
Abstract
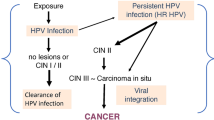
HPVs transforming activities represent the viral replication strategy that is driven to replicate viral genomes and to establish long-term maintenance in a tissue. High-risk-HPV-infected cells and carcinogenesis progression are terminal events, since cancer cells contain integrated HPV genomes and do produce viral progeny. High-risk HPV (HR-HPV) genome integration indeed represents a consequence of HPV E6/E7- induced genomic instability. HR-HPV E6 and E7 proteins critically contribute to viral life cycle and transforming activity. HR-HPV E7 proteins bind to pRB and decreased efficiency. HR-HPV E6 proteins efficiently interact with TP53 and promote for TP53 degradation. High-risk HPVs can frequently persist for decades in an infected host cell at a low number of copies. One of the events of HPV-induced carcinogenesis is the HPV genome integration into a host chromosome, and it is probably a failed viral mechanism. High Risk-HPV E6 proteins and E7 contribute to immortalization of primary human epithelial cells by induction of telomerase activity. Data evidences suggest that microbial dysbiosis is associated with malignant transformation, but future discussion and direction for microbiome in cancer research (oncobioma) and particularly in HPV-associated human cancer could be evaluated as causative causes that modulate initiation, progression, or cancer metastasis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- HPV
- High risk-HPV
- Neoplastic transformation
- HPV genome integration
- C-myc
- E6
- pRb
- E7
- p53
- Telomerase
- Tumor metabolome
- Dysbiosis
- Oncobioma
3.1 HPV Generalities, Life Cycle and Genome
“To avoid criticism, do nothing, say nothing, be nothing” Elbert Hubbard (writer)
Human papillomaviruses (HPVs) are members of the papovaviridae family. The viral structure consists of a 72-capsomere capsid; capsomeres are two structural proteins: 57 kD late protein L1 (80% of the viral particle) and 43–53 kD minor capsid protein L2 [1]. The HPV absence of envelope makes them stable and let them remain infectious for months in hostile environments [2]. The HPVs are present in higher vertebrates, however, they show a species-specificity pattern but the horizontal transmission from non-primates to humans has not been reported. HPV causes a local infection in the stratified epithelia and induces a productive replication with differentiation in a non-acute one, but produces a chronic disease where viral spread and/or viremia do not occur. The life cycle of HPV is associated with the differentiation program of epithelial cells. In normal epithelial cells, the only actively dividing cells are present in the basal layers of the stratified epithelium, which is basically formed by “transit amplifying cells” (TAC) and stem cells. Cells that are proliferating and can terminally differentiate are TAC and cells, which have the potential to proliferate indefinitely, are stem cells, although they divide infrequently in order to replenish the TAC pool [3]. After viral infection, HPVs deposit their double strand DNA genome into nuclei of infected cells and establishes as extra-chromosomal plasmids or episomes [4]. HPV gains entry to cells in the basal layer of the epithelium that becomes exposed through micro-abrasions [4]. The HPV genome has three genomic regions E and L genes, which are numbered according to the size: 4-kb early (E) region that encodes nonstructural proteins, 3-kb late (L) region that encodes two capsid proteins (L1, L2 genes), and 1-kb noncoding long control region (LCR) that regulates viral replication and gene expression (Fig. 3.1). Papillomavirus life cycle is linked to the differentiation program of infected epithelium cells and infects basal epithelial cells in the sole cell layer in the epithelium actively dividing. Although integrin-6 has been related, HPV receptor(s) has not been characterized [5]. Viral HPV-DNA is in the nuclei of infected host cells in a low copy number and lately undergoes differentiation moving toward the epithelium surface. The mechanism changes when the HPV-DNA is present in terminally differentiated cells; the virus replicates in a high number of copies, late genes are expressed, and progeny virus is produced [6]. The HPVs are not lytic viruses and the progeny virus is shed into the environment as a cargo within epithelial squamae. This review discusses the various mechanisms of transformation and the roles HPV plays in cervical carcinogenesis.

HPV Genome linear arrangement. HPV genome is small (8 kb), circular and with double strand DNA. There are eight open reading frames (ORFs) expressed from a single polycistronic transcript transcribed from a single strand of DNA. The Upstream Regulatory Region (URR) is in the non-coding region and regulates viral transcription and replication. HPV genes are regulated during differentiation by the virus early promoter (P97), the differentiation late-dependent promoter (P742), and tow Polyadenylation signals (PolyA). E6 and E7 are oncogenes responsible of the replication competence. E1 and E2 are genes of viral DNA replication and regulation of viral transcription. E4 and E5 are genes for late viral functions and L1 and L2 are genes for protein capsids
3.2 Transforming Mechanisms of HPV Viral Oncoproteins
HPVs potential to promote malignant transformation is the key for the low- and high-risk classification created from observations of HPV types found in cancers as high-risk (HR-HPV) and the ones found in benign lesions as low-risk (LR-HPV), and by experimental evidence that demonstrated their abilities to modify proliferation and genome stability. The general molecular aspects and functions of human papillomavirus proteins are shown in Table 3.1. The most important oncogenic proteins are E6 and E7, because of their immortalizing and transforming high potential, both in animal models and ‘in vitro’ models. The hallmarks of dysplasia lesions (low and high grade) as precursor of cervical cancer are the expression of HR-HPV E6/E7 genes; the expression of both genes contributes to genomic stability and malignant progression [7]. HPV E6 is a 150-amino acid protein containing two metal binding motifs (PDZ protein-binding motif), which acts as molecular organizing center for cellular signal transduction pathways [8]. There is a cellular defense mechanism that induces synthesis of aberrant and/or viral DNA into differentiated keratinocytes, which eliminates cells by selective and type-specific processes such as apoptosis, differentiation and senescence; the name of such mechanism is “trophic sentinel response” (TSR) [8]. One of the molecular mechanisms of E6 protein to promote malignancy in epithelial cells is the induction of TP53 ubiquitination and proteasome degradation by retargeting E6-AP [9]. A considerable number of cellular proteins have been reported to associate with E6 (see Table 3.1). HR-HPV E6 proteins eliminate the TSR triggered by E7 expression through inactivation of TP53 [10]. The HPV16 E6/E7 ORF cassette is regulated by the epidermal growth factor (EGF) pathway; there is a natural gradient of EGF and EGFR expression in the stratified epithelium, and it is the reason to assume that EGF modulates E6/E7 splicing during the viral life cycle and transformation [11]. HPV E7 is a low-molecular-weight protein of approximately 100 amino acids without intrinsic enzymatic activities. HPV-16 E7 oncoprotein has an amino-terminal 37-amino-acid residue similar to sequences of CR1 and to CR2 of Adenovirus E1A protein (Ad E1A). CR1 sequences are responsible for the retinoblastoma tumor suppressor protein (pRB) degradation and cellular transformation; CR2 sequences are the pRB-binding site (LXCXE), necessary for cellular transformation. E7 carboxyl terminus contains a metal binding motif for association with host cellular proteins, which include histone-modifying enzymes, in order to contribute toward malignant transformation. Such as AdE1A and SV40 T antigen, the HPV E7 protein interacts with several host cellular proteins (see Table 3.1). The ability of HPV E7, Ad E1A, and SV40 T antigen to associate with pRB is basic for the viral genome replication. HR-HPV-derived E7 proteins interact with pRB more efficiently than the E7 proteins encoded by LR-HPVs [12]. E7-interacting proteins, including transcription factors, cell cycle regulators, and metabolic enzymes, appear to associate with carboxyl-terminal E7 sequences [13]. The HPV E7 amino-terminal pRB binding site protein has been implicated in histone deacetylase binding, a necessary event for the HPV viral life cycle [14]. HPV-16 E6 and E7 oncoproteins over-regulate the TGF-beta1 promoter in cervical tumor cells [15]. The HPV oncoproteins E6 and E7 have been implicated in the regulation of the Wnt/β-catenin pathway [16].
3.3 Genomic Integration of HPV and Host Genomic Instability Induction as Basic Steps toward Malignant Transformation by HPV
Cellular signal transduction pathways are dysfunctional in human solid tumors [17], and it has been proposed the minimally oncogenic steps necessary to generate ‘in vitro’ transformed human epithelial cells. The expression of simian virus 40 (SV40) large tumor antigen (T), SV40 small tumor antigen (t), inactivates TP53 and pRb tumor suppressors, just like the HPV E6 and E7 oncoproteins work; the catalytic subunit of human telomerase (hTERT) which HPV E6 can activate transcriptionally, and the activated H-Ras oncogene are required to transform primary human epithelial cells [18]. Hence, the expression of HR-HPV E6/E7 oncogenes provides the minimal carcinogenic hits for primary human epithelial cells transformation [19]. HPV infects differentiated squamous epithelial cells (growth arrested) incompetent to support genome synthesis, but the HPV genome encodes functions that create and/or maintain a genome replication competence in differentiated keratinocytes. During the HPV life cycle, it is established a mechanism of long-term viral persistence into the squamous epithelia. HR-HPVs have evolved specific molecular mechanisms to maintain the host immune evasion and escape to guaranty viral progeny and not to induce an oncogenic process, which is not the natural function of the HPV infection. One of the events of HPV-induced carcinogenesis is the HPV genome integration into a host chromosome, and it is probably a failed viral mechanism. The HPV genome integration occurs into common fragile sites of the human genome [20], but there are not apparent hot spots for integration and no evidence for insertional mutagenesis [21]. Papillomavirus E1 and E2 proteins play a role in viral replication. The papillomavirus E2 protein works: (i) as a DNA binding transcription factor interacting with specific motifs (ACCN6GGT) in the LCR region [22]; (ii) as a transcriptional activator or transcriptional repressor in keratinocytes [23]; (iii) associated with viral DNA helicase E1 to modulate viral gene expression, in order to increase the recognition of the origin and the viral genome replication [24]; (iv) playing a role in viral genome segregation during cell division by tethering viral genomes to mitotic chromosomes [25]; (v) in association with mitotic chromosomes by interaction with the human bromo-domain protein Brd4 [26]. HPV genome integration to host genome follows a major specific pattern regarding the HPV genome function, and the consequence is the consistently maintained expression of the viral E6 and E7 genes, whereas other DNA viral genome regions (such as E2 region) are deleted and/or their expression is disturbed [27]. The E2 loss expression is significant and results in deregulated E6 and E7 expression; when it happens with HPV-16 an increased E6/E7 expression and stability after genome integration occurs [28], and host cellular specific alterations of gene expression appears [29]. Infected cells with integrated HPV genome that expresses E6/E7 have a selective growth advantage in comparison with infected cells harboring episomal HPV genome. The continued E6/E7 expression in cervical cancer cells is an obligated process for transformed phenotype maintenance [30]. HPV 16 and 18 integrations in high-grade lesions are accompanied by chromosomal abnormalities [31]. HR-HPV genomes are integrated into the host genome in the most invasive cancers, an increased ability of HR-HPV types to integrate into host DNA compared to those with low-risk types [32]. Extra-chromosomal HPV DNA is found in benign and low-grade lesions and HPV integration can be found in premalignant lesions grade 2/3 such as cervical intraepithelial neoplasia (CIN2/3). LR-HPV types are rarely found integrated in tumors, which was demonstrated by the absence of full-length E2 transcripts studies of tissue specimens from patients with a history of benign early-onset recurrent respiratory papillomatosis developing laryngeal cancer [33]. HPV integration disrupts the E2 gene [34], so determination of absent amplification of E2 sequences has been considered a molecular marker of integration or progression in cervical cancer; unfortunately, the results obtained are ambiguous. Detection of early gene transcripts by reverse-transcription PCR is more sensitive in cancers as well as in benign or dysplastic cervical samples, in which the presence of integrated genomes correlates with the severity of the disease, especially for HPV 18 [35]. HPV genomes integration is a failed step that affects both viral and host gene expression. Increased E7 protein synthesis correlate with viral DNA integration where integrated viral DNA confers growth advantages and phenotypic cellular changes with high-grade neoplasia compared to extrachromosomal viral DNA cells. The disruption pattern in the viral genome does not occur in the host genome, in contrast, HPV DNA sequences integration uses preferential sites of human chromosomes and suggests a non-random pattern of integration, for example, in cervical carcinomas it has been observed HPV integration into and around the hTERT gene, which resulted in an increase in hTERT expression [36], or HPV 18 DNA has been found integrated in the proximity of c-myc gene in several cervical cancers, but surprisingly not up-regulation of endogenous proto-oncogene expression was observed [37]. HPV 16 and 18 DNA sequences have been found integrated in particular chromosomal loci known as common fragile sites in cervical cancers [37]. An association between the loss of fragile histidine tetrads (FHIT) expression and progression of HPV 16-positive CIN has been demonstrated [38]. Invasive cervical cancers expressing HR-HPV E6 and E7 transcripts contain normal FHIT transcription, while low amount of viral transcripts were detected when FHIT was abnormally expressed, which suggests that E6 and E7 could be repressed in the presence of FHIT aberrations [39]. An intensive review of HPV integration sites in cervical dysplasia and cancer concluded that integration is randomly distributed over the whole host genome with genomic fragile sites predilection [40]. Modification of host cell genes that interfere with the expression or function of viral genes will eventually contribute to immune evasion; the tumor progression and invasion are an important event for malignant cellular transformation [41]. We have to continue looking for the physical and functional relevance of viral and cellular genes in the HPV-mediated transforming mechanisms, since experimental evidence could be an indication that the major function of HPV integration is the conservation and stabilization of HPV gene expression. Human carcinogenesis is considered a genomic instability disease [42]. Human solid tumors display aneuploidy, however; transformed human cells generated ‘in vitro’ maintain their genome stability [43]. Therefore, genomic instability is not a generic manifestation of oncogenic transformation but represents a tumor cell characteristic to acquire genetic alterations necessary for the survival and clonal expansion within the emergent neoplasia changing microenvironment [44]. Recently, it has been demonstrated that beyond HPV-induced immortalization, the chromosomal aberrations are inversely related to the HPV type immortalization capacity, which means that HR-HPV types with reduced immortalization capacity, needs more genetic host cell aberrations to facilitate immortalization and these could explain the differences in HPV-type prevalence in cervical cancers [45]. The combined expression of HR-HPV E6 and E7 proteins in cervical cancer cells causes inactivation of p53 and pRb tumor suppressor pathways and induces telomerase activation; these signal transduction pathways are disrupted in the majority of human solid tumors and they constitute a minimal subset of oncogenic hits to generate transformed ‘in vitro’ human cells [17]; complementary oncogenic events as E6/E7 expression are necessary to ‘in vivo’ and ‘in vitro’ transformation. Cervical carcinomas show chromosomal abnormalities [41], such as a specific gain at chromosome 3q for transition from HR-HPV-associated severe dysplasia to invasive carcinoma [46]. HPV-16 E7 oncoprotein contributes to genomic instability by the induction of centrosome duplication errors and generation of mitotic defects and aneuploidy in normal human epithelial cells, and also the characteristic multipolar mitoses in cervical lesions [47]. Centrosomic abnormalities emerge as a consequence of cytokinesis and/or cell division defects, thus occurring mostly in cells that have also accumulated nuclear abnormalities [48], also associated mitotic defects are present in cells that express episomal HPV-16 at a low number of copies, similarly to low-grade HPV-associated lesions [49], but the incidence of these alterations increases in cells when HPV genome is integrated to the host genome [50]. HPV E7 expression induces primary centrosome and centriole duplication errors in normal diploid cells but the mechanism remains absent of an explanation [51]. HPV E7 expression has the ability to target pRB family members, and it can explain the reason why the expression of HPV-16 E7 causes an increased incidence of centrosome abnormalities in mouse embryo fibroblasts that lack of pRB, p107, and p130 expression [48]. Centrosome abnormalities have also been detected in cervical lesions [49]. HPV-16 E7 expression works as mitotic mutator due to increased mitotic errors each round of cell division, inducing the genomic plasticity for the acquisition of cellular mutations that contribute to malignant progression [50]. The presence of double-strand DNA breaks in HPV-16 E6/E7- expressing cells is a mechanistic rationalization of what is facilitating HPV genome integration and why it is accompanying malignant progression [52]. However, integration of oncogenic HPV genomes in cervical lesions is a consequence rather than the cause of chromosomal instability induced by deregulated HR-HPV E6-E7 oncogene expression [53], and there is a gain of human telomerase gene TERC as important associated genetic event during the progression of dysplasia to cervical cancer [54].
3.4 Telomerase Activation as Molecular Transformation and Immortalization Mechanisms by HPV
Telomere shortening is a cell-autonomous mechanism that restricts the proliferative capacity of normal somatic cells. The hTERT expression of the catalytic telomerase subunit, in primary human cells, causes life span extension and immortalization. Cell types that must undergo a large number of cell divisions such as stem cells, express telomerase, a ribonucleoprotein that prevents telomere erosion. Many human tumor cells express actively telomerase, suggesting that aberrant telomerase activity is critical for human tumorigenesis. hTERT expression is considered one of the obligatory components for the generation of human tumor-like cells ‘in vitro’ [55]. HR-HPV E6 proteins and E7 contribute to immortalization of primary human epithelial cells by induction of telomerase activity [56]. HPVs have been shown to integrate in the proximity of c-myc gene, which justifies the search for alterations of this proto-oncogene in HPV-associated lesions. Ocadiz et al. [57] described for the first time the amplification of a human oncogene in samples of cervical cancers, such oncogene was c-myc. Recently, another group described it but compared it with benign and premalignant cervical lesions [58]. Moreover, a significant association between c-myc amplification and HPV 16 infection was observed. Elevated levels of c-myc have been found in several HPV-positive cervical carcinoma cell lines [59]. However, the significance of these events in HPV-mediated transformation remains unclear. The involvement of the c-Myc protein in HPV-induced immortalization was recently addressed [60]. HR-HPV E6 was shown to associate with c-Myc complexes (Myc/Max) and activate the hTERT promoter. The specific c-Myc antagonist, Mad, represses E6-transactivation of hTERT. HR-E6 proteins induce hTERT expression at a transcriptional level [61]. The minimal E6 responsive hTERT promoter fragment contains c-myc responsive E-boxes that contribute to E6-mediated transcriptional activation, but E6 does not markedly affect c-myc expression or the composition of c-Myc transcription factor complexes [62]. In HR-HPV expressing E6, the direct interaction with c-myc oncogene form, a c-Myc/E6 complex that activates hTERT expression [63]. An alternative hypothesis that tries to explain forward is that E6 relieves the telomerase promoter repression by inducing NFX1–91 degradation, which is a transcriptional repressor [64]. In E6-positive cells the telomerase activity increases when they become immortalized, although E6 expression levels do not change [65], meaning that other factors are participating in telomerase activation. Other experiments have revealed the E6 immortalization potential in mammary epithelial cells and keratinocytes by inactivation of TP53 [66]. Nowadays there is a scientific evidence that oncogenic types specifically activate the hTERT promoter (a limited set of viruses within the Alphapapillomavirus genus are oncogenic), while non-oncogenic types do not, which that means activation of the hTERT promoter is associated with oncogenic types [67].
3.5 Metabolic Tumor Adaptations as HPV Transforming Mechanisms
Although long-term information is stored almost exclusively in the genome, the proteome is crucial for short-term information storage; and the transcription factor-controlled information retrieval is strongly influenced by the state of the metabolome. The elementary building blocks in the proteome are proteins, but metabolomes are constituted by proteins and metabolites that form different interacting network called metabolic pathways [68, 69]. Nowadays is well knows that tumor cells metabolism (tumor metabolome) is characterized by a high concentration of glycolytic enzymes. About these scope there is an interesting report, where they characterized the metabolism of non-transformed rat kidney cells (NRK cells), showing a high glutaminolytic flux rate and a low (ATP + GTP):(CTP + UTP) ratio, whereas fructose 1,6-biphosphate (FBP) levels and pyruvate kinase isoenzyme type M2 (M2-PK) activity was very low. When a stable oncogenic ras and HPV-16 E7 expression were stablish in the NRK cells, an FBP up-regulation and M2-PK activity was detected, these results suggest, that oncogenic ras and E7 protein are the perfect conditions to create the ideal tumor metabolome as generally found in tumor cells [70,71,72]. Folic acid is necessary for the synthesis of S-adenosylmethionine, an elementary sustrate in the DNA methylation [73], but low folate levels increase the fragile sites on DNA which also decrease the DNA repair [74] and the DNA methylation process [75, 76], these conditions enhance the risk of DNA attacks by virus and carcinogens [77, 78], which also include HPV [79]. The global DNA methylation increase in the cervical tissue, increase the grade of cervical dysplasia, suggesting that the methylation status is an early event in the cervical transformation mechanisms. About genital HPV types, DNA methylation in the regulatory region, regulate ‘in vitro’ HR-HPV expression [80, 81]. One of these ‘in-vitro’ studies demonstrated that methylation of CpG sites in the HPV 18 enhancer region resulted in a down-regulation of transcriptional activity [80]. Other study demonstrates the methylation was found to be more at CpGs within E2 binding sites proximal to the P97 promoter, which means that the E2 binding site methylation in presence of intact E2, cause to loss of E2 repressor activity [82]. The reactive oxygen species (ROS) and their down-regulation by anti-oxidants is the other metabolic point that have relevance during the process of HPV infection. Activation of AP-1 (main transcription factor for the expression of E6 and E7 proteins of HR-HPVs) is down regulated by antioxidants in ‘in vitro’ models [83,84,85] and recently by other dietary molecules such as Curcumin (diferuloylmethane), which is an active component of the perennial herb turmeric and a potent antioxidant and is well-known for its anti-inflammatory and anti-carcinogenic activity [86]. Experimental evidence demonstrated that pyrrolidine-dithiocarbamate (antioxidant) selectively inhibit AP-1-induced by HPV 16 gene expression in immortalized human keratinocytes, suggesting that redox potential manipulation could be a therapeutic approach to interfere with the HR-HPVs transformation mechanisms [87]. Surprisingly, other study demonstrates that using curcumin (diferuloylmethane) in HeLa cells culture it was possible to modulate the transcription of AP-1 and HPV [88, 89]. Retinoic acid indirectly reduces HPV mRNA levels by modification of AP-1 activity [90] and/or transforming growth factor β (TGFβ) expression [91]. The property of cell growth suppression by retinoic acid is lost in the latest stages of HPV 16-induced transformation in cervical tumor cell lines and keratinocytes, the mechanism includes loss of growth inhibition and TGF β sensitivity [92], continued growth stimulation [93, 94] and loss of retinoid receptor expression [95]. In the serum, All Trans Retinoic Acid (ATRA) level highly influences the progression of cervical lesions to invasive cancer [96]. The therapy with retinoic acid does not reduce recurrence rates of advanced cervical cancer [97,98,99] or the cervical intra-neoplasia grade 3 (CIN3) regression [100], even when is combined with chemotherapy and immunotherapy [101]. All these data suggest that retinoic acid could be effective only in the early stages of cervical dysplasia lesions, modulating the clearance and persistence of HPV. The sequence of events required for the establishment of the tumor metabolome in cervical cancer is presently unknown, but it is clear the participation of specific metabolic pathways, specific modulation of metabolites and the HPV infection event during the malignant transformation mechanisms of the cervical epithelium.
3.6 Cervical Host Infections Patterns (Oncobioma) of Malignant Precursor Lesions and Tumor Microenvironment as Promoters and Enhancers of HPV Transforming Mechanisms
Microbiome research has presented an unprecedented growth over the last decade, due to the great developments in the new DNA sequencing technologies such as Next Generation Sequencing (NGS) [102]. The human microbiome study has been focused on health and disease, the clinical interpretation; however, the ability to understand these studies in the context of disease is less straightforward. Pathological conditions such as cancer have seen an increase in research focused on the microbiome pathogenic role, but the clinical value to interpret and/or use these scientific findings are not still translated. The purpose of this chapter section is to provide an introduction for clinicians to learn about how microbiome research and HPV positive cervical cancer could be associated. Microbiota considers a wide variety of microorganisms (bacteria, viruses, protozoa, fungi, and archaea) and the eclectic ecosystem of every individual, creating a commensal, symbiotic, and pathobiont (microorganisms that normally behave in a symbiotic manner with their host but exhibit pathogenic potential based on changes in their abundance or environmental conditions) relationship that has generated focus on its role in carcinogenesis [103]. The new scientific research focused on the interplay between the human microbiome and cancer development, has been termed the ‘oncobiome’ (the intricate interplay and study of the human microbiome and its influence on cancer development) [103]. It is clear, that these preliminary studies have demonstrated associative relationships rather than causative ones. But the question of whether this emerging field of research is a ‘landscape’ without a clear picture yet or it represents a new paradigm for cancer research such as other authors and we refer [104]. We propose the scientific evidence to answer the question and to push the new paradigm forward to bring a new perspective to understand and treat cancer. The mechanisms proposed in which infectious agents are suggested as associated co-factors in HPV malignant transformation is by direct biological interactions, such as modification of HPV replication and transcription, and/or indirect effects, such as inflammation and damage to the epithelial barrier that protects against HPV infection to facilitate the virus access to target epithelial cells. In the 1970’s laboratory studies demonstrated the ability of Herpes simplex virus-1 (HSV-1) and Herpes simplex virus-2 (HSV-2) to transform hamster cells [105]. The inconsistent HSV DNA detection in human cervical cancer samples created the ‘hit and run’ hypotheses, which means that a virus may be involved in the initiation or promotion of malignant transformation, but is not required for the maintenance of the transformed phenotype [106]. HSV-2 is an infectious agent that has been studied as a potential co-factor for cervical cancer. Several studies have demonstrated an interaction between HSV-2 and HPV in ‘in vitro’ transformation [107]. HSV-2 can suppress HPV gene expression [108]. HSV induce tumorigenic clones in keratinocytes that had been immortalized by HPV [108]. HSV is a co-factor in HPV-associated cervical transforming mechanisms, not an etiological agent. Thus, laboratory data and epidemiological data are not consistent with a possible interaction of HSV-2 in HPV-associated epithelial transformation. A study of 200 human cervical cancer specimens failed to detect HSV-2 sequences using sensitive PCR methods [110]. HSV infection downregulate the secretory leukocyte protease inhibitor (SLPI) levels and may impart a greater susceptibility for HPV16 infection by the annexin A2 heterotetramer cell receptor (A2t), providing a mechanism to explain the etiological link between HSV and HPV-associated cancers [111]. Other herpesviruses are reported to infect the cervix and have been demonstrated to transform epithelial cells in ‘in vitro’ models, which include cytomegalovirus (CMV) [112], human herpesvirus 6 (HHV-6) [113] and Epstein-Barr virus (EBV) [114], the co-infection with herpesviruses, especially CMV and EBV, may be involved in the integration of the HPV-16 genome and may contribute to the development of cervical cancer [115], but unfortunately there is no strong evidence of the potential role of these viruses in the HPV transforming mechanisms. On the other hand, Adeno-associated virus (AAV) may have a protective effect against HPV-associated transforming mechanisms. AAV is a helper-dependent parvovirus that needs for its replication the co-infection with other DNA viruses, such as adenovirus [116]. In ‘in vitro’ models, AAV inhibits the transforming effect of HPV and furthermore, HPV can support replication of AAV, a finding that is consistent with possible HPV/AAV co-infection in nature [117]. Scientific reports found that AAV suppressed papillomavirus replication by its protein Rep 78 (an AAV major non-structural regulatory protein), and interferes with transcription factors and HPV promoter activity [118]. Nevertheless, AAV high levels decreased HPV replication, low levels increased it, and certain conditions increase the HPV transforming capacity [117]. By direct interaction between AAV proteins and cellular genes, AAV induces tumor cell differentiation, down-regulates c-Fos and c-Myc genes, inhibits cell proliferation and reduces carcinogen-induced mutagenicity [119,120,121]. Finally, extensive experimental and limited epidemiological evidence suggests that adeno-associated viruses (AAV) may have anti-oncogenic activity and has also anti-neoplastic effects that are independent of its proposed biological interaction with HPV [122]. Cervical cancer is increased in women who have human immunodeficiency virus (HIV) [123]; HIV-positive patient biomarkers, such as, HIV RNA level and CD4+ T-cell count, are associated with HPV infection risk and cervical cancer. It has been demonstrated by ‘in vitro’ models that epithelial cells can be infected by HIV [124, 125], and also have shown that HIV TAT protein can co-activate HPV [126, 127]; but unfortunately, there is not the same correlation when it is used an ‘in vivo’ infection model system [128,129,130]. The association between multiple HPV infection, low CD4 count and cytological abnormalities supports the interplay of virological and immunological factors in cervical cancer pathogenesis [131, 132]. HPV infection may predispose to HIV infection and facilitate its progression probably by interaction with HIV proteins enhancing effectiveness of HPV proteins, and perhaps contributing to cell cycle disruption [133]. At this time, it looks improbable that HPV-infected cervical epithelial cells could be co-infected with HIV, which limits the idea that both viruses interact biologically at a molecular level. A microbial agent that has been associated with HPV infected cervical cancer patients by epidemiological studies is Chlamydia trachomatis (C. trachomatis). Different proposed mechanisms by which C. trachomatis increases the risk for cervical cancer have been described such as: (i) an anti-apoptotic effect, since infected cells are resistant to apoptosis by the C. trachomatis persistent infection [134, 135]. These anti-apoptotic effects result in an epithelial cells persistence and resistance that HPV co-infection follows the development of chromosomal abnormalities and increase the cervical dysplasia grade [136, 137]; (ii) its infection causes human cervical epithelial cells separate from each other due to the breakdown of the N-cadherin/β-catenin complex junctions in the epithelium and increases the basal cells exposure to HPV [138]; (iii) its infection is associated with squamous metaplasia and hypertrophic ectopy, which is a cervical neoplasia risk factor [139]; (iv) its persistent infection increases the HPV risk infection due to modulate immune factors like the inhibition of interferon (IFN) γ-inducible major histocompatibility complex (MHC) class II, as well as MHC class I expression [140, 141]; (v) by inhibition of NK cell function, decreasing the NK cells lytic capability, reducing TNFα and IFNγ production by NK cells and thereby decreasing antibody-dependent cellular cytotoxicity [142]; (vi) its chronic infection is associated with a predominantly T-helper (Th2) (humoral immune) cytokine pattern, whereas Th1 (cellular immune) cytokines participate in the control of intracellular microbes such as C. trachomatis and HPV [143]. A meta-analysis of HPV and C. thracomatis co-infection demonstrated that individuals infected with C. trachomatis have a higher risk of cervical cancer [144]. Based on this scientific evidence, it could be considered that it occurs an adaptive immune response to facilitate the HPV infection. MHC class I quantitative or qualitative alterations due to the presence of viral antigens can result in stimulation of natural killer (NK) cells that can kill a broad range of intracellular microbial infected cells without prior sensitization. During cervical inflammation, the immune response to microbial infection plays a role in HPV-associated tumorigenesis and explains the associations of precursor lesions and cervical cancer with a wide spectrum of pathogens, which include herpesviruses, C. trachomatis, Trichomonas vaginalis, Neisseria gonorrhoeae, Candida albicans and others [145]. The mechanisms by which inflammation might cause an increased risk for cervical cancer have been best described for C. trachomatis. Many of the cytokines that are secreted during C. trachomatis infection, including TNFα and IFNγ, could cause tissue damage by inducing apoptosis of uninfected cells; infiltrating macrophages releasing reactive oxygen species causes a mayor tissue damage [135, 145]. These effects result in partial disruption of the tissue barrier and exposure of basal cells to HPV infection. Furthermore, the ROS released by infiltrating macrophages could cause host cell DNA damage and increase the risk for malignant precursor lesions and cervical cancer [146, 147], these proposed mechanisms have evolved from the association between inflammatory host responses and oxidative DNA damage [148]. There is no scientific evidence to have a direct effect of C. trachomatis on host DNA or on the transcription of HPV genes though. A Korean group recently report that the predominance of A. vaginae, G. vaginalis and L. iners with a concomitant paucity of L. crispatus in the cervical microbiota was associated with CIN risk, suggesting that bacterial dysbiosis and its combination with oncogenic HPV may be a risk factor for cervical neoplasia [149]. The following is one of the last observations of the associated mechanisms of HPV multiple infections we want to refer to and what is the potential roles of the microbiome in cervicovaginal diseases. A few years ago, in our institutions we were evaluating an anti-HPV topical drug and using the most advanced methods, we had to detect the HPV pattern infection of these patients; we found a complex association of multiple HR-HPV infections in patients according to the dysplasia grade, which means higher dysplasia grade higher number of HR-HPV types associated with the lesion [150]. We discuss the interpretation of the scientific evidence in a biological and clinical context, for analysis and future discussion and direction for microbiome in cancer research and particularly in human cancer associated to viral pathogens. The genomic medicine for the routine clinical use should be seen as a blueprint for the microbiome or better understood as ‘oncobioma’. These scientific evidences suggest that microbial dysbiosis (a biological state whereby host microbial composition is unbalanced toward other micro-organisms compared to ‘healthy’ host composition) is associated with malignant transformation. Whether these associations are causative and can therefore modulate initiation, progression, or cancer metastasis at this moment remains unclear.
References
Pfister H, Fuchs PG. Anatomy, taxonomy and evolution of papillomaviruses. Intervirology. 1994;37:143–9.
Orth G, Jablonska S, Breitburd F, Favre M, Croissant O. The human papillomaviruses. Bull Cancer. 1978;65:151–64.
Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 2011;19(1):33–9.
Moody CA, Laimins LA. Human Papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–60.
Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. Identification of the 6 integrin as a candidate receptor for papillomaviruses. J Virol. 1997;71:2449–56.
Stubenrauch F, Laimins LA. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol. 1999;9:379–86.
McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci U S A. 1988;85:7169–73.
Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–8.
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505.
Jones DL, Münger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71:2905–12.
Rosenberger S, De-Castro Arce J, Langbein L, Steenbergen RD, Rösl F. Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc Natl Acad Sci U S A. 2010;107(15):7006–11.
Münger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–21.
Münger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888–98.
Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18:2449–58.
Peralta-Zaragoza O, Bermudez-Morales V, Gutierrez-Xicotencatl L, Alcocer-Gonzalez J, Recillas-Targa F, Madrid-Marina V. E6 and E7 oncoproteins from human papillomavirus type 16 induce activation of human transforming growth factor beta1 promoter throughout Sp1 recognition sequence. Viral Immunol. 2006;19(3):468–80.
Bello JO, Nieva LO, Paredes AC, Gonzalez AM, Zavaleta LR, Lizano M. Regulation of the Wnt/β-catenin signaling pathway by human papillomavirus E6 and E7 oncoproteins. Viruses. 2015;7(8):4734–55.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–41.
Astudillo de la Vega H, Benítez-Bribiesca L. Is it possible to create human malignant cells in the laboratory? Gac Med Mex. 2000;136(2):173–4.
Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22:1225–37.
Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, von Knebel Doeberitz M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene. 2003;22:3977–84.
McBride AA, Romanczuk H, Howley PM. The papillomavirus E2 regulatory proteins. J Biol Chem. 1991;266:18411–4.
Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol. 1997;71:9343–9.
Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci U S A. 1992;89:5799–803.
Skiadopoulos MH, McBride AA. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J Virol. 1998;72:2079–88.
You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117(3):349–60.
Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and translational analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol. 1987;61:962–71.
Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995;92:1654–8.
Alazawi W, Pett M, Arch B, Scott L, Freeman T, Stanley MA, Coleman N. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res. 2002;62:6959–65.
Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–8.
Hopman AH, Smedts F, Dignef W, Ummelen M, Sonke G, Mravunac M, Vooijs GP, Speel EJ, Ramaekers FC. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J Pathol. 2004;202(1):23–33.
Kessis TD, Connolly DC, Hedrick L, Cho KR. Expression of HPV16 E6 or E7 increases integration of foreign DNA. Oncogene. 1996;13:427–31.
Reidy PM, Dedo HH, Rabah R, Field JB, Mathog RH, Gregoire L, Lancaster WD. Integration of human papillomavirus type 11 in recurrent respiratory papilloma-associated cancer. Laryngoscope. 2004;114(11):1906–9.
Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314(6006):111–4.
Hudelist G, Manavi M, Pischinger KI, Watkins-Riedel T, Singer CF, Kubista E, Czerwenka KF. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol Oncol. 2004;92(3):873–80.
Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, Nagorney DM, Gostout BS, Burgart LJ, Boix L, Bruix J, McMahon BJ, Cheung TH, Chung TK, Wong YF, Smith DI, Roberts LR. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22(24):3813–20.
Ferber MJ, Thorland EC, Brink AA, Rapp AK, Phillips LA, McGovern R, Gostout BS, Cheung TH, Chung TK, Fu WY, Smith DI. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene. 2003;22(46):7233–42.
Butler D, Collins C, Mabruk M, Leader MB, Kay EW. Loss of Fhit expression as a potential marker of malignant progression in preinvasive squamous cervical cancer. Gynecol Oncol. 2002;86(2):144–9.
Segawa T, Sasagawa T, Yamazaki H, Sakaike J, Ishikawa H, Inoue M. Fragile histidine triad transcription abnormalities and human papillomavirus E6-E7 mRNA expression in the development of cervical carcinoma. Cancer. 1999;85(9):2001–10.
Wentzensen N, Vinokurova S, von Knebel DM. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004;64(11):3878–84.
zur Hausen H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin Cancer Biol. 1999;9:405–11.
Klausner RD. The fabric of cancer cell biology—weaving together the strands. Cancer Cell. 2002;1:3–10.
Zimonjic D, Brooks MW, Popescu N, Weinberg RA, Hahn WC. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001;61:8838–44.
Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and Darwinian selection in tumours. Trends Cell Biol. 1999;9:M57–60.
Schütze DM, Krijgsman O, Snijders PJ, Ylstra B, Weischenfeldt J, Mardin BR, Stütz AM, Korbel JO, de Winter JP, Meijer CJ, Quint WG, Bosch L, Wilting SM, Steenbergen RD. Immortalization capacity of HPV types is inversely related to chromosomal instability. Oncotarget. 2016;7:37608–21. doi:10.18632/oncotarget.8058.
Habermann JK, Hellman K, Freitag S, Heselmeyer-Haddad K, Hellstrom AC, Shah K, Auer G, Ried T. A recurrent gain of chromosome arm 3q in primary squamous carcinoma of the vagina. Cancer Genet Cytogenet. 2004;148:7–13.
Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54.
Duensing S, Münger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331–5.
Riley RR, Duensing S, Brake T, Münger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71.
Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109:157–62.
Duensing S, Duensing A, Crum CP, Münger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356–60.
Thomas JT, Laimins LA. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J Virol. 1998;72:1131–7.
Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, Kisseljov F, Dürst M, Schneider A, von Knebel DM. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008;68(1):307–13.
Hopman AH, Theelen W, Hommelberg PP, Kamps MA, Herrington CS, Morrison LE, Speel EJ, Smedts F, Ramaekers FC. Genomic integration of oncogenic HPV and gain of the human telomerase gene TERC at 3q26 are strongly associated events in the progression of uterine cervical dysplasia to invasive cancer. J Pathol. 2006;210(4):412–9.
Blasco MA, Hahn WC. Evolving views of telomerase and cancer. Trends Cell Biol. 2003;13:289–94.
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–8.
Ocadiz R, Sauceda R, Cruz M, Graef AM, Gariglio P. High correlation between molecular alterations of the c-myc oncogene and carcinoma of the uterine cervix. Cancer Res. 1987;47(15):4173–7.
Abba MC, Laguens RM, Dulout FN, Golijow CD. The c-myc activation in cervical carcinomas and HPV 16 infections. Mutat Res. 2004;557(2):151–8.
Dürst M, Croce CM, Gissmann L, Schwarz E, Huebner K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc Natl Acad Sci U S A. 1987;84(4):1070–4.
Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci U S A. 2003;100(14):8211–6.
McMurray HR, McCance DJ. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J Virol. 2003;77(18):9852–61.
Gewin L, Galloway DA. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J Virol. 2001;75(15):7198–201.
Liu X, Dakic A, Zhang Y, Dai Y, Chen R, Schlegel R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc Natl Acad Sci U S A. 2009;106(44):18780–5.
Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004;18(18):2269–82.
Fu B, Quintero J, Baker CC. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res. 2003;63(22):7815–24.
McMurray HR, McCance DJ. Degradation of p53, not telomerase activation, by E6 is required for bypass of crisis and immortalization by human papillomavirus type 16 E6/E7. J Virol. 2004;78(11):5698–706.
Van Doorslaer K, Burk RD. Association between hTERT activation by HPV E6 proteins and oncogenic risk. Virology. 2012;433(1):216–9.
Oltvai ZN, Barabási AL. Systems biology. Life's complexity pyramid. Science. 2002;298(5594):763–4.
Hughes TR, Robinson MD, Mitsakakis N, Johnston M. The promise of functional genomics: completing the encyclopedia of a cell. Curr Opin Microbiol. 2004;7(5):546–54.
Mazurek S, Zwerschke W, Jansen-Dürr P, Eigenbrodt E. Metabolic cooperation between different oncogenes during cell transformation: interaction between activated ras and HPV-16 E7. Oncogene. 2001;20(47):6891–8.
Mazurek S, Eigenbrodt E. The tumor metabolome. Anticancer Res. 2003;23(2A):1149–54.
Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43(7):969–80.
Poirier LA. The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J Nutr. 2002;132(8 Suppl):2336S–9S.
Ames BN, Wakimoto P. Are vitamin and mineral deficiencies a major cancer risk? Nat Rev Cancer. 2002;2(9):694–704.
Wainfan E, Kilkenny M, Dizik M. Comparison of methyltransferase activities of pair-fed rats given adequate or methyl-deficient diets. Carcinogenesis. 1988;9(5):861–3.
Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, Selhub J, Mason JB. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer. 1994;74(3):893–9.
Hsieh LL, Wainfan E, Hoshina S, Dizik M, Weinstein IB. Altered expression of retrovirus-like sequences and cellular oncogenes in mice fed methyl-deficient diets. Cancer Res. 1989;49(14):3795–9.
Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med (Maywood). 2004;229(10):988–95.
Flatley JE, Sargent A, Kitchener HC, Russell JM, Powers HJ. Tumour suppressor gene methylation and cervical cell folate concentration are determinants of high-risk human papillomavirus persistence: a nested case control study. BMC Cancer. 2014;14:803. doi:10.1186/1471-2407-14-803.
Rösl F, Arab A, Klevenz B, zur Hausen H. The effect of DNA methylation on gene regulation of human papillomaviruses. J Gen Virol. 1993;74(Pt 5):791–801.
Thain A, Jenkins O, Clarke AR, Gaston K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J Virol. 1996;70(10):7233–5.
Bhattacharjee B, Sengupta S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology. 2006;354(2):280–5.
Cripe TP, Alderborn A, Anderson RD, Parkkinen S, Bergman P, Haugen TH, Pettersson U, Turek LP. Transcriptional activation of the human papillomavirus-16 P97 promoter by an 88-nucleotide enhancer containing distinct cell-dependent and AP-1-responsive modules. New Biol. 1990;2(5):450–63.
Offord EA, Beard P. A member of the activator protein 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. J Virol. 1990;64(10):4792–8.
Velazquez Torres A, Gariglio Vidal P. Possible role of transcription factor AP1 in the tissue-specific regulation of human papillomavirus. Rev Invest Clin. 2002;54(3):231–42.
Mishra A, Kumar R, Tyagi A, Kohaar I, Hedau S, Bharti AC, Sarker S, Dey D, Saluja D, Das B. Curcumin modulates cellular AP-1, NF-kB, and HPV16 E6 proteins in oral cancer. Ecancermedicalscience. 2011;9:525.
Rösl F, Das BC, Lengert M, Geletneky K, zur Hausen H. Antioxidant-induced changes of the AP-1 transcription complex are paralleled by a selective suppression of human papillomavirus transcription. J Virol. 1997;71(1):362–70.
Prusty BK, Das BC. Constitutive activation of transcription factor AP-1 in cervical cancer and suppression of human papillomavirus (HPV) transcription and AP-1 activity in HeLa cells by curcumin. Int J Cancer. 2005;113(6):951–60.
Date AA, Destache CJ. Natural polyphenols: potential in the prevention of sexually transmitted viral infections. Drug Discov Today. 2016;21(2):333–41.
Schüle R, Rangarajan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM. Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci U S A. 1991;88(14):6092–6.
Batova A, Danielpour D, Pirisi L, Creek KE. Retinoic acid induces secretion of latent transforming growth factor beta 1 and beta 2 in normal and human papillomavirus type 16-immortalized human keratinocytes. Cell Growth Differ. 1992;3(11):763–72.
Borger DR, Mi Y, Geslani G, Zyzak LL, Batova A, Engin TS, Pirisi L, Creek KE. Retinoic acid resistance at late stages of human papillomavirus type 16-mediated transformation of human keratinocytes arises despite intact retinoid signaling and is due to a loss of sensitivity to transforming growth factor-beta. Virology. 2000;270(2):397–407.
Sizemore N, Choo CK, Eckert RL, Rorke EA. Transcriptional regulation of the EGF receptor promoter by HPV16 and retinoic acid in human ectocervical epithelial cells. Exp Cell Res. 1998;244(1):349–56.
Rorke EA, Zhang D, Choo CK, Eckert RL, Jacobberger JW. TGF-beta-mediated cell cycle arrest of HPV16-immortalized human ectocervical cells correlates with decreased E6/E7 mRNA and increased p53 and p21(WAF-1) expression. Exp Cell Res. 2000;259(1):149–57.
Bartsch D, Boye B, Baust C, zur Hausen H, Schwarz E. Retinoic acid-mediated repression of human papillomavirus 18 transcription and different ligand regulation of the retinoic acid receptor beta gene in non-tumorigenic and tumorigenic HeLa hybrid cells. EMBO J. 1992;11(6):2283–91.
Berlin Grace VM, Niranjali Devaraj S, Radhakrishnan Pillai M, Devaraj H. HPV-induced carcinogenesis of the uterine cervix is associated with reduced serum ATRA level. Gynecol Oncol. 2006;103(1):113–9.
Wadler S, Schwartz EL, Haynes H, Rameau R, Quish A, Mandeli J, Gallagher R, Hallam S, Fields A, Goldberg G, McGill F, Jennings S, Wallach RC, Runowicz CD. All-trans retinoic acid and interferon-alpha-2a in patients with metastatic or recurrent carcinoma of the uterine cervix: clinical and pharmacokinetic studies. New York Gynecologic Oncology Group. Cancer. 1997;79(8):1574–80.
Weiss GR, Liu PY, Alberts DS, Peng YM, Fisher E, Xu MJ, Scudder SA, Baker Jr LH, Moore DF, Lippman SM. 13-cis-retinoic acid or all-trans-retinoic acid plus interferon-alpha in recurrent cervical cancer: a Southwest Oncology Group phase II randomized trial. Gynecol Oncol. 1998;71(3):386–90.
Keefe KA, Schell MJ, Brewer C, McHale M, Brewster W, Chapman JA, Rose GS, McMeeken DS, Lagerberg W, Peng YM, Wilczynski SP, Anton-Culver H, Meyskens FL, Berman ML. A randomized, double blind, Phase III trial using oral beta-carotene supplementation for women with high-grade cervical intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev. 2001;10(10):1029–35.
Meyskens Jr FL, Surwit E, Moon TE, Childers JM, Davis JR, Dorr RT, Johnson CS, Alberts DS. Enhancement of regression of cervical intraepithelial neoplasia II (moderate dysplasia) with topically applied all-trans-retinoic acid: a randomized trial. J Natl Cancer Inst. 1994;86(7):539–43.
Braud AC, Gonzague L, Bertucci F, Genre D, Camerlo J, Gravis G, Goncalves A, Moutardier V, Viret F, Maraninchi D, Viens P. Retinoids, cisplatin and interferon-alpha in recurrent or metastatic cervical squamous cell carcinoma: clinical results of 2 phase II trials. Eur Cytokine Netw. 2002;13(1):115–20.
Ash C, Mueller K. Manipulating the Microbiota. Science. 2016;352(6285):530–1.
Thomas RM, Jobin C. The microbiome and cancer: is the ‘Oncobiome’ mirage real? Trends Cancer. 2015;1(1):24–35.
de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15.
Duff R, Rapp F. Properties of hamster embryo fibroblasts transformed in vitro after exposure to ultraviolet-irradiated herpes simplex virus type 2. J Virol. 1971;8(4):469–77.
Galloway DA, McDougall JK. The oncogenic potential of herpes simplex viruses: evidence for a ‘hit-and-run’ mechanism. Nature. 1983;302(5903):21–4.
Dhanwada KR, Garrett L, Smith P, Thompson KD, Doster A, Jones C. Characterization of human keratinocytes transformed by high risk human papillomavirus types 16 or 18 and herpes simplex virus type 2. J Gen Virol. 1993;74(Pt 6):955–63.
Fang L, Ward MG, Welsh PA, Budgeon LR, Neely EB, Howett MK. Suppression of human papillomavirus gene expression in vitro and in vivo by herpes simplex virus type 2 infection. Virology. 2003;314(1):147–60.
DiPaolo JA, Woodworth CD, Popescu NC, Koval DL, Lopez JV, Doniger J. HSV-2-induced tumorigenicity in HPV16-immortalized human genital keratinocytes. Virology. 1990;177(2):777–9.
Tran-Thanh D, Provencher D, Koushik A, Duarte-Franco E, Kessous A, Drouin P, Wheeler CM, Dubuc-Lissoir J, Gauthier P, Allaire G, Vauclair R, Dipaolo JA, Gravitt P, Franco E, Coutlée F. Herpes simplex virus type II is not a cofactor to human papillomavirus in cancer of the uterine cervix. Am J Obstet Gynecol. 2003;188(1):129–34.
Skeate JG, Porras TB, Woodham AW, Jang JK, Taylor JR, Brand HE, Kelly TJ, Jung JU, Da Silva DM, Yuan W, Kast WM. Herpes simplex virus downregulation of secretory leukocyte protease inhibitor enhances human papillomavirus type 16 infection. J Gen Virol. 2016;97(2):422–34.
Doniger J, Muralidhar S, Rosenthal LJ. Human cytomegalovirus and human herpesvirus 6 genes that transform and transactivate. Clin Microbiol Rev. 1999;12:367–82.
Kashanchi F, Araujo J, Doniger J, Muralidhar S, Hoch R, Khleif S, Mendelson E, Thompson J, Azumi N, Brady JN, Luppi M, Torelli G, Rosenthal LJ. Human herpesvirus 6 (HHV-6) ORF-1 transactivating gene exhibits malignant transforming activity and its protein binds to p53. Oncogene. 1997;14:359–67.
Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin Cancer Res. 2004;10:803–21.
Szostek S, Zawilinska B, Kopec J, Kosz-Vnenchak M. Herpesviruses as possible cofactors in HPV-16-related oncogenesis. Acta Biochim Pol. 2009;56(2):337–42.
Leonard CJ, Berns KI. Adeno-associated virus type 2: a latent life cycle. Prog Nucleic Acid Res mol Biol. 1994;48:29–52.
Meyers C, Alam S, Mane M, Hermonat PL. Altered biology of adeno-associated virus type 2 and human papillomavirus during dual infection of natural host tissue. Virology. 2001;287:30–9.
Prasad CK, Meyers C, Zhan DJ, You H, Chiriva-Internati M, Mehta JL, Liu Y, Hermonat PL. The adeno-associated virus major regulatory protein Rep78-c-Jun-DNA motif complex modulates AP-1 activity. Virology. 2003;314:423–31.
Bantel-Schaal U. Growth properties of a human melanoma cell line are altered by adenoassociated parvovirus type 2. Int J Cancer. 1995;60:269–74.
Walz C, Schlehofer JR. Modification of some biological properties of HeLa cells containing adeno-associated virus DNA integrated into chromosome 17. J Virol. 1992;66:2990–3002.
Schlehofer JR, Heilbronn R. Infection with adeno-associated virus type 5 inhibits mutagenicity of herpes simplex virus type 1 or 4-nitroquinoline-1-oxide. Mutat Res. 1990;244:317–20.
Al-Daraji WI, Smith JH. Infection and cervical neoplasia: facts and fiction. Int J Clin Exp Pathol. 2009;2(1):48–64.
Mbulaiteye SM, Biggar RJ, Goedert JJ, Engels EA. Immune deficiency and risk for malignancy among persons with AIDS. J Acquir Immune Defic Syndr. 2003;32:527–33.
Moore JS, Rahemtulla F, Kent LW, Hall SD, Ikizler MR, Wright PF, Nguyen HH, Jackson S. Oral epithelial cells are susceptible to cell-free and cell-associated HIV-1 infection in vitro. Virology. 2003;313:343–53.
Yeaman GR, Howell AL, Weldon S, Demian DJ, Collins JE, O'Connell DM, Asin SN, Wira CR, Fanger MW. Human immunodeficiency virus receptor and coreceptor expression on human uterine epithelial cells: Regulation of expression during the menstrual cycle and implications for human immunodeficiency virus infection. Immunology. 2003;109:137–46.
Vernon SD, Hart CE, Reeves WC, Icenogle JP. The HIV-1 tat protein enhances E2dependent human papillomavirus 16 transcription. Virus Res. 1993;27:133–45.
Buonaguro FM, Tornesello ML, Buonaguro L, Del Gaudio E, Beth-Giraldo E, Giraldo G. Role of HIV as cofactor in HPV oncogenesis: in vitro evidences of virus interactions. Antibiot Chemother. 1994;46:102–9.
Greenhead P, Hayes P, Watts PS, Laing KG, Griffin GE, Shattock RJ. Parameters of human immunodeficiency virus infection of human cervical tissue and inhibition by vaginal virucides. J Virol. 2000;74:5577–86.
Miller CJ, Shattock RJ. Target cells in vaginal HIV transmission. Microbes Infect. 2003;5:59–67.
Wu Z, Chen Z, Phillips DM. Human genital epithelial cells capture cell-free human immunodeficiency virus type 1 and transmit the virus to CD4+ cells: Implications for mechanisms of sexual transmission. J Infect Dis. 2003;188:1473–82.
Garbuglia AR, Piselli P, Lapa D, Sias C, Del Nonno F, Baiocchini A, Cimaglia C, Agresta A, Capobianchi MR. Frequency and multiplicity of human papillomavirus infection in HIV-1 positive women in Italy. J Clin Virol. 2012;54(2):141–6.
Strickler HD, Burk RD, Fazzari M, Anastos K, Minkoff H, Massad LS, Hall C, Bacon M, Levine AM, Watts DH, Silverberg MJ, Xue X, Schlecht NF, Melnick S, Palefsky JM. Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J Natl Cancer Inst. 2005;97(8):577–86.
Clarke B, Chetty R. Postmodern cancer: the role of human immunodeficiency virus in uterine cervical cancer. Mol Pathol. 2002;55(1):19–24.
Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. Inhibition of apoptosis in chlamydia-infected cells: Blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med. 1998;187:487–96.
Perfettini JL, Hospital V, Stahl L, Jungas T, Verbeke P, Ojcius DM. Cell death and inflammation during infection with the obligate intracellular pathogen, Chlamydia. Biochimie. 2003;85:763–9.
Lorenzato M, Clavel C, Masure M, Nou JM, Bouttens D, Evrard G, Bory JP, Maugard B, Quereux C, Birembaut P. DNA image cytometry and human papillomavirus (HPV) detection help to select smears at high risk of high-grade cervical lesions. J Pathol. 2001;194:171–6.
Melsheimer P, Klaes R, von Knebel-Doeberitz M, Bastert G. Prospective clinical study comparing DNA flow cytometry and HPV typing as predictive tests for persistence and progression of CIN I/II. Cytometry. 2001;46:166–71.
Prozialeck WC, Fay MJ, Lamar PC, Pearson CA, Sigar I, Ramsey KH. Chlamydia trachomatis disrupts N-cadherin-dependent cell–cell junctions and sequesters β-catenin in human cervical epithelial cells. Infect Immun. 2002;70:2605–13.
Moscicki AB, Burt VG, Kanowitz S, Darragh T, Shiboski S. The significance of squamous metaplasia in the development of low grade squamous intraepithelial lesions in young women. Cancer. 1999;85:1139–44.
Zhong G, Liu L, Fan T, Fan P, Ji H. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon γ-inducible major histocompatibility complex class I expression in chlamydia-infected cells. J Exp Med. 2000;191:1525–34.
Hook CE, Telyatnikova N, Goodall JC, Braud VM, Carmichael AJ, Wills MR, Gaston JSH. Effects of Chlamydia trachomatis infection on the expression of natural killer (NK) cell ligands and susceptibility to NK cell lysis. Clin Exp Immunol. 2004;138:54–60.
Mavoungou E, Poaty-Mavoungou V, Touré FS, Sall A, Delicat A, Yaba P, Mandeme Y, Nabias R, Lansoud-Soukate J. Impairment of natural killer cell activity in Chlamydia trachomatis infected individuals. Trop Med Int Health. 1999;4:719–27.
Stephens RS. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol. 2003;11:44–51.
Zhu H, Shen Z, Luo H, Zhang W, Zhu X. Chlamydia Trachomatis Infection-Associated Risk of Cervical Cancer: A Meta-Analysis. Medicine (Baltimore). 2016;95(13):e3077.
Castle PE, Giuliano AR. Genital tract infections, cervical inflammation, and antioxidant nutrients — Assessing their roles as human papillomavirus cofactors. J Natl Cancer Inst Monogr. 2003;31:29–34.
Gravitt PE, Castle PE. Chlamydia trachomatis and cervical squamous cell carcinoma (Letter to the Editor). JAMA. 2001;285:1703–4.
Smith JS, Bosetti C, Muñoz N, Herrero R, Bosch FX, Eluf-Neto J, Meijer CJLM, van den Brule AJC, Franceschi S, Peeling RW. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case–control study. Int J Cancer. 2004;111:431–9.
Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124(5):1408–19.
Oh HY, Kim BS, Seo SS, Kong JS, Lee JK, Park SY, Hong KM, Kim HK, Kim MK. The association of uterine cervical microbiota with an increased risk for cervicalintraepithelial neoplasia in Korea. Clin Microbiol Infect. 2016;21(7):674.e1–9.
Munoz D, Cantu D, Gonzalez A, Meneses A, Mohar A, Astudillo-de la Vega H, Nguyen B. A phase II trial of the use of 4, 4'-dihydroxybenzophenone-2, 4-dinitrophenyl-hydrazone (A-007) topical gel in the treatment of high-grade squamous intraepithelial lesions (HSIL) of the cervix. ASCO Annual Meeting Proceedings. J Clin Oncol. 2006;25(18_suppl):5593.
Acknowledgment
The contributors of this chapter want to pay homage to Luis Benitez-Bribiesca M.D. who worked in our Institutions as Professor, Mentor, Colleague and Editor for over 40 years. We are extremely grateful to him for all his scientific efforts, teachings, advice and critiques, which were the translational pathway to take our scientific works from basic research to the clinic and patient care, such as he always claimed.
In Memoriam

Luis S. Benitez-Bribiesca M.D.
(1934–2015)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Astudillo-de la Vega, H., Ruiz-Garcia, E., Lopez-Camarillo, C., de la Garza-Salazar, J.G., Meneses-Garcia, A., Benitez-Bribiesca, L. (2017). Malignant Transforming Mechanisms of Human Papillomavirus. In: de la Garza-Salazar, J., Morales-Vásquez, F., Meneses-Garcia, A. (eds) Cervical Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-45231-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-45231-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45230-2
Online ISBN: 978-3-319-45231-9
eBook Packages: MedicineMedicine (R0)