
Abstract
Adult mammalian hearts lack significant regenerative potential, partially explaining why cardiomyopathies are a major cause of human death in the world. By contrast, adult lower vertebrates and neonatal mice can regenerate their heart after ischemic or physical injury. Significant efforts over the last several decades have led to advances in the understanding of cardiac biology and mechanisms of natural regeneration. For example, genetic lineage tracing evidence has shown that regenerative species use the expansion of pre-existing differentiated cardiomyocytes as a source of new myocardium. However, despite promising developments in basic science, therapeutic outcomes are modest with currently available therapy, underscoring the need for further discovery and translational research. Here, we review recent progress in the pursuit of human heart regeneration with a focus on cellular approaches. Highlights include somatic cell reprogramming, improved cardiac differentiation protocols, and a plethora of clinical trials, with some showing improvement in functional recovery after myocardial infarction.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Heart
- Regeneration
- Stem cell
- Cardiovascular disease
- Cardiac myocyte
- Heart failure
- Development
- Differentiation
6.1 Introduction
6.1.1 Human Heart Failure
Cardiomyopathies are a major cause of death throughout the world, due in part to the inability of the human heart to significantly regenerate. Improvements in the management of acute myocardial infarction (MI) have led to drastic improvements in short-term mortality rates since the 1960s [1]. However, due to a scarcity of effective long-term therapeutic options, the 5-year survival after diagnosis of heart failure is only 50 % [2]. Thus, heart failure remains an incurable condition and a major cause of death.
The etiology of heart failure is complex, but the syndrome is characterized by cardiac output that is insufficient to meet the metabolic demands of the body. A central complication of heart failure in general is the loss of cardiomyocytes through various cell death mechanisms (reviewed in [3]). In acute myocardial infarction, catastrophic cell death is incurred due to the occlusion of coronary vasculature, which deprives the infarcted region of oxygen and nutrient rich blood. Cardiomyocytes die from both apoptosis and necrosis, though the percent contribution of each death mechanism is unclear. Necrotic myocardium is eventually replaced by scar tissue, which lacks the contractile and elastic properties needed for optimal heart function. Ischemic reperfusion is thought to contribute to cell death [4] through inflammation [5], radical oxygen species generation [6], and abnormal calcium handling [7]. Strategies to mitigate peripheral myocardial cell death could potentially be implemented during surgical reperfusion [8, 9]. However, due to the acute lack of blood supply, reperfusion therapy is typically too late to save the dying infarcted myocardium, and fibrotic remodeling follows.
In chronic heart failure, cell death is thought to slowly contribute to deterioration of the ventricular myocardium, thus reducing its ability to effectively contract. This is further complicated in many cases by several aspects of remodeling, such as proliferation of fibroblasts, conversion to myofibroblasts [10], and accompanying alterations in extracellular matrix composition [11]. The re-expression of fetal-specific genes during heart failure has been described by several groups, including a switch from α-myosin heavy chain to β-myosin heavy chain (reviewed in [12]). Metabolic remodeling of cardiomyocytes is also seen in heart failure, such as a shift from fatty acid oxidation to glycolysis (reviewed in [13]). Collectively, these aspects of myocardial remodeling can result in gross morphological changes and associated alterations in tissue mechanics, such as myocardial stiffening, thickening or thinning of the ventricular myocardium, and ventricular dilation, as well as alterations in calcium handling and contractility; all of which can severely affect heart function and feedback on disease progression.
6.1.2 Species Variability in Heart Regeneration
Although adult mammals exhibit an insufficient natural ability to repair damaged myocardium, several lower vertebrates, such as zebrafish, newt, and axolotl, maintain a remarkable regenerative capacity, even in later stages of life. These species-specific differences in regenerative capacity (reviewed in [14, 15]) are an important topic of study in the pursuit of human regeneration. Due to the availability of transgenic models, zebrafish is the best characterized of these species. Mechanistically, genetic lineage tracing experiments show that zebrafish heart regeneration relies primarily on the dedifferentiation and expansion of pre-existing differentiated cardiomyocytes [16, 17]. Poss and colleagues showed this myocardial dedifferentiation involves re-expression of early developmental markers such as gata4 with an accompanying reduction in myocardial conduction velocity at the injury site [16]. Furthermore, a cryoinjury model demonstrated enhanced cell cycling in a fraction of cardiomyocytes expressing embryonic cardiac myosin heavy chain [18]. Epicardial signaling seems to play a role in the regenerative response to injury [19, 20], but myocyte contributions from epicardial cells directly are apparently limited. The role of a dynamic extracellular matrix was shown to be important in mediating zebrafish heart regeneration [21]. Specifically, fibronectin was upregulated in the myocardium following injury and was required for regeneration. Interestingly, fibronectin deposition in adult mammalian hearts has also been observed post-injury [22, 23], but may signal a fibrotic response in this context [24–26].
Several reports have also demonstrated a strong regenerative ability in adult newt [27–30] and axolotl hearts [31] using various injury models. Due to a lack of lineage tracing transgenic tools in these organisms, the source of new myocardium has not been definitively shown. However, Braun and colleagues showed a reduction in contractile protein expression after injury [32], reminiscent of the cardiomyocyte dedifferentiation observed in zebrafish heart regeneration [16, 17], suggesting a possible common mechanism. Not surprisingly, changes in extracellular matrix protein expression were also shown to accompany adult newt heart regeneration. Of particular interest, tenascin C was found to increase newt cardiomyocyte cell cycle re-entry in vitro [33]. However, evidence for cytokinesis was not shown. Interestingly, matrix production and remodeling enzymes were shown to change along with differentiation of immortalized CPCs in vitro, providing a direct link between the state of cardiomyocyte maturation and extracellular matrix remodeling [34].
Some reports have suggested that accelerated lower vertebrate regeneration is a consequence of cellular plasticity . For example, adult newt cardiomyocytes have been shown to transdifferentiate toward skeletal myocyte or chondrocyte lineages after transplantation into regenerating limb blastema [32]. Conversely, transdifferentiation was not observed during in vitro culture or after transplantation into intact limbs. It would be interesting to see if adult mammalian cardiomyocytes can be transdifferentiated by amphibian blastema ; this would indicate a conserved intrinsic regenerative program within vertebrate cardiomyocytes and a non-conserved extrinsic tissue response to injury.
Although adult mammalian hearts do not efficiently regenerate, Olson and colleagues showed in 2011 that neonatal mice (up to postnatal day 7) can regenerate their heart after apical resection [35]. Genetic lineage tracing experiments showed that, similar to zebrafish, the cardiomyocytes are repopulated by pre-existing cardiomyocytes. Immunostaining with anti-Troponin antibodies demonstrated sarcomeric disassembly in myocytes, again suggesting dedifferentiation and expansion of resident cardiomyocytes as a driver of regeneration. Notably, there has been some controversy over the extent of neonatal cardiac regeneration, where it has been suggested that neonatal hearts heal by scarring after apical resection [36]. However, several investigators report the reproducibility of neonatal heart regeneration in an apical resection model and have suggested technical differences as a source of variability [37]. Furthermore, it is not surprising that the severity of injury influences the efficiency of regeneration [38].
Whether or not neonatal hearts exhibit complete regeneration in response to injury, their apparent neomyogenic capacity is a major point of focus that could potentially be used clinically if similar mechanisms can be exploited in the adult myocardium. Thus, it is important to critically evaluate not only the functional recovery after MI, but also the extent of new cardiomyocyte generation in neonatal mice. To that end, cell cycle re-entry of neonatal cardiomyocytes has been thoroughly demonstrated. Soonpaa et al. used tritiated thymidine to demonstrate a spike in S-phase DNA synthesis in neonatal murine cardiomyocytes, beginning near birth and persisting throughout the first week of life [39]. The fraction of binucleated cardiomyocytes increased steadily during this period as the cells lost the ability to complete cytokinesis.

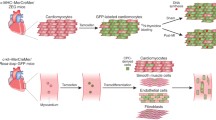
Autologous cellular approaches to cardiac regeneration. Promising sources of autologous patient cells for therapeutic cardiac regeneration include dermal fibroblasts and bone marrow cells, which can be delivered to the infarct via intracoronary (IC) or intramyocardial (IM) injection. Bone marrow cells are thought to act via paracrine effects to encourage regeneration. Fibroblasts can be converted directly to cardiomyocyte-like cells via GHMT or small molecules (SCPF) and Oct4. An expandable population of cardiac progenitors can be created using cell activated and signaling-directed (CASD) lineage conversion. CPCs and cardiomyocytes can also be created via embryonic stem cells created using somatic cell nuclear transfer (SCNT). (Inset) In vivo reprogramming can be used to convert resident cardiac fibroblasts into cardiomyocyte-like cells in situ using GHMT factors
In contrast to S-phase re-entry, the study of cell division is currently more technically challenging. Cytokinesis has traditionally been evaluated using antibodies against cleavage furrow markers such as Aurora B kinase. These techniques can be difficult to interpret with in vivo or in vitro samples, since staining in closely associated non-cardiomyocytes could contribute to false-positive results. This has led investigators to explore alternative methods, such as mosaic analysis with double markers (MADM) , to genetically trace divided cardiomyocytes [40]. Interestingly, pulsing of MADM transgenic mice with tamoxifen between postnatal day 2 and 8 revealed that 5 % of labeled MYH6-expressing cardiomyocytes had undergone cytokinesis, giving rise to single labeled (GFP+ or RFP+) cells. Due to differential sorting of chromosomes, as well as non-sortable labeling in G0/G1, this figure likely underestimates the actual rate of cytokinesis in labeled cardiomyocytes. Furthermore, it is unclear whether Cre-mediated interchromosomal recombination is unbiased with respect to different cellular states in the heterogeneous cardiomyocyte population. Thus, at this time it is difficult to quantify the actual rate of cardiomyocyte cell division. Nonetheless, it is generally accepted that a significant proportion of neonatal cardiomyocytes have the ability complete cell division and contribute to cardiac regeneration. However, by postnatal day 7, murine cardiomyocytes have mostly exited the cell cycle [39] and lost their ability to regenerate injured myocardium [35].
Interestingly, it has been suggested that altered cardiac circulation accompanies newt heart regeneration, where blood is shunted away from the left ventricle [41]. This is reminiscent of enhanced cardiomyocyte cell cycle and myocardial remodeling in patients with ventricular assist device [42, 43], where a reduction in load may allow partial induction of a regenerative response. It would be interesting to see if neonatal mice exhibit a similar phenomenon during cardiac regeneration. For example, although functional closure of the ductus arteriosus occurs within 3 h post-birth in mice, remodeling takes place over several weeks [41]. Thus, additional studies would be prudent to evaluate the possibility of compensatory shunting of circulation during ventricular regeneration in neonatal mice.
6.1.3 Developments in Induced Heart Regeneration
Despite significant progress in understanding regenerative processes in lower vertebrates and in neonatal mice, it is still unclear how many of these findings can be applied to induce cardiac regeneration in adult mammals. The observation that neonatal mouse hearts can regenerate cardiac injuries is alluring, but there are major differences between neonates and adults with respect to cardiac physiology at the cellular, tissue, and neurohumoral levels. A modest degree of cell cycle re-entry has been observed in adult human and mouse cardiomyocytes [39, 44–46], but evidence for cardiomyocyte cell division in adult mammals is scant. To estimate human cardiomyocyte turnover, Bergmann et al. took advantage of a period of nuclear bomb testing in the 1950s and 1960s, which resulted in a pulse of atmospheric 14C eventually being incorporated into newly synthesized DNA in human cardiomyocytes [44, 45]. They found that less than 1 % cardiomyocytes were turned over annually in adult humans. Additionally, they showed that DNA content increased in the first 10 years of human life, until most cardiomyocytes were tetraploid [44]. In contrast to mice, most adult human cardiomyocytes are mononucleate [47]. Together, these results indicate that most human cardiomyocytes terminally exit the cell cycle before karyokinesis, whereas mouse cardiomyocytes tend to exit the cell cycle after karyokinesis, but before cytokinesis [48].
Although measurement of cell division in human cardiomyocytes is extremely difficult, recent advances in lineage tracing technology have enabled definitive labeling of divided cardiomyocytes in mice. A recent study using mosaic analysis with double markers [49] showed that approximately 1 % of labeled adult cardiomyocytes had undergone cell division after 2 weeks of daily tamoxifen induction [40]. However, as discussed above, potential bias of interchromosomal recombination could obscure quantification of cell division. Importantly, myocardial infarction prior to labeling did not increase cell division, indicating a lack of regeneration in adult mouse hearts. Still, the immense burden on human health has warranted an abundance of investigations seeking the ultimate feat of cardiovascular medicine: induced adult human heart regeneration.
Numerous strategies have been devised to induce adult mammalian heart regeneration and typically rely on mouse models of myocardial infarction , such as permanent left anterior descending (LAD) artery ligation [50, 51]. Ischemia-reperfusion (IR) models [52] are an even better representation of human myocardial infarction, due to post-MI surgical intervention [8, 9]. Large animal models [53, 54] are useful to translate findings in mice and to test regenerative strategies that are difficult in rodent models due to differences in anatomy, physiology or scalability.
Here, we discuss various therapeutic approaches (summarized in Fig. 6.1) to induce mammalian heart regeneration, including strategies that augment endogenous cardiac regeneration, or supply an exogenous source of cardiomyocyte replacement, consisting of allografts or the re-introduction of modified autologous cells.
6.1.4 Cardiac Progenitor Cells
Attempts to stimulate endogenous heart regeneration and replenish lost cardiomyocytes has been in part motivated by the hypothetical existence of a population of resident or non-resident cardiac progenitor cells (CPCs), which were thought to be a renewable source of committed cardiomyogenic cells. In theory, either autologous or allogeneic CPCs could conceivably be grafted into ischemic injuries to facilitate cardiac regeneration. However, several supposed CPC cell types have ultimately been found to represent at best a very rare contributor to new cardiomyocytes in vivo. For example, Lin − c-kit + CPCs initially showed promise for adult mammalian heart regeneration [55]. However, these cells were later reported to have limited utility in induced adult mammalian heart regeneration, despite their potential to support regeneration in neonates [56, 57]. A recent article confirmed the lack of significant direct contribution by cardiac resident c-kit+ progenitors to new cardiomyocytes [58]. Specifically, c-kit + cells did not co-express Nkx2.5 or sarcomeric proteins at any stage, but were consistently found to co-express the endothelial marker CD31. Furthermore, endothelial-specific Tie2-driven expression of Cre completely abolished a c-kit driven floxed LacZ reporter. Thus, despite the observation of c-kit+ cells in both the developing and adult heart, they were found to contribute mostly to endothelial cells, rather than cardiomyocytes. As an exogenous cell therapy for heart regeneration [59], it seems likely that any potential benefit of c-kit + progenitor cells to cardiac function would be indirect, for example through paracrine signaling. Other potential endogenous adult murine CPCs have been described, such as Sca1 + cells [60, 61]. However ectopic Cre-expression may have confounded initial interpretations of Sca1 + CPCs, and the lack of a human ortholog limits the application to human heart failure therapy (reviewed in [62]).
By contrast, Isl1 + cells are a true cardiomyocyte progenitor population derived from the second heart field and have been shown to give rise to a majority of cardiomyocytes in the developing mouse heart [63, 64]. Cre-based lineage tracing experiments showed that by embryonic day 9.75, Isl1 + progenitor cells generated nearly all cells in the outflow tract and right ventricle, as well as 65 % of the left atria and 20 % of the left ventricle [63]. Moretti et al. showed that Isl1 + precursors are multipotent and could give rise to smooth muscle and endothelial lineages in addition to cardiomyocytes [65]. They also demonstrated that Isl1 + cells could be differentiated in vitro from ES cells and propagated on cardiac mesenchyme feeder layers, indicating a potential source of therapeutic progenitor cells for heart failure. A majority of the remaining heart, including the left ventricle, is derived from Isl1 − progenitors from the primary heart field, characterized by expression of early developmental markers such as GATA4, NKX2.5, and TBX5 (reviewed in [66]).
The persistence of a clinically useful population of resident CPCs in adult mammalian hearts has been an elusive and ongoing pursuit. However, more tangible applications of developmental CPC research in heart regeneration have come through the use of CPC markers to identify potential alternative therapeutic cellular sources of neomyogenesis. Such induced CPCs can now be obtained by pretreatment of ES and iPS cells, as discussed below. Furthermore, the understanding of fetal heart development on the molecular level has led to the discovery of fetal gene re-expression during heart failure [12], which could represent failed attempts to regenerate the adult heart through developmental recapitulation.
6.1.5 Bone-Marrow Derived Cells
Bone marrow-derived cells (BMCs) represent an attractive source of regenerative therapy, since autologous donor tissue can be easily and safely obtained . Initial promise came from an early study that showed 5-azacytidine treatment could induce cardiomyocyte differentiation from immortalized BMCs in vitro [67]. Subsequently, it was shown that autologous BMCs could improve recovery after myocardial infarction in rats [68, 69]. A 2001 study showed a low rate of myocardial engraftment in an ischemia-reperfusion model after bone marrow transplantation of supposed multipotent CD34 -/low, c-kit +, sca1 + side population (SP) cells, obtained from Rosa26-lacZ donor mice [70]. The purity of the SP cells was high at 91 %, but a even a low rate of contamination by other cell types could confound the interpretation that SP cells themselves give rise to cardiomyocytes. Nevertheless, the observation that bone marrow derived cells could contribute to endothelial cells and cardiomyocytes at all was encouraging for future developments.
Numerous other pre-clinical and clinical studies have investigated the safety and efficacy of bone marrow-derived cell therapy on acute myocardial infarction and heart failure. Results from some individual clinical trials have been positive [71], but large-scale meta-analyses have shown either modest or no benefit on cardiac function or mortality [72, 73]. Looking forward, it will be interesting to see the results of an ongoing large scale phase III clinical trial testing the efficacy of intracoronary delivery of autologous BMCs [74].
6.1.6 Embryonic Stem Cells
Human embryonic stem (hES) cells can be obtained from sperm-fertilized blastocysts [75] or, more conveniently, produced from adult fibroblasts by somatic cell nuclear transfer into oocytes [76, 77]. Being pluripotent, ES cells have the ability to give rise to all three germ layers, including all cell types of the heart. Thus, ES cells are a promising source of cardiomyocyte replacement in the failing heart. However, teratoma formation from direct ES cell injection demonstrates that neither normal nor failing myocardium lacks the developmental signals for faithful differentiation into myocardial lineages [78, 79]. ES cell-derived cardiomyocytes (ES-CMs) can be differentiated from hES cells in vitro by treatment with activin A and BMP4 [80]. In an athymic rat IR model, it was shown that infarcted myocardium could be grafted with hES-CMs by direct cardiac injection [80]. Importantly, a pro-survival cocktail (containing cell adhesion promoting Matrigel, mitochondrial death inhibitors Bcl-KL peptide and cyclosporine A, vasodilator pinacidil, AKT activator IGF-1, and caspase inhibitor ZVAD-fmk) was used to improve graft survival and functional recovery.
Despite the initial excitement for ES-CM treatment, a later study showed that although both allogeneic undifferentiated ES cell and ES-CM treatment provided improvements to ejection fraction in infarcted mouse myocardium, the ES-CM treated groups had an increased risk of cardiac arrhythmia and death [81]. This observation was presumably due to incomplete maturity of in vitro differentiated hES-CMs, or alternatively to the mismatch in normal heart rate between human and mouse cardiomyocytes. A subsequent study using an immunocompromised guinea pig cryoinjury model showed engraftment by hES-derived cardiomyocytes with reduced arrhythmia [82]. However, a non-human primate model of the more relevant IR injury again showed significant arrhythmia after engraftment of hES-CMs [83].
These exciting developments in ES-derived myocardial grafts show promise for future heart failure treatments. However, there is a clear need to better understand cardiomyocyte differentiation and to develop protocols to create more mature cardiomyocyte grafts that can recapitulate native pacing. In that light, a recent study showed that 1 year old in vitro differentiated ES-CMs are more similar to mature myocardial tissue in vivo and that the let-7 miR family plays an important role in the maturation process [84]. Furthermore, an earlier study showed that forced expression of connexin 43 improved conduction not only in embryonic cardiomyocyte grafts, but even in skeletal myoblast grafts in infarcted mouse hearts [85].
Despite the use of ES cells as a powerful research tool, and the promising results of preclinical heart regeneration studies, reluctance to enter clinical trials hinges in part on their potential for immune rejection and tumorigenesis [86], not to mention ethical constraints. It will be interesting to see if future developments in autologous ES cell creation [76] and refinements in differentiation and purification protocols will change these perspectives.
6.1.7 Induced Pluripotent Stem Cells
In 2006, Takahashi and Yamanaka reported that adult fibroblasts could be reprogrammed to become induced pluripotent stem (iPS) cells [87]. By forced expression of Oct3/4, Sox2, c-Myc, and Klf4, adult mouse fibroblasts became competent for teratoma formation and differentiation into all three germ layers. However, it was still not clear whether the same protocol could be used with human cells. The following year, the same group reported that iPS cells could be generated using human fibroblasts [88]. This was a landmark development in regenerative medicine because it indicated that dispensable autologous adult donor tissue could be used to potentially regenerate any tissue, including the heart.
Although iPS cells theoretically should avoid complications due to immune rejection when using reprogrammed autologous cells, some evidence has suggested otherwise [89]. Furthermore, the tumorigenic risk of retrovirus-reprogrammed cells has led others to pursue chemical or protein-mediated derivation of reprogrammed cells [90, 91]. Still, the pluripotency of iPS cells necessitates a better understanding of differentiation and the development of robust progenitor purification before clinical applications can safely use iPS cells. Nonetheless, iPS cells have become an invaluable research tool and will continue to change the face of regenerative research.
6.1.8 Direct Reprogramming
The discovery of iPS cell reprogramming and the risk of teratoma/tumor formation from the use of pluripotent stem cells quickly led others to pursue alternative approaches to cellular reprogramming. Related approaches were then used to directly reprogram fibroblasts into induced cardiomyocyte-like (iCM) cells without a pluripotent intermediate. The motivation for this type of reprogramming lies in the abundance of fibroblasts in the infarcted myocardium that could serve as a source of new cardiomyocytes. A key observation that led to the discovery of direct reprogramming approaches was the recognition that several core transcription factors (GATA4, HAND2, MEF2C, MESP1, NKX2-5, and TBX5) play a major role in heart development and differentiation. In 2010, a subset of these factors, GMT (GATA4, MEF2C, and TBX5), was used to directly reprogram mouse cardiac and dermal fibroblasts into iCM cells in vitro [92]. Subsequently, in vivo reprogramming was achieved with either GMT or GHMT (GMT + HAND2), yielding improved cardiac function after myocardial infarction in mice [93, 94]. Co-injection of thymosin β4 with GMT reprogramming improved myocardial function after MI [93, 95]. Ding and colleagues showed that small molecules SCPF (SB431542, CHIR99021, parnate, and forskolin) and Oct4 alone could achieve direct reprogramming in vitro [96]. Alternative reprogramming formulations have since been developed, including a microRNA cocktail that effectively converts adult cardiac fibroblasts [97]. Importantly, Olson and colleagues reported a cardiac reprogramming cocktail that works in human cells [98]. Recently, it was shown that Akt1/protein kinase B enhances GHMT conversion efficiency and iCM maturity, including increased polynucleation [99].
In contrast to iPS cells , direct reprogramming offers a source of cardiomyocyte replacement that bypasses the teratoma-competent pluripotent stage. However, more efficient methods to convert and target cardiac fibroblasts need to be developed to move forward in the clinic [100]. In addition, the use of safe vectors or chemical approaches for reprogramming factors would expedite clinical utility of direct reprogramming [96, 100]. Furthermore, despite its promising direction, the tradeoff of reprogramming fibroblasts into cardiomyocytes must still be critically evaluated with respect to the loss of fibroblast function in the failing heart [101]. Perhaps the recent discovery of expandable induced cardiomyocyte-like progenitors [102] will lead to similar strategies that can address concerns of a fibroblast-cardiomycote tradeoff for in vivo conversion.
6.1.9 Dedifferentiated Adult Cardiomyocytes
Dedifferentiation of adult cardiomyocytes can be seen through the re-expression of fetal gene programs in heart failure [12]. Thus, it should not be surprising that adult mammalian cardiomyocytes can dedifferentiate to some degree in culture [103, 104]. Still, evidence for true adult cardiomyocyte cell division, even in the far-removed in vitro environment, is scarce. This suggests that despite varying degrees of dedifferentiation of adult cardiomyocytes in vitro and in vivo, there may exist an inherent block to actually complete cell division. This idea is further supported by the rarity of cardiomyocyte-derived cancers. Nevertheless, rare examples of significantly proliferating adult mammalian cardiomyocytes have been reported, such as rat cardiomyocytes showing high levels of bromodeoxyuridine (BrdU), Ki67 and phosphohistone 3 (PH3) staining in vitro [104]. Recently, the dedifferentiation process of these cultured myocytes was shown to be regulated by epigenomic reprogramming [105].
Fascinatingly, explanted cardiac tissue, cultured under non-adhesive conditions, has been shown to recapitulate a stem cell-like niche that apparently contributes to myocardial repair [106]. The cell preparations derived from such cultures, deemed cardiosphere-derived cells (CDCs) are now being evaluated for the treatment of heart failure in humans. Phase I clinical trials have shown positive results with an increase in viable mass and a reduction in scar size [107, 108]. Interestingly, it was recently shown that exosomes from CDCs may help mediate their regenerative effects [109]. It will be interesting to see how ongoing clinical trials could potentially improve patient outcome [110].
6.1.10 Stimulation of Adult Cardiomyocyte Proliferation
The induction of cardiomyocyte proliferation through cell cycle re-entry and true cell division has been a heavily sought goal of research, with the ultimate goal of adult human heart regeneration through the expansion and replenishment of endogenous cardiomyocytes. Numerous reports have demonstrated induced re-entry into S-phase by adult mammalian cardiomyocytes, for example by cell cycle activators Cyclin A2 [111] and E2F [112]. Although cytokinetic figures have been observed, robust cardiomyocyte cell division has been difficult to achieve. Immortalization with SV40TAg indicated that it is possible to induce persistent cell division in adult rat ventricular myocytes [113]. However, it is unclear what percentage of adult cardiomyocytes have the capacity to divide without apoptosis even under oncogenic conditions. Since the risk of tumorigenesis precludes serious consideration of SV40Tag in the clinic, the search for regulated stimulation of cardiomyocyte proliferation continues. Various approaches have since been used to increase cardiomyocyte proliferation and enhance MI repair, such as those involving miRNAs [114–116] and neuregulin [117, 118] signaling. The Hippo pathway has recently become an intense subject of investigation in heart regeneration due to its role in organ size control [119]. Modulation of the Hippo pathway has been shown to extend the developmental window of cardiomyocyte proliferation and offer modest improvements when administered after MI in several reports [120–122]. Despite promising results from many of these studies, the major cell cycle blocks in adult mammalian cardiomyocytes are largely not well understood. Furthermore, definitive regeneration in adult mammals is still an active pursuit with room for improvement.
6.1.11 Tissue Mechanics
As mentioned earlier, mechanical stiffness has been associated with reduced ventricular function and progressive heart failure. Recombinant elastin production by transduced endothelial cell transplants reduced infarct size and improved cardiac function after myocardial infarction in rats [123]. This result corroborates observations of progressive heart malfunction as a result of mechanically mediated myofibroblast conversion and runaway fibrosis accompanied by cardiomyocyte cell death (reviewed in [124]). Tissue mechanics has been shown to be important in several aspects of cardiomyocyte biology, such as contractility [125], development [126–128], differentiation [129], and maturation [130]. Recently, a collagen matrix patch containing FSTL1 was used to promote myocardial repair in a porcine myocardial infarction model [131]. It was found that therapeutic effect was influenced not only by the location of FSTL1 secretion, but also by the elasticity of the collagen patch. Thus, it is becoming increasingly clear that tissue/matrix mechanics plays an important role in cardiac disease and remodeling and should be carefully considered in future efforts to induce heart regeneration.
6.1.12 Engraftment
Engraftment of exogenous cells into the heart has been a challenging hurdle to treat heart disease via cellular approaches. The dynamic mechanical demands of the human heart, forcefully pumping at approximately 1 Hz, likely pose a thermodynamic barrier to cell attachment and integration within the dense extracellular matrix. Not surprisingly, there may be an age-dependence on the success of donor cell engraftment, as shown by higher engraftment of fetal and neonatal rat cardiomyocytes into injured and non-injured adult rat hearts when compared to adult cardiomyocyte engraftment [132]. Despite a higher rate of engraftment for younger donor tissue, engraftment cell survival is typically very low, even for stem and progenitor cell grafts [133]. Nevertheless, an enormous body of work describes various attempts to achieve therapeutic benefit from exogenous cell therapy in heart injury models, as reviewed above. Concurrent developments are underway to increase cell engraftment in the heart and other tissues, including cell adhesive matrices [134, 135] as well as cell pretreatment to increase cardiac homing (reviewed in [136]).
6.2 Conclusions
The field of regenerative biology has made enormous progress in understanding some of the species differences in cardiac regeneration and in the discovery of several therapeutic strategies that have shown some effect on mitigating the effects of human heart failure. However, the ultimate therapeutic endpoint is still out of reach, and further work will be required to obtain a better basic understanding of myocardial biology, including the molecular nature of adult cardiomyocyte cell cycle block, the role of tissue mechanics in heart disease, and the interplay between fibrosis and cardiomyocyte health. Exciting clinical and preclinical developments in cellular and molecular therapies utilizing cardiospheres or miRNA and Hippo signaling could be revealing in the oncoming years. Still, it will be crucial to continue the pursuit of basic discovery in cardiomyocyte biology and the refinement of drug, gene, and cell delivery approaches to maximize progress toward human heart regeneration.
References
Braunwald E (1997) Shattuck lecture-cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med 337(19):1360–1369. doi:10.1056/NEJM199711063371906
Askoxylakis V, Thieke C, Pleger ST et al (2010) Long-term survival of cancer patients compared to heart failure and stroke: a systematic review. BMC Cancer 10(1):105. doi:10.1186/1471-2407-10-105
Konstantinidis K, Whelan RS, Kitsis RN (2012) Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 32(7):1552–1562. doi:10.1161/ATVBAHA.111.224915
Farb A, Kolodgie FD, Jenkins M et al (1993) Myocardial infarct extension during reperfusion after coronary artery occlusion: pathologic evidence. J Am Coll Cardiol 21(5):1245–1253. doi:10.1016/0735-1097(93)90253-W
Minezaki KK, Suleiman MS, Chapman RA (1994) Changes in mitochondrial function induced in isolated guinea-pig ventricular myocytes by calcium overload. J Physiol 476(3):459–471
Ferrari R, Alfieri O, Curello S et al (1990) Occurrence of oxidative stress during reperfusion of the human heart. Circulation 81(1):201–211. doi:10.1161/01.CIR.81.1.201
Opie LH (1991) Role of calcium and other ions in reperfusion injury. Cardiovasc Drugs Ther 5(Suppl 2):237–247
Beyersdorf F (2009) The use of controlled reperfusion strategies in cardiac surgery to minimize ischaemia/reperfusion damage. Cardiovasc Res 83(2):262–268, 10.1093/cvr/cvp110
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123(1):92–100. doi:10.1172/JCI62874
Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC (2013) Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 10(1):15–26. doi:10.1038/nrcardio.2012.158
Rysä J, Leskinen H, Ilves M, Ruskoaho H (2005) Distinct upregulation of extracellular matrix genes in transition from hypertrophy to hypertensive heart failure. Hypertension 45(5):927–933, 10.1093/cvr/cvp110
Dirkx E, da Costa Martins PA, De Windt LJ (2013) Regulation of fetal gene expression in heart failure. Biochim Biophys Acta 1832(12):2414–2424. doi:10.1016/j.bbadis.2013.07.023
Kolwicz SC, Purohit S, Tian R (2013) Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113(5):603–616. doi:10.1161/CIRCRESAHA.113.302095
Jaźwińska A, Sallin P (2016) Regeneration versus scarring in vertebrate appendages and heart. J Pathol 238(2):233–246. doi:10.1002/path.4644
Judd J, Xuan W, Huang GN (2015) Cellular and molecular basis of cardiac regeneration. Turk J Biol 40(2):265–275
Kikuchi K, Holdway JE, Werdich AA et al (2010) Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464(7288):601–605. doi:10.1038/nature08804
Jopling C, Sleep E, Raya M et al (2010) Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464(7288):606–609. doi:10.1038/nature08899
Sallin P, de Preux Charles A-S, Duruz V, Pfefferli C, Jaźwińska A (2015) A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Dev Biol 399(1):27–40. doi:10.1016/j.ydbio.2014.12.002
Kikuchi K, Holdway JE, Major RJ et al (2011) Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell 20(3):397–404. doi:10.1016/j.devcel.2011.01.010
Lepilina A, Coon AN, Kikuchi K et al (2006) A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127(3):607–619, 10.1016/j.cell.2006.08.052
Wang J, Karra R, Dickson AL, Poss KD (2013) Fibronectin is deposited by injury-activated epicardial cells and is necessary for zebrafish heart regeneration. Dev Biol 382(2):427–435. doi:10.1016/j.ydbio.2013.08.012
Willems IE, Arends JW, Daemen MJ (1996) Tenascin and fibronectin expression in healing human myocardial scars. J Pathol 179(3):321–325. doi:10.1007/s12265-012-9406-3
Knowlton AA, Connelly CM, Romo GM et al (1992) Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. J Clin Invest 89(4):1060–1068. doi:10.1172/JCI115685
Altrock E, Sens C, Wuerfel C et al (2015) Inhibition of fibronectin deposition improves experimental liver fibrosis. J Hepatol 62(3):625–633
Serini G, Bochaton-Piallat M-L, Ropraz P et al (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J Cell Biol 142(3):873–881. doi:10.1016/j.jhep.2014.06.010
Bhattacharyya S, Tamaki Z, Wang W et al (2014) FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 6(232):232ra50. doi:10.1126/scitranslmed.3008264
Bettencourt-Dias M, Mittnacht S, Brockes JP (2003) Heterogeneous proliferative potential in regenerative adult newt cardiomyocytes. J Cell Sci 116(Pt 19):4001–4009. doi:10.1242/jcs.00698
Witman N, Murtuza B, Davis B, Arner A, Morrison JI (2011) Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol 354(1):67–76. doi:10.1016/j.ydbio.2011.03.021
Oberpriller JO, Oberpriller JC (1974) Response of the adult newt ventricle to injury. J Exp Zool 187(2):249–253
Piatkowski T, Mühlfeld C, Borchardt T, Braun T (2013) Reconstitution of the myocardium in regenerating newt hearts is preceded by transient deposition of extracellular matrix components. Stem Cells Dev 22(13):1921–1931. doi:10.1089/scd.2012.0575
Cano-Martínez A, Vargas-González A, Guarner-Lans V et al (2010) Functional and structural regeneration in the axolotl heart (Ambystoma mexicanum) after partial ventricular amputation. Arch Cardiol Mex 80(2):79–86
Laube F, Heister M, Scholz C, Borchardt T, Braun T (2006) Re-programming of newt cardiomyocytes is induced by tissue regeneration. J Cell Sci 119(Pt 22):4719–4729. doi:10.1242/jcs.03252
Mercer SE, Odelberg SJ, Simon H-G (2013) A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev Biol 382(2):457–469. doi:10.1016/j.ydbio.2013.08.002
Bax NAM, van Marion MH, Shah B et al (2012) Matrix production and remodeling capacity of cardiomyocyte progenitor cells during in vitro differentiation. J Mol Cell Cardiol 53(4):497–508. doi:10.1016/j.yjmcc.2012.07.003
Porrello ER, Mahmoud AI, Simpson E et al (2011) Transient regenerative potential of the neonatal mouse heart. Science 331(6020):1078–1080. doi:10.1126/science.1200708
Andersen DC, Ganesalingam S, Jensen CH, Sheikh SP (2014) Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports 2(4):406–413. doi:10.1016/j.stemcr.2014.02.008
Sadek HA, Martin JF, Takeuchi JK et al (2014) Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Reports 3(1):1. doi:10.1016/j.stemcr.2014.06.009
Bryant DM, O’Meara CC, Ho NN et al (2015) A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol 79:315–918. doi:10.1016/j.yjmcc.2014.12.011
Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 271(5 Pt 2):H2183–H2189
Ali SR, Hippenmeyer S, Saadat LV et al (2014) Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A 111(24):8850–8855. doi:10.1073/pnas.1408233111
Tada T, Kishimoto H (1990) Ultrastructural and histological studies on closure of the mouse ductus arteriosus. Acta Anat 139(4):326–334
Canseco DC, Kimura W, Garg S et al (2015) Human ventricular unloading induces cardiomyocyte proliferation. J Am Coll Cardiol 65(9):892–900. doi:10.1016/j.jacc.2014.12.027
Dandel M, Weng Y, Siniawski H et al (2011) Heart failure reversal by ventricular unloading in patients with chronic cardiomyopathy: criteria for weaning from ventricular assist devices. Eur Heart J 32(9):1148–1160. doi:10.1093/eurheartj/ehq353
Bergmann O, Bhardwaj RD, Bernard S et al (2009) Evidence for cardiomyocyte renewal in humans. Science 324(5923):98–102. doi:10.1126/science.1164680
Bergmann O, Zdunek S, Felker A et al (2015) Dynamics of cell generation and turnover in the human heart. Cell 161(7):1566–1575. doi:10.1016/j.cell.2015.05.026
Soonpaa MH, Field LJ (1994) Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol 266(4 Pt 2):H1439–H1445
Olivetti G, Cigola E, Maestri R et al (1996) Aging, cardiac hypertrophy and ischemic cardiomyopathy do not affect the proportion of mononucleated and multinucleated myocytes in the human heart. J Mol Cell Cardiol 28(7):1463–1477. doi:10.1006/jmcc.1996.0137
Engel FB, Schebesta M, Keating MT (2006) Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol 41(4):601–612. doi:10.1016/j.yjmcc.2006.06.012
Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L (2005) Mosaic analysis with double markers in mice. Cell 121(3):479–492. doi:10.1016/j.cell.2005.0
Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA (2009) Surgical models for cardiac regeneration in neonatal mice. Nat Protoc 9(2):305–311. doi:10.1038/nprot.2014.021
Curaj A, Simsekyilmaz S, Staudt M, Liehn E (2015) Minimal invasive surgical procedure of inducing myocardial infarction in mice. J Vis Exp 99, e52197. doi:10.3791/52197
Xu Z, Alloush J, Beck E, Weisleder N (2015) A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J Vis Exp 86, e51329. doi:10.3791/51329
Chong JJH, Murry CE (2014) Cardiac regeneration using pluripotent stem cells–progression to large animal models. Stem Cell Res 13(3 Pt B):654–665. doi:10.1016/j.scr.2014.06.005
Dixon JA, Spinale FG (2009) Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2(3):262–271. doi:10.1161/CIRCHEARTFAILURE.108.814459
Beltrami AP, Barlucchi L, Torella D et al (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114(6):763–776. doi:10.1016/S0092-8674(03)00687-1
Zaruba M-M, Soonpaa M, Reuter S, Field LJ (2010) Cardiomyogenic potential of C-kit(+)-expressing cells derived from neonatal and adult mouse hearts. Circulation 121(18):1992–2000. doi:10.1161/CIRCULATIONAHA.109.909093
Jesty SA, Steffey MA, Lee FK et al (2012) C-kit + precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci U S A 109(33):13380–13385. doi:10.1073/pnas.1208114109
Sultana N, Zhang L, Yan J et al (2015) Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun 6:8701. doi:10.1038/ncomms9701
Bolli R, Chugh AR, D’Amario D et al (2011) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378(9806):1847–1857. doi:10.1016/S0140-6736(11)61590-0
Oh H, Bradfute SB, Gallardo TD et al (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A 100(21):12313–12318. doi:10.1073/pnas.2132126100
Uchida S, De Gaspari P, Kostin S et al (2013) Sca1-derived cells are a source of myocardial renewal in the murine adult heart. Stem Cell Reports 1(5):397–410. doi:10.1016/j.stemcr.2013.09.004
van Berlo JH, Molkentin JD (2014) An emerging consensus on cardiac regeneration. Nat Med 20(12):1386–1393. doi:10.1038/nm.3764
Cai C-L, Liang X, Shi Y et al (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5(6):877–889. doi:10.1016/S1534-5807(03)00363-0
Laugwitz K-L, Moretti A, Lam J et al (2005) Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433(7026):647–653. doi:10.1038/nature03215
Moretti A, Caron L, Nakano A et al (2006) Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127(6):1151–1165. doi:10.1016/j.cell.2006.10.029
Xin M, Olson EN, Bassel-Duby R (2013) Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol 14(8):529–541. doi:10.1038/nrm3619
Makino S, Fukuda K, Miyoshi S et al (1999) Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest 103(5):697–705
Tomita S, Li RK, Weisel RD et al (1999) Autologous transplantation of bone marrow cells improves damaged heart function. Circulation 100(Suppl II):247–256. doi:10.1161/01.CIR.100.suppl_2.II-247
Kocher AA, Schuster MD, Szabolcs MJ et al (2001) Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med 7(4):430–436. doi:10.1038/86498
Jackson KA, Majka SM, Wang H et al (2001) Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest 107(11):1395–1402
Mathiasen AB, Qayyum AA, Jørgensen E et al (2015) Bone marrow-derived mesenchymal stromal cell treatment in patients with severe ischaemic heart failure: a randomized placebo-controlled trial (MSC-HF trial). Eur Heart J 36(27):1744–1753. doi:10.1093/eurheartj/ehv136
Nowbar AN, Mielewczik M, Karavassilis M et al (2014) Discrepancies in autologous bone marrow stem cell trials and enhancement of ejection fraction (DAMASCENE): weighted regression and meta-analysis. Br Med J 348:g2688. doi:10.1136/bmj.g2688
Gyöngyösi M, Wojakowski W, Lemarchand P et al (2015) Meta-Analysis of Cell-based CaRdiac stUdiEs (ACCRUE) in patients with acute myocardial infarction based on individual patient data. Circ Res 116(8):1346–1360. doi:10.1161/CIRCRESAHA.116.304346
Mathur A. BAMI. The effect of intracoronary reinfusion of Bone Marrow-derived Mononuclear Cells (BM-MNC) on all cause mortality in acute myocardial infarction. https://clinicaltrials.gov/ct2/show/nct01569178. Accessed 5 Apr 2016
Thomson JA (1998) Embryonic stem cell lines derived from human blastocysts. Science 282(5391):1145–1147. doi:10.1126/science.282.5391.1145
Tachibana M, Amato P, Sparman M et al (2013) Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 154(2):1228–1238. doi:10.1016/j.cell.2013.05.006
Chung YG, Eum JH, Lee JE et al (2014) Human somatic cell nuclear transfer using adult cells. Cell Stem Cell 14(6):777–780. doi:10.1016/j.stem.2014.03.015
Nussbaum J, Minami E, Laflamme MA et al (2007) Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J 21(7):1345–1357. doi:10.1096/fj.06-6769com
Swijnenburg R-J, Tanaka M, Vogel H et al (2005) Embryonic stem cell immunogenicity increases upon differentiation after transplantation into ischemic myocardium. Circulation 112(Suppl 9):I166–I172. doi:10.1161/CIRCULATIONAHA.104.525824
Laflamme MA, Chen KY, Naumova AV et al (2007) Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25(9):1015–1024. doi:10.1038/nbt1327
Liao S-Y, Liu Y, Siu C-W et al (2010) Proarrhythmic risk of embryonic stem cell-derived cardiomyocyte transplantation in infarcted myocardium. Heart Rhythm 7(12):1852–1859. doi:10.1016/j.hrthm.2010.09.006
Shiba Y, Fernandes S, Zhu W-Z et al (2012) Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489(7415):322–325. doi:10.1038/nature11317
Chong JJH, Yang X, Don CW et al (2014) Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510(7504):273–277. doi:10.1038/nature13233
Kuppusamy KT, Jones DC, Sperber H et al (2015) Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc Natl Acad Sci U S A 112(21):E2785–E2794. doi:10.1073/pnas.1424042112
Roell W, Lewalter T, Sasse P et al (2007) Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature 450(7171):819–824. doi:10.1038/nature06321
Sanganalmath SK, Bolli R (2013) Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res 113(6):810–834. doi:10.1161/CIRCRESAHA.113.300219
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. doi:10.1016/j.cell.2006.07.024
Takahashi K, Tanabe K, Ohnuki M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872. doi:10.1016/j.cell.2007.11.019
Zhao T, Zhang Z-N, Rong Z, Xu Y (2011) Immunogenicity of induced pluripotent stem cells. Nature 474(7350):212–215. doi:10.1038/nature10135
Xie M, Cao N, Ding S (2014) Small molecules for cell reprogramming and heart repair: progress and perspective. ACS Chem Biol 9(1):34–44. doi:10.1021/cb400865w
Zhou H, Wu S, Joo JY et al (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4(5):381–384. doi:10.1016/j.stem.2009.04.005
Ieda M, Tsuchihashi T, Ivey KN et al (2009) Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell 16(2):233–244. doi:10.1016/j.devcel.2008.12.007
Qian L, Huang Y, Spencer CI et al (2012) In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485(7400):593–598. doi:10.1038/nature11044
Song K, Nam Y-J, Luo X et al (2012) Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485(7400):599–604. doi:10.1038/nature11139
Srivastava D, Ieda M, Fu J, Qian L (2012) Cardiac repair with thymosin β4 and cardiac reprogramming factors. Ann N Y Acad Sci 1270:66–72. doi:10.1111/j.1749-6632.2012.06696.x
Wang H, Cao N, Spencer CI et al (2014) Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4. Cell Rep 6(5):951–960. doi:10.1016/j.celrep.2014.01.038
Jayawardena TM, Egemnazarov B, Finch EA et al (2012) MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res 110(11):1465–1473. doi:10.1161/CIRCRESAHA.112.269035
Nam Y-J, Song K, Luo X et al (2013) Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A 110(14):5588–5593. doi:10.1073/pnas.1301019110
Zhou H, Dickson ME, Kim MS, Bassel-Duby R, Olson EN (2015) Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc Natl Acad Sci U S A 112(38):11864–11869. doi:10.1073/pnas.1516237112
Zhao Y, Londono P, Cao Y et al (2015) High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat Commun 6:8243. doi:10.1038/ncomms9243
Nagalingam RS, Safi HA, Czubryt MP (2015) Gaining myocytes or losing fibroblasts: challenges in cardiac fibroblast reprogramming for infarct repair. J Mol Cell Cardiol 93:108–114. doi:10.1016/j.yjmcc.2015.11.029
Zhang Y, Cao N, Huang Y et al (2016) Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell 18(3):368–381. doi:10.1016/j.stem.2016.02.010
Nag AC, Cheng M (1986) Biochemical evidence for cellular dedifferentiation in adult rat cardiac muscle cells in culture: expression of myosin isozymes. Biochem Biophys Res Commun 137(2):855–862
Zhang Y, Li T-S, Lee S-T et al (2010) Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One 5(9), e12559. doi:10.1371/journal.pone.0012559
Zhang Y, Zhong JF, Qiu H et al (2015) Epigenomic reprogramming of adult cardiomyocyte-derived cardiac progenitor cells. Sci Rep 5:17686. doi:10.1038/srep17686
Li T-S, Cheng K, Lee S-T et al (2010) Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells 28(11):2088–2098. doi:10.1002/stem.532
Malliaras K, Makkar RR, Smith RR et al (2014) Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived autologous stem cells to reverse ventricUlar dySfunction). J Am Coll Cardiol 63(2):110–122. doi:10.1016/j.jacc.2013.08.724
Makkar RR, Smith RR, Cheng K et al (2012) Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379(9819):895–904. doi:10.1016/S0140-6736(12)60195-0
Ibrahim AG-E, Cheng K, Marbán E (2014) Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2(5):606–619. doi:10.1016/j.stemcr.2014.04.006
Allogeneic heart stem cells to achieve myocardial regeneration—full text view. [Internet]. Available from: https://clinicaltrials.gov/ct2/show/NCT01458405. Accessed 6 Apr 2016
Chaudhry HW, Dashoush NH, Tang H et al (2004) Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 279(34):35858–35866. doi:10.1074/jbc.M404975200
Ebelt H, Hufnagel N, Neuhaus P et al (2005) Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ Res 96(5):509–517. doi:10.1161/01.RES.0000159705.17322.57
Miller C, Rulfs J, Jaspers SR, Buckholt M, Miller TB (1994) Transformation of adult ventricular myocytes with the temperature sensitive A58 (tsA58) mutant of the SV40 large T antigen. Mol Cell Biochem 136(1):29–34
Eulalio A, Mano M, Dal Ferro M et al (2012) Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492(7429):376–381. doi:10.1038/nature11739
Tian Y, Liu Y, Wang T et al (2015) A microRNA-hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med 7(279):279ra38. doi:10.1126/scitranslmed.3010841
Aguirre A, Montserrat N, Zachiggna S et al (2014) In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15(5):589–604. doi:10.1016/j.stem.2014.10.003
Polizzotti BD, Ganapathy B, Walsh S et al (2015) Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med 7(281):281ra45. doi:10.1126/scitranslmed.aaa5171
Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138(2):257–270. doi:10.1016/j.cell.2009.04.060
Dong J, Feldmann G, Huang J et al (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130(6):1120–1133. doi:10.1016/j.cell.2007.07.019
Lin Z, von Gise A, Zhou P et al (2014) Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res 115(3):354–363. doi:10.1161/CIRCRESAHA.115.303632
Heallen T, Morikawa Y, Leach J et al (2013) Hippo signaling impedes adult heart regeneration. Development 140(23):4683–4690. doi:10.1242/dev.102798
Xin M, Kim Y, Sutherland LB et al (2013) Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A 110(34):13839–13844. doi:10.1073/pnas.1313192110
Mizuno T, Yau TM, Weisel RD, Kiani CG, Li R-K (2005) Elastin stabilizes an infarct and preserves ventricular function. Circulation 112(Suppl 9):I81–I88. doi:10.1073/pnas.1313192110
Piek A, de Boer RA, Silljé HHW (2016) The fibrosis-cell death axis in heart failure. Heart Fail Rev 21(2):199–211. doi:10.1007/s10741-016-9536-9
Engler AJ, Carag-Krieger C, Johnson CP et al (2008) Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci 121(Pt 22):3794–3802. doi:10.1242/jcs.029678
Patwari P, Lee RT (2008) Mechanical control of tissue morphogenesis. Circ Res 103(3):234–343. doi:10.1161/CIRCRESAHA.108.175331
Hove JR, Köster RW, Forouhar AS et al (2003) Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421(6919):172–177. doi:10.1038/nature01282
Jacot JG, Martin JC, Hunt DL (2010) Mechanobiology of cardiomyocyte development. J Biomech 43(1):93–98. doi:10.1016/j.jbiomech.2009.09.014
Arshi A, Nakashima Y, Nakano H et al (2013) Rigid microenvironments promote cardiac differentiation of mouse and human embryonic stem cells. Sci Technol Adv Mater 14(2):pii:025003. doi:10.1088/1468-6996/14/2/025003
Young JL, Kretchmer K, Ondeck MG, Zambon AC, Engler AJ (2014) Mechanosensitive kinases regulate stiffness-induced cardiomyocyte maturation. Sci Rep 14:6425. doi:10.1038/srep06425
Wei K, Serpooshan V, Hurtado C et al (2015) Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 525(7570):479–485. doi:10.1038/nature15372
Reinecke H, Zhang M, Bartosek T, Murry CE (1999) Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation 100(2):193–202. doi:10.1161/01.CIR.100.2.193
Hong KU, Guo Y, Li Q-H, Cao P et al (2014) C-kit+ Cardiac stem cells alleviate post-myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS One 9(5), e96725. doi:10.1371/journal.pone.0096725
Santhakumar R, Vidyasekar P, Verma RS (2014) Cardiogel: a nano-matrix scaffold with potential application in cardiac regeneration using mesenchymal stem cells. PLoS One 9(12), e114697. doi:10.1371/journal.pone.0114697
Hasan A, Khattab A, Islam MA et al (2015) Injectable hydrogels for cardiac tissue repair after myocardial infarction. Adv Sci 2:1500122. doi:10.1002/advs.201500122
Chavakis E, Koyanagi M, Dimmeler S (2010) Enhancing the outcome of cell therapy for cardiac repair: progress from bench to bedside and back. Circulation 121(2):325–335. doi:10.1161/CIRCULATIONAHA.109.901405
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Judd, J., Huang, G.N. (2016). Cellular Approaches to Adult Mammalian Heart Regeneration. In: Wilson-Rawls, J., Kusumi, K. (eds) Innovations in Molecular Mechanisms and Tissue Engineering. Stem Cell Biology and Regenerative Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-44996-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-44996-8_6
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-44994-4
Online ISBN: 978-3-319-44996-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)