination Action for Research Activities on Life in Extreme Environments), and Japan (JAMSTEC Frontier Research System for Extremophiles program) (Jamieson 2015; Rothschild and Mancinelli 2001).
During this period, environmental and marine research initiatives and programs have seen an impressive increase in scope, reach, complexity, and dimension. Many of these projects have a global scale and include a very wide variety of measured parameters. A non-extensive list of more visible initiatives would include the Census of Marine Life ( http://www.coml.org ), Global Ocean Sampling (GOS; http://www.jcvi.org/cms/research/projects/gos/overview , MaCuMBA ( http://www.macumbaproject.eu ), Malaspina ( http://www.expedicionmalaspina.es ), MAMBA ( http://mamba.bangor.ac.uk ), TARA Oceans ( http://www.embl.de/tara-oceans ), and Micro B3 ( http://www.microb3.eu ).
Several other initiatives target general genomic and metagenomic data generation, frequently involved in filling current gaps in our understanding of specific environments or phylogenetic groups. Noteworthy examples include the Earth Microbiome Project (EMP; www.earthmicrobiome.org ), the Genomic Encyclopedia of Bacteria and Archaea (GEBA; www.jgi.doe.gov/programs/GEBA ), and the Marine Microbial Genome Sequencing Project ( http://camera.calit2.net/microgenome ).
Microbial Biological Resource Centers (mBRCs) and culture collections also play an important role, fueling the bio-economy as sources of microbiological resources, data, and expertise. It is worth noting the current programs that are moving towards regional integration of mBRCs, and the promotion of a more active interaction with industry (e.g. the EU-funded Microbial Research Infrastructure; www.mirri.org ). Closer interactions between industrial and research institutions are further highlighted by the recent wave of clusters formed within the Bioindustrie 2021 initiative, funded by the Bundes Ministerium für Bildung und Forschung in Germany ( http://www.bioindustrie2021.eu ), and looking into fostering new innovations in bioproducts (e.g. biofuels, biopolymers, and biocatalysts).
5.7 Overview and Conclusions
Archaea were originally perceived as evolutionary oddities with restricted importance. However, a significant shift in our understanding of their diversity, ecology, and impact is currently under way. Increased exploration efforts in multiple environments, and the continued development, and application of new methodologies for cultivation, molecular-based studies, and in silico approaches will further promote this shift, and are expected to lead the way towards a wave of new discoveries. Furthermore, correct annotation of genomes still remains one of the major challenges in genomic data mining. Different strategies are evolving and improved algorithms together with experimental data established in the laboratory are poised to handle these challenges.
Halophilic archaea are a prime example of the increasing reach and range of applications and are perceived as rising stars for industrial biotechnology (e.g. biocatalysis, bioengineering, biofuel, pharmaceuticals). Further bioprospecting initiatives will foster new innovations in bioproducts, and help to fuel the bio-economy.
References
Akolkar AV, Durai D, Desai AJ (2010) Halobacterium sp. SP1(1) as a starter culture for accelerating fish sauce fermentation. J Appl Microbiol 109:44–53
Al-Mailem DM, Al-Awadh H, Sorkhoh NA et al (2011) Mercury resistance and volatilization by oil utilizing haloarchaea under hypersaline conditions. Extremophiles 15:39–44
Alam I, Antunes A, Kamau AA et al (2013) INDIGO – Integrated data warehouse of microbial genomes with examples from the red sea extremophiles. PLoS One 8(12):e82210. doi:10.1371/journal.pone.0082210
Allers T, Barak S, Liddell S et al (2010) Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl Environ Microbiol 76(6):1759–1769. doi:10.1128/AEM.02670-09
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. doi:10.1016/S0022-2836(05)80360-2
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59(1):143–169
Amoozegar MA, Makhdoumi-Kakhki A, Shahzadeh Fazeli SA et al (2012) Halopenitus persicus gen. nov., sp. nov., an archaeon from an inland salt lake. Int J Syst Evol Microbiol 62:1932–1936
Amoozegar MA, Malekzadeh F, Malik KA (2003) Production of amylase by newly isolated moderate halophile, Halobacillus sp. strain MA-2. J Microbiol Methods 52(3):353–359
Amoozegar MA, Salehghamari E, Khajeh K et al (2008) Production of an extracellular thermohalophilic lipase from a moderately halophilic bacterium, Salinivibrio sp. strain SA-2. J Basic Microbiol 48(3):160–167
Antunes A, Rainey F, Wanner G et al (2008) A new lineage of halophilic, wall-less, contractile bacteria from a brine-filled Deep of the Red Sea. J Bacteriol 190:3580–3587
Antunes A, Ngugi DK, Stingl U (2011a) Microbiology of the Red Sea (and other) deep-sea anoxic brine lakes. Environ Microbiol Rep 3:416–433
Antunes A, Alam I, Bajic VB, Stingl U (2011b) Genome sequence of Salinisphaera shabanensis, a gammaproteobacterium from the harsh, variable environment of the brine-seawater interface of the Shaban Deep in the Red Sea. J Bacteriol 193(17):4555–4556
Antunes A, Alam I, Simões MF et al (2015) First insights on the viral communities of the deep-sea anoxic brines of the Red Sea. Genomics Proteomics Bioinformatics (accepted)
Arico S, Salpin C (2005) Bioprospecting of genetic resources in the deep seabed: scientific, legal and policy aspects. UNU-IAS Report pp 1–72. http://www.ias.unu.edu
Ashby R, Solaiman D, Foglia T (2002) Poly(ethylene glycol)-mediated molar mass control of short-chain- and medium-chain-length poly(hydroxyalkanoates) from Pseudomonas oleovorans. Appl Microbiol Biotechnol 60:154–159
Atanasova NS, Pietilä MK, Oksanen HM (2013) Diverse antimicrobial interactions of halophilic archaea and bacteria extend over geographical distances and cross the domain barrier. MicrobiologyOpen 2(5):811–825. doi:10.1002/mbo3.115
Baker M (2012) De novo genome assembly: what every biologist should know. Nat Methods 9(4):333–337. doi:10.1038/nmeth.1935
Balch WE, Magrum LJ, Fox GE et al (1977) An ancient divergence among the bacteria. J Mol Evol 9(4):305–311
Bardavid RE, Mana L, Oren A (2007) Haloplanus natans gen. nov., sp. nov., an extremely halophilic, gas-vacuolate archaeon isolated from Dead Sea–Red Sea water mixtures in experimental outdoor ponds. Int J Syst Evol Microbiol 57:780–783
Barns SM, Delwiche CF, Palmer JD, Pace NR (1996) Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc Natl Acad Sci U S A 93(17):9188–9193
Benson DA, Clark K, Karsch-Mizrachi I et al (2015) GenBank. Nucleic Acids Res, 43(Database issue):D30–D35. doi:10.1093/nar/gku1216
Bini E (2010) Archaeal transformation of metals in the environment. FEMS Microbiol Ecol 73:1–16
Bodaker I, Beja O, Sharon I et al (2009) Archaeal diversity in the Dead Sea: microbial survival under increasingly harsh conditions. Nat Resour Environ Issues 15(1):25
Bougouffa S, Yang JK, Lee OO et al (2013) Distinctive microbial community structure in highly stratified deep-sea brine water columns. Appl Environ Microbiol 79(11):3425–3437
Boutaiba S, Bhatnagar T, Hacene H et al (2006) Preliminary characterisation of a lipolytic activity from an extremely halophilic archaeon, Natronococcus sp. J Mol Catal B Enzym 41(1):21–26
Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P (2008) Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol 6(3):245–252
Brochier C, Gribaldo S, Zivanovic Y et al (2005) Nanoarchaea: representatives of a novel archaeal phylum or a fast-evolving euryarchaeal lineage related to Thermococcales? Genome Biol 6(5):R42
Burns DG, Janssen PH, Itoh T et al (2007) Haloquadratum walsbyi gen. nov., sp. nov., the square haloarchaeon of Walsby, isolated from saltern crystallizers in Australia and Spain. Int J Syst Evol Microbiol 57:387–392
Burns DG, Janssen PH, Itoh T, Kamekura M et al (2010) Halonotius pteroides gen. nov., sp. nov., an extremely halophilic archaeon recovered from a saltern crystallizer. Int J Syst Evol Microbiol 60:1196–1199
Cao Y, Liao L, Xu XW et al (2008) Characterization of alcohol dehydrogenase from the haloalkaliphilic archaeon Natronomonas pharaonis. Extremophiles 12(3):471–476. doi:10.1007/s00792-007-0133-7
Castelle CJ, Wrighton KC, Thomas BC et al (2015) Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25(6):690–701
Castillo AM, Gutiérrez MC, Kamekura M et al (2006a) Halostagnicola larsenii gen. nov., sp. nov., an extremely halophilic archaeon from a saline lake in Inner Mongolia, China. Int J Syst Evol Microbiol 56:1519–1524
Castillo AM, Gutiérrez MC, Kamekura M et al (2006b) Halovivax asiaticus gen. nov., sp. nov., a novel extremely halophilic archaeon isolated from Inner Mongolia, China. Int J Syst Evol Microbiol 56:765–770
Chaga G, Porath J, Illéni T (1993) Isolation and purification of amyloglucosidase from Halobacterium sodomense. Biomed Chromatogr 7(5):256–261
Chanprateep S (2010) Current trends in biodegradable polyhydroxyalkanoates. J Biosci Bioeng 110:621–632
Chen GQ (2010) Introduction of bacterial plastics PHA, PLA, PBS, PE, PTT, and PPP. In: Chen GQ (ed) Plastics from bacteria: natural functions and applications. Springer, Berlin/Heidelberg, pp. 1–16
Choi J, Lee SY (1999) Factors affecting the economics of poly- hydroxyalkanoate production by bacterial fermentation. Appl Microbiol Biotechnol 51:13–21
Connaris H, West SM, Hough DW, Danson MJ (1998) Cloning and overexpression in Escherichia coli of the gene encoding citrate synthase from the hyperthermophilic Archaeon Sulfolobus solfataricus. Extremophiles 2(2):61–66
Cui HL, Qiu XX (2014) Salinarubrum litoreum gen. nov., sp. nov.: a new member of the family Halobacteriaceae isolated from Chinese marine solar salterns. Antonie van Leeuwenhoek 105:135–141
Cui HL, Zhang WJ (2014) Salinigranum rubrum gen. nov., sp. nov., a member of the family Halobacteriaceae isolated from a marine solar saltern. Int J Syst Evol Microbiol 64:2029–2033
Cui HL, Gao X, Yang X, Xu XW (2010a) Halorussus rarus gen. nov., sp. nov., a new member of the family Halobacteriaceae isolated from a marine solar saltern. Extremophiles 14:493–499
Cui HL, Gao X, Sun FF et al (2010b) Halogranum rubrum gen. nov., sp. nov., a halophilic archaeon isolated from a marine solar saltern. Int J Syst Evol Microbiol 60:1366–1371
Cui HL, Li XY, Gao X et al (2010c) Halopelagius inordinatus gen. nov., sp. nov., a new member of the family Halobacteriaceae isolated from a marine solar saltern. Int J Syst Evol Microbiol 60:2089–2093
Cui HL, Yang X, Mou YZ (2011a) Salinarchaeum laminariae gen. nov., sp. nov.: a new member of the family Halobacteriaceae isolated from salted brown alga Laminaria. Extremophiles 15:625–631
Cui HL, Gao X, Yang X, Xu XW (2011b) Halolamina pelagica gen. nov., sp. nov., a new member of the family Halobacteriaceae. Int J Syst Evol Microbiol 61:1617–1621
Cui HL, Yang X, Gao X, Xu XW (2011c) Halobellus clavatus gen. nov., sp. nov. and Halorientalis regularis gen. nov., sp. nov., two new members of the family Halobacteriaceae. Int J Syst Evol Microbiol 61:2682–2689
Cui HL, Mou YZ, Yang X et al (2012) Halorubellus salinus gen. nov., sp. nov. and Halorubellus litoreus sp. nov., novel halophilic archaea isolated from a marine solar saltern. Syst Appl Microbiol 35:30–34
Cunningham F, Amode MR, Barrell D et al (2015) Ensembl 2015. Nucleic Acids Res 43(Database issue):D662–D669. doi:10.1093/nar/gku1010
Daffonchio D, Borin S, Brusa T et al (2006) Stratified prokaryote network in the oxic–anoxic transition of a deep-sea halocline. Nature 440(7081):203–207
Das D, Salgaonkar BB, Mani K, Bragança JM (2014) Cadmium resistance in extremely halophilic archaeon Haloferax strain BBK2. Chemosphere 112:385–392
DasSarma S, Arora P (2001) Halophiles. In: Encyclopedia of life sciences. Nature Publishing Group, London. http://www.els.net
DasSarma P, Coker JA, Huse V, DasSarma S (2010) Halophiles, industrial applications. In: Flickinger MC (ed) Encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology. Wiley, Hoboken, pp. 1–43
Delgado-García M, Valdivia-Urdiales B, Aguilar-González CN et al (2012) Halophilic hydrolases as a new tool for the biotechnological industries. J Sci Food Agric 92:2575–2580
de Macario EC, Macario AJ (2009) Methanogenic archaea in health and disease: a novel paradigm of microbial pathogenesis. Int J Med Microbiol 299(2):99–108
Demirjian DC, Moris-Varas F, Cassidy CS (2001) Enzymes from extremophiles. Curr Opin Chem Biol 5(2):144–151
Divya G, Achana T, Manzano RA (2013) Polyhydroxinates, a sustainable alternative to petro-based plastics. J Pet Environ Biotechnol 4(3):1000143
Echigo A, Minegishi H, Shimane Y et al (2013) Halomicroarcula pellucida gen. nov., sp. nov., a non-pigmented, transparent-colony-forming, halophilic archaeon isolated from solar salt. Int J Syst Evol Microbiol 63:3556–3562
Elazari-Volcani B (1957) Genus XII. Halobacterium Elazari-Volcani, 1940. In: Breed RS, Murray EGD, Smith NR (eds) Bergey’s manual of determinative bacteriology, 7th edn. Williams & Wilkins, Baltimore, pp. 207–212
Eme L, Doolittle WF (2015) Microbial diversity: a bonanza of phyla. Curr Biol 25(6):R227–R230
Ferrera I, Takacs-Vesbach CD, Reysenbach AL (2008) Archaeal ecology. In: Encyclopedia of life sciences (eLS). Wiley, Chichester
Fish SA, Shepherd TJ, McGenity TJ, Grant WD (2002) Recovery of 16S ribosomal RNA gene fragments from ancient halite. Nature 417(6887):432–436
Fox GE, Magrum LJ, Balch WE et al (1977) Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proc Natl Acad Sci U S A 74(10):4537–4541
Galinski EA, Trüper HG (1994) Microbiol behaviour in salt-stressed ecosystems. FEMS Microbiol Rev 15:95–108
Giménez MI, Studdert CA, Sánchez JJ, De Castro RE (2000) Extracellular protease of Natrialba magadii: purification and biochemical characterization. Extremophiles 4(3):181–188
Good WA, Hartman PA (1970) Properties of the amylase from Halobacterium halobium. J Bacteriol 104(1):601–603
Grötzinger SW, Alam I, Ba Alawi WB et al (2014) Mining a database of single amplified genomes from red sea brine pool extremophiles-improving reliability of gene function prediction using a profile and pattern matching algorithm (PPMA). Front Microbiol 5:134. doi:10.3389/fmicb.2014.00134
Guan Y, Hikmawan T, Antunes A et al (2015) Diversity of methanogens and sulfate-reducing bacteria in the interfaces of five deep-sea anoxic brines of the Red Sea. Res Microbiol (accepted)
Gupta RS, Naushad S, Baker S (2015) Phylogenomic analyses and molecular signatures for the class Halobacteria and its two major clades: a proposal for division of the class Halobacteria into an emended order Halobacteriales and two new orders, Haloferacales ord. nov. and Natrialbales ord. nov. Int J Syst Evol Microbiol 65:1050–1069
Gutiérrez MC, Castillo AM, Kamekura M et al (2007) Halopiger xanaduensis gen. nov., sp. nov., an extremely halophilic archaeon isolated from saline Lake Shangmatala in Inner Mongolia, China. Int J Syst Evol Microbiol 57:1402–1407
Guy L, Ettema TJ (2011) The archaeal ‘TACK’superphylum and the origin of eukaryotes. Trends Microbiol 19(12):580–587
Hampp N (2000) Bacteriorhodopsin as a photochromic retinal protein for optical memories. Chem Rev 100:1755–1776
Han J, Lu Q, Zhou L et al (2007) Molecular characterization of the phaECHm genes, required for biosynthesis of poly (3-hydroxybutyrate) in the extremely halophilic archaeon Haloarcula marismortui. Appl Environ Microbiol 73(19):6058–6065
Han J, Hou J, Liu H et al (2010) Wide distribution among halophilic Archaea of a novel polyhydroxyalkanoate synthase subtype with homology to bacterial type III synthases. Appl Environ Microbiol 76:7811–7819
Hezayen FF, Tindall BJ, Steinbüchel A, Rehm BH (2002) Characterization of a novel halophilic archaeon, Halobiforma haloterrestris gen. nov., sp. nov., and transfer of Natronobacterium nitratireducens to Halobiforma nitratireducens comb. nov. Int J Syst Evol Microbiol 52:2271–2280
Holmes ML, Scopes RK, Moritz RL et al (1997) Purification and analysis of an extremely halophilic β-galactosidase from Haloferax alicantei. BBA-Protein Struct M 1337(2):276–286
Huang TY, Duan KJ, Huang SY, Chen CW (2006) Production of polyhydroxyalkanoates from inexpensive extruded rice bran and starch by Haloferax mediterranei. J Ind Microbiol Biotechnol 33:701–706
Huber H, Hohn MJ, Rachel R et al (2002) A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 417(6884):63–67
Inoue K, Itoh T, Ohkuma M, Kogure K (2011) Halomarina oriensis gen. nov., sp. nov., a halophilic archaeon isolated from a seawater aquarium. Int J Syst Evol Microbiol 61:942–946
Itoh T, Yamaguchi T, Zhou P, Takashina T (2005) Natronolimnobius baerhuensis gen. nov., sp. nov. and Natronolimnobius innermongolicus sp. nov., novel haloalkaliphilic archaea isolated from soda lakes in Inner Mongolia, China. Extremophiles 9:111–116
Izotova LS, Strongin AY, Chekulaeva LN et al (1983) Purification and properties of serine protease from Halobacterium halobium. J Bacteriol 155(2):826–830
Jamieson A (2015) The hadal zone: life in the deepest oceans. Cambridge University Press, Cambridge
Jones P, Binns D, Chang HY et al (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240. doi:10.1093/bioinformatics/btu031
Joye SB, Samarkin VA, MacDonald IR et al (2009) Metabolic variability in seafloor brines revealed by carbon and sulphur dynamics. Nat Geosci 2(5):349–354
Kakhki AM, Amoozegar MA, Khaledi EM (2011) Diversity of hydrolytic enzymes in haloarchaeal strains isolated from salt lake. Int J Environ Sci Technol 8(4):705–714
Kamekura M, Dyall-Smith ML (1995) Taxonomy of the family Halobacteriaceae and the description of two new genera Halorubrobacterium and Natrialba. J Gen Appl Microbiol 41:333–350
Kamekura M, Dyall-Smith ML, Upasani V et al (1997) Diversity of alkaliphilic halobacteria: proposals for transfer of Natronobacterium vacuolatum, Natronobacterium magadii, and Natronobacterium pharaonis to Halorubrum, Natrialba, and Natronomonas gen. nov., respectively, as Halorubrum vacuolatum comb. nov., Natrialba magadii comb. nov., and Natronomonas pharaonis comb. nov., respectively. Int J Syst Bacteriol 47:853–857
Kamekura M, Seno Y (1990) A halophilic extracellular protease from a halophilic archaebacterium strain 172 P1. Biochem Cell Biol 68(1):352–359
Kamekura M, Seno Y, Holmes ML, Dyall-Smith ML (1992) Molecular cloning and sequencing of the gene for a halophilic alkaline serine protease (halolysin) from an unidentified halophilic archaea strain (172P1) and expression of the gene in Haloferax volcanii. J Bacteriol 174(3):736–742
Karan R, Capes MD, DasSarma P, DasSarma S (2013) Cloning, overexpression, purification, and characterization of a polyextremophilic beta-galactosidase from the Antarctic haloarchaeon Halorubrum lacusprofundi. BMC Biotechnol 13:3. doi:10.1186/1472-6750-13-3
Karan R, Capes MD, DasSarma S (2012) Function and biotechnology of extremophilic enzymes in low water activity. Aquatic Biosystems 8(4) www.aquaticbiosystems.org/content/8/1/4
Karner MB, DeLong EF, Karl DM (2001) Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409(6819):507–510
Kashtan N, Roggensack SE, Rodrigue S et al (2014) Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344(6182):416–420. doi:10.1126/science.1248575
Kharroub K, Gomri MA, Aguilera M, Monteoliva-Sanchez M (2014) Diversity of hydrolytic enzymes in haloarchaea isolated from Algerian sabkhas. Afr J Microbiol Res 8(52):3992–4001
Kis-Papo T, Oren A (2000) Halocins: are they involved in the competition between halobacteria in saltern ponds? Extremophiles 4(1):35–41
Kobayashi T, Kanai H, Hayashi T et al (1992) Haloalkaliphilic maltotriose-forming alpha-amylase from the archaebacterium Natronococcus sp. strain Ah-36. J Bacteriol 174(11):3439–3444
Kobayashi T, Kanai H, Aono R et al (1994) Cloning, expression, and nucleotide sequence of the alpha-amylase gene from the haloalkaliphilic archaeon Natronococcus sp. strain Ah-36. J Bacteriol 176(16):5131–5134
Kottmann R, Gray T, Murphy S et al (2008) A standard MIGS/MIMS compliant XML schema: toward the development of the Genomic Contextual Data Markup Language (GCDML). OMICS 12(2):115–121. doi:10.1089/omi.2008.0A10
Krishnan G, Altekar W (1991) An unusual class I (Schiff base) fructose-1,6-bisphosphate aldolase from the halophilic archaebacterium Haloarcula vallismortis. Eur J Biochem 195(2):343–350
Land M, Hauser L, Jun SR et al (2015) Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15(2):141–161. doi:10.1007/s10142-015-0433-4
Land ML, Hyatt D, Jun SR et al (2014) Quality scores for 32,000 genomes. Stand Genomic Sci 9:20. doi:10.1186/1944-3277-9-20
Lasken RS (2007) Single-cell genomic sequencing using multiple displacement amplification. Curr Opin Microbiol 10(5):510–516
Law JH, Slepecky RA (1961) Assay of poly-β-hydroxybutyric acid. J Bacteriol 82:33–36
Lee H-S (2013) Diversity of halophilic Archaea in fermented foods and human intestines and their application. J Microbiol Biotechnol 23(12):1645–1653
Legat A, Gruber C, Zangger K et al (2010) Identification of polyhydroxyalkanoates in Halococcus and other haloarchaeal species. Appl Microbiol Biotechnol 87:1119–1127
Leininger S, Urich T, Schloter M et al (2006) Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442(7104):806–809
Leinonen R, Sugawara H, Shumway M (2011) The sequence read archive. Nucleic Acids Res 39(Database issue):D19–D21. doi:10.1093/nar/gkq1019
Li Y, Xiang H, Liu J et al (2003) Purification and biological characterization of halocin C8, a novel peptide antibiotic from Halobacterium strain AS7092. Extremophiles 7(5):401–407
Litchfield CD (2011) Potential for industrial products from the halophilic Archaea. J Ind Microbiol Biotechnol 38(10):1635–1647
Liszka MJ, Clark ME, Schneider E, Clark DS (2012) Nature versus nurture: developing enzymes that function under extreme conditions. Ann Rev Chem Biol Eng 3:77–102
LPSN-List of Prokaryotic Names with Standing in Nomenclature (2015) List of prokaryotic names with standing in nomenclature. www.bacterio.net
Madern D, Ebel C, Zaccai G (2000) Halophilic adaptation of enzymes. Extremophiles 4:91–98
Makhdoumi-Kakhki A, Amoozegar MA, Bagheri M et al (2012a) Haloarchaeobius iranensis gen. nov., sp. nov., an extremely halophilic archaeon isolated from a saline lake. Int J Syst Evol Microbiol 62:1021–1026
Makhdoumi-Kakhki A, Amoozegar MA, Ventosa A (2012b) Halovenus aranensis gen. nov., sp. nov., an extremely halophilic archaeon from Aran-Bidgol salt lake. Int J Syst Evol Microbiol 62:1331–1336
Mapelli F, Borin S, Daffonchio D (2012) Microbial diversity in deep hypersaline anoxic basins. In: Stan-Lotter H, Fendrihan S (eds) Adaption of microbial life to environmental extremes. Springer, Wien/New York, pp. 21–36
Margesin R, Schinner F (2001) Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles 5:73–83
Marhuenda-Egea F, Bonete MJ (2002) Extreme halophilic enzymes in organic solvents. Curr Opin Biotechnol 13:385–389
McGenity TJ, Grant WD (1995) Transfer of Halobacterium saccharovorum, Halobacterium sodomense, Halobacterium trapanicum NRC 34021 and Halobacterium lacusprofundi to the genus Halorubrum gen. nov., as Halorubrum saccharovorum comb. nov., Halorubrum sodomense comb. nov., Halorubrum trapanicum comb., nov., and Halorubrum lacusprofundi comb. nov. Syst Appl Microbiol 18:237–243
McGenity TJ, Gemmell RT, Grant WD (1998) Proposal of a new halobacterial genus Natrinema gen. nov., with two species Natrinema pellirubrum nom. nov. and Natrinema pallidum nom. nov. Int J Syst Bacteriol 48:1187–1196
McGenity TJ, Gemmell RT, Grant WD, Stan-Lotter H (2000) Origins of halophilic microorganisms in ancient salt deposits. Environ Microbiol 2(3):243–250
Meknaci R, Lopes P, Servy C et al (2014) Agar-supported cultivation of Halorubrum sp. SSR, and production of halocin C8 on the scale-up prototype platotex. Extremophiles 18(6):1049–1055
Meng J, Xu J, Qin D et al (2014) Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J 8(3):650–659
Mick E, Sorek R (2014) High-resolution metagenomics. Nat Biotechnol 32(8):750–751. doi:10.1038/nbt.2962
Minegishi H, Echigo A, Nagaoka S et al (2010) Halarchaeum acidiphilum gen. nov., sp. nov., a moderately acidophilic haloarchaeon isolated from commercial solar salt. Int J Syst Evol Microbiol 60:2513–2516
Minegishi H, Kamekura M, Kitajima-Ihara T et al (1998) Halogeometricum borinquense gen. nov., sp. nov., a novel halophilic archaeon from Puerto Rico. Int J Syst Bacteriol 48:1305–1312
Mitchell A, Chang HY, Daugherty L et al (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43(Database issue):D213–D221. doi:10.1093/nar/gku1243
Mohamed YM, Ghazy MA, Sayed A et al (2013) Isolation and characterization of a heavy metal-resistant, thermophilic esterase from a Red Sea brine pool. Sci Rep 3:3358. doi:10.1038/srep03358
Moshfegh M, Shahverdi AR, Zarrini G, Faramarzi MA (2013) Biochemical characterization of an extracellular polyextremophilic α-amylase from the halophilic archaeon Halorubrum xinjiangense. Extremophiles 17(4):677–687
Mou YZ, Qiu XX, Zhao ML et al (2012) Halohasta litorea gen. nov. sp. nov., and Halohasta litchfieldiae sp. nov., isolated from the Daliang aquaculture farm, China and from Deep Lake, Antarctica, respectively. Extremophiles 16(6):895–901
Namwong S, Tanasupawat S, Visessanguan W et al (2007) Halococcus thailandensis sp. nov., from fish sauce in Thailand. Int J Syst Evol Microbiol 57:2199–2203
Nielsen HB, Almeida M, Juncker AS et al (2014) Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol 32(8):822–828. doi:10.1038/nbt.2939
Nunoura T, Takaki Y, Kakuta J et al (2010) Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Ac Res 1–20. doi:10.1093/nar/gkq1228
O’Connor EM, Shand RF (2002) Halocins and sulfolobicins: the emerging story of archaeal protein and peptide antibiotics. J Ind Microbiol Biotechnol 28:23–31
Ollivier B, Caumette P, Garcia J-L, Mah R (1994) Anaerobic bacteria from hypersaline environments. Microbiol Rev 58(1):27–38
Ondov B, Bergman N, Phillippy A (2014) Krona: interactive metagenomic visualization in a web browser. In: Nelson KE (ed) Encyclopedia of metagenomics. Springer, New York, pp. 1–8
Onishi H, Mori T, Takeuchi S et al (1983) Halophilic nuclease of a moderately halophilic Bacillus sp.: production, purification, and characterization. Appl Environ Microbiol 45(1):24–30
Oren A (1983) A thermophilic amyloglucosidase from Halobacterium sodomense, a halophilic bacterium from the Dead Sea. Curr Microbiol 8(4):225–230
Oren A (2000) Life at high salt concentrations. In: The prokaryotes: an evolving electronic resource for the microbiological community, 3rd edn, release 3.1, Springer, New York. http://link.springer-ny.com/link/service/books/10125
Oren A (2002) Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol 28:56–63
Oren A (2010) Industrial and environmental applications of halophilic microorganisms. Environ Technol 31:825–834
Oren A, Gurevich P, Gemmell RT, Teske A (1995) Halobaculum gomorrense gen. nov., sp. nov., a novel extremely halophilic archaeon from the Dead Sea. Int J Syst Bacteriol 45:747–754
Oren A, Elevi R, Watanabe S et al (2002) Halomicrobium mukohataei gen. nov., comb. nov., and emended description of Halomicrobium mukohataei. Int J Syst Evol Microbiol 52:1831–1835
Ostle AG, Holt J (1982) Nile blue A as a fluorescent stain for poly-beta-hydroxybutyrate. Appl Environ Microbiol 44:238–241
Park SH, Lee JH, Lee HK (2000) Purification and characterization of chitinase from a marine bacterium, Vibrio sp. 98CJ11027. J Microbiol 38:224–229
Paula DP, Gleny A, Martha H et al (2013) Kinetics of arsenite removal by halobacteria from a highly and Andean Chilean Salar. Aquat Biosyst 9:8. www.aquaticbiosystems.org/content/9/1/8 .
Pérez-Pomares F, Bautista V, Ferrer J et al (2003) α-amylase activity from the halophilic archaeon Haloferax mediterranei. Extremophiles 7(4):299–306
Preston CM, Wu KY, Molinski TF, DeLong EF (1996) A psychrophilic crenarchaeon inhabits a marine sponge: cenarchaeum symbiosum gen. nov., sp. nov. Proc Natl Acad Sci U S A 93(13):6241–6246
Price LB, Shand RF (2000) Halocin S8: a 36-amino-acid microhalocin from the haloarchaeal strain S8a. J Bacteriol 182(17):4951–4958
Probst AJ, Auerbach AK, Moissl-Eichinger C (2013) Archaea on human skin. PLoS One 8(6):e65388. doi:10.1371/journal.pone.0065388
Probst AJ, Moissl-Eichinger C (2015) “Altiarchaeales”: uncultivated Archaea from the subsurface. Life 5(2):1381–1395
Quillaguamán J, Guzmán H, Van-Thuoc D, Hatti-Kaul R (2010) Synthesis and production of polyhydroxyalkanoates by halophiles: current potential and future prospects. Appl Microbiol Biotechnol 85:1687–1696
Rai R, Keshavarz T, Roether JA et al (2011) Medium chain length polyhydroxyalkanoates, promising new biomedical materials for the future. Mater Sci Eng R Rep 72:29–47
Reed CJ, Lewis H, Trejo E et al (2013) Protein adaptations in archaeal extremophiles. Archaea 2013. http://dx.doi.org/10.1155/2013/373275
Reddy TB, Thomas AD, Stamatis D et al (2015) The genomes onLine database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res 43(Database issue):D1099–D1106. doi:10.1093/nar/gku950
Riley MA, Wertz JE (2002) Bacteriocins: evolution, ecology, and application. Ann Rev Microbiol 56(1):117–137
Rinke C, Schwientek P, Sczyrba A et al (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499(7459):431–437
Rodríguez-Valera F (1988) Characteristics and microbial ecology of hypersaline environments. In: Rodríguez-Valera F (ed) Halophilic bacteria. CRC Press, Boca Raton, pp. 3–30
Rothschild LJ, Mancinelli RL (2001) Life in extreme environments. Nature 409(6823):1092–1101
Ryu K, Kim J, Dordick JS (1994) Catalytic properties and potential of an extracellular protease from an extreme halophile. Enzym Microb Technol 16(4):266–275
Sagar S, Esau L, Hikmawan T et al (2013) Cytotoxic and apoptotic evaluations of marine bacteria isolated from brine-seawater interface of the red sea. BMC Complement Altern Med 13(1):29
Salgaonkar BB, Bragança JM (2015) Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Halogeometricum borinquense strain E3. Int J Biol Macromol 78:339–346
Salgaonkar BB, Das D, Bragança JM (2015) Resistance of extremely halophilic archaea to zinc and zinc oxide nanoparticles. Appl Nanosci. doi:10.1007/s13204-015-0424-8
Salgaonkar BB, Kabilan M, Nair A et al (2012) Interspecific interactions among members of family Halobacteriaceae from natural solar salterns. Probiotics Antimicrob Proteins 4(2):98–107
Sánchez RJ, Schripsema J, da Silva LF et al (2003) Medium-chain-length polyhydroxyalkanoic acids (PHAmcl) produced by Pseudomonas putida IPT 046 from renewable sources. Eur Polym J 39:1385–1394
Savage KN, Krumholz LR, Oren A, Elshahed MS (2007) Haladaptatus paucihalophilus gen. nov., sp. nov., a halophilic archaeon isolated from a low-salt, sulfide-rich spring. Int J Syst Evol Microbiol 57:19–24
Savage KN, Krumholz LR, Oren A, Elshahed MS (2008) Halosarcina pallida gen. nov., sp. nov., a halophilic archaeon from a low-salt, sulfide-rich spring. Int J Syst Evol Microbiol 58:856–860
Sayed A, Ghazy MA, Ferreira AJ (2014) A novel mercuric reductase from the unique deep brine environment of Atlantis II in the Red Sea. J Biol Chem 289(3):1675–1687
Schoop G (1935) Halococcus litoralis, ein obligat halphiler Farbstoffbildner. Dtsch Tierarztl Wochenschr 43:817–820
Shimane Y, Hatada Y, Minegishi H et al (2010) Natronoarchaeum mannanilyticum gen. nov., sp. nov., an aerobic, extremely halophilic archaeon isolated from commercial salt. Int J Syst Evol Microbiol 60:2529–2534
Shimane Y, Hatada Y, Minegishi H et al (2011) Salarchaeum japonicum gen. nov., sp. nov., an aerobic, extremely halophilic member of the Archaea isolated from commercial salt. Int J Syst Evol Microbiol 61:2266–2270
Siam R, Mustafa GA, Sharaf H et al (2012) Unique prokaryotic consortia in geochemically distinct sediments from Red Sea Atlantis II and Discovery Deep brine pools. PLoS One 7(8):e42872. doi:10.1371/journal.pone.0042872
Skerman VBD, McGowan V, Sneath PHA et al (1980) Approved lists. Int J Syst Bacteriol 30:225–420
Soares MM, Silva RD, Gomes E (1999) Screening of bacterial strains for pectinolytic activity: characterization of the polygalacturonase produced by Bacillus sp. Rev Microbiol 30(4):299–303
Spang A, Saw JH, Jørgensen SL et al (2015) Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521(7551):173–179
Spiekermann P, Rehm BH, Kalscheuer R (1999) A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch Microbiol 171:73–80
Srivastava P, Bragança J, Ramanan SR, Kowshik M (2013) Synthesis of silver nanoparticle synthesis using haloarchaeal isolate Halococcus salifodinae BK3. Extremophiles 17:821–831
Srivastava P, Bragança J, Kowshik M (2014) In vivo synthesis of selenium nanoparticles by Halococcus salifodinae BK18 and their anti-proliferative properties against HeLa cell line. Biotechnol Prog 30:1480–1487
Song HS, Cha IT, Yim KJ et al (2014) Halapricum salinum gen. nov., sp. nov., an extremely halophilic archaeon isolated from non-purified solar salt. Antonie Van Leeuwenhoek 105:979–986
Sorek R, Cossart P (2010) Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet 11(1):9–16. doi:10.1038/nrg2695
Stein L (2001) Genome annotation: from sequence to biology. Nat Rev Genet 2(7):493–503. doi:10.1038/35080529
Stepanov VM, Rudenskaya GN, Revina LP et al (1992) A serine proteinase of an archaebacterium, Halobacterium mediterranei. A homologue of eubacterial subtilisins. Biochem J 285:281–286
Studdert CA, De Castro RE, Seitz KH, Sánchez JJ (1997) Detection and preliminary characterization of extracellular proteolytic activities of the haloalkaliphilic archaeon Natronococcus occultus. Arch Microbiol 168(6):532–535
Tan GY, Chen CL, Li L et al (2014) Start a research on biopolymer polyhydroxyalkanoate (PHA): a review. Polymers 6:706–754
Tapingkae W, Tanasupawat S, Itoh T et al (2008) Natrinema gari sp. nov., a halophilic archaeon isolated from fish sauce in Thailand. Int J Syst Evol Microbiol 58:2378–2383
Tindall BJ, Ross HNM, Grant WD (1984) Natronobacterium gen. nov. and Natronococcus gen. nov., two new genera of haloalkaliphilic archaebacteria. Syst Appl Microbiol 5:41–57
Torreblanca M, Rodriguez-Valera F, Juez G et al (1986) Classification of non-alkaliphilic halobacteria based on numerical taxonomy and polar lipid composition, and description of Haloarcula gen. nov. and Haloferax gen. nov. Syst Appl Microbiol 8:89–99
Torreblanca M, Meseguer I, Ventosa A (1994) Production of halocin is a practically universal feature of archaeal halophilic rods. Lett Appl Microbiol 19(4):201–205
Trivedi S, Choudhary OP, Gharu J (2011) Different proposed applications of bacteriorhodopsin. Recent Pat DNA Gen Seq 5:35–40
van den Burg B (2003) Extremophiles as a source for novel enzymes. Curr Opin Microbiol 6:213–218
van der Wielen PW, Bolhuis H, Borin S et al (2005) The enigma of prokaryotic life in deep hypersaline anoxic basins. Nature 307:121–123
Ventosa A, Gutiérrez MC, Kamekura M, Dyall-Smith ML (1999) Proposal to transfer Halococcus turkmenicus, Halobacterium trapanicum JCM 9743 and strain GSL-11 to Haloterrigena turkmenica gen. nov., comb. nov. Int J Syst Bacteriol 49:131–136
Ventosa A, Nieto JJ (1995) Biotechnological applications and potentialities of halophilic microorganisms. World J Microbiol Biotechnol 11:85–94
Ventosa A, Nieto JJ, Oren A (1998) Biology of moderately halophilic aerobic bacteria. Microbiol Mol Biol Rev 62:504–544
Ventosa A, Sánchez-Porro C, Martín S, Mellado E (2005) Halophilic archaea and bacteria as a source of extracellular hydrolytic enzymes. In: Adaptation to life at high salt concentrations in Archaea, Bacteria, and Eukarya. Springer, Dordrecht, pp. 337–354
Vidyasagar M, Prakash S, Litchfield C, Sreeramulu K (2006) Purification and characterization of a thermostable, haloalkaliphilic extracellular serine protease from the extreme halophilic archaeon Halogeometricum borinquense strain TSS101. Archaea 2(1):51–57
von Mering C, Hugenholtz P, Raes J et al (2007) Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 315(5815):1126–1130. doi:10.1126/science.1133420
Vreeland RH, Rosenzweig WD, Powers DW (2000) Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature 407(6806):897–900
Vreeland RH, Straight S, Krammes J et al (2002) Halosimplex carlsbadense gen. nov., sp. nov., a unique halophilic archaeon, with three 16S rRNA genes, that grows only in defined medium with glycerol and acetate or pyruvate. Extremophiles 6:445–452
Wainø M, Tindall BJ, Ingvorsen K (2000) Halorhabdus utahensis gen. nov., sp. nov., an aerobic, extremely halophilic member of the Archaea from Great Salt Lake, Utah. Int J Syst Evol Microbiol 50:183–190
Wainø M, Ingvorsen K (2003) Production of β-xylanase and β-xylosidase by the extremely halophilic archaeon Halorhabdus utahensis. Extremophiles 7(2):87–93
Wang Y, Yang J, Lee OO et al (2011) Hydrothermally generated aromatic compounds are consumed by bacteria colonizing in Atlantis II Deep of the Red Sea. ISME J 5(10):1652–1659
Waters E, Hohn MJ, Ahel I et al (2003) The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc Natl Acad Sci U S A 100(22):12984–12988
Wejse PL, Ingvorsen K, Mortensen KK (2003) Purification and characterisation of two extremely halotolerant xylanases from a novel halophilic bacterium. Extremophiles 7(5):423–431
Williams GP, Gnanadesigan M, Ravikumar S (2013) Biosorption and bio-kinetic properties of solar saltern halobacterial strains for managing Zn2+, As2+ and Cd2+ metals. Geomicrobiol J 30:497–500
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74(11):5088–5090
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 87(12):4576–4579
Wu CS, Liao HT (2014) The mechanical properties, biocompatibility and biodegradability of chestnut shell fibre and polyhydroxyalkanoate composites. Polym Degrad Stab 99:274–282
Xu Y, Zhou P, Tian X (1999) Characterization of two novel haloalkaliphilic archaea Natronorubrum bangense gen. nov., sp. nov. and Natronorubrum tibetense gen. nov., sp. nov. Int J Syst Bacteriol 49:261–266
Xue Y, Fan H, Ventosa A et al (2005) Halalkalicoccus tibetensis gen. nov., sp. nov., representing a novel genus of haloalkaliphilic archaea. Int J Syst Evol Microbiol 55:2501–2505
Yatsunami R, Ando A, Yang Y et al (2014) Identification of carotenoids from the extremely halophilic archaeon Haloarcula japonica. Front Microbiol 5:100. doi:10.3389/fmicb.2014.00100
Yooseph S, Sutton G, Rusch DB et al (2007) The sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol 5(3):e16. doi:10.1371/journal.pbio.0050016
Yu TX (1991) Protease of haloalkaliphiles. In: Horikoshi K, Grant WD (eds) Superbugs: microorganisms in extreme environments. Springer, New York, pp. 76–83
Acknowledgments
The authors of this publication were partially supported by competitive research funding from King Abdullah University of Science and Technology (KAUST), and by KAUST baseline research funds to VBB.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Antunes, A., Simões, M.F., Grötzinger, S.W., Eppinger, J., Bragança, J., Bajic, V.B. (2017). Bioprospecting Archaea: Focus on Extreme Halophiles. In: Paterson, R., Lima, N. (eds) Bioprospecting. Topics in Biodiversity and Conservation, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-319-47935-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-47935-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47933-0
Online ISBN: 978-3-319-47935-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)