
Abstract
Toxin-antitoxin systems are widespread in the bacterial kingdom, including in pathogenic species, where they allow rapid adaptation to changing environmental conditions through selective inhibition of key cellular processes, such as DNA replication or protein translation. Under normal growth conditions, type II toxins are inhibited through tight protein-protein interaction with a cognate antitoxin protein. This toxin-antitoxin complex associates into a higher-order macromolecular structure, typically heterotetrameric or heterooctameric, exposing two DNA binding domains on the antitoxin that allow auto-regulation of transcription by direct binding to promoter DNA. In this chapter, we review our current understanding of the structural characteristics of type II toxin-antitoxin complexes in bacterial cells, with a special emphasis on the staggering variety of higher-order architecture observed among members of the VapBC family. This structural variety is a result of poor conservation at the primary sequence level and likely to have significant and functional implications on the way toxin-antitoxin expression is regulated.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
14.1 Introduction
Toxin-antitoxin (TA) systems are characterised by being non-essential for normal growth of bacterial cells, but essential for survival during stress, where they provide a means of rapid adaptation via an adjustment of the overall metabolic rate. Stress conditions include changes in humidity, temperature, nutrient supply, oxidative state, the presence of antibiotics and bacteriophages or other competing organisms (Hayes 2003; Sat et al. 2003; Hazan et al. 2004; Sat et al. 2001; Hazan and Engelberg-Kulka 2004). TA systems are widespread in both bacteria and archaea and are often found in large numbers, a surprising fact given the small size of most prokaryotic genomes (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007; Makarova et al. 2009). Free-living prokaryotes appear to contain an especially large number of TA systems, whereas obligate intracellular prokaryotes contain few or even no TA systems (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007; Makarova et al. 2009). First discovered in the 1980s as loci that conferred plasmid maintenance (Gerdes et al. 1986b; Ogura and Hiraga 1983), TA systems were later identified also on chromosomes of free-living prokaryotes (Masuda et al. 1993; Gerdes 2000), and in the genomes of obligate intracellular organisms (Ogata et al. 2005). TA systems are characterised by two components, a stable toxin and an unstable antitoxin that inhibits the toxin under normal cellular conditions by a range of different mechanisms (Pandey and Gerdes 2005; Gerdes et al. 2005; Van Melderen and Saavedra De Bast 2009). During cellular stress, the antitoxin is inactivated, in some cases by proteases, releasing the active toxin. Once activated, toxins inhibit essential cellular processes, including DNA replication, protein synthesis, peptidoglycan formation and cell division, but the full range of targets has not yet been elucidated as many putative TA systems remain functionally uncharacterised (Gerdes et al. 2005; Unterholzner et al. 2013).
The overall physiological roles of TA systems range widely, from plasmid maintenance, programmed cell death , and stress-response, to the formation of persister cells (Magnuson 2007). The ccdAB locus was initially identified on the mini-F plasmid of Escherichia coli as a segment promoting plasmid maintenance during cell division (Ogura and Hiraga 1983). Shortly after, other TA systems with similar functions were identified and the observed function was coined post-segregational killing (Gerdes et al. 1986b; Jaffe et al. 1985). According to this principle, daughter cells stochastically receive either a plasmid containing the TA system during cell division, or not. In daughter cells without plasmid, toxin and antitoxin molecules will still be present, but will not be replenished. Therefore, due to the instability of the antitoxin, toxin molecules will gradually be released and activated, resulting in cell death (Van Melderen and Saavedra De Bast 2009). Consequently, plasmids containing the TA system (TA+) are able to outcompete plasmids devoid of the TA system (TA-), since a loss of the TA+ plasmid will lead to cell death (Van Melderen and Saavedra De Bast 2009). A similar function was described for some chromosome-encoded TA systems and termed programmed cell death (Arcus et al. 2011). In this situation, a population of cells is believed to benefit from suicide of a specific subpopulation by activation of the toxin (Yarmolinsky 1995; Jensen and Gerdes 1995). For example, the E. coli MazEF system (Aizenman et al. 1996) was shown to play a part in mediating programmed cell death under a variety of stress conditions, such as phage infection, high temperature, DNA damage, oxidative stress , and the presence of antibiotics (Hazan and Engelberg-Kulka 2004; Hazan et al. 2004; Sat et al. 2001). However, other studies have found that the MazF toxin improves survival of cells by inducing a static condition where cells are viable, but unable to proliferate during unfavourable conditions. This bacteriostatic effect (as opposed to bactericidal) is reversible and can be relieved upon expression of MazE antitoxin at any later time (Pedersen et al. 2002). TA systems have also been suggested to act as quality control elements at the post-translational level: Under conditions of limited growth, a cell can save energy by optimising its gene expression quality so only functional transcripts are expressed (Makarova et al. 2009). Both the E. coli RelBE and MazEF TA systems have been shown to inhibit translation by cleavage of mRNA during nutritional stress and could thus be thought of as quality control factors (Van Melderen and Saavedra De Bast 2009; Gerdes and Wagner 2007; Hayes 2003).
Finally, it has recently become clear that TA systems likely play a role in the formation of bacterial persister cells. Persister cells are a subset of a bacterial population that have entered a dormant state and remain viable after treatment with e.g. antibiotics (Dorr et al. 2010; Brantl and Jahn 2015; Wang and Wood 2011). Characteristic of persister cells is that they do not acquire inheritable resistance and revert to normal growing cells upon further culturing in absence of the selective pressure (Singh et al. 2009). The detailed mechanism of persister cell formation remains somewhat of an enigma, but it has been suggested that TA systems play an important role by down-regulating essential metabolic functions during stress, which could cause a dormant state (Wang and Wood 2011). The first TA system to be implicated in persister cell formation was the E. coli HipBA system (Keren et al. 2004), where overexpression of the HipA toxin can be shown to correlate with increased persister cell formation and, conversely, deletion of the entire HipBA locus leads to a decrease in persister cell formation. Since this discovery, numerous other TA systems have been associated with persister cell formation, including TisAB, MqsRA, RelBE, HigAB, MazEF, DinJ-YafQ, and YefM-YoeB (Kim and Wood 2010; Dorr et al. 2010; Wang and Wood 2011; Lewis 2010; Brantl and Jahn 2015).
14.2 Functional and Genetic Organisation of TA Systems
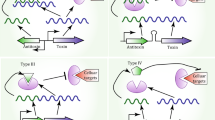
A TA system is defined by the presence of both a toxin and an antitoxin component, and the group has been subdivided into six distinct types (I-VI) based on the nature of the antitoxin, which can be either RNA (types I and III) or protein (types II, IV, V and VI), and the mode of action of the toxin, which is always protein (Fig. 14.1) (Unterholzner et al. 2013; Pandey and Gerdes 2005). By definition, the antitoxin blocks the cellular function of the toxin under normal growth conditions in what is called the inhibited state. The toxin and antitoxin are often encoded in a bicistronic locus, nearly always with the antitoxin preceding the toxin and under the control of a unique TA promoter. This genetic organisation presumably ensures a high antitoxin:toxin ratio and a solid inhibition of the toxin under normal growth conditions (Pandey and Gerdes 2005). Finally, the antitoxin often regulates the transcription from the TA operon by binding directly to the promoter region at a pseudo-palindromic site, which provides a means of auto-regulating TA levels in the cell for any given locus (see Fig. 14.1 for an overview). In the following, we will describe the characteristics of each type of TA system, highlighting in each case the regulation and function of the active toxin.

Overview of the six types of TA systems found in bacteria. In all cases, the toxin (T) and antitoxin (A) are expressed from a common promoter (P), but in some cases (type III) interrupted by a terminator (Tm). Type I. The T and A transcripts are partly complementary and base-pair in the inhibited state. Upon degradation of the A transcript, T is expressed and forms a membrane pore. Type II. Both T and A are expressed as proteins which form a tight complex in the inhibited state that regulates transcription through operator binding. Upon degradation of A, T is released and is active as an RNA endonuclease with specific cellular targets. Type III. The A transcript consists of a number of repeats and is expressed as RNA only. T is expressed as a protein which binds to the repeats of the A RNA in the inhibited state. The activated T is an RNase capable of degrading A. Type IV. Both T and A are expressed as proteins as in Type II, however, A functions by binding to the target of T in the inhibited state, thus preventing toxin action (red cross). Upon degradation of A, T becomes active and can bind its target. Type V. During the inhibited state, A is expressed as a protein that is able to cleave the T transcript. During stress, T is expressed and forms a membrane pore like in Type I. Type VI. Both T and A are expressed as proteins as in Type II, but A has the ability to guide T to the cellular protein degradation machinery, thus inactivating the toxin by promoting its degradation. When A is not active, T is not degraded and can exert its function as a toxin
Type I TA Systems
In type I TA systems, transcription of the antitoxin results in a small antisense RNA, which is complementary to a part of the toxin mRNA. Under normal growth conditions, the antitoxin transcript anneals to the toxin mRNA, resulting in a very stable dsRNA complex that is readily degraded, thus preventing expression of the toxin (Fig. 14.1, Type I) (Fozo et al. 2008a; Gerdes and Wagner 2007; Fozo et al. 2008b). The toxin protein product is often a small, hydrophobic polypeptide that folds into a single α-helix which locates to the inner cell membrane. The transmembrane helix then polymerises, forming a pore that destroys the membrane potential and inhibits ATP synthesis (Fozo et al. 2008a; Fozo et al. 2008b; Yamaguchi et al. 2011). The Hok-Sok system was the first type I TA system to be characterised, identified in 1986 in a screen for plasmid-stabilising genes (Gerdes et al. 1986a). The hok-sok locus contains three genes: hok, mok, and sok, with sok in the antisense direction preceding the hok open reading frame. Translation of the gene encoding the toxin, hok, results in production of a 52-residue transmembrane polypeptide that irreversibly damages the cell membrane through loss of the membrane potential. The mok open reading frame overlaps with both sok and hok and is required for translation of hok, while the sok gene specifies a small antisense RNA that blocks translation of mok, thereby indirectly inhibiting hok translation (Gerdes et al. 1986a; Thisted and Gerdes 1992). During normal growth, the sok antisense RNA, which is transcribed from a downstream promoter on the reverse strand, is expressed in excess over toxin mRNA and therefore inhibits its translation by formation of a dsRNA complex, which is rapidly degraded by RNase III (Gerdes et al. 1990; Gerdes et al. 1992). However, the sok transcript is unstable and when degraded, the hok mRNA is translated producing the toxin protein product. Several other type I loci have since been discovered, including rdlD-ldrA (Kawano et al. 2002), symR-symE (Kawano et al. 2007), and istR-tisB (Vogel et al. 2004). Due to their small size, type I TA systems are very difficult to identify and their distribution in the bacterial kingdom is therefore not known in detail.
Type II TA Systems
The type II TA systems are by far the most well-characterised and most abundant in prokaryotes (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007; Makarova et al. 2009). In type II systems, both toxin and antitoxin are proteins and form a tight complex under normal growth conditions thereby keeping the toxin inactive (Fig. 14.1, Type II). At the genomic level, the TA genes are arranged in a bicistronic operon, usually with the antitoxin gene preceding the toxin gene. An exception to this rule is the higAB and hicAB families, where the toxin precedes the antitoxin (Tian et al. 1996a; Jorgensen et al. 2009; Makarova et al. 2006). In operons where the antitoxin gene precedes the toxin gene, overlaps of 1–4 base pairs between the open reading frames of the antitoxin and toxin are common, reflecting a translational coupling that presumably helps secure a stable 1:1 expression ratio of the proteins (Pandey and Gerdes 2005). The operon is tightly controlled by negative auto-regulation of transcription via a DNA-binding domain on the antitoxin (Gerdes et al. 1986a), except for the ω-ε-ζ and paaR-paaA-parE families, which are three-component systems for which the additional ω (or PaaR, respectively) protein is the transcriptional repressor (de la Hoz et al. 2000; Hallez et al. 2010). The antitoxin binds to the promoter region of the operon either in complex with the toxin or alone, and both toxin and DNA binding stabilises the antitoxin, which is otherwise disordered and prone to proteolysis (Hayes 2003). The type II TA complex often binds to the promoter region more tightly than the isolated antitoxin, and in these cases the toxin is a co-repressor of its own transcription (Gerdes et al. 2005). During stress, the antitoxin is degraded by cellular proteases and the toxin is released and activated (Gerdes et al. 2005; Yamaguchi et al. 2011; Unterholzner et al. 2013; Tian et al. 1996b). The type II TA systems have been subdivided into families (VapBC, CcdAB, MazEF, Phd/Doc, ParDE, HigAB, RelBE, HipBA, HicAB and ω-ε-ζ) based on the mode of action of the toxin as well as conserved structural domains (Pandey and Gerdes 2005; Ogura and Hiraga 1983; Masuda et al. 1993; Lehnherr et al. 1993; Roberts et al. 1994; Tian et al. 1996b; Gotfredsen and Gerdes 1998; Black et al. 1991; de la Hoz et al. 2000; Jorgensen et al. 2009).
Type III TA Systems
For type III TA systems, the antitoxin gene is expressed as an RNA that directly binds and inhibits the toxin protein (Fig. 14.1, Type III). At the genomic level, the operon is bicistronic with the antitoxin gene, which consists of a tandem array of direct repeats, preceding the toxin gene. Two short, inverted repeats are located between the two genes where they function as transcriptional terminators to regulate the relative amounts of antitoxin and toxin RNA (Fineran et al. 2009). Binding of antitoxin RNA to the toxin protein results in the formation of a heterohexameric complex consisting of three toxin proteins and three antitoxin RNAs (Unterholzner et al. 2013). The free toxin is an active RNase targeting the bicistronic TA mRNA at the sites of the direct repeats, thus abolishing the function of the antitoxin (Blower et al. 2012). The first characterised type III system was ToxIN from the Pectobacterium carotovoum plasmid pECA1039 (Fineran et al. 2009). Recent studies have shown that the 125 type III TA systems identified to date can be assigned to three families based on sequence similarity, ToxIN, CptIN, and TenpIN (Blower et al. 2012).
Type IV TA Systems
For type IV TA systems, the antitoxin is a protein that suppresses the toxicity of the toxin by functioning as an antagonist that blocks the toxin’s target (Fig. 14.1, Type IV) (Masuda et al. 2012a). At the genomic level, the two proteins are expressed from a single operon where the antitoxin precedes the toxin. At the protein level, the antitoxin does not interfere with the toxin, but binds directly to the target of it, thereby preventing binding of the toxin. Only two type IV TA systems are known, E. coli yeeU-cbtA and Streptococcus agalactiae abiEi-abiEii (Masuda et al. 2012b; Dy et al. 2014). In the yeeU-cbtA system, the genes are separated by 89 base pairs in the bicistronic operon, which might play a role in protein expression. The toxin, CbtA, targets cell division and morphology by inhibiting polymerisation of the proteins MreB and FtsZ (Masuda et al. 2012a), homologues of the eukaryotic cytoskeleton proteins, actin and tubulin (van den Ent et al. 2001; Erickson 1997). The antitoxin, YeeU, acts as an antagonist of CbtA by binding MreB and FtsZ, enhancing their polymerisation, thus counteracting the effect of CbtA (Masuda et al. 2012a). In the abiEi-abiEii system, the two genes overlap by four base pairs at the 5′-end of the abiEii, reflecting translational coupling. The toxin, AbiEii, is a GTP-specific NTase that transfers a nucleotide to a yet unknown target, resulting in growth inhibition (Dy et al. 2014). The antitoxin, AbiEi, negatively auto-regulates transcription via binding to operator DNA and counteracts the toxicity of the toxin, probably by interfering with the toxin target. However, the exact mechanism of how AbiEi inhibits AbiEii is still unclear and the designation of the TA system as type IV is thus somewhat tentative (Dy et al. 2014).
Type V TA Systems
In type V TA systems, the antitoxin is a protein that masks the toxicity of the toxin by cleaving its mRNA (Fig. 14.1, Type V). To date only one type V TA system has been identified, namely E. coli ghoS-ghoT (Wang et al. 2012). At the genomic level, the antitoxin and the toxin are encoded by a single operon, again with the antitoxin preceding the toxin, and a distance of 27 base pairs between the open reading frames in the case of ghoS-ghoT. The protein products are the antitoxin GhoS and the toxin GhoT for which the antitoxin is a small protein with sequence-specific endoribonuclease activity related to the CAS2 CRISPR RNase family. GhoS directly targets the GhoT mRNA, thus preventing translation (Wang et al. 2012). Unusually, however, the GhoS antitoxin is stable and remains in the cell during stressful periods and moreover, the antitoxin does not regulate its own transcription through DNA binding (Wang et al. 2012). The toxin is a small (57 residues), hydrophobic protein with two transmembrane regions (Hofmann and Stoffel 1993) that causes persistence and ghost cell formation by disrupting the membrane, upon activation, hence the name (Wang et al. 2012).
Type VI TA Systems
A putative type VI TA system has been described relatively recently in which the antitoxin is a protein that promotes the degradation of the toxin (Fig. 14.1, Type VI) (Aakre et al. 2013; Markovski and Wickner 2013). To date, only one type VI TA system has been identified, namely Caulobacter crescentus socAB. At the genomic level, the two proteins are expressed from a single operon where the antitoxin precedes the toxin. At the protein level, the unstable toxin, SocB, is constitutively degraded by the ClpXP protease by a mechanism where the antitoxin, SocA, acts as a proteolytical adaptor by bringing the toxin to the protease. During stress, when the toxin protein is not degraded, it functions to disrupt DNA replication elongation by binding to the β-sliding clamp of DNA polymerase and thereby outcompeting the other clamp-binding proteins.
TA
TA loci, even of different types, tend to cluster in the bacterial genomes, probably reflecting a tendency towards horizontal gene transfer and intra-genomic recombination, and for this reason they are generally considered mobile genetic elements (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007). Genome-wide prediction of TA loci has further suggested that some bacteria contain toxin genes that are located in proximity to unrelated antitoxins (Gerdes et al. 2005; Hayes 2003). Furthermore, some toxins (including RelE, VapC, and MazF) have also been found as solitary genes, not accompanied by antitoxins (Pandey and Gerdes 2005). Some organisms, including major pathogens, have been found to contain a surprisingly large number of TA systems in their genomes (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007; Makarova et al. 2009). This is particularly true for Mycobacterium tuberculosis, which harbours at least 88 TA systems (Ramage et al. 2009), and E. coli K12, which has at least 33 known TA systems (Yamaguchi et al. 2011). On the other hand, there are also prokaryotes that have few or even no TA systems. For instance M. smegmatis, a non-pathogenic Mycobacterium species related to M. tuberculosis, only harbours three identified TA systems (Pandey and Gerdes 2005; Robson et al. 2009). It is still debated what the benefits of such a high number of TA systems are and several studies have investigated possible correlations between a large number of loci and various phenotypic features, such as life style, phylum, growth rate, cell shape, respiratory system, GC content, presence of one or several replicons, generation time, and genome size, but no correlation has been found yet (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007). As mentioned above, a link between TA loci and the ability to form persister cells, which are critical to pathogenicity, is currently being investigated. Presumably, a large number of TA loci would be particularly important for pathogenic species that have to survive in a challenging host-pathogen environment. It is possible that the plethora of toxins allow pathogenic bacteria to specifically target several cellular pathways at the same time and thus allow for a greater variety in adaption to stressful situations. Finally, it is worth noting that the cellular levels of both toxins and antitoxins are kept at stable steady-state levels during normal growth, which allows cells to very quickly respond to environmental changes through toxin activation, also under conditions where gene expression is slow or inhibited.
14.3 Structural Hierarchy of the VapBC TA Systems
The VapBC (Virulence associated proteins B and C) system constitutes the largest subgroup of the type II TA systems (Pandey and Gerdes 2005), and vapBC loci are found in surprisingly large numbers in some prokaryotes and archaea. For example, M. tuberculosis contains at least 47 vapBC loci and these loci have also been found in large numbers in most hyper thermophilic archaea (Pandey and Gerdes 2005; Sevin and Barloy-Hubler 2007). The specific, biological advantage of having so many homologous loci is presently unknown, but it is possible that it allows organisms to deliver a very fine-grained response towards external stress conditions, like antibiotic pressure or the immune system of a host organism in the case of human pathogens. The vapBC operon is an archetypical type II TA system, and as such bicistronic at the genomic level, with the vapB antitoxin gene preceding the vapC toxin gene, allowing expression levels of the antitoxin to exceed those of the toxin (Gerdes et al. 2005) (Fig. 14.1, Type II). During normal growth conditions, the VapC and VapB proteins form a tight protein complex , in which the VapB antitoxin wraps its C-terminal tail around and inhibits VapC (Gerdes et al. 2005). This inhibited VapBC complex negatively auto-regulates transcription of the operon by binding directly to the promoter region using a DNA-binding domain located in the N-terminus of VapB (Gerdes et al. 2005; Wilbur et al. 2005; Robson et al. 2009). During cellular stress, the labile and highly flexible VapB antitoxin is degraded, releasing the active VapC. Due to the high prevalence of vapBC loci in pathogenic bacteria and the prospects of understanding human infectious diseases at the molecular level, a lot of research has recently gone into deciphering the molecular architecture and mechanisms of the VapBC TA systems. To date, crystal structures of seven distinct VapBC-type complexes have been determined, and together these highlight the great structural diversity of the family (Mattison et al. 2006; Miallau et al. 2009; Dienemann et al. 2011; Mate et al. 2012; Min et al. 2012; Das et al. 2014; Lee et al. 2015) (Table 14.1).
The VapC Toxin
All VapC toxins belong to the conserved PIN (PilT N-terminal domain) domain family, which is a relatively small (app. 130 residues), compact domain with bona fide Mg2+/Mn2+-dependent endoribonuclease activity (Arcus et al. 2011; Levin et al. 2004). PIN domains are found in all domains of life and are involved in cleavage of a range of specific RNA targets in both sequence and structure specific fashions (Arcus et al. 2011). However, in prokaryotes, the vast majority of PIN domains are associated with TA systems (Arcus et al. 2011). While the overall PIN domain fold is highly conserved there is surprisingly low overall primary sequence conservation and alignments are most precise when carried out structurally with tools like PROMALS3D (Pei and Grishin 2014). There are currently 11 different structures of prokaryotic and archaeal PIN domains in the PDB that have been functionally classified as VapC toxins, many of which have been determined in complex with their cognate VapB antitoxin. These structures include A. fulgidus VapC-9 (DAF0591) (Levin et al. 2004), M. tuberculosis VapC-3, VapC-5, VapC-15, and VapC-30 (Min et al. 2012; Lee et al. 2015; Das et al. 2014; Miallau et al. 2009), N. gonorrhoeae FitB, a VapC homologue (Mattison et al. 2006), P. aerophilum VapC-3 and Vap-C9 (Bunker et al. 2008; Arcus et al. 2004), P. horikoshii VapC-4 (Jeyakanthan et al. 2005), R. felis VapC2 (Mate et al. 2012), and a VapC from S. flexneri virulence plasmid pMYSH6000 (Dienemann et al. 2011). In addition to these, the PDB currently contains two structures of homologous proteins from A. fulgidus (AF1683, PDB ID 1W8I) and P. furiosus (Pfu-367848-001, PDB ID 1Y82) with no associated publications, which are not considered here due to their putative assignment as VapC toxins. Structural alignment of the 11 VapC proteins using PROMALS3D reveals that the sequence identity varies between 10 and 36% among the structurally characterised VapC toxins, with the R. felis and S. flexneri orthologues being the most similar proteins (Fig. 14.2). When grouping amino acids based on similar functionality (F, Y, W = aromatic; V, I, L = aliphatic; R, K, H = charged positive; D, E = charged negative; S, T = alcohol, and N, Q = polar), the VapC orthologues are between 23 and 61% similar, again with the R. felis and S. flexneri toxins as the most closely related proteins. Most PIN domain proteins are active as Mg2+/Mn2+-dependent endoribonucleases and share between three and five conserved, acidic residues that are required for metal ion binding and consequently RNase activity. Of these, three (D-E-D) are universally conserved (Fig. 14.2, dashed boxes) and these residues can be used as a signature sequence for locating PIN domain proteins, which can otherwise be difficult to identify. For bacterial TA systems, the genetic organisation with the antitoxin preceding the PIN domain toxin is also a strong constraint for locating such loci (Pandey and Gerdes 2005).

Structural alignment of bacterial and archaeal VapC toxins. Structural alignment of M. tuberculosis VapC-15 (PDB ID 4CHG) (Das et al. 2014), VapC-5 (PDB ID 3DBO) (Miallau et al. 2009), VapC-30 (PDB ID 4XGQ) (Lee et al. 2015), and VapC-3 (PDB ID 3H87) (Min et al. 2012), N. gonorrhoeae FitB (PDB ID 1H1O) (Mattison et al. 2006), S. flexneri pMYSH6000 VapC (PDB ID 3TND) (Dienemann et al. 2011), R. felis VapC2 (PDB ID 3ZVK) (Mate et al. 2012), P. aerophilum VapC-9 (PDB ID 1V8P) (Arcus et al. 2004) and VapC-3 (PDB ID 2FE1) (Bunker et al. 2008), A. fulgidus VapC-9 (PDB ID 1O4W) (Levin et al. 2004), and P. horikoshii VapC-5 (PDB ID 1 V96) (Jeyakanthan et al. 2005) coloured by conservation from low (purple) to high (red) as shown by the inset colour bar. Secondary structure elements are shown and annotated and the five conserved active site residues are indicated (1st through 5th)
The PIN Domain
The PIN domain fold itself consists of a α/β/α sandwich core with alternating α-helices and β-strands that organise the conserved acidic residues in close proximity in the active site. In this chapter, we will use a standard nomenclature to denote the architecture of toxins, antitoxins, and their complexes: TnAm, where n is the number of toxin (T) molecules and m the number of antitoxin (A) molecules in any given complex. Using this nomenclature, the toxin monomer can be referred to as T1 and its core PIN domain fold is shown in Fig. 14.3. Accumulating evidence indicates that VapC PIN domain RNases are able to target specific RNAs in bacterial cells, including unique tRNA and rRNA species (Winther et al. 2013; Winther and Gerdes 2011; Cruz et al. 2015; Sharp et al. 2012). However, there are yet no structures of PIN domains bound to their cognate RNA targets that would allow us to establish a detailed molecular basis of recognition. The mechanism of cleavage, on the other hand, is likely related to T4 RNase H, which is a nuclease specific for RNA-DNA duplexes (Xu et al. 2016). As such structures exhibit a conformation similar to A-form RNA they may be quite similar to the structured RNA targets of the PIN domains. An intriguing observation is that even though each PIN domain appears to contain a complete active site, all structures of bacterial and archaeal VapCs determined so far have shown the protein as a homodimer. This is unlikely to be a result of crystallisation as several of the VapC toxins display the same behaviour in solution as judged by gel filtration chromatography (Dienemann et al. 2011; Xu et al. 2013; Xu et al. 2016). The VapC dimer is formed by tight packing of the third α-helix (α3, Fig. 14.2), the β-strand following (β3) and the penultimate α-helix (α5), and sometimes the fourth and ultimate α-helices (α4 and α6). Although there are typically both a number of strong hydrogen bonds as well as hydrophobic interactions between the chains, dimerisation appears to be driven largely by shape complementarity between the two PIN domains folds. This mode of dimerisation is observed among all structures of bacterial and archaeal VapC proteins determined to date, either as a direct dimer in the crystallographic asymmetric unit or in a few instances, as the results of a crystallographic two-fold symmetry (such as in the cases of M. tuberculosis VapC-5, PDB ID 3DBO (Miallau et al. 2009) and P. aerophilum VapC-3, PDB ID 2FE1 (Bunker et al. 2008)). The only departure from this rule is the putative VapC homologue from A. fulgidus (AF1683, PDB ID 1W8I), for which the dimer is similar, but skewed compared to the canonical structures. Since there are presently no structures available of bacterial PIN domains bound to their cognate RNA targets, it is not known if the dimer represents the active conformation of the toxin or somehow mimics the higher-order structure of the antitoxin-inhibited state (see below). Nevertheless, two active sites are placed in relative proximity in the T2 dimer (Fig. 14.3, T2, red spheres), so if the dimer does indeed represent the active conformation of the toxin it would lead to a whole new range of questions concerning substrate specificity and activity such as whether both active sites are engaged simultaneously in RNA cleavage or both bind RNA, but only one is catalytically active.

Hierarchical architecture of TA systems. Overview of the hierarchical structure of the TA systems. The figure uses the TnAm nomenclature to indicate the complex architecture, where n is the number of toxin (T) molecules and m the number of antitoxin (A) molecules in the complex. The small schematics illustrate the overall architecture of each complex, with green and blue spheres representing toxin molecules, red and orange the antitoxin DNA-binding domain (rounded squares) and C-terminal extension (lines), and grey lines representing DNA. T 1 , Fig. 14.3 (continued) the isolated toxin structure showing the PIN domain fold (PDB ID 5ECW) in green cartoon (Xu et al. 2016). Acidic residues in the active site are shown in sticks and the expected positions of the bound Mn2+ and Mg2+ ions are shown with purple and green spheres, respectively, according to their position in M. tuberculosis VapC-15 (4CHG) (Das et al. 2014). T 2 , the VapC toxin dimer (blue and green cartoons) as observed in nearly all crystal structures with residues interacting at the interface (sticks) and the location of the two active sites (red circles) shown (PDB ID 5ECW) (Xu et al. 2016). T 2 A 2 , two orthogonal views of the heterotetramer formed in the absence of the antitoxin DNA-binding domains. Colouring as above with the antitoxin molecules in red and orange (PDB ID 3DBO) (Miallau et al. 2009). T 4 A 4 , the heterooctameric structure formed upon dimerisation of the antitoxin DNA-binding domains (red and orange) (PDB ID 3TND) (Dienemann et al. 2011). T 4 A 4 -DNA, interaction of the heterooctamer with duplex DNA at the operator binding site (PDB ID 3ZVK) (Mate et al. 2012). Organisms abbreviations: Ngo, Neisseria gonorrhoeae; Sfl, Shigella flexneri; Rfe, Rickettsia felis; Mtu, Mycobacterium tuberculosis. All structure figures in this chapter were created with PyMol (Schrodinger 2010)
The VapB Antitoxin
The VapB antitoxin serves two main purposes in VapBC TA complexes, (1) it inhibits the activity of the toxin by binding directly to its active site, and (2) it carries a DNA-binding domain that promotes binding of the TA complex to operator DNA and confers auto-inhibition of transcription from the TA locus. These functions are maintained by two distinct domains in the antitoxins, an N-terminal DNA-binding domain and a C-terminal intrinsically disordered region. VapB toxins are typically around 80 amino acids in length, of which the first approx. 50 dimerise to form a DNA-binding domain while the last approx. 30 amino acids contain a flexible “tail” that wraps around the toxin and physically inhibits it by directly binding the active site. Four types of DNA-binding domains have been identified so far in VapB toxins, namely the AbrB-type, the ribbon-helix-helix (RHH) domain, the helix-turn-helix (HTH), and the Phd/YefM-type domain (Gerdes et al. 2005). The VapB C-terminal tail is intrinsically disordered in the isolated antitoxin and prone to proteolysis, and presumably only forms correct secondary structure upon binding to its cognate toxin molecule. Consequently, all VapB structures have been determined in the context of the VapBC TA complex, except in a few cases, such as isolated DNA-binding domains (R. equi VapB, PDB ID 4CV7 (Geerds et al. 2014)) or putative VapB antitoxins (M. tuberculosis VapB-49, Rv2018, PDB ID 5AF3). As for the VapBC complexes, there are currently seven unique structures available in the PDB (Table 14.1). In all these cases the flexible C-terminal domain of the VapB antitoxin is visible. The N-terminal DNA binding domain is visible in some of these structures, but has also been removed in others (Mattison et al. 2006; Miallau et al. 2009; Dienemann et al. 2011; Mate et al. 2012; Min et al. 2012; Das et al. 2014; Lee et al. 2015).
The Mechanism of VapB Inhibition
A conserved feature of VapBC TA systems (and type II TA systems in general) is that the antitoxin blocks the cellular function of the toxin by formation of a tight protein complex . This function is maintained by the flexible C-terminal domain of VapB which contains functional residues that appear to be widely conserved within the VapBC family (Dienemann et al. 2011). The seven available VapBC structures reveal that the C-terminal domain of the VapB proteins most often consists of an α-helix followed by an extended tail with no secondary structure that wraps around the toxin (Fig. 14.3, T2A2). Exceptions to this include M. tuberculosis VapB-3, which has of a loop followed by an α-helix (Min et al. 2012) and M. tuberculosis VapB-5 (Miallau et al. 2009) and VapB-15 (Das et al. 2014), which both contain two α-helices. The interaction of VapB with VapC can be separated into two sites, which we term the lateral and groove sites (Fig. 14.3, T2A2). The first site, the lateral site, occurs on the side of the TA complex and usually involves an α-helix of the antitoxin that makes hydrophobic interactions with residues of nearby α-helices (α2 and α4) of the toxin, as well as a few specific hydrogen bonds involving both side chains and protein backbone of both partners (Mattison et al. 2006; Dienemann et al. 2011; Mate et al. 2012; Lee et al. 2015) (Fig. 14.3, T2A2, and Fig. 14.5d). The remaining, extended tail of the antitoxin is located in a large continuous groove formed by the dimerisation of the toxins on the “top” of the complex, which also contains the two actives sites of the T2 dimer. Outside the active sites, this groove is lined with hydrophobic residues from the core of the protein as well as some charged residues (Mate et al. 2012; Min et al. 2012). The tail of the antitoxin runs along the groove, where hydrophobic residues from both proteins interact with each other, tightly anchoring the antitoxin to the surface of the toxin (Fig. 14.3, T2A2, and Fig. 14.5d). Although the antitoxin tails show a large degree of variability, both with respect to length and amino acid content, some of these interactions appear to be conserved across the VapBC family (Dienemann et al. 2011).
14.4 Overall TA Complex Architecture
Despite the relatively small size and conserved folds of their components, intact TA complexes display a surprising structural and architectural variety. In all VapBC and isolated VapC structures determined to date, the VapC toxin exhibits the conserved PIN domain structure, which appears as a rigid domain with only minor conformational changes observed between the VapB-bound and free toxin states (Xu et al. 2016). The structural complexity of TA pairs therefore appears to be largely due to the presence of one or more VapB antitoxin molecules, which contain both flexible regions and a variable DNA-binding domain. Since both toxins and antitoxins form homodimers, most intact VapBC complexes have a heterooctameric T4A4 architecture that can be described as (TA-AT)2 or in other words, homodimers of homodimers of heterodimers (Fig. 14.3, T4A4). This complex presents two antitoxin DNA-binding homodimers (A2) on one side of the complex, which are able to interact with two adjacent major grooves on operator DNA (Fig. 14.3, T4A4-DNA). There are presently seven unique VapBC-type TA structures in the PDB, which include N. gonorrhoeae (N. gonorrhoeae) FitAB (a VapBC orthologue) (Mattison et al. 2006), S. flexneri (S. flexneri) pMYSH6000 VapBC (Dienemann et al. 2011), R. felis (R. felis) VapBC2 (Mate et al. 2012), and four complexes from M. tuberculosis (M. tuberculosis), VapBC-3 (Min et al. 2012), VapBC-5 (Miallau et al. 2009), VapBC-30 (Lee et al. 2015), and VapBC-15 (Das et al. 2014) (Fig. 14.4). Two of these structures, N. gonorrhoeae FitAB and R. felis VapBC2 where determined as bound to their cognate operator DNA sequence, but not in isolation. Since much structural variation is observed between various VapBC orthologues this means that we still do not know exactly whether DNA binding induces conformational changes in the TA complex. R. felis VapBC2 was reported to form a heterohexamer in solution, however, this result is based on size exclusion chromatography making it somewhat uncertain (Mate et al. 2012). In fact, the most commonly observed stoichiometry of TA complexes in absence of DNA is a heterooctamer with four antitoxin molecules and four toxin molecules in a T4A4 architecture (Table 14.1 and Fig. 14.4, N. gonorrhoeae FitAB, S. flexneri VapBC, R. felis VapBC2, and M. tuberculosis VapBC-3) (Mattison et al. 2006; Dienemann et al. 2011; Mate et al. 2012; Min et al. 2012; Lee et al. 2015). M. tuberculosis VapBC-30 was also reported as a heterooctamer, however, the interaction between T2A2 heterotetramers in the crystallographic asymmetric unit is weak and not likely to be physiological (Fig. 14.4e) (Lee et al. 2015). Likewise, M. tuberculosis VapBC-5 was reported as a heterodimer, but appears more likely to form a heterotetrameric assembly in the crystal (Fig. 14.4g) (Miallau et al. 2009). Adding to this complexity, the structure of M. tuberculosis VapBC-15 contains both a heterotrimeric T2A1 and a heterotetrameric T2A2 complex in the crystallographic asymmetric unit (Fig. 14.4f). Finally, even for identical stoichiometries, large differences are observed in complex organisation, such as N. gonorrhoeae FitAB, for example, which displays a very loose arrangement of domains, whereas most of the other structures adopt a much more compact arrangement (Fig. 14.4). Due to these discrepancies and since apparent oligomeric state can be affected by crystal packing interactions, we decided to reinvestigate all known VapBC (and homologous) complex structures using the protein interaction server PISA to assess the extent and strength of reported and observed homodimer and heterotetramer interaction interfaces (Krissinel and Henrick 2007). Tables 14.1 and 14.2 provide an overview of all crystal structures of VapBC-type TnAm complexes, along with their respective PDB codes and their overall architecture, as predicted by PISA analysis. Most correspond well with what was reported in the original papers, except M. tuberculosis VapBC-5 and M. tuberculosis VapBC-30, which differ from the reported as mentioned above. Such characteristics become evident when investigating the heterotetramer interaction areas and energies (ΔG interaction ) listed in Table 14.2 (T2A2:T2A2). We find that stable heterooctamers typically display interface sizes in the 3–4000 Å2 range, while it for M. tuberculosis VapBC-30, for example, is only 542 Å2 suggesting this complex is more likely on heterotetrameric form. Likewise, ΔG interaction values tend to lie in the −20 to −40 kcal/mol range for heterooctamers while for VapBC-30 we get a value of only −9.5 kcal/mol. The other columns of the table list the corresponding values for the TA-interactions (“T2A (high)” and “T2A (low)”, see below), and the toxin (T:T) and antitoxin (A:A) homodimers. It is interesting to note that toxin and antitoxin dimerisation take place using comparable interaction areas and interaction energies, possibly reflecting that they are both equally important for formation of the heterooctamer during DNA-binding. Values for the antitoxin dimerisation are missing for those structures where the DNA-binding domain was left out of the structure or not visible (M. tuberculosis VapBC-5, VapBC-15, and VapBC-30).

VapBC higher-order complex structure. Overview of known VapBC complex structures shown as cartoon with toxins in green and antitoxins in orange. (a) N. gonorrhoeae FitAB heterooctamer bound to DNA (PDB ID 2H1O) (Mattison et al. 2006). (b) S. flexneri pMYSH6000 VapBC heterooctamer (PDB ID 3TND) (Dienemann et al. 2011). (c) R. felis VapBC2 heterooctamer bound to DNA (PDB ID 3ZVK) (Mate et al. 2012). (d) M. tuberculosis VapBC-3 heterooctamer (PDB ID 3H87) (Min et al. 2012). (e) M. tuberculosis VapBC-30 heterotetramer (PDB ID 4XGR) (Lee et al. 2015). (f) M. tuberculosis VapBC-15 T2A1 heterotrimer (left) and heterotetramer (right) (PDB ID 4CHG) (Das et al. 2014). (g) M. tuberculosis VapBC-5 heterotetramer (PDB ID 3DBO) (Miallau et al. 2009)
14.5 The Stoichiometry of VapB Inhibition
In the canonical VapBC complexes, each VapC toxin is inhibited by a single VapB antitoxin, which wraps around it and inhibits its active site by directly interfering with the conserved, charged residues (see Active site inhibition below). Both VapC and VapB form homodimers giving rise to a circular (AT-TA)2 structure, which is most clearly observed for N. gonorrhoeae FitAB due to the distance between the heterotetramers (Fig. 14.4a). In the most common case, the VapB antitoxin interferes with the active site of its nearby or cognate VapC toxin in a binding mode, which we term 1:1 proximal binding. This mode of inhibition has been observed in the structures of N. gonorrhoeae FitAB, S. flexneri VapBC, and M. tuberculosis VapBC-5 (Table 14.1) (Mattison et al. 2006; Miallau et al. 2009; Dienemann et al. 2011). Only about ten residues of VapB (typically residues 60–70 out of about 80) are required for the interaction at the groove site, where the antitoxin polypeptide has a fully extended conformation (Fig. 14.5a). This leaves about ten residues in the C-terminus of VapB unaccounted for, which are presumed to be pointing into solution as the last few visible residues tend to point away from VapC (e.g. in S. flexneri VapBC) (Dienemann et al. 2011). However, as first observed in the structure of R. felis VapBC2, these residues can also in some instances continue across the combined groove of the VapC homodimer and block the adjacent active site, thus one antitoxin interferes with two VapC active sites, a phenomenon that has been termed 1:2 binding of antitoxin to toxin (Fig. 14.5b and Table 14.1) (Mate et al. 2012). Consequently, the VapB tail associated with the other VapC of the toxin dimer in the canonical interaction must detach and in such cases only interacts with VapC at the lateral site. This results in two distinct modes of toxin-antitoxin interaction in a single, two-fold symmetrical heterooctamer, which we term T 2 A (high) and T 2 A (low), corresponding to high affinity (VapB bound at the lateral site and across the groove sites of both toxins) and low affinity (VapB bound exclusively to the lateral site), respectively. This distinction becomes especially clear when looking at the interaction areas and energies as listed in Table 14.2, where complexes displaying 1:2 binding display larger interaction areas for the high affinity interaction (e.g. R. felis VapBC2) as well as a much larger difference between the values for the high and low affinity sites. M. tuberculosis VapBC-15 is an interesting case as the crystal contains both a T2A2 heterotetramer and a T2A1 heterotrimer in the asymmetric unit for which the heterotetramer displays 1:1 binding and the heterotrimer 1:2 binding. Based solely on energetics, we cannot say whether the observed 1:2 interaction in the trimer is a result of the missing VapC molecule or vice versa, that the VapC molecule is missing due to the 1:2 interaction, as the T2:A energies are comparable to the T:T energies for this complex. It is also not known at present whether there are any significant, biological consequences of the ability of VapB to bind two VapC molecules simultaneously, nor whether it interaction is affected by (or affects) DNA binding by the TA complex. Another variation in VapBC toxin-antitoxin interaction has been observed for M. tuberculosis VapBC-3 and VapBC-30, which both on the surface display 1:1 binding in that the VapB tails only cover the nearby VapC toxin groove. However, closer inspection of the structures revealed that the very last part of the antitoxin that is structurally organised actually reaches across in both cases and interferes with the active site of the adjacent VapC rather than the cognate one to which it is bound. We term the resulting crossed-over or swapped inactivation mode 1:1 distal binding (Fig. 14.5c and Table 14.1) (Min et al. 2012; Lee et al. 2015).

Inhibition modes of the VapB antitoxin. (a) 1:1 inhibition of the proximal toxin as observed in the S. flexneri pMYSH6000 VapBC complex (PDB ID 3TND) (Dienemann et al. 2011). (b) 1:2 inhibition of the toxin dimer as observed in the R. felis VapBC2 complex (PDB ID 3ZVK) (Mate et al. 2012). (c) 1:1 inhibition of the distal toxin in the M. tuberculosis VapBC-3 complex (PDB ID 3H87) (Min et al. 2012). (d) Overview and details of the toxin-antitoxin interactions at the “lateral” and “groove” sites exemplified by the S. flexneri VapBC complex (PDB ID 3TND) (Dienemann et al. 2011). Interactions at the groove site (upper inset) are both hydrophilic at the active site (top) and hydrophobic (bottom). The surface of VapC is shown coloured by hydrophobicity, with white representing the most hydrophobic areas. Interactions at the lateral site (lower inset) are primarily hydrophobic
14.6 Active Site Inhibition
The VapC active sites are located in two negatively charged cavities along a continuous groove on the toxin homodimer surface. In the inhibited state, electrostatic and hydrogen bond interactions between specific residues of the antitoxin and the toxin active site(s) effectively prevent binding of potential substrates (Table 14.1 and Fig. 14.6). In N. gonorrhoeae FitAB and M. tuberculosis VapBC-5, which both exhibit the canonical 1:1 proximal binding, and VapBC-3, in which the distal VapB is involved, a positively charged arginine residue from the antitoxin points directly into the active site, where its guanidinium group interacts electrostatically with the carboxyl groups of several of the conserved acidic residues of the toxin (Fig. 14.6a) (Mattison et al. 2006; Miallau et al. 2009; Min et al. 2012). In addition, in M. tuberculosis VapBC-3, a bulky tryptophan side chain from the antitoxin buries itself in the crevice between VapC monomers, which runs perpendicular to the groove, thus anchoring the antitoxin across to the next monomer (Fig. 14.6e) (Min et al. 2012). In the S. flexneri VapBC complex, which also displays 1:1 proximal binding, both an arginine (Arg64) and a glutamine residue (Gln66) from VapB interferes with the active site where they interact with the conserved acidic residues via electrostatic and hydrogen bond interactions (Fig. 14.6b) (Dienemann et al. 2011). Intriguingly, the VapB interactions differ between the four VapC active sites in the R. felis VapBC2 structure, which displays 1:2 binding as described above, where one antitoxin straddles across both VapC molecules of the toxin homodimer. In one VapC dimer of the heterooctamer, two VapB arginine residues (Arg66/Arg74) block the two active sites through electrostatic interactions with the conserved acidic residues, and this is also the case for one VapC of the other homodimer (Arg66). However, in the last active site, the Arg has shifted away and a tyrosine residue (Tyr76) appears to have taken its place (Mate et al. 2012) (Fig. 14.6d). In the M. tuberculosis VapBC-15 structure, several configurations of the active site were also observed in that two active sites were found to bind divalent metal ions (one Mg2+ and one Mn2+) while the two other sites were empty, despite all VapC toxins being in complex with antitoxin. In the active site containing bound ions, a glutamate residue (Glu67) from the antitoxin interacts with both ions (Fig. 14.6c), whereas in the active site without divalent cations, in analogy with the previously described pattern, an arginine residue (Arg74) and a lysine residue (Lys75) point into the active site where they interact with the conserved, acidic residues (Das et al. 2014). As if to highlight the great diversity in VapB inhibition modes, no direct interactions take place between the conserved, acidic residues in active site and the C-terminal tail of VapB in M. tuberculosis VapBC-30. Rather, several, likely specific hydrogen bonds are formed between residues close to the active site of VapC and VapB (Fig. 14.6f). In this case, the TA complex is further stabilised by a few hydrophobic interactions between the C-terminal tails of two VapB antitoxins (Lee et al. 2015).

Details of VapB-VapC active site interactions. (a) In N. gonorrhoeae FitA, Arg68 inhibits the FitB active site through electrostatic interactions and possibly displacement of divalent metal ions (PDB ID 2H1O) (Mattison et al. 2006). (b) In S. flexneri, VapB inhibits the Fig. 14.6 (continued) VapC active site using both Arg64 and Gln66 (PDB ID 3TND) (Dienemann et al. 2011). (c) In M. tuberculosis VapBC-15, VapB-15 inhibits the active site of VapC-15 using a single glutamate Glu67 (PDB ID 4CHG) (Das et al. 2014). (d) In R. felis VapBC2, VapB2 inhibits the active site of VapC2 by placing Tyr76 in the active site (PDB ID 3ZVK) (Mate et al. 2012). (e) In M. tuberculosis VapBC-3, Trp72 of the antitoxin locates in the toxin dimer interface (PDB ID 3H87) (Min et al. 2012). (f) Inhibition of the active site of M. tuberculosis VapC-30 (PDB ID 4XGQ) (Das et al. 2014). Toxins are shown as green and teal cartoons and antitoxins are in orange and red. Relevant residues are shown as sticks
14.7 DNA Binding by VapBC Complexes
All characterised type II TA systems, including VapBC, display auto-regulation of transcription through direct binding of the TA complex to operator DNA. As described above, this is achieved through presentation of the two DNA-binding antitoxin dimers on one side of the TA heterooctamer, where they are in a position to interact with two adjacent major grooves on DNA (Fig. 14.3, T4A4-DNA) (Gerdes et al. 2005). Interestingly, VapB antitoxins contain one of at least four different types of DNA binding domains, namely the helix-turn-helix (HTH) motif, the ribbon-helix-helix (RHH) motif, the Phd/YefM domain, or the AbrB domain (Table 14.1) (Gerdes et al. 2005). This supports the general notion that TA systems have evolved through active gene shuffling by swapping modules required for DNA binding and transcriptional regulation . Apart from the VapBC complex, the VapB antitoxin can also bind to the promoter region on its own, but biochemical analyses have shown that the TA complex binds with much higher affinity than the antitoxin alone due to the stabilisation conferred by effective cross-linking of VapB dimers by VapC in the heterooctamer. Consequently, the toxin can therefore be regarded as a co-repressor of its own transcription (Wilbur et al. 2005; Gerdes et al. 2005). Only two structures of VapBC complexes bound to their promoter region exist to date, namely the N. gonorrhoeae FitAB (PDB IDs 2BSQ, 2H1O, and 2H1C) (Mattison et al. 2006; Wilbur et al. 2005) and R. felis VapBC2 (PDB ID 3ZVK) complexes (Mattison et al. 2006; Mate et al. 2012). Furthermore, despite DNA being absent, the VapB DNA-binding domains are present in what appears to be approximately the correct orientation in both the S. flexneri VapBC (PDB ID 3TND) and M. tuberculosis VapB-3 (PDB ID 3H87) heterooctamer structures (Dienemann et al. 2011; Min et al. 2012).
Among the known VapBC structures, two of the four types of DNA binding domains are represented; the RHH motif, which is found in N. gonorrhoeae FitA and M. tuberculosis VapB-3 (Mattison et al. 2006; Min et al. 2012), and the AbrB motif, found in S. flexneri VapB and R. felis VapB2 (Dienemann et al. 2011; Mate et al. 2012). The RHH domain was originally identified in the phage P22 Arc repressor and consists of a β/α motif with a β-strand followed by two α-helices (Raumann et al. 1994b, 1994a). The DNA-binding unit is a homodimer of the β/α motif and thus has an antiparallel β-sheet surrounded by four α-helices (Somers and Phillips 1992). Of these, the β-sheet inserts into the DNA major groove and is responsible for specific sequence recognition, whereas the α-helices primarily are responsible for dimerisation (Somers and Phillips 1992; Raumann et al. 1994a). The transcription of many RHH-containing proteins is found to be auto-regulated by the protein itself through binding to pseudo-palindromic, inverted repeats in the promoter region (Wilbur et al. 2005). The N. gonorrhoeae FitAB antitoxin, FitA, belongs to the RHH family (Wilbur et al. 2005) and consequently adopts the conserved β/α motif (Fig. 14.7a) (Mattison et al. 2006). As for other type II TA systems, the FitAB complex was found to bind to its promoter DNA with higher affinity than FitA alone due to the formation of the heterooctameric superstructure, which results in increased stability (Wilbur et al. 2005). Two RHH DNA-binding domains were observed in the crystal structure of FitAB bound to a 36 base pair cognate operator DNA fragment (Fig. 14.7b) (Mattison et al. 2006). The two RHH domains in the heterooctamer bind to two adjacent inverted repeat sequences on operator DNA. The domains bind on the same side of the DNA and make few specific contacts, and those there are, are all mediated through the β-sheet. The two binding sites on DNA are separated by a 14 base pair thymine/adenine-rich sequence, which adopts a straight and rigid structure with a compressed minor groove and shorter helical repeat compared to standard B-DNA (Mattison et al. 2006). In the crystal structure of M. tuberculosis VapBC-3, the VapB-3 antitoxin was also found to contain a RHH motif and although DNA was not present in this crystal form, two complete DNA-binding domains were found to protrude from the heterooctameric structure (Fig. 14.7c) (Min et al. 2012).

DNA binding by VapBC complexes. (a) The ribbon-helix-helix DNA binding domain of N. gonorrhoeae FitA (PDB ID 2H1O). (b). The N. gonorrhoeae FitAB complex forms two DNA binding sites. (c) The ribbon-helix-helix DNA binding domain of M. tuberculosis VapB-3 (PDB ID 3H87). (d) The AbrB DNA binding domain of R. felis VapB2 (PDB ID 3ZVK). (e) The R. felis VapBC2 complex forms two DNA binding sites. (f) The AbrB DNA binding domain of Shigella flexneri VapB (PDB ID 3TND). For (a), (b) and (c) toxins are coloured green and teal, and antitoxins are coloured red and pink. For (d), (e) and (f), toxins are coloured green and teal, and antitoxins are coloured orange and sand
The AbrB-type DNA binding domain was first identified in B. subtilis as a transition-state regulator that binds promoter DNA via its N-terminal domain (Xu and Strauch 2001; Strauch et al. 1989). It consists of a β/α/β motif with two β-strands forming a β-hairpin followed by a short α-helix and two more β-strands, which form another hairpin. The two hairpins interleave each other in the dimer, and curve upwards to form a deep DNA binding cleft (Coles et al. 2005). Like seen for the RHH domain, dimerisation is important for stability and binding to DNA (Xu and Strauch 2001; Sullivan et al. 2008). Adjacent to the β-sheet, which locates in the DNA major groove, the two short α-helices interact with DNA on the rim next to the major groove (Sullivan et al. 2008). Four arginine residues located throughout the protein sequence appear to be critical for the ability to bind DNA, which involves both electrostatic and hydrogen bond interactions (Sullivan et al. 2008). Like the RHH domain, the AbrB domain binds to pseudo-palindromic thymine/adenine-rich sequences in promoter regions. For TA operator sequences, this feature represents a hallmark (Klein and Marahiel 2002; Bailey and Hayes 2009). The R. felis VapB2 antitoxin contains an AbrB-type motif in its N-terminal domain that dimerises with a neighbouring VapB monomer using the β-strands to form a complete DNA binding domain. However, the VapB DNA-binding domains do not contain the short α-helices seen in the original AbrB domain. Due to the heterooctameric VapBC architecture, there are two DNA binding sites in the intact structure (Fig. 14.7d, e) (Mate et al. 2012). Sequence analysis identified two potential pseudo-palindromic binding sites in the promoter region of the R. felis vapBC2 operon that subsequently led to the design of a 27 base pair pseudo-palindromic dsDNA sequence that could be stably bound to the protein. The crystal structure of R. felis VapBC2 bound to its cognate operator DNA revealed that binding of the protein complex in two adjacent major grooves results in perturbation of the widths of both minor and major grooves, with major grooves spanning 15.5 Å, somewhat expanded compared to normal B-DNA (11.7 Å) (Neidle 2002). Inside the DNA, the R. felis VapB2 AbrB domain interacts with the concave surface of the major groove floor, but the two domains in the heterooctamer approach the DNA double helix from opposite sides. Contacts to the backbone are thought to open up the major groove where there are electrostatic interactions with the phosphates of the DNA backbone (Mate et al. 2012). DNA is specifically recognised in two ways, both through direct base readout, involving direct hydrogen bonds between protein and specific atoms of the nucleobases of DNA, and indirect readout, where the affinity is based on shape complementarity between the AbrB domain and the DNA major groove (Mate et al. 2012). S. flexneri VapB from the pMYSH6000 virulence plasmid VapBC complex also contains an AbrB-type motif in its N-terminus, but the only available structure was determined in absence of nucleic acids (Dienemann et al. 2011). The S. flexneri VapB displays the classical swapped β-hairpin fold but lacks the two short α-helices like seen for R. felis VapBC2 (Fig. 14.7f). The S. flexneri VapBC complex was shown biochemically to bind specifically to two binding sites within the promoter region of the pMYSH6000 plasmid from where it is expressed, of the operon but the DNA-bound structure has not yet been determined (Dienemann et al. 2011).
14.8 Discussion
Despite their relatively small size, bacterial toxin-antitoxin complexes display an impressive array of large, higher-order structures. Through dimerisation of both the toxin and antitoxin components; dimers, tetramers and even octamers are frequently observed and these structures are known to be central for maintaining steady cellular TA levels during normal growth. Although some TA complexes have been reported as heterotetramers, structural and energetic considerations reveal that this conclusion is usually either due to the DNA-binding domain being absent from the antitoxin or incomplete analysis of crystal packing. Therefore, for the type II TA complexes as exemplified here by the VapBC group, the canonical architecture is a heterooctamer consisting of four toxin and four antitoxin molecules. The heterooctamer is obviously required for placing two DNA-binding motifs into adjacent major grooves, so the question arises of whether the TA complexes are heterotetramers off DNA and only assemble when bound to the operator region? Several lines of evidence speak against the free TA complex as being present on tetramer form. Firstly, the interaction energies involved in toxin-toxin and antitoxin-antitoxin dimerisation are comparable (Table 14.2) and secondly, all intact VapBC structures determined so far, i.e. in presence of the DNA-binding domain, show the complex on heterooctameric form. Together these data suggest that the heterooctamer exists in solution and approaches the DNA operator as such.
Intriguingly, several types of DNA-binding domains are observed in type II TA systems, the HTH motif, the RHH motif, the Phd/YefM domain, and the AbrB domain (Table 14.1) (Gerdes et al. 2005). Two of these, the RHH and AbrB domains, are observed among the available VapBC structures. Likewise, several types of antitoxin inhibition, including 1:1 proximal, 1:1 distal, and 1:2 have been described for members of the VapBC family. This has led to speculations as to whether there is a correlation between the type of DNA-binding domain and the mode of inhibition, for example, it has been proposed that the AbrB-type DNA-binding domain would correlate with 1:2 binding as observed in the R. felis VapBC-2 structure (Mate et al. 2012). However, such a simple correlation does not seem to be the case as the structure of the S. flexneri VapB also has the AbrB domain but interacts with VapC via 1:1 proximal binding (Table 14.1). Furthermore, there is some evidence that the toxin-antitoxin interaction mode might be flexible and vary depending on the cellular context of the complex. For example, in M. tuberculosis VapBC-3, an example of 1:1 distal binding, both antitoxins lie in the continuous groove formed by the dimerization of the toxins and appear to physically clash in the crystal structure near their termini. The authors therefore modelled the structure with static main chain disorder at this location reflecting a situation whereby the VapB tail can be either in one or the other conformation at any given time (Min et al. 2012). Currently, no VapBC structures are available both on and off DNA, so it is possible that some of this inherent flexibility relates to DNA binding.
When wrapping around their cognate VapC toxins, the VapB antitoxins in most cases place one or more charged residues in the active site, presumably to inactive the toxin. As described in this chapter, both positively charged residues (arginine and lysine) and negatively charged residues (glutamate) have been observed. The VapC PIN domain is known to function as divalent ion-depending endonucleases when activated, but usually no metal ions are observed in the inhibited, VapB-bound state. This suggests that the charged residues, in particular arginine and lysine residues, may function to displace ions from the active site by compensating for the loss of positive charge. The involvement of a glutamate residue is much more debatable and somewhat enigmatic. In the structure of M. tuberculosis VapBC-15 structure, a glutamate residue from VapB-15 was found in one of four active sites, which also contained two metal ions. This suggests that VapB inactivation functions by a charge-reversal principle, in which the presence of ions dictates the charge of the residue required for inactivation, or vice versa. However, neither the extent of this phenomenon, not its functional and regulatory consequences are known at present. However, the observations suggest that active site inhibition of VapC toxins by VapB antitoxins follows a complex and multipronged approach, in which both acidic and basic residues can take part. It is likely that in those cases where acidic residues of VapB are used, that these compete with the RNA substrate for binding to the active site. It is not known whether the metal ions are present in the VapC active sites in the absence of RNA, but if that is the case, it is likely that interaction with the positive charged arginine or lysine would physically displace the ions (Dienemann et al. 2011).
References
Aakre CD, Phung TN, Huang D, Laub MT (2013) A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol Cell 52(5):617–628. doi:10.1016/j.molcel.2013.10.014
Aizenman E, Engelberg-Kulka H, Glaser G (1996) An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci U S A 93(12):6059–6063
Arcus VL, Backbro K, Roos A, Daniel EL, Baker EN (2004) Distant structural homology leads to the functional characterization of an archaeal PIN domain as an exonuclease. J Biol Chem 279(16):16471–16478. doi:10.1074/jbc.M313833200
Arcus VL, McKenzie JL, Robson J, Cook GM (2011) The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng Des Sel 24(1–2):33–40. doi:10.1093/protein/gzq081
Bailey SE, Hayes F (2009) Influence of operator site geometry on transcriptional control by the YefM-YoeB toxin-antitoxin complex. J Bacteriol 191(3):762–772. doi:10.1128/JB.01331-08
Black DS, Kelly AJ, Mardis MJ, Moyed HS (1991) Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 173(18):5732–5739
Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, Luisi BF, Salmond GP (2012) Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res 40(13):6158–6173. doi:10.1093/nar/gks231
Brantl S, Jahn N (2015) sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol Rev 39(3):413–427. doi:10.1093/femsre/fuv003
Bunker RD, McKenzie JL, Baker EN, Arcus VL (2008) Crystal structure of PAE0151 from Pyrobaculum aerophilum, a PIN-domain (VapC) protein from a toxin-antitoxin operon. Proteins 72(1):510–518. doi:10.1002/prot.22048
Coles M, Djuranovic S, Soding J, Frickey T, Koretke K, Truffault V, Martin J, Lupas AN (2005) AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi beta barrels. Structure 13(6):919–928. doi:10.1016/j.str.2005.03.017
Cruz JW, Sharp JD, Hoffer ED, Maehigashi T, Vvedenskaya IO, Konkimalla A, Husson RN, Nickels BE, Dunham CM, Woychik NA (2015) Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat Commun 6:7480. doi:10.1038/ncomms8480
Das U, Pogenberg V, Subhramanyam UK, Wilmanns M, Gourinath S, Srinivasan A (2014) Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J Struct Biol 188(3):249–258. doi:10.1016/j.jsb.2014.10.002
de la Hoz AB, Ayora S, Sitkiewicz I, Fernandez S, Pankiewicz R, Alonso JC, Ceglowski P (2000) Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc Natl Acad Sci U S A 97(2):728–733
Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE (2011) Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J Mol Biol 414(5):713–722. doi:10.1016/j.jmb.2011.10.024
Dorr T, Vulic M, Lewis K (2010) Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8(2):e1000317. doi:10.1371/journal.pbio.1000317
Dy RL, Przybilski R, Semeijn K, Salmond GP, Fineran PC (2014) A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res 42(7):4590–4605. doi:10.1093/nar/gkt1419
Erickson HP (1997) FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol 7(9):362–367. doi:10.1016/S0962-8924(97)01108-2
Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP (2009) The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 106(3):894–899. doi:10.1073/pnas.0808832106
Fozo EM, Hemm MR, Storz G (2008a) Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev 72(4):579–589. Table of Contents. doi:10.1128/MMBR.00025–08
Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, Jones KL, Ocampo A, Rudd KE, Storz G (2008b) Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 70(5):1076–1093. doi:10.1111/j.1365-2958.2008.06394.x
Geerds C, Wohlmann J, Haas A, Niemann HH (2014) Structure of Rhodococcus equi virulence-associated protein B (VapB) reveals an eight-stranded antiparallel beta-barrel consisting of two Greek-key motifs. Acta Crystallogr F Struct Biol Commun 70(Pt 7):866–871. doi:10.1107/S2053230X14009911
Gerdes K (2000) Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J Bacteriol 182(3):561–572
Gerdes K, Wagner EG (2007) RNA antitoxins. Curr Opin Microbiol 10(2):117–124. doi:10.1016/j.mib.2007.03.003
Gerdes K, Bech FW, Jorgensen ST, Lobner-Olesen A, Rasmussen PB, Atlung T, Boe L, Karlstrom O, Molin S, von Meyenburg K (1986a) Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J 5(8):2023–2029
Gerdes K, Rasmussen PB, Molin S (1986b) Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci U S A 83(10):3116–3120
Gerdes K, Thisted T, Martinussen J (1990) Mechanism of post-segregational killing by the hok/sok system of plasmid R1: sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol Microbiol 4(11):1807–1818
Gerdes K, Nielsen A, Thorsted P, Wagner EG (1992) Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable hok, srnB and pndA effector messenger RNAs. J Mol Biol 226(3):637–649
Gerdes K, Christensen SK, Lobner-Olesen A (2005) Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3(5):371–382. doi:10.1038/nrmicro1147
Gotfredsen M, Gerdes K (1998) The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol Microbiol 29(4):1065–1076
Hallez R, Geeraerts D, Sterckx Y, Mine N, Loris R, Van Melderen L (2010) New toxins homologous to ParE belonging to three-component toxin-antitoxin systems in Escherichia coli O157:H7. Mol Microbiol 76(3):719–732. doi:10.1111/j.1365-2958.2010.07129.x
Hayes F (2003) Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301(5639):1496–1499. doi:10.1126/science.1088157
Hazan R, Engelberg-Kulka H (2004) Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics 272(2):227–234. doi:10.1007/s00438-004-1048-y
Hazan R, Sat B, Engelberg-Kulka H (2004) Escherichia coli mazEF-mediated cell death is triggered by various stressful conditions. J Bacteriol 186(11):3663–3669. doi:10.1128/JB.186.11.3663-3669.2004
Hofmann K, Stoffel W (1993) TMBASE – a database of membrane spanning protein segments. Biol Chem 374(166)
Jaffe A, Ogura T, Hiraga S (1985) Effects of the ccd function of the F plasmid on bacterial growth. J Bacteriol 163(3):841–849
Jensen RB, Gerdes K (1995) Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol Microbiol 17(2):205–210
Jeyakanthan J, Inagaki E, Kuroishi C, Tahirov TH (2005) Structure of PIN-domain protein PH0500 from Pyrococcus horikoshii. Acta Crystallogr Sect F Struct Biol Cryst Commun 61(Pt 5):463–468. doi:10.1107/S1744309105012406
Jorgensen MG, Pandey DP, Jaskolska M, Gerdes K (2009) HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J Bacteriol 191(4):1191–1199. doi:10.1128/JB.01013-08
Kawano M, Oshima T, Kasai H, Mori H (2002) Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol Microbiol 45(2):333–349
Kawano M, Aravind L, Storz G (2007) An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64(3):738–754. doi:10.1111/j.1365-2958.2007.05688.x
Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186(24):8172–8180. doi:10.1128/JB.186.24.8172-8180.2004
Kim Y, Wood TK (2010) Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem Biophys Res Commun 391(1):209–213. doi:10.1016/j.bbrc.2009.11.033
Klein W, Marahiel MA (2002) Structure-function relationship and regulation of two Bacillus subtilis DNA-binding proteins, HBsu and AbrB. J Mol Microbiol Biotechnol 4(3):323–329
Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372(3):774–797. doi:10.1016/j.jmb.2007.05.022
Lee IG, Lee SJ, Chae S, Lee KY, Kim JH, Lee BJ (2015) Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: implications for the design of novel antimicrobial peptides. Nucleic Acids Res 43(15):7624–7637. doi:10.1093/nar/gkv689
Lehnherr H, Maguin E, Jafri S, Yarmolinsky MB (1993) Plasmid addiction genes of bacteriophage P1: doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J Mol Biol 233(3):414–428. doi:10.1006/jmbi.1993.1521
Levin I, Schwarzenbacher R, Page R, Abdubek P, Ambing E, Biorac T, Brinen LS, Campbell J, Canaves JM, Chiu HJ, Dai X, Deacon AM, Di Donato M, Elsliger MA, Floyd R, Godzik A, Grittini C, Grzechnik SK, Hampton E, Jaroszewski L, Karlak C, Klock HE, Koesema E, Kovarik JS, Kreusch A, Kuhn P, Lesley SA, McMullan D, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Quijano K, Reyes R, Rezezadeh F, Robb A, Sims E, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, von Delft F, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA (2004) Crystal structure of a PIN (PilT N-terminus) domain (AF0591) from Archaeoglobus fulgidus at 1.90 A resolution. Proteins 56(2):404–408. doi:10.1002/prot.20090
Lewis K (2010) Persister cells. Annu Rev Microbiol 64:357–372. doi:10.1146/annurev.micro.112408.134306
Magnuson RD (2007) Hypothetical functions of toxin-antitoxin systems. J Bacteriol 189(17):6089–6092. doi:10.1128/JB.00958-07
Makarova KS, Grishin NV, Koonin EV (2006) The HicAB cassette, a putative novel, RNA-targeting toxin-antitoxin system in archaea and bacteria. Bioinformatics 22(21):2581–2584. doi:10.1093/bioinformatics/btl418
Makarova KS, Wolf YI, Koonin EV (2009) Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 4:19. doi:10.1186/1745-6150-4-19
Markovski M, Wickner S (2013) Preventing bacterial suicide: a novel toxin-antitoxin strategy. Mol Cell 52(5):611–612. doi:10.1016/j.molcel.2013.11.018
Masuda Y, Miyakawa K, Nishimura Y, Ohtsubo E (1993) chpA and chpB, Escherichia coli chromosomal homologs of the pem locus responsible for stable maintenance of plasmid R100. J Bacteriol 175(21):6850–6856
Masuda H, Tan Q, Awano N, Wu KP, Inouye M (2012a) YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol Microbiol 84(5):979–989. doi:10.1111/j.1365-2958.2012.08068.x
Masuda H, Tan Q, Awano N, Yamaguchi Y, Inouye M (2012b) A novel membrane-bound toxin for cell division, CptA (YgfX), inhibits polymerization of cytoskeleton proteins, FtsZ and MreB, in Escherichia coli. FEMS Microbiol Lett 328(2):174–181. doi:10.1111/j.1574-6968.2012.02496.x
Mate MJ, Vincentelli R, Foos N, Raoult D, Cambillau C, Ortiz-Lombardia M (2012) Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res 40(7):3245–3258. doi:10.1093/nar/gkr1167
Mattison K, Wilbur JS, So M, Brennan RG (2006) Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J Biol Chem 281(49):37942–37951. doi:10.1074/jbc.M605198200
Miallau L, Faller M, Chiang J, Arbing M, Guo F, Cascio D, Eisenberg D (2009) Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J Biol Chem 284(1):276–283. doi:10.1074/jbc.M805061200
Min AB, Miallau L, Sawaya MR, Habel J, Cascio D, Eisenberg D (2012) The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg(2)(+) ion in the active site and a putative RNA-binding site. Protein Sci 21(11):1754–1767. doi:10.1002/pro.2161
Neidle S (2002) Nucleic acid structure and recognition, 1st edn. Oxford University Press, Oxford
Ogata H, Renesto P, Audic S, Robert C, Blanc G, Fournier PE, Parinello H, Claverie JM, Raoult D (2005) The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol 3(8):e248. doi:10.1371/journal.pbio.0030248
Ogura T, Hiraga S (1983) Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 80(15):4784–4788
Pandey DP, Gerdes K (2005) Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 33(3):966–976. doi:10.1093/nar/gki201
Pedersen K, Christensen SK, Gerdes K (2002) Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol Microbiol 45(2):501–510
Pei J, Grishin NV (2014) PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol Biol 1079:263–271. doi:10.1007/978-1-62703-646-7_17
Ramage HR, Connolly LE, Cox JS (2009) Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet 5(12):e1000767. doi:10.1371/journal.pgen.1000767
Raumann BE, Brown BM, Sauer RT (1994a) Major groove DNA recognition by β-sheets: the ribbon-helix-helix family of gene regulatory proteins. Curr Opin Struct Biol 4(1):36–43. doi:10.1016/s0959-440x(94)90057-4
Raumann BE, Rould MA, Pabo CO, Sauer RT (1994b) DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature 367(6465):754–757. doi:10.1038/367754a0
Roberts RC, Strom AR, Helinski DR (1994) The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J Mol Biol 237(1):35–51. doi:10.1006/jmbi.1994.1207
Robson J, McKenzie JL, Cursons R, Cook GM, Arcus VL (2009) The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J Mol Biol 390(3):353–367. doi:10.1016/j.jmb.2009.05.006
Sat B, Hazan R, Fisher T, Khaner H, Glaser G, Engelberg-Kulka H (2001) Programmed cell death in Escherichia coli: some antibiotics can trigger mazEF lethality. J Bacteriol 183(6):2041–2045. doi:10.1128/JB.183.6.2041-2045.2001
Sat B, Reches M, Engelberg-Kulka H (2003) The Escherichia coli mazEF suicide module mediates thymineless death. J Bacteriol 185(6):1803–1807
Schrodinger, LLC (2010) The PyMOL molecular graphics system, version 1.3r1
Sevin EW, Barloy-Hubler F (2007) RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol 8(8):R155. doi:10.1186/gb-2007-8-8-r155
Sharp JD, Cruz JW, Raman S, Inouye M, Husson RN, Woychik NA (2012) Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. J Biol Chem 287(16):12835–12847. doi:10.1074/jbc.M112.340109
Singh R, Ray P, Das A, Sharma M (2009) Role of persisters and small-colony variants in antibiotic resistance of planktonic and biofilm-associated Staphylococcus aureus: an in vitro study. J Med Microbiol 58(Pt 8):1067–1073. doi:10.1099/jmm.0.009720-0
Somers WS, Phillips SE (1992) Crystal structure of the met repressor-operator complex at 2.8 A resolution reveals DNA recognition by beta-strands. Nature 359(6394):387–393. doi:10.1038/359387a0
Strauch MA, Spiegelman GB, Perego M, Johnson WC, Burbulys D, Hoch JA (1989) The transition state transcription regulator abrB of Bacillus subtilis is a DNA binding protein. EMBO J 8(5):1615–1621
Sullivan DM, Bobay BG, Kojetin DJ, Thompson RJ, Rance M, Strauch MA, Cavanagh J (2008) Insights into the nature of DNA binding of AbrB-like transcription factors. Structure 16(11):1702–1713. doi:10.1016/j.str.2008.08.014
Thisted T, Gerdes K (1992) Mechanism of post-segregational killing by the hok/sok system of plasmid R1. Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. J Mol Biol 223(1):41–54
Tian QB, Hayashi T, Murata T, Terawaki Y (1996a) Gene product identification and promoter analysis of hig locus of plasmid Rts1. Biochem Biophys Res Commun 225(2):679–684. doi:10.1006/bbrc.1996.1229
Tian QB, Ohnishi M, Tabuchi A, Terawaki Y (1996b) A new plasmid-encoded proteic killer gene system: cloning, sequencing, and analyzing hig locus of plasmid Rts1. Biochem Biophys Res Commun 220(2):280–284. doi:10.1006/bbrc.1996.0396
Unterholzner SJ, Poppenberger B, Rozhon W (2013) Toxin-antitoxin systems: biology, identification, and application. Mob Genet Elements 3(5):e26219. doi:10.4161/mge.26219
van den Ent F, Amos LA, Lowe J (2001) Prokaryotic origin of the actin cytoskeleton. Nature 413(6851):39–44. doi:10.1038/35092500
Van Melderen L, Saavedra De Bast M (2009) Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet 5(3):e1000437. doi:10.1371/journal.pgen.1000437
Vogel J, Argaman L, Wagner EG, Altuvia S (2004) The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 14(24):2271–2276. doi:10.1016/j.cub.2004.12.003
Wang X, Wood TK (2011) Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl Environ Microbiol 77(16):5577–5583. doi:10.1128/AEM.05068-11
Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK (2012) A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat Chem Biol 8(10):855–861. doi:10.1038/nchembio.1062
Wilbur JS, Chivers PT, Mattison K, Potter L, Brennan RG, So M (2005) Neisseria gonorrhoeae FitA interacts with FitB to bind DNA through its ribbon-helix-helix motif. Biochemistry 44(37):12515–12524. doi:10.1021/bi0511080
Winther KS, Gerdes K (2011) Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci U S A 108(18):7403–7407. doi:10.1073/pnas.1019587108
Winther KS, Brodersen DE, Brown AK, Gerdes K (2013) VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat Commun 4:2796. doi:10.1038/ncomms3796
Xu K, Strauch MA (2001) DNA-binding activity of amino-terminal domains of the Bacillus subtilis AbrB protein. J Bacteriol 183(13):4094–4098. doi:10.1128/JB.183.13.4094-4098.2001
Xu K, Dedic E, Cob-Cantal P, Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE (2013) Protein expression, crystallization and preliminary X-ray crystallographic analysis of the isolated Shigella flexneri VapC toxin. Acta Crystallogr Sect F Struct Biol Cryst Commun 69(Pt 7):762–765. doi:10.1107/S1744309113014012
Xu K, Dedic E, Brodersen DE (2016) Structural analysis on the active site architecture of the VapC toxin from Shigella flexneri. Proteins. doi:10.1002/prot.25002
Yamaguchi Y, Park JH, Inouye M (2011) Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet 45:61–79. doi:10.1146/annurev-genet-110410-132412
Yarmolinsky MB (1995) Programmed cell death in bacterial populations. Science 267(5199):836–837
Acknowledgements
We are thankful to Kristoffer Winther and Kenn Gerdes for insightful comments and critical reading of the manuscript. This work was supported by Lundbeck Foundation (grant no. R173-2014-1182) and the Danish National Research Foundation Centre for Bacterial Stress Response and Persistence (DNRF120).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bendtsen, K.L., Brodersen, D.E. (2017). Higher-Order Structure in Bacterial VapBC Toxin-Antitoxin Complexes. In: Harris, J., Marles-Wright, J. (eds) Macromolecular Protein Complexes. Subcellular Biochemistry, vol 83. Springer, Cham. https://doi.org/10.1007/978-3-319-46503-6_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-46503-6_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46501-2
Online ISBN: 978-3-319-46503-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)