
Abstract
Clinical diagnostic pathways in patients presenting with acute/subacute or chronic para- or tetraparesis routinely include imaging workup (MRI, CT, myelography) to identify spinal cord compression and cerebrospinal fluid (CSF) analysis to identify inflammatory/infectious causes of spinal cord injury. Once these diagnostic procedures have been performed without a clear hint toward the etiology of spinal cord disease, other causes have to be considered. Metabolic, toxic, hereditary (hereditary spastic paraplegia, HSP), and other rare causes, which in most instances do not present as acute onset paraparesis/tetraparesis, are likely candidates to explain the clinical phenotype. In this chapter the clinical presentation of respective entities, the specific diagnostic workup, therapy, and prognosis will be discussed. Other rare causes such as epidural lipomatosis, flexion myelopathy, and conversion (dissociative) paraplegia will be presented as well.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Methylene Blue
- Cytosine Arabinoside
- Hereditary Spastic Paraplegia
- Autosomal Recessive
- Autosomal Dominant
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The first step toward a successful identification of the specific etiology is to recognize clinical symptoms, which indicate the presence of spinal cord disease. In case a patient presents with bilateral sensorimotor dysfunction in combination with autonomic dysfunction (most relevant in the acute situation is neurogenic bladder dysfunction), spinal cord disease has to be considered unless it can be excluded based on additional exams such as imaging (spine CT/MRI), CSF analysis, and neurophysiology.
In case spinal cord compression can be excluded, CSF is unremarkable, and the spinal cord parenchyma does not show detectable structural changes in a properly conducted spinal MRI, diagnosis becomes more difficult. As almost always, thorough evaluation of the past medical history may give hints in respect to the correct diagnosis. In particular in cases of subacute or slowly progressive diseases, it might become particularly difficult to link the clinical presentation with a specific disease-promoting process.
Overall there appears to be a distinct “core” clinical pattern, which is typical, however not specific, for metabolic versus toxic versus hereditary causes of spinal cord disease. Of course, in each disease category, there are additional symptoms, which go beyond this “core” phenotype. In metabolic causes, which are dominated by cobalamin and copper deficiency, the dorsal columns are predominantly affected with related sensory disturbances characterized as spinal ataxia. In addition, metabolic diseases are frequently associated with polyneuropathies. Systemically incorporated toxic substances, which target specifically the spinal cord, are rare. In this category only recreational drugs are discussed, which can present clinically in a rather variable fashion. Much more uniform is the clinical picture of accidentally or intentionally intrathecally administered substances. Patients develop a mostly subacutely progressing and ascending para- and tetraplegia, which is heavily focused on the motor system, more precisely on the lower motoneuron. In addition, autonomic function is frequently affected with relative sparing of the sensory system. Finally, hereditary spinal cord disease represented by hereditary spastic paraplegia (HSP) is characterized by slowly progressive upper motoneuron degeneration with a slowly ascending spastic paraplegia. Here, the signs of autonomic and sensory dysfunction are usually mild to moderate.
2 Metabolic Causes
2.1 Subacute Combined Degeneration
2.1.1 Vitamin B12 (Cobalamin Deficiency)
Pathophysiology
Adenosylcobalamin is required as a cofactor for the conversion of methylmalonyl coenzyme A (CoA) to succinyl CoA. Lack of adenosylcobalamin may lead to accumulation of methylmalonyl CoA, causing a decrease in normal myelin synthesis [1]. Causes are atrophic gastritis with consecutive cobalamin malabsorption, gastric surgery, acid reduction therapy, parasitic infestation by fish tapeworm, hereditary enzymatic defect, and rarely strict vegetarianism. Often exact causes cannot be identified.
Clinical Presentation
Macrocytic anemia is the most common clinical finding. The macrocytosis may precede the anemia by months. Symptoms related to myelopathy are as follows: gait disturbance, hypesthesia, dysesthesia, impairment of vibration and position sense. Moreover, autonomic dysfunction represented by constipation, erectile dysfunction, and urinary frequency have been described. Tendon reflexes can be hyperactive with pyramidal signs; affection of the motor system presents with a typically mild para- or tetraparesis. Facultative findings are cognitive impairment/dementia [2]. Often an inverse relationship between hematologic and neurological abnormalities is observed [3].
Diagnostics
Analysis of serum cobalamin represents the most widely used screening test. However, sensitivity and specificity are poor. Serum methylmalonic acid and plasma total homocysteine (methylmalonic acid more specific, homocysteine more sensitive) are employed as ancillary tests. Serum methylmalonic acid and plasma total homocysteine are good monitoring tools and should be measured annually.
To determine the causes of cobalamin deficiency means to determine the causes of malabsorption. In this respect the so-called Schilling test is considered obsolete. Elevated serum gastrin and decreased pepsinogen have a limited specificity, whereas positive anti-intrinsic factor antibodies are more specific [3].
With spinal MRI hyperintense signal changes in the posterior and lateral columns with facultative contrast enhancement, and spinal cord atrophy can be observed [4, 5]. In some instances anterior columns show similar signal changes. Neurophysiologically, tibial nerve sensory evoked potentials (SEPs) are found frequently to be abnormal, whereas delayed P37 responses are observed in median nerve SEP. Motor evoked potentials (MEPs) are abnormal only in a subset of patients showing prolonged central motor conduction times [2]. Cobalamin deficiency can promote concomitant or even exclusive neuropathic changes in peripheral nerves. Respective nerve conduction and electromyogramm (EMG)-studies indicate an axonal polyneuropathy [6]; however, abnormal findings are not specific for cobalamin deficiency-related polyneuropathy.
Therapy
For immediate effectiveness cobalamin (1000 μg cobalamin) should be administered initially intramuscularly. Thereafter, 8–10 injections over 3 months followed by monthly injections are recommended. Since 1.2 % of any oral dose of cobalamin is absorbed unrelated to intrinsic factor, 1000 μg of oral cobalamin is also effective for substitution in cases of malabsorption. As a first treatment effect within 1 week after substitution, reticulocytosis can be detected [3]. Mean corpuscular volume (MCV) normalizes within 8 weeks after cobalamin substitution (once daily for 4 weeks, then once weekly for 1 year, then once per month) [2]. Neurological symptoms start to improve as early as 1 week and continue to improve until 3 months after cobalamin substitution. In parallel, nerve conduction velocities and median nerve SEP normalize. Signal changes in the spinal cord detected with MRI may disappear over time.
2.1.2 Copper Deficiency
Pathophysiology
Balanced oral food intake covers the required copper intake. Copper is absorbed in the stomach and duodenum. There, zinc and iron can inhibit copper absorption. Following uptake copper becomes transported in the bloodstream via ceruloplasmin and subsequently integrated into a variety of key enzymes such as oxidoreductases and monooxygenases. Ceruloplasmin, which has not bound copper, becomes rapidly degraded. Acquired copper deficiency (serum copper levels below 0.1 μg/ml) is typically caused by upper gastrointestinal surgery or zinc overload. Gastrointestinal surgery can be divided into non-bariatric surgery (mostly partial gastrectomy for peptic ulcer disease) and bariatric interventions (weight loss surgery) [7]. Malabsorption of copper can also be caused by celiac disease. More likely excessive zinc intake competes with copper uptake in the gut and thus reduces copper serum levels. Zinc overload is typically induced by zinc-containing denture creams [8] or zinc supplementation, e.g., to prevent or treat common colds. Rarely, a lack of appropriate intake represents the single cause of copper deficiency.
Clinical Presentation
The interval between upper gastrointestinal surgery and first symptoms ranges between 5 and 46 years. Clinical symptoms such as spastic gait and sensory ataxia related to a myelopathy or myeloneuropathy develop subacutely. Cases of combined copper and cobalamin deficiency have been described [9].
Diagnostics
Anemia, leukopenia, and ringed sideroblasts are common hematological manifestations. Similar to subacute combined degeneration, T2 MRI shows hyperintense signal changes in the dorsal and lateral columns. Besides abnormalities in SEPs, nerve conduction studies and EMG may reveal axonal loss in peripheral nerves. Serum copper and ceruloplasmin are found decreased. In case of a non-compressive myelopathy of unknown origin and related clinical symptoms, serum copper and ceruloplasmin levels should be analyzed.
Therapy
If possible, the underlying cause for copper deficiency needs to be treated. For example, excessive zinc uptake due to denture creams or oral zinc supplementation needs to be stopped. Oral supplementation of 8 mg of elemental copper per day is sufficient to restore copper storages. For obvious reasons care should be taken to avoid combinatorial preparations of copper and zinc. In case copper serum level and hematological findings do not normalize, parenteral copper substitution may be required. Neurological symptoms will improve invariably [9].
2.1.3 Hepatic Myelopathy
Pathophysiology
As opposed to hepatic myelopathy, hepatic encephalopathy is a well-recognized neurological disease entity, which has been linked pathophysiologically to portacaval shunts with consecutive increase of nitrogenous products such as ammonia [10]. However, blood ammonia-lowering therapies, which are effective in hepatic encephalopathy, have not been shown to improve signs of hepatic myelopathy. Nutritional factors such as cobalamin deficiency may be relevant and, however, have not been positively confirmed as underlying cause. Structural changes, which have been obtained from postmortem analysis, include demyelination of pyramidal tracts with only occasional affection of the dorsal columns. Axonal loss is rather mild if present at all.
Clinical Presentation
Approximately 90 cases are described in the literature. Patients typically present with subacute onset of spastic paraparesis showing a slow progression over several years. Sensory and autonomous deficits are in most instances minimal. In the majority of cases, hepatic encephalopathy precedes the manifestation of myelopathy.
Diagnostics
Confirmation of myelopathy by means of ancillary exams is difficult. Imaging – in particular MRI – usually does not show any abnormalities along the spinal cord. Likewise CSF analysis does not provide any relevant clues. Therefore, the proper diagnosis can only be confirmed by excluding all relevant differential diagnosis of non-compressive spinal cord disease. Neurophysiological analysis (evoked potentials) reveals prolonged latencies or even absent motor or sensory responses, which are of course not specific for hepatic myelopathy.
Therapy
As mentioned above blood ammonia-lowering therapies do not ameliorate symptoms or the disease course of hepatic myelopathy. Single cases suggest that occlusion of a splenorenal shunt and liver transplantation promote neurological improvement in a hepatic myelopathy patient [11, 12].
3 Toxic Causes
3.1 Nitrous Oxide (N2O)
N2O inactivates cobalamin through irreversible oxidation of the cobalt core. Exposure to N2O (laughing gas), which is commonly used as inhalational anesthetic agent or abused for its euphoriant properties, can lead to an acute myelopathy, typically in elderly patients with an unrecognized cobalamin deficiency [13, 14]. Myelopathy can develop already after single exposure to N2O. Besides myelopathy neuropathies and mental status changes have been described. MRI in typical cases shows T2 signal changes in the dorsal and lateral columns [15]. Routine cobalamin and MCV testing is advised before surgical procedures involving N2O exposure in the elderly are performed. In case of low levels, cobalamin should be substituted prophylactically.
3.2 Recreational Drugs (Heroin, Ecstasy, Cocaine)
Pathophysiology
Heroin and cocaine are the main recreationally used drugs, which have been described to cause myelopathy with subsequent sensorimotor and autonomous dysfunction. In case of heroin, it is still unclear which pathomechanisms are responsible for the observed myelopathy. Direct neurotoxicity, hypotension, vasculitis, and a hypersensitivity reaction have been discussed as underlying mechanisms. Hypersensitivity has been favored in some cases, since onset of disease followed a heroin abstinent period with subsequent i.v. or intranasal insufflation of heroin [16]. In another study, clinical and neurophysiological evaluation indicated selective damage to spinal ventral horn motoneurons suggesting a neurotoxic mechanism [17]. Ecstasy has been described only in a single case report, where the drug was inhaled together with heroin. Therefore, it is unknown whether ecstasy contributed at all to the observed myelopathy [17]. In terms of cocaine, ischemia is the likely underlying mechanism in cases of myelopathy paralleling the likely pathomechanism for cerebral lesions [18].
Clinical Presentation
In a number of heroin cases, symptoms developed after drug administration and a subsequent symptom-free interval of several hours. Thereafter, flaccid paraplegia with urinary retention and diminished rectal tone has been described [16, 17]. Cocaine use related to myelopathy has been reported within several hours after drug inhalation. The myelopathy is confined to the ventral portion of the cervical spinal cord presenting similar to central cord syndrome with pronounced upper extremity weakness and rather moderate involvement of the lower extremities [18]. In case of myelopathy confined to the dorsal columns, paresthesias and variable degrees of deep sensation loss may represent the only clinical signs [19]. Neurological recovery over time is variable.
Diagnostics
Depending on the clinical presentation, spinal MRI indicates the affected rostrocaudal and cross-sectional extent of the lesion. Electrophysiological workups (EMG, motor nerve conduction studies) are recommended to confirm lower motoneuron involvement.
Therapy
Besides rehabilitative interventions, no specific treatments have been described to be effective. Even in cases where hypersensitivity has been proposed as potential underlying mechanism [16], immunomodulatory therapies such as the application of steroids have not been tested.
3.3 Therapeutic Intrathecal Drug Administration: Cytosine Arabinoside (ara-C) and Methotrexate (MTX)
For prophylaxis and treatment of intrathecal spread of tumor disease (e.g., leukemia, glioblastoma), several cytotoxic agents are routinely administered via the intrathecal route. Thus far, cytosine arabinoside (ara-C) and methotrexate (MTX) represent the most commonly used cytotoxic agents. According to a recent review, more than 30 pediatric patients with paraplegia in the course of intrathecal chemotherapy have been reported [20].
Pathophysiology
There is sparse information regarding the mechanism underlying CNS damage due to intrathecal chemotherapy. Postmortem analysis revealed primary neuronal damage with secondary myelin breakdown and axon degeneration. In particular spinal cord areas, which are in direct contact with CSF, are at highest risk to be damaged by the cytotoxic agent [21].
Clinical Presentation
Patients typically present within hours after intrathecal injection of ara-C or MTX in a prodromal stage with signs of meningeal irritation such as headache, diffuse pain, nausea, vomiting, and fever. Within days up to several weeks, a progressive ascending weakness in the lower extremities and neurogenic bladder dysfunction follow. Sensory deficits are mostly mild if not absent [20]. In severe cases, cranial nerve palsies, visual impairment, and progressive decline of consciousness develop. In a series of 23 patients, ten patients survived with persistent paraplegia, three remained permanently respirator dependent, and eight patients died. Two patients recovered completely [22].
Diagnostics
Early radiological diagnostics including spinal MRI usually reveal unremarkable results. In some instances diffuse swelling with hyperintense signal changes in T2-weighted images can be seen. Besides elevated protein level, standard CSF analysis is normal. As a specific marker for myelin breakdown, the myelin basic protein level is elevated.
Therapy
Once patients present with a progressive paraparesis, there is no specific therapy available. Currently it has yet to be defined which patients will be at risk to develop such a complication in the course of an intrathecal chemotherapy. It appears that preceding CNS radiation therapy and combined intrathecal chemotherapy (MTX together with ara-C) represent risk factors for a respective complication. Prophylactic treatment with concomitant steroids intrathecally, immunoglobulins, or vitamin substitution has not shown to protect against chemotherapy-induced paraplegia.
3.4 Diagnostic Intrathecal Drug Administration: Methylene Blue
In the past, methylene blue was routinely injected intrathecally to detect CSF leakage.
Pathophysiology
The mechanism underlying the toxic effects of methylene blue circulating in the CSF after lumbar intrathecal injection is still unknown. Postmortem analysis in a subject surviving the injection more than 10 years revealed spinal cord necrosis [23].
Clinical Presentation
A patient who received a methylene blue injection into a protruded disc at thoracic level for easier identification during spine surgery was transferred to our center. Within minutes the patient developed a rapidly progressing tetraplegia, respiratory failure, cranial nerve palsy, and hydrocephalus malresorptivus. Sensory and autonomous functions were only mildly affected. Over time he regained minimal motor function showing diffuse lower motoneuron damage.
Diagnostics
The temporal sequence of a progressive tetraplegia in the course of an intrathecal injection or an injection close to the intrathecal compartment clearly explains the etiology relevant for the clinical picture.
Therapy
There is no known therapy which may help to reduce damage to the spinal neural tissue. More than 40 years ago, clear warnings were published, which strongly disencourage intrathecal injections of methylene blue for its known above-described complications [24, 25]. Therefore, one could think that the description of methylene blue injections causing severe neurological dysfunction should be rather added to a history chapter. Unfortunately, the case description from our patient and a recent report indicating that methylene blue is still being used to identify the location of CSF leakage [26] demonstrate that methylene blue is still considered for diagnostic purposes directly in the CSF or in close proximity. Even worse, a recent randomized placebo-controlled clinical trial reported that intradiscal methylene blue injections dramatically reduce symptoms in chronic lower back pain patients [27]. Based on the above-described complications, any kind of methylene blue injection in close proximity to the nervous system is strongly disencouraged [24, 25, 28].
3.5 Diagnostic or Therapeutic Intrathecal Drug Administration: Spinal Anesthesia
Spinal anesthesia represents a procedure, which is applied worldwide millions of times each year and is considered to be safe. Nevertheless, numerous case reports describe the occurrence of progressive paraplegia in the course of spinal and epidural anesthesia [29].
Pathophysiology
So far it is unknown what ultimately causes the clinical picture of progressive paraplegia. It can only be speculated that either a hemorrhage in the course of the required lumbar puncture, the local anesthetic drug itself, unknown contaminants, or administered disinfectants cause the adhesive arachnoiditis, which subsequently affects the spinal cord parenchyma and nerve roots with respective neurological dysfunction. Respective factors induce a severe inflammatory reaction of the pia and arachnoidea followed by adhesion and tethering of nerve roots and spinal cord parenchyma. The blockade of CSF circulation can induce extensive syringomyelia and hydrocephalus malresorptivus.
Clinical Presentation
The interval between spinal/epidural anesthesia and presentation of neurological dysfunction varies greatly. Initial symptoms such as back pain, nausea, and headache are rather unspecific and rarely lead to confirmation of the diagnosis. More specific symptoms such as paraparesis and bladder and bowel dysfunction can appear within a few hours up to several months.
Diagnostics
Once clinical signs reflecting spinal cord disease become apparent, spinal MRI should be performed. MRI findings are characteristic for adhesive arachnoiditis showing conglomerations of adherent nerve roots residing centrally within the thecal sac, nerve roots adherent peripherally, and soft tissue masses replacing the subarachnoid space [30].
Therapy
In case of compression of neural structures (spinal cord, radices), decompressive laminectomy might be a strategy to slow down disease progression. Careful neurosurgical lysis of adherent meningeal structures can represent an additional treatment option. However, outcome following respective interventions is not favorable in most instances [31].
3.6 Accidental Intrathecal Drug Administration
The majority of reported cases of inadvertent intrathecal drug administration with the consequence of para- or tetraplegia are related to chemotherapeutic agents. The most notoriously accidentally intrathecally administered drug is vincristine. Thus far, 31 cases worldwide have been reported, where vincristine was injected into the intrathecal space with a mostly fatal outcome [32]. All individuals surviving the injection suffered from severe paraparesis. Besides vincristine intrathecal injections have been reported for the chemotherapeutic agents vindesine, asparaginase, bortezomib, daunorubicin, and dactinomycin. However, the majority of related case reports describe fatal cases. None of these cases survived in the long term with para- or tetraparesis.
Pathophysiology
Vincristine has a strong affinity to tubulin and neurofilament, thus destructing the cytoskeleton of neurons. Histologically, severe degeneration of myelin and axons with pseudocystic transformation most prominently in the lumbosacral and thoracic spinal cord has been described [33].
Clinical Presentation
Within hours to few days after intrathecal injection of vincristine, patients present with rapidly progressing paraparesis, urinary retention, and loss of anal sphincter function.
Diagnostics
Once the history of accidental vincristine injection is available, no further diagnostics are required. Spinal MRI shows a perimedullar enhancement around the conus medullaris. Neurophysiological analysis reveals acute denervation of affected muscles indicating predominant damage to the lower motoneuron (ventral horn, radix).
Therapy
Only patients, where measures to dilute the drug in the cerebrospinal fluid are applied immediately, survived, however, with the consequence of severe neurological deficits. The most successful therapy seems to be the simultaneous placement of an external ventricular and a lumbar drain with consecutive craniocaudal irrigation of ringer solution. In order to enhance clearing of the drug through the infused solution, fresh frozen plasma has been added [33].
4 Hereditary Causes (Hereditary Spastic Paraplegia)
Neurologic syndromes, in which progressive bilateral lower extremity weakness and spasticity (each of variable degree) are the predominant manifestations and for which gene mutations are proven or assumed as the major causative factor, are designated as hereditary spastic paraplegia (HSP). First described by Adolf von Strümpell in 1880 [34], HSP constitutes a clinically and genetically heterogeneous group of neurodegenerative disorders, historically divided into “pure” and “complicated” forms [35, 36]. Many HSPs are caused by mutations in genes encoding for proteins involved in the maintenance of corticospinal tract neurons, causing distal axonopathy of the longest corticospinal tract axons [37]. Nevertheless, HSP represents with a strong genetic heterogeneity. To date, 72 different spastic gait disease loci have been identified [38], and 54 spastic paraplegia genes (SPG) have already been cloned; other HSP causative genes are not in the SPG classification yet [39]. HSPs are transmitted in an autosomal dominant (AD), autosomal recessive (AR), X-linked (XL), or mitochondrial manner [40]. Moreover, sporadic HSP due to true de novo mutations or reduced penetrance of AD mutations as well as single cases in unrecognized AR kindred are common [41]. Due to the fact that sole clinical parameters are not reliable to differentiate the SPG subtypes, molecular testing is essential, despite the fact that around 30 % of affected patients have no genetic diagnosis [39]. The prevalence of all forms of HSP ranges from 4.1 to 9.8/100,000 [42–44]. In the European populations, AD HSP are more frequent than other forms [45], and 40–45 % of those have mutations in the SPAST gene [46]. In contrast, AR HSP are more frequent in consanguineous populations with a prevalence of up to 5.7/100,000 [42]. Mutations in KIAA1840/SPG11 represent the most common cause of AR HSP [47]. Sporadic HSP cases are frequent in clinical practice, with a prevalence of 1.3/100,000 in Norway [44]. Moreover, some of these cases could be explained by mutations in known genes, e.g., in SPG4 [39, 48].
Clinical Presentation
In most patients, HSP initially presents with progressive gait impairment due to lower extremity weakness and spasticity. Urinary urgency is common in HSP and occasionally may be an early feature. Symptoms may be first evident at any age, from early childhood through senescence. In general, subjects with pure HSP report progressive difficulty walking, often requiring canes, walkers, or wheelchairs. Besides their lower limb weakness and spasticity, they often present with diminished vibration and joint position sensation on the lower limbs [36]. The severity of spasticity and lower limb weakness varies between patients. Patients have a normal life expectancy and do not experience loss of dexterity, strength, or coordination in the upper extremities, dysarthria, dysphagia, or any other “complicating” neurological symptom. Age of onset in pure HSP is highly variable; progression of symptoms is usually slow over the years, without abrupt worsening or remissions [49].
Complicated HSP is characterized by neurological or non-neurological features in addition to the pure phenotype. Respective patients can present with spasticity in the upper extremities, cognitive impairment, epilepsy, extrapyramidal disturbances (parkinsonism, chorea, dystonia), dysphagia, dysarthria, cerebellar abnormalities (ataxia, nystagmus, tremor), spinal cord atrophy, muscle wasting, peripheral polyneuropathy, thin corpus callosum, or white matter lesions, in the absence of coexisting disorders [40]. Non-neuronal manifestations of complicated HSP include gastroesophageal reflux, orthopedic abnormalities (foot deformity, scoliosis, maxilla hypoplasia), retinopathy, macular degeneration, optic atrophy, cataract, facial dysmorphism, deafness, or skin lesions. Occasionally, complicated HSP might present with a pure phenotype in early phases of the disease, while complicating symptoms develop later on.
Importantly, marked clinical variability does not allow generalizations that apply to all HSP types and patients: Age of symptom onset ranges from infancy through late adulthood. The gait disturbance ranges from non-disabling, subtle forward-shifted heel strike to severe spastic paraplegia resulting in wheelchair dependence. Lower extremity spasticity is often but not always accompanied by weakness of the legs, primarily affecting iliopsoas, hamstring, and tibialis anterior muscles. Sometimes, symptoms begin asymmetrically, while over time both legs are more or less similarly affected. Frequent urinary bladder disturbance typically manifests as urinary urgency; defecation might be affected as well [49]. In general, there is marked clinical variability between different genetic types of HSP, as well as between individuals with the same genetic type. In addition, the course of HSP is also quite variable. Early-onset HSP may not worsen significantly over many years; adult-onset HSP can present with very slow progression over decades as well as more profound deterioration over 10–15 years. Onset and progression of symptoms over days to months is not typical for HSP and suggests alternative disorders [49].
Diagnostics
HSP can be suspected when patients present with slowly progressing spastic paraplegia of lower limbs, potential urinary urgency, and subtle sensory disturbances, usually in the context of suspicious family history. Nevertheless, several conditions need to be ruled out (Table 8.1) [49].
Diagnostic steps include individual and family history to detect the type of transmission. Moreover, a careful neurological examination, neurophysiological testing (nerve conduction studies, EMG, MEP, somatosensory evoked potential (SSEP), visual evoked potential (VEP)), imaging studies (spinal MRI, CT), and laboratory tests are needed for diagnosis, in particular to identify pure and complicated forms prior to genetic testing (Table 8.1). Screening for SPG mutations is usually carried out by direct Sanger sequencing and needs to be coupled with multiplex ligation-dependent probe amplification [39]. Recent improvements analyzing large panels of SPG genes by next-generation sequencing (NGS) permit confirmation of diagnosis in many patients; nevertheless this approach does not cover all SPG genes and may not identify all gene copy variants, including exon deletions [51]. Genetic testing is most useful to confirm a clinical diagnosis of HSP, and results of genetic testing need to be interpreted in the light of the clinical context (Table 8.2). Importantly, HSP gene variations with unknown clinical significance need careful consideration, in particular when the clinical diagnosis does not conform to HSP [49]. Genetic counseling is recommended for affected families, and the gene test results must consider mode of inheritance, and the possible degree of genetic penetrance, which is not very well known in most types of HSP [52].
Genetic counseling aims to inform affected patients and unaffected family members at risk about the nature and inheritance of the disorder. In case of AD transmission, most affected individuals have an affected parent, depending on genetic penetrance. Nevertheless, the frequency of de novo mutations causing AD HSP is unknown. In general, caution must be exercised in providing genetic counseling and prognosis for many HSP types, due to unclear genetic penetrance and insufficient information about the full phenotypic spectrum. In particular, the clinical description of the majority of genetic types of HSP is limited to one or a few families (Table 8.2).
Therapy
Currently, no specific treatments to prevent, halt, or reverse the pathological processes underlying HSP are available. Treatment options are exclusively symptomatic. Spasticity is managed by regular physical therapy, occupational therapy, assistive walking devices, orthotics, or drugs reducing muscle tone, in particular baclofen or tizanidine [41]. Chemodenervation with botulinum toxin A or B can be an alternative option [53, 54], as well as intrathecal baclofen therapy in selected cases. Secondary complications, such as tendon contractures, scoliosis, or foot deformities, may be delayed or even prevented by intense and regular physical therapy. Urinary urgency can be treated with anticholinergic drugs. Pain, a quite common symptom in HSP, should be treated according to general guidelines. Neuropathic pain benefits from gabapentin and pregabalin. In complicated forms patients with cognitive decline or dementia may profit from cholinergic drugs, while epilepsy should be treated according to established guidelines. If parkinsonism is a feature of the clinical phenotype, L-DOPA or dopamine-receptor agonists may be a treatment option. If dystonia is a prominent presentation of the HSP, botulinum toxin or even deep brain stimulation may be beneficial. In patients with SPG1 who develop hydrocephalus, shunt implantation is required [39, 40]. Regular clinical reevaluations of patients once or twice yearly are recommended to identify complications and progression of the disease.
5 Rare Cases
5.1 Cervical Flexion Myelopathy
A number of cases of spinal cord disease due to protracted fixed cervical spine positions – predominantly in young individuals – have been reported in the course of surgeries requiring a flexed cervical spine position, unconsciousness due to medication overdose, or after an assault forcing the victim into a flexed cervical spine position for a prolonged period of time [55].
Pathophysiology
In the literature, the term cervical flexion myelopathy is reserved for a chronic disease condition also known as Hirayama syndrome [56]. In case of Hirayama syndrome, the hypermobile dura compresses the cord microcirculation repeatedly for a short duration. As a consequence only highly susceptible neural cells within the spinal cord namely motoneurons, in the ventral horn are affected with isolated lower motoneuron signs and minimal MRI changes. In contrast, in case of subacute cervical flexion myelopathy, the shift of the dura over longer periods of time (hours to days) may compromise larger-caliber blood vessels (posterior spinal artery, epidural veins), causing more extensive circulatory problems affecting the white matter structures predominantly in the dorsal half of the spinal cord.
Clinical Presentation
After a protracted period of fixed cervical spine position, patients present with tetraparesis/tetraplegia meaning bilateral sensorimotor dysfunction in the upper and lower extremities including bladder and bowel dysfunction.
Diagnostics
If a subacute flexion myelopathy is suspected, spinal MRI should be performed to exclude compressive causes of spinal cord disease, in particular intraspinal hemorrhage. Within the cord parenchyma MRI longitudinally extending, dorsally accentuated signal changes with signs of cord swelling can be observed in T2-weighted sequences.
Therapy
Effective treatment options are not available. The existence of such a pathomechanisms potentially leading to irreversible neurological dysfunction should raise the awareness to check the necessity for surgeries requiring intra- or postoperative flexed cervical spine positions very carefully, particularly in young individuals, who are predominantly affected by subacute cervical flexion myelopathy. In cases where respective positions cannot be avoided, intra-/postoperative neuromonitoring should be considered to detect spinal cord dysfunction before irreversible damage to neural tissue occurs [55].
5.2 Epidural Lipomatosis
The spinal epidural lipomatosis is a rare entity. A majority of male patients and a mean manifestation at the age of 43 are described in the literature. Most of the cases are associated with obesity, chronic use of steroids, Cushing’s syndrome, and other endocrinopathies. Only a small group of patients without relevant concomitant diseases (idiopathic spinal epidural lipomatosis) are described so far. In all these cases, manifestation of the lipomatosis was restricted to the thoracic and lumbar segments of the spinal cord especially in the dorsal and lateral parts of the myelin [57].
Pathophysiology
Excess adipose tissue deposits in the spinal canal cause radiculopathy or spinal cord compression.
Clinical Presentation
Main symptoms are back pain, followed by a slowly progressive weakness of the legs, sensory loss, and sometimes a radicular manifestation or a claudication mainly seen with a lumbar affection of the spinal cord. An autonomic dysfunction with bowel and urinary incontinence is not typical.
Diagnostics
MRI shows a peculiar signal intensity in the T1-weighted sequences. Myelography is helpful to detect an obstruction in the CSF circulation. Often associated with the spinal epidural lipomatosis are degenerative processes of the spine [58]. CSF analysis typically reveals high protein levels with mild pleocytosis reflecting compromised CSF circulation.
Therapy
Due to the small amount of cases, no evidence-based therapeutic strategies are available. In general a conservative therapy is proposed with minor symptoms, e.g., weight reduction and reduction of steroid intake. On the other hand, a surgical intervention to reduce epidural fat should be preferred with major functional deficits. The success rate meaning complete functional recovery varies between 20 and 60 % dependent on localization and etiology.
In conclusion, spinal epidural lipomatosis should be regarded as a possible differential diagnosis when a patient presents with the typically slowly progressive para-/tetraparesis and sensory deficits especially if predisposing factors are present (see chapter 7). Without specific signal enhancements indicating a spinal cord injury in the MRI, the relevant compression caused by epidural fat can be elucidated by myelography in combination with a below-block CSF analysis.
5.3 Conversion (dissociative) Paraplegia
The term dissociative paraplegia refers to an alteration or loss of function in the lower limbs without an anatomical or physiological explanation. This type of illness belongs to the group of conversion disorders. The average prevalence of conversion disorders in the general population is estimated at 5–22 cases per 100,000 persons. In the absence of mental state abnormalities, dissociative paraplegia remains a diagnostic and therapeutic challenge on the borderline between neurology and psychiatry [59].
Clinical Presentation
The disease condition is commonly observed in young female patients. Neurological examination yields findings, which are not congruent with a lesion of the central or peripheral nervous system. Respective patients typically present with complete paralysis of the lower extremities. In case of an “incomplete” paraparesis, flexor and extensor muscles are equally affected contradicting an expected central paresis pattern, where flexor muscles are more severely affected than extensor muscles. While examining the patient in a supine position, lifting the legs off the bench is not possible. However, walking on tiptoe or heels is possible. The Hoover sign represents another test to unmask a nonorganic origin of the presented symptoms: direct testing of the hip extension reveals weakness in the respective muscle. While testing contralateral hip flexion against resistance, ipsilateral hip extension becomes suddenly strong. Patients, who are still able to walk frequently, display a dragging gait with external or internal hip rotation. Tendon reflexes are usually unremarkable. Structured clinical interviews usually do not identify any acute or chronic psychiatric comorbidity.
Diagnostics
Appropriate diagnostic tests have to be performed to rule out a somatic cause for the disease. Therefore, imaging studies – ideally MRI of the complete neuraxis (spine and brain) – have to rule out spinal cord compression or a non-compressive lesion of the spinal cord, cauda equina, or relevant areas in the brain (e.g., parasagittal cortical syndrome due to anterior cerebral artery infarction or meningioma located in the falx). Once respective studies do not show pathological changes, inflammatory/infectious causes are addressed with CSF analysis. The integrity of descending motor and ascending sensory pathways can be objectively assessed with MEP and SSEP workup.
Therapy
To prevent disease chronification, it is suggested that patients should be informed early about the diagnosis and treated subsequently. Therefore, it is necessary to conduct a multifunctional approach therapy, including psychotherapy, relaxation techniques, autogenic training, and intensive physical therapy. Physical therapy especially might help the patient to give up symptoms without losing face and consequently improve bodily experience, movement ability, interpersonal attunement, and social well-being. There is also some evidence suggesting that patient education regarding the underlying mechanisms is helpful, but special attention must be paid to possible stigmatization and labeling of the patient. Emphasis should be placed on the reversibility and good prognosis of the disorder [60]. A major challenge can be to protect patients with dissociative paraplegia in the long run from potentially harmful surgical interventions.
Conclusion
This chapter provided an overview about rare spinal cord disease entities, which are in many instances challenging to diagnose. Careful compilation of the past medical history including family history and clinical neurological examination represent key instruments leading to the correct diagnosis. Specific ancillary tests (e.g., specific blood chemistry analysis, genetic testing) besides imaging studies and CSF analysis are needed to confirm the suspected diagnosis, in particular metabolic causes (copper/cobalamin deficiency).
References
Beck WS (1991) Diagnosis of megaloblastic anemia. Annu Rev Med 42:311–322
Hemmer B, Glocker FX, Schumacher M, Deuschl G, Lucking CH (1998) Subacute combined degeneration: clinical, electrophysiological, and magnetic resonance imaging findings. J Neurol Neurosurg Psychiatry 65(6):822–827
Carmel R (2008) How I treat cobalamin (vitamin B12) deficiency. Blood 112(6):2214–2221
Bassi SS, Bulundwe KK, Greeff GP, Labuscagne JH, Gledhill RF (1999) MRI of the spinal cord in myelopathy complicating vitamin B12 deficiency: two additional cases and a review of the literature. Neuroradiology 41(4):271–274
Locatelli ER, Laureno R, Ballard P, Mark AS (1999) MRI in vitamin B12 deficiency myelopathy. Can J Neurol Sci 26(1):60–63
Saperstein DS, Barohn RJ (2002) Peripheral neuropathy due to cobalamin deficiency. Curr Treat Options Neurol 4(3):197–201
Kumar N (2014) Neurologic complications of bariatric surgery. Continuum (Minneap Minn) 20(3 Neurology of Systemic Disease):580–597
Shammaa Y, Rodgers J (2012) Denture fixative cream and the potential for neuropathy. Dent Update 39(8):575–577
Jaiser SR, Winston GP (2010) Copper deficiency myelopathy. J Neurol 257(6):869–881
Nardone R, Holler Y, Storti M, Lochner P, Tezzon F, Golaszewski S, Brigo F, Trinka E (2014) Spinal cord involvement in patients with cirrhosis. World J Gastroenterol 20(10):2578–2585
Caldwell C, Werdiger N, Jakab S, Schilsky M, Arvelakis A, Kulkarni S, Emre S (2010) Use of model for end-stage liver disease exception points for early liver transplantation and successful reversal of hepatic myelopathy with a review of the literature. Liver Transpl 16(7):818–826
Wang MQ, Liu FY, Duan F (2012) Management of surgical splenorenal shunt-related hepatic myelopathy with endovascular interventional techniques. World J Gastroenterol 18(47):7104–7108
Kinsella LJ, Green R (1995) ‘Anesthesia paresthetica’: nitrous oxide-induced cobalamin deficiency. Neurology 45(8):1608–1610
Ng J, O’Grady G, Pettit T, Frith R (2003) Nitrous oxide use in first-year students at Auckland University. Lancet 361(9366):1349–1350
Marie RM, Le Biez E, Busson P, Schaeffer S, Boiteau L, Dupuy B, Viader F (2000) Nitrous oxide anesthesia-associated myelopathy. Arch Neurol 57(3):380–382
McCreary M, Emerman C, Hanna J, Simon J (2000) Acute myelopathy following intranasal insufflation of heroin: a case report. Neurology 55(2):316–317
Riva N, Morana P, Cerri F, Gerevini S, Amadio S, Formaglio F, Comi G, Comola M, Del Carro U (2009) Acute myelopathy selectively involving lumbar anterior horns following intranasal insufflation of ecstasy and heroin. BMJ Case Rep 2009. doi:10.1136/bcr.08.2008.0669
Schreiber AL, Formal CS (2007) Spinal cord infarction secondary to cocaine use. Am J Phys Med Rehabil 86(2):158–160
Luigetti M, Cianfoni A, Conte A, Colosimo C, Tonali PA, Sabatelli M (2010) Posterior ischaemic myelopathy associated with cocaine abuse. Intern Med J 40(10):732–733
Rolf N, Boehm H, Kaindl AM, Lauterbach I, Suttorp M (2006) Acute ascending motoric paraplegia following intrathecal chemotherapy for treatment of acute lymphoblastic leukemia in children: case reports and review of the literature. Klin Padiatr 218(6):350–354
von der Weid NX, de Crousaz H, Beck D, Deonna T, Miklossy J, Janzer RC (1991) Acute fatal myeloencephalopathy after combined intrathecal chemotherapy in a child with acute lymphoblastic leukemia. Med Pediatr Oncol 19(3):192–198
Watterson J, Toogood I, Nieder M, Morse M, Frierdich S, Lee Y, Moertel CL, Priest JR (1994) Excessive spinal cord toxicity from intensive central nervous system-directed therapies. Cancer 74(11):3034–3041
Sharr MM, Weller RO, Brice JG (1978) Spinal cord necrosis after intrathecal injection of methylene blue. J Neurol Neurosurg Psychiatry 41(4):384–386
Mameghani A (2011) [Intrathecal administration of methylene blue is obsolete]. Unfallchirurg 114(6):549
Schultz P, Schwarz GA (1970) Radiculomyelopathy following intrathecal instillation of methylene blue. A hazard reaffirmed. Arch Neurol 22(3):240–244
Schulz N, Kolenda H, Thiel A, Vestring T, Schulte M (2010) Subarachnoid pleural fistula and subsequent pneumocephalus as complication of vertebral body replacement of the thoracic spine. Unfallchirurg 113(11):951–956
Peng B, Pang X, Wu Y, Zhao C, Song X (2010) A randomized placebo-controlled trial of intradiscal methylene blue injection for the treatment of chronic discogenic low back pain. Pain 149(1):124–129
Levine R, Richeimer SH (2011) Spinal methylene blue is hazardous. Pain 152(4):952–953; author reply 953
Killeen T, Kamat A, Walsh D, Parker A, Aliashkevich A (2012) Severe adhesive arachnoiditis resulting in progressive paraplegia following obstetric spinal anaesthesia: a case report and review. Anaesthesia 67(12):1386–1394
Ross JS, Masaryk TJ, Modic MT, Delamater R, Bohlman H, Wilbur G, Kaufman B (1987) MR imaging of lumbar arachnoiditis. AJR Am J Roentgenol 149(5):1025–1032
Kane RE (1981) Neurologic deficits following epidural or spinal anesthesia. Anesth Analg 60(3):150–161
Gilbar PJ (2014) Intrathecal chemotherapy: potential for medication error. Cancer Nurs 37(4):299–309
Qweider M, Gilsbach JM, Rohde V (2007) Inadvertent intrathecal vincristine administration: a neurosurgical emergency. Case report J Neurosurg Spine 6(3):280–283
Engmann B, Wagner A, Steinberg H (2012) Adolf von Strumpell: a key yet neglected protagonist of neurology. J Neurol 259(10):2211–2220
Harding AE (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1(8334):1151–1155
Harding AE (1993) Hereditary spastic paraplegias. Semin Neurol 13(4):333–336
Blackstone C, O’Kane CJ, Reid E (2011) Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci 12(1):31–42
Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, Masri A, Kayserili H, Al-Aama JY, Abdel-Salam GM, Karminejad A, Kara M, Kara B, Bozorgmehri B, Ben-Omran T, Mojahedi F, Mahmoud IG, Bouslam N, Bouhouche A, Benomar A, Hanein S, Raymond L, Forlani S, Mascaro M, Selim L, Shehata N, Al-Allawi N, Bindu PS, Azam M, Gunel M, Caglayan A, Bilguvar K, Tolun A, Issa MY, Schroth J, Spencer EG, Rosti RO, Akizu N, Vaux KK, Johansen A, Koh AA, Megahed H, Durr A, Brice A, Stevanin G, Gabriel SB, Ideker T, Gleeson JG (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343(6170):506–511
Lo Giudice TF, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A (2014) Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 261C:518–539
Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 318(1-2):1–18
Fink JK (1993) Hereditary spastic paraplegia overview. In: Pagon RA et al (eds) GeneReviews(R). [internet]. Seattle (WA), 1993–2016
Boukhris A, Stevanin G, Feki I, Denora P, Elleuch N, Miladi MI, Goizet C, Truchetto J, Belal S, Brice A, Mhiri C (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75(6):527–536
Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J, Tuna A, Barbot C, Guimaraes J, Alonso I, Silveira I, Sequeiros J, Marques Neves J, Serrano P, Silva MC (2013) Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 70(6):746–755
Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM (2009) Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain J Neurol 132(Pt 6):1577–1588
Reid E (1997) Pure hereditary spastic paraplegia. J Med Genet 34(6):499–503
Beetz C, Nygren AO, Schickel J, Auer-Grumbach M, Burk K, Heide G, Kassubek J, Klimpe S, Klopstock T, Kreuz F, Otto S, Schule R, Schols L, Sperfeld AD, Witte OW, Deufel T (2006) High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology 67(11):1926–1930
Siri L, Battaglia FM, Tessa A, Rossi A, Rocco MD, Facchinetti S, Mascaretti M, Santorelli FM, Veneselli E, Biancheri R (2010) Cognitive profile in spastic paraplegia with thin corpus callosum and mutations in SPG11. Neuropediatrics 41(1):35–38
Depienne C, Tallaksen C, Lephay JY, Bricka B, Poea-Guyon S, Fontaine B, Labauge P, Brice A, Durr A (2006) Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet 43(3):259–265
Fink JK (2014) Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol 34(3):293–305
Fink JK (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 126(3):307–328
Schlipf NA, Schule R, Klimpe S, Karle KN, Synofzik M, Schicks J, Riess O, Schols L, Bauer P (2011) Amplicon-based high-throughput pooled sequencing identifies mutations in CYP7B1 and SPG7 in sporadic spastic paraplegia patients. Clin Genet 80(2):148–160
Gasser T, Finsterer J, Baets J, Van Broeckhoven C, Di Donato S, Fontaine B, De Jonghe P, Lossos A, Lynch T, Mariotti C, Schols L, Spinazzola A, Szolnoki Z, Tabrizi SJ, Tallaksen CM, Zeviani M, Burgunder JM, Harbo HF, Efns (2010) EFNS guidelines on the molecular diagnosis of ataxias and spastic paraplegias. Eur J Neurol 17(2):179–188
Geva-Dayan K, Domenievitz D, Zahalka R, Fattal-Valevski A (2010) Botulinum toxin injections for pediatric patients with hereditary spastic paraparesis. J Child Neurol 25(8):969–975
Hecht MJ, Stolze H, Auf dem Brinke M, Giess R, Treig T, Winterholler M, Wissel J (2008) Botulinum neurotoxin type A injections reduce spasticity in mild to moderate hereditary spastic paraplegia – report of 19 cases. Mov Disord 23(2):228–233
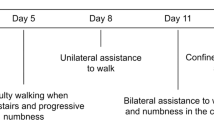
Fehre KS, Weber MA, Hensel C, Weidner N (2016) Tetraparesis as clinical correlate of subacute cervical flexion myelopathy. J Spinal Cord Med 39(3):359–362
Hirayama K, Tokumaru Y (2000) Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. Neurology 54(10):1922–1926
Al-Khawaja D, Seex K, Eslick GD (2008) Spinal epidural lipomatosis – a brief review. J Clin Neurosci 15(12):1323–1326
Pinkhardt EH, Sperfeld AD, Bretschneider V, Unrath A, Ludolph AC, Kassubek J (2008) Is spinal epidural lipomatosis an MRI-based diagnosis with clinical implications? A retrospective analysis. Acta Neurol Scand 117(6):409–414
Stone J, Carson A, Sharpe M (2005) Functional symptoms in neurology: management. J Neurol Neurosurg Psychiatry 76(Suppl 1):i13–i21
Hirjak D, Thomann PA, Wolf RC, Weidner N, Wilder-Smith EP (2013) Dissociative paraplegia after epidural anesthesia: a case report. J Med Case Rep 7:56
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Weidner, N., Kohl, Z. (2017). Metabolic, Toxic, Hereditary, and Rare Causes of Spinal Cord Disease. In: Weidner, N., Rupp, R., Tansey, K. (eds) Neurological Aspects of Spinal Cord Injury. Springer, Cham. https://doi.org/10.1007/978-3-319-46293-6_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-46293-6_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46291-2
Online ISBN: 978-3-319-46293-6
eBook Packages: MedicineMedicine (R0)