
Abstract
Since the introduction of Doxil® on the market nearly 20 years ago, a number of nanomedicines have become part of treatment regimens in the clinic. With the exception of antibody-drug conjugates, these nanomedicines are all devoid of targeting ligands and rely solely on their physicochemical properties and the (patho)physiological processes in the body for their biodistribution and targeting capability. At the same time, many preclinical studies have reported on nanomedicines exposing targeting ligands, or ligand-targeted nanomedicines, yet none of these have been approved at this moment. In the present review, we provide a concise overview of 13 ligand-targeted particulate nanomedicines (ligand-targeted PNMs) that have progressed into clinical trials. The progress of each ligand-targeted PNM is discussed based on available (pre)clinical data. Main conclusions of these analyses are that (a) ligand-targeted PNMs have proven to be safe and efficacious in preclinical models; (b) the vast majority of ligand-targeted PNMs is generated for the treatment of cancer; (c) contribution of targeting ligands to the PNM efficacy is not unambiguously proven; and (d) targeting ligands do not cause localization of the PNM within the target tissue, but rather provide benefits in terms of target cell internalization and target tissue retention once the PNM has arrived at the target site. Increased understanding of the in vivo fate and interactions of the ligand-targeted PNMs with proteins and cells in the human body is mandatory to rationally advance the clinical translation of ligand-targeted PNMs. Future perspectives for ligand-targeted PNM approaches include the delivery of drugs that are unable or inefficient in passing cellular membranes, treatment of drug resistant tumors, targeting of the tumor blood supply, the generation of targeted vaccines and nanomedicines that are able to cross the blood-brain barrier.
*Author contributed equally with all other contributors.
Published from Advanced Drug Delivery Reviews 65: 1284–1298 (2013) in slightly modified form with kind permission of © Elsevier 2016.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nanomedicines
- Targeting ligand
- Clinical translation
- Particulate nanocarrier
- Liposome
- Polymeric nanoparticle
- Bacterial-derived minicell
- Retrovector
1 Introduction
Nanomedicine is the science and application of nanotechnology for diagnosis, monitoring, prevention, treatment and understanding of disease to ultimately gain clinical benefit (European Medical Research Counsils 2004). The focus of the current review is on targeted nanomedicines developed to generate therapeutics that are more effective and/or less harmful to patients compared to conventional drugs. Interdisciplinary pioneering research over the last few decades that focused on colloidal systems, polymer chemistry and antibody technology, has led to the introduction of the term “nanomedicine”(Drexler et al. 1991), and has facilitated the rapid evolvement of the drug targeting and delivery field and subsequent clinical translation of targeted nanomedicines (Duncan and Gaspar 2011; Kamaly et al. 2012; Allen and Cullis 2013). The exploitation of nanocarriers for drug delivery has many potential advantages:
(1) improve unfavorable pharmacokinetics and tissue distribution of many drugs, (2) increase therapeutic efficacy by achieving higher accumulation of a drug in the target tissue, (3) reduce (dose-limiting) adverse effects by minimizing drug exposure to non-target tissues, (4) feasibility of combination therapy by targeted delivery of multiple therapeutic agents in one nanomedicine, (5) ability to manipulate the nanocarrier surface with a range of molecules such as targeting moieties for increased target specificity or polymers to reduce interactions with plasma proteins and blood cells to improve circulation kinetics.
Targeted nanomedicines, either marketed or under development, are designed for the treatment of a broad range of indications such as infections (Huh and Kwon 2011), cardiovascular diseases (Lobatto et al. 2011), central nervous system diseases (Srikanth and Kessler 2012) and inflammatory diseases (Crielaard et al. 2012). The primary emphasis is however on the development of nanomedicines for the treatment of (mostly solid) malignancies (Peer et al. 2007; Davis et al. 2008; Jain and Stylianopoulos 2010). The discovery that macromolecules accumulate in solid tumors over time by virtue of the enhanced retention and permeability (EPR) effect (Box 7.1) (Matsumura and Maeda 1986), has greatly advanced the development and clinical translation of anti-cancer nanomedicines.
With the exception of antibody-drug conjugates (ADCs), all currently marketed nanomedicines are devoid of targeting ligands and their pharmacokinetic properties and biodistribution rely solely on physicochemical properties of the nanomedicine and subsequent interactions in the circulation and at tissue sites including the site of disease. Ligand-targeted particulate nanomedicines (ligand-targeted PNMs) (Box 7.1) are equipped with targeting ligands to increase the target specificity of ligand-lacking particulate nanomedicines (ligand-lacking PNMs). Additionally, ligand-targeted PNMs can be applied to target diseases where the EPR effect is not present.
Box 7.1 Definitions
-
Particulate nanomedicines
Particulate nanomedicines (PNMs) are drug-loaded submicrometer size delivery vehicles designed to improve the pharmacokinetic and biodistribution profiles of the encapsulated molecules. These molecules can be adsorbed, entrapped or dissolved in particulate nanocarriers via non-covalent interactions or via degradable or non-degradable covalent linkers (Petros and DeSimone 2010). Nanomedicines discussed in this review are defined as particulate nanocarriers developed to deliver therapeutic agents to sites of disease. Over the last few decades, many particulate nanocarriers have been developed for the delivery of therapeutics including liposomes, polymer-drug conjugates, micelles, polymeric nanoparticles, dendrimers and albumin nanoparticles. Currently, a few dozen first generation nanomedicines are routinely used in the clinic and it is estimated that approximately 250 nanomedicines are under (pre)clinical investigation (Svenson 2012; Etheridge et al. 2013).
-
PEGylation
Polyethylene glycol (PEG) is a hydrophilic polymer that has been widely used for the development of drug-polymer conjugates because it can improve protein solubility, stability and pharmacokinetic parameters (Abuchowski et al. 1977; Knop et al. 2010). In addition, coating the surface of PNMs with PEG provides ‘stealth’ properties by inhibiting blood protein adsorption. This effect inhibits subsequent clearance of PNMs from the circulation by the mononuclear phagocyte system (MPS). The discovery that PEGylation could greatly enhance the circulation time of nanocarriers such as polymeric nanoparticles (Gref et al. 1994) and liposomes (Blume and Cevc 1990; Klibanov et al. 1990) has greatly advanced the clinical translation of nanomedicines. Although the majority of PNMs clinically approved or under evaluation contains PEG, several issues regarding PEGylation remain such as decrease of drug release and cell uptake (‘PEG dilemma’) (Romberg et al. 2008), activation of the complement system (Moghimi et al. 2010) and accelerated blood clearance of consecutive administered doses (Dams et al. 2000; Laverman et al. 2001).
-
EPR effect
The enhanced permeability and retention (EPR) effect was first proposed by Matsumura and Maeda (Matsumura and Maeda 1986) and describes the phenomenon that macromolecules accumulate in tumors over time. Tumor vasculature is characterized by poorly developed leaky vasculature containing inter-endothelial gaps which allow for the extravasation of PNMs. In addition, tumors often fail to drain extravasated PNMs due to an impaired lymphatic system (Maeda et al. 2013). The EPR effect is exploited by most anti-cancer nanomedicines as it is expected to increase the therapeutic efficacy of chemotherapeutics due to the relative improvement in tumor accumulation of PNMs compared to small molecules.
-
Targeted drug delivery
In the field of nanomedicine, targeting refers to the design of therapeutic nanocarriers with the intention to increase accumulation at sites of disease in the body. This is fundamentally different from molecularly targeted drugs that are intended to specifically interact with a certain protein, but have not been designed to localize at specific sites in the body (Kamaly et al. 2012). Historically, the terms ‘passive’ and ‘active’ targeting were implemented to distinguish between nanomedicines without or equipped with targeting ligands, respectively. Passive targeting primarily refers to anti-cancer nanomedicines that accumulate in tumors due to a combination of the physicochemical properties of the PNMs and prolonged circulation half life, extravasation from the blood circulation and the pathophysiology of the tumor contributing to the EPR effect. Active targeting (also described as ligand-targeting or receptor-mediated targeting) involves the attachment of ligands to the surface of PNMs that bind to proteins overexpressed on diseased cells. Although in theory this can potentially improve PNM target specificity and improve therapeutic activity, it is believed that in the case of many pathologies, ligand-targeted nanomedicines are subjected to the same physiological localization as ligand-lacking nanomedicines and therefore have comparable biodistribution and accumulation profiles. However, targeting ligands may offer advantages at in terms of target cell uptake once arrived at the target site. It is increasingly recognized that the terms ‘passive’ and ‘active’ targeting are not correctly representing the real-life situation (Bae and Park 2011; Kwon et al. 2012; Lammers et al. 2012). We have therefore decided for the sake of clarity to use the terms ‘ligand-lacking nanomedicines’ (ligand-lacking PNMs) and ‘ligand-targeted nanomedicines’ (ligand-targeted PNMs). A third targeting strategy based on stimuli-responsive PNMs referred to as triggered drug release is currently receiving much attention but is beyond the scope of this manuscript (for review see (Ganta et al. 2008)).
Whereas several ligand-lacking PNMs have become part of treatment regimens in the clinic, only a small number of ligand-targeted PNMs have progressed into (early) clinical evaluation and none of them have thus far been approved (Svenson 2012). The aim of this review is to reveal and discuss the evidence for added delivery benefits of conjugating targeting ligands to PNMs currently undergoing clinical evaluation based on analysis of available (pre)clinical data (Table 7.1). The aim is not to provide a complete (historical) overview, discuss basic scientific issues regarding targeted drug delivery and/or perspectives of nanomedicines in general, which has already been discussed in several excellent reviews (Hoffman 2008; Petros and DeSimone 2010; Duncan and Gaspar 2011; Cheng et al. 2012; Kamaly et al. 2012; Svenson 2012; Etheridge et al. 2013). The ligand-targeted PNMs discussed in this review are defined by three components: the particulate nanocarrier, targeting ligands and therapeutic agent. This review therefore does not focus on other nanomedicines such as ligand-lacking PNMs (Svenson 2012), ADCs (Adair et al. 2012; Casi and Neri 2012; Sievers and Senter 2013) and stimuli-responsive nanomedicines (Ganta et al. 2008). The review consists of an objective presentation of available evidence for target localization, safety and efficacy of ligand-targeted PNMs. Based on these data, a scoring table was prepared which summarizes the main characteristics and research outcomes of the evaluated nanomedicines (Table 7.2).
2 Ligand-Targeted Particulate Nanomedicines Under Clinical Evaluation
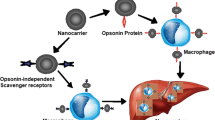
Up to date, 13 ligand-targeted particulate nanomedicines (PNMs) have progressed into clinical trials (Table 7.1). These systems include lipid- and polymer-based delivery vehicles, a retroviral vector and bacterially-derived minicells (Fig. 7.1).

Schematic overview of ligand-targeted nanomedicines undergoing clinical evaluation. The discussed ligand-targeted nanomedicines (ligand-targeted PNMs) are defined by three components: the particulate nanocarrier, targeting ligands and therapeutic agent. Utilized particulate delivery systems include lipid- and polymer based nanocarriers, bacterially-derived minicells and a retroviral vector. Targeting ligands conjugated to PNM include antibodies or antibody fragments, protein (transferrin) or small molecules. Therapeutically active cargo of the ligand-targeted PNMs includes chemotherapeutics, small interfering RNA, plasmid DNA or antigens and adjuvants
2.1 Lipid-Based Nanomedicines
Originally discovered by Bangham and colleagues (Bangham et al. 1965), liposomes were one of the first particulate nanocarriers utilized for the generation of nanomedicines. Liposomes are vesicular structures which consist of an aqueous core surrounded by a lipid bilayer. Doxorubicin (DOX) encapsulated in long-circulating PEGylated liposomes (Doxil®/Caelyx) was approved in 1995 and has been used in the clinic since then (Barenholz 2012). The lipid-based ligand-targeted PNMs discussed in this review feature either liposomes or formulations based on lipids such as lipoplexes.
2.1.1 MBP-426
MBP-426 (Mebiopharm) is a liposome loaded with oxaliplatin (L-OHP) currently undergoing phase Ib/II trials for treatment of second line gastric, gastroesophageal or esophageal adenocarcinomas in combination with leucovorin and fluorouracil (Mebiopharm Co. Ltd.). The liposome is conjugated to transferrin (Tf) for tumor targeting. Platinum binds irreversibly to plasma proteins and erythrocytes and encapsulation of L-OHP in nanocarriers can reduce these interactions thereby improving tumor accumulation and circulation time (Graham et al. 2000). Initial studies with empty PEGylated liposomes showed increased Tf-specific cell association and internalization in Tf-overexpressing murine colon carcinoma (Colon 26) cells of Tf-PEG-liposomes compared to ligand-lacking formulations (Ishida et al. 2001). Tf-PEG-liposomes loaded with L-OHP (EC50 8 μg/mL L-OHP) were more cytotoxic compared to PEG-liposomes devoid of Tf (EC50 18 μg/mL L-OHP) in Colon 26 cells. The cytotoxicity of L-OHP encapsulated in Tf-PEG liposomes could be inhibited by adding an excess of free Tf, indicating that that the cytotoxic effects were mediated by Tf-specific delivery (Suzuki et al. 2008). Studies in mice bearing Colon 26 tumors revealed similar plasma clearance values and biodistribution for ligand-targeted and ligand-lacking L-OHP-loaded PEG-liposomes indicating that the conjugation of Tf did not influence circulation times or uptake by the mononuclear phagocyte system (MPS). Although biodistribution for the ligand-lacking and ligand-targeted formulation was similar, L-OHP concentration in tumors 72 h after injection was ~2.5 times higher in mice treated with L-OHP encapsulated in Tf-PEG-liposomes when compared to PEG-liposomes (Suzuki et al. 2008). Tf-PEG-liposomes loaded with L-OHP significantly suppressed tumor growth compared to L-OHP encapsulated in ligand-lacking liposomes (Suzuki et al. 2008). Based on these results, Mebiopharm further developed this formulation for clinical evaluation. The original formulation was optimized and N-glutaryl-phosphatidylethanolamine (NGPE) is used to couple Tf. The use of NGPE causes the liposome to collapse in environments with low pH such as the endosome. In this way, MBP-426 releases L-OHP upon receptor mediated endocytosis and endosomal localization. In mice bearing human pancreas xenograft tumors, additive tumor growth inhibiting effects were observed when MBP-426 treatment was combined with either gemcitabine or erlotinib (Izbicka et al. 2007). Phase I studies in 39 patients with advanced solid or metastatic solid tumors revealed thrombocytopenia as dose limiting toxicity and a dose of 226 mg/m2 was recommend for further studies (Mebiopharm Co. Ltd., Sankhala et al. 2009). Results of phase Ib trials in nine patients reported 170 mg/m2 (versus free L-OHP 85 mg/m2) as recommended dose for phase II studies and potential activity was observed in two L-OHP-resistant patients (Mebiopharm Co. Ltd., Senzer et al. 2009).
2.1.2 SGT-53 and SGT-94
SGT-53 (SynerGene Therapeutics) is a nanomedicine developed for the treatment of solid tumors. The formulation consists of cationic lipids that are complexed with plasmid DNA encoding wild-type p53 tumor suppressor protein. SGT-53 is targeted to the Tf receptor (TfR) on tumor cells via a single-chain antibody fragment (scFv) to achieve intracellular delivery of the plasmid DNA (Xu et al. 2001). Initial formulations contained Tf as targeting ligand (Xu et al. 1997) but the scFv has a smaller size than the Tf molecule and it allows large scale recombinant production and stricter quality control (Xu et al. 2002). TfRscFv-lipoplexes were shown to associate specifically with head and neck and prostate tumor cells (Xu et al. 2001). Using reporter assays and Western blotting it was demonstrated that transfection of tumor cells by TfRscFv-lipoplexes resulted in functional exogenous p53 expression in vitro and in vivo (Xu et al. 2001; Xu et al. 2002). Importantly, in a mouse tumor metastasis model treatment with TfRscFv-p53-lipoplexes combined with docetaxel (DTXL) resulted in a significant increase in survival compared to non-targeted p53-lipolexes combined with DTXL (Xu et al. 2001). Although these results were promising, rapid clearance of the TfRscFv-lipoplexes was observed. A sterically stabilized PEGylated lipoplex was designed to optimize circulation times in vivo (Yu et al. 2004). Although PEGylation of the lipoplexes resulted in reduced transfection efficiency in vitro, in a human xenograft prostate tumor model it was demonstrated that the targeted PEGylated lipoplexes induced approximately 7-fold more protein expression in tumors 96 h after treatment than non-PEGylated targeted lipoplexes, indicating the importance of lipoplex stability and circulation time (Yu et al. 2004). Recently reported results of a phase I trial with SGT-53 as a single agent in 11 patients with advanced solid tumors demonstrated no dose limiting toxicities and dose-dependent levels of the transgene were present in tumor biopsies of three patients. After 6 weeks of treatment, 7 of 11 patients had stable disease (Senzer et al. 2013). As SGT-53 is intended to be used in combination with standard radio/chemotherapy, it is now undergoing phase Ib trials to evaluate the safety of combinational therapy with DTXL and to establish a recommended dose for further studies (Synergene Therapeutics Inc.).
SGT-94 utilizes the same TfR-targeted platform as SGT-53 but its cargo consists of the gene that encodes the tumor suppressor protein RB94 (Pirollo et al. 2008). RB94 has broad anti-tumor activity and up to date no cytotoxicity with normal human cells or tumor cell resistance to RB94 has been observed (Xu et al. 1994; Xu et al. 1996; Zhang et al. 2003). In vitro cytotoxicity studies revealed that Tf-decorated RB94 lipoplexes increased chemosensitization of human bladder cancer cells 30-fold to gemcitabine and >55-fold to cisplatin compared to ligand-lacking formulations. Treatment of normal human endothelial cells did not result in significant sensitization which indicates that Tf mediated tumor cell specificity (Pirollo et al. 2008). RB94 protein expression was detected in tumors derived from mice injected with Tf-RB94-lipoplexes and TfRscFv-RB94-lipoplexes but not in mice injected with control formulations. Importantly, no detectable RB94 expression in the liver was observed as determined by WB, immunohistochemistry and DNA PCR (Pirollo et al. 2008). In efficacy studies with mice bearing human bladder carcinoma xenografts, treatment of mice with TfRscFv-RB94-lipoplexes combined with gemcitabine significantly inhibited tumor growth compared to ligand-lacking RB-94-lipoplexes and gemcitabine, and targeted formulations with a control vector combined with gemcitabine (Pirollo et al. 2008). SGT-94 has entered phase I trials to evaluate its safety and maximum tolerated dose (MTD) and to find evidence of RB94 expression in tumors after systemic administration (Synergene Therapeutics Inc.).
2.1.3 MM-302
MM-302 (Merrimack Pharmaceuticals) is a HER2-targeted nanomedicine that consists of PEGylated liposomes loaded with DOX and has progressed into phase I trials (Merrimack Pharmaceuticals Inc.). Tumor targeting is achieved by the attachment of HER2-targeted scFv antibody fragments to the surface of the liposomes (Park et al. 2001; Park et al. 2004, Nellis et al. 2005a, b). Since the first reports on HER2-targeted immunoliposomes (ILs) loaded with DOX emerged (Park et al. 1995; Kirpotin et al. 1997), many parameters of the formulation have been optimized such as liposomal composition, antibody construct and conjugation method (Park et al. 2001). These early studies have described the increase in HER2-positive (HER2+) cell binding and internalization of anti-HER2 liposomes compared to control liposomes. Increased cell association could be reversed by addition of free anti-HER2 antibody fragments confirming HER2-mediated interactions of the ligand-targeted PNM. In addition, a HER2-negative cell line did not show detectable uptake of anti-HER2 liposomes (Park et al. 1995; Kirpotin et al. 1997). Importantly, biodistribution studies in vivo revealed that conjugation of anti-HER2 antibody fragments did not increase radiolabeled liposomal tumor accumulation of the nanomedicine compared to PEGylated liposomal DOX (PLD) (Kirpotin et al. 2006). At the same time, gold-labeled anti-HER2 liposomes localized intracellularly while ligand-lacking liposomes primarily distributed to the extracellular tumor stroma. In a HER2-negative xenograft model the intratumoral distribution of ligand-targeted and ligand-lacking liposomes was similar indicating that both formulations accumulate in the tumor but anti-HER2 ILs associated directly with tumor cells (Kirpotin et al. 2006). Pharmacokinetic (PK) studies in rats showed comparable circulation times for HER2-targeted ILs and control formulations indicating that the presence of an antibody fragment on the liposomes did not alter clearance rates or induced accelerated clearance after multiple doses (Park et al. 2001; Park et al. 2002). Anti-tumor efficacy of anti-HER2 ILs-DOX has been extensively evaluated in multiple studies in four different human HER2+ breast cancer xenograft models. Although liposomal formulations varied between studies with regards to PEGylation, antibody fragment and conjugation method, pooled results of all eight studies demonstrate that treatment with anti-HER2 ILs-DOX significantly inhibited tumor growth when compared to PLD. In one of the xenograft studies, anti-HER2 ILs-DOX demonstrated cure rates up to 50 % (Park et al. 2001; Park et al. 2002). Additionally, anti-HER2 ILs-DOX treatment also showed superior efficacy in a xenograft model when compared to combination treatment with either free DOX or PLD and trastuzumab. In a xenograft model expressing low levels of HER2, treatment with anti-HER2 ILs-DOX and PLD induced only modest anti-tumor effects, confirming anti-HER2 ILs-DOX in vivo selectivity and the requirement of a receptor density or activity threshold for effective drug delivery (Park et al. 2001; Park et al. 2002). These studies have resulted in an optimized formulation used for clinical evaluation that consists of the anti-HER2 scFv F5 conjugated to PEG-PE micelles which are incorporated into PLD (Park et al. 2001; Park et al. 2004, Nellis et al. 2005a, b). The last few years, updates were presented at conferences on the progress of MM-302 including cardiosafety, efficacy and PK studies in preclinical models (Wickham et al. 2010; Geretti et al. 2011; Klinz et al. 2011). Recently, preliminary data of the ongoing phase I trials were presented. So far, 34 patients with HER2+ advanced breast cancer have enrolled of which 12 patients achieved stable disease and two patients have achieved partial response. MM-302 is tolerable in patients up to 40 mg/m2 and plasma pharmacokinetics are similar to ligand-lacking PLD (Wickham and Futch 2012).
2.1.4 Anti-EGFR ILs-DOX
Generated by the same original developers as MM-302 (Hermes Biosciences), ILs loaded with DOX that target epidermal growth factor receptor (EGFR) overexpressing tumors via coupling of Fab’ fragments of the anti-EGFR mAb cetuximab have also progressed into clinical trials (Noble et al. 2004). In vitro studies showed superior cell association and internalization of anti-EGFR ILs-DOX compared to ligand-lacking control formulations. For example, quantitative studies performed with pH-sensitive-loaded liposomes demonstrated ~30-fold more EGFR-positive cell internalization of anti-EGFR ILs compared to non-targeted PEGylated liposomes. In addition, cytotoxicity studies in EGFR-positive MDA-MB-468 cells showed that anti-EGFR ILs-DOX were 29-fold more effective than PLD (Mamot et al. 2003). Studies in rats showed similar pharmacokinetics of ligand-targeted and ligand-lacking liposomal DOX indicating that conjugation of antibody fragments did not alter liposomal stability or circulation time (Mamot et al. 2005). As observed with MM-302, biodistribution studies in mice showed no differences in tumor accumulation for EGFR-targeted liposomes and ligand-lacking formulations. However, quantitative flow cytometry analysis demonstrated that cellular accumulation of anti-EGFR liposomes was 6-fold higher when compared to ligand-lacking liposomes in tumor cells derived from mice (Mamot et al. 2005). In two EGFR-overexpressing tumor xenograft models, anti-EGFR ILs-DOX significantly inhibited tumor growth when compared to PLD (Mamot et al. 2005). Interestingly, in a drug resistant tumor xenograft model anti-EGFR ILs-DOX could significantly inhibit tumor growth when compared to PLD, suggesting that anti-EGFR ILs-DOX can overcome multidrug resistance (MDR) (Mamot et al. 2012a). In a recently finished phase I trial (University Hospital Basel Switzerland), 26 patients with EGFR-overexpressing advanced solid tumors were enrolled and treated with escalating doses of anti-EGFR ILs-DOX. One patient showed complete response, one partial response and ten patients had stable disease lasting 2–12 months. A recommended dose of 50 mg DOX per m2 was recommended for phase II trials (Mamot et al. 2012b).
2.1.5 2B3-101
2B3-101 (to-BBB Technologies) is liposome loaded with DOX designed to cross the blood-brain barrier (BBB) for the treatment of glioma. The BBB is a physical, transport and metabolic barrier that poses challenges for drug delivery to the brain. Important criteria related to the BBB, nanocarrier and clinical translation have been proposed for the development of nanomedicines to treat central nervous system (CNS) diseases (Gaillard et al. 2012b). 2B3-101 makes use of glutathione (GSH) as a targeting ligand, with the aim to cross the BBB via glutathione transporters without disrupting the neuroprotective function of the BBB. In proof-of-concept studies in rats, it was demonstrated that increasing amounts of GSH conjugated to PEGylated liposomes loaded the antiviral drug ribavirin resulted in higher amounts of free ribavirin in the brain (Rip et al. 2010). In preclinical studies in rats, 2B3-101 showed similar PK values and toxicity profile as compared to PLD (Gaillard et al. 2012a). However, DOX retention in the brain of rats was significantly higher after repeated administrations of 2B3-101 compared to PLD (Gaillard et al. 2012a). In a human breast cancer xenograft model in mice, both 2B3-101 and PLD demonstrated significant anti-tumor efficacy. In mice bearing intracranial U87 xenograft tumors, treatment with 2B3-101 given at the MTD prolonged survival up to 60 % compared to controls (Gaillard et al. 2012a). 2B3-101 is currently undergoing phase I/IIa trials to determine the safety and PK of the ligand-targeted PNM as a single agent or in combination with trastuzumab (to-BBB technologies B.V.). Accessible and/or available (pre)clinical data of 2B3-101 has been mostly confined to conference abstracts and the developers’ website.
2.1.6 MCC-465
MCC-465 (Mitsubishi Tanabe Pharma) is a DOX-loaded PEGylated liposome targeted to tumor cells via the conjugation of F(ab’)2 of the human GAH antibody (Hosokawa et al. 2003). Although its target antigen has not been characterized, selective binding of GAH antibody was demonstrated as staining of viable tumor tissues and tissue sections stained positively while no staining was observed on non-cancerous tissues (Hamaguchi et al. 2004; Hosokawa et al. 2004). Confocal microscopy studies showed that fluorescently labeled GAH-conjugated ILs loaded with DOX internalized in human stomach cancer cells via GAH-mediated interactions, as the addition of free GAH in combination with GAH-ILs-DOX prevented cell uptake. Ligand-lacking control formulations were hardly internalized by the tumor cells (Hosokawa et al. 2003; Hosokawa et al. 2004). In a pulse-chase assay in vitro, GAH-ILs-DOX induced significantly stronger dose-dependent cytotoxicity in human gastric tumor cells compared to PLD. No significant cytotoxicity of GAH-ILs-DOX was observed in human endothelial cells. The anti-tumor efficacy of GAH-ILs-DOX in various human xenograft models in mice was significantly higher than ligand-lacking control PLD (Hosokawa et al. 2003; Hamaguchi et al. 2004; Hosokawa et al. 2004; Shimada et al. 2005). No significant anti-tumor efficacy was observed in xenograft studies with GAH-negative cell lines and it was suggested that GAH-ILs-DOX can overcome DOX resistance of tumor cells (Hosokawa et al. 2003; Hamaguchi et al. 2004). Results from a phase I study indicated that MCC-465 was well tolerated with an MTD of 45.5 mg/m2 and a dose of 32.5 mg/m2 in an equivalent amount of DOX was recommended for phase II studies. No anti-tumor effects were observed but stable disease was observed in 10 of 18 patients (Matsumura et al. 2004). Recent updates on MCC-465 are not available and it is uncertain whether development is discontinued.
2.1.7 Lipovaxin-MM
Lipovaxin-MM (Lipotek) is a lipid-based vaccine for immunotherapy of malignant melanoma. Lipovaxin-MM does not directly target melanoma cells, but instead its strategy is based on delivering melanoma antigens to dendritic cells (DC) which in turn activate tumor-specific CD8+ cytotoxic T cells (CTL) (Altin and Parish 2006). The melanoma antigens in Lipovaxin-MM are derived from the membrane fraction of lysed MM200 melanoma cells. MM200 plasma derived membrane vesicles are isolated and subsequently fused with liposomes containing cytokines such as interferon-gamma (IFN-γ) or lipopolysaccharide (LPS) that provide a DC “danger” or maturation signal. The vaccine is targeted to DCs via engraftment of the domain antibody DMS5000 which is highly specific for DC-specific intracellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) (Altin et al., Altin and Parish 2006). In proof of concept studies, cell association of DC-targeted vaccines in vitro was 4 to 8-fold higher than ligand-lacking control formulations. This effect could be reversed by pre-incubation of the cells with free targeting ligand demonstrating specific interactions of the ligand-targeted PNM. DC-targeting in vivo was demonstrated by determining the number of fluorescent-positive cells in a draining lymph node after injection with ligand-targeted and ligand-lacking formulations. DC-targeted vesicles induced 4-fold more fluorescent cells than ligand-lacking formulations (van Broekhoven et al. 2004). In addition, in a B16-OVA melanoma model immunization of mice with targeted vaccines induced strong CTL responses in splenic T cells, induced protective immunity against tumors and could inhibit tumor growth (van Broekhoven et al. 2004). According to the patent application, Lipovaxin-MM used for studies in non-human primates consists of 4 pre-mix components (MM200 membrane vesicles, lyophilized liposomes, IFN-gamma and DMS5000) that are formulated prior to administration (Altin et al.). Treatment of macaques with Lipovaxin-MM resulted in production of vaccine-specific antibodies but it is not certain if this effect is caused specifically by the antigens as a ligand-lacking control was not included in this study (Altin et al.). A phase I study in 12 melanoma patients to determine adverse events, immunogenicity and efficacy of Lipovaxin-MM was recently completed but results have not yet been made available (Lipotek Pty. Ltd.).
2.2 Polymer-Based Nanomedicines
The potential of polymers for drug delivery was demonstrated by pioneering work in the 1970s (Couvreur et al. 1979; Gros et al. 1981). Polymer-based particulate nanocarriers such as polymeric nanoparticles are produced by self-assembly or cross-linking of polymeric building blocks to obtain nanoparticles with favorable physicochemical characteristics.
2.2.1 BIND-014
BIND-014 (BIND Biosciences) is a polymeric nanoparticle developed for the treatment of solid tumors. BIND-014 is composed of poly(d,l-lactide) (PLA) and PEG block copolymers to form a hydrophobic core for the encapsulation of DTXL, and a hydrophilic surface for prolonged circulation (Hrkach et al. 2012). The ligand-targeted PNM is targeted to prostate-specific membrane antigen (PSMA) expressing cells using the small-molecule S,S-2-[3-[5-amino-1-carboxypentyl]-ureido]-pentanedioic acid (ACUPA) as targeting ligand (Maresca et al. 2009; Hrkach et al. 2012). PSMA is expressed by prostate tumor cells and additionally, by the neovasculature of other types of solid tumors but not on normal vasculature (Chang et al. 1999). BIND-014 was developed by a novel strategy in which a library was composed of more than 100 self-assembling nanoparticles to obtain a single ligand-targeted PNM with optimized physicochemical properties (Gu et al. 2008; Shi et al. 2011; Hrkach et al. 2012). Initial in vitro studies performed with the PSMA-targeting RNA aptamer A10 (Lupold et al. 2002) as targeting ligand demonstrated a 77-fold increase in cell association of PSMA-targeted formulations compared to ligand-lacking formulations. No cell association to PSMA-negative cells was observed for either of the formulations (Farokhzad et al. 2004). In mice bearing human PSMA-positive prostate xenograft tumors, targeted poly(lactic-co-glycolic acid) (PLGA)-based nanoparticles delivered 3.77-fold more chemotherapeutic agent to tumors compared to ligand-lacking control nanoparticles after 24 h (Cheng et al. 2007). PSMA-targeted nanoparticles loaded with DTXL were significantly more cytotoxic in vitro compared to control DTXL nanoparticles without targeting ligand. In xenograft studies, ligand-targeted PNM loaded with DTXL significantly inhibited tumor growth and increased survival compared to ligand-lacking DTXL nanoparticles (Farokhzad et al. 2006). In later preclinical studies, optimized BIND-014 treatment caused significant tumor growth inhibition in a mouse xenograft prostate tumor model compared to ligand-lacking controls. In contrast, no difference in anti-tumor effect was observed in PSMA-negative xenograft models (Hrkach et al. 2012). BIND-014 is currently undergoing a phase I clinical trial to determine the safety in patients with advanced or metastatic cancer (BIND Biosciences). Interim data in three patients demonstrated that DTXL plasma levels are two orders of magnitude higher when administered as BIND-014 compared to solvent-based DTXL. Preliminary signs of BIND-014 anti-tumor efficacy were observed in two patients (Hrkach et al. 2012). Full phase I results with BIND-014 in patients with advanced solid tumors were recently presented which included anti-tumor response in 9 out of 28 patients and a MTD of 60 mg/m2 (Von Hoff et al. 2013). Phase II studies to evaluate the safety and efficacy of BIND-014 in patients with metastatic castration-resistant prostate cancer or as second-line therapy for patients with lung cancer have recently been initiated (BIND Biosciences, BIND Biosciences).
2.2.2 CALAA-01
CALAA-01 (Calando Pharmaceuticals) is a polymeric nanoparticle for siRNA-mediated treatment of solid tumors. This nanomedicine based on the RONDEL™ platform, which consists of four components that are mixed together and self-assemble into nanoparticles prior to administration: a linear cyclodextrin-containing polymer (CDP) backbone, adamantane-conjugated polyethylene glycol (AD-PEG), Tf-conjugated AD-PEG (Tf-PEG-AD) and siRNA (Davis 2009). CALAA-01 induces knockdown of the M2 subunit of ribonucleotide reductase (RRM2), which catalyzes the formation of deoxyribonucleotides from ribonucleotides for DNA synthesis (Cerqueira et al. 2005, Heidel et al. 2007a). Tf-nanoparticles were shown to associate with HeLa cells in a ligand density-dependent manner and cell uptake studies in the presence of free Tf demonstrated TfR-mediated cell internalization (Bartlett and Davis 2007). A multimodality imaging approach revealed no differences in tumor accumulation and tissue distribution between ligand-targeted PNM and ligand-lacking PNM siRNA formulations (Bartlett et al. 2007). However, using reporter assays it was shown that Tf-targeted nanoparticles did exhibit enhanced transfection efficiency in tumor bearing mice compared to ligand-lacking formulations (Bartlett et al. 2007). Increased inhibition of tumor growth in mice by Tf-siRNA-nanoparticles compared to ligand-lacking formulations was demonstrated in a mouse model of metastatic Ewing’s sarcoma (Hu-Lieskovan et al. 2005). In addition, in mice bearing head and neck cancer xenografts, CALAA-01 treatment reduced RRM2 mRNA and protein levels resulting in significant inhibition of tumor growth compared to nanoparticles with control siRNA (Rahman et al. 2012). Multiple systemic doses of CALAA-01 in non-human primates were well-tolerated and no significant signs of toxicity were observed at siRNA doses up to 8 mg/kg (Heidel et al. 2007b). Phase I trials evaluating CALAA-01 are ongoing (Calando Pharmaceuticals) and early results in three patients with solid tumors showed dose-dependent intracellular localization in tumor cells but not in the adjacent epidermis. Decreased protein expression of RRM2 in the tumor was observed in at least one patient, suggesting evidence for RNAi in humans (Davis et al. 2010).
2.2.3 SEL-068
SEL-068 (Selecta Biosciences) is a nicotine vaccine developed for treatment of tobacco dependence (Goniewicz and Delijewski 2013). The self-assembling synthetic polymeric nanoparticle (Gu et al. 2008) contains encapsulated toll-like receptor agonist to reduce the production of inflammatory cytokines, encapsulated universal T-helper cell peptide to evoke T-cell responses and nicotine covalently conjugated to the surface of the nanoparticle as a B-cell antigen (Kishimoto et al. 2012; Pittet et al. 2012). Administration of SEL-068 in mice and cynomolgus monkeys induced high titers of anti-nicotine antibodies with high affinity (Kishimoto et al. 2012; Pittet et al. 2012). In this way, addictive effects of smoking are counteracted by largely preventing nicotine in the circulation to cross the blood brain barrier and bind to nicotine receptors. Although SEL-068 is currently undergoing phase I clinical trials to evaluate the safety in smokers and non-smokers (Selecta Biosciences Inc.), available and/or accessible data is largely limited to conference abstracts and the website of Selecta Biosciences.
2.3 Bacterially-Derived Minicells
A relatively new NC platform utilizes bacterially-derived minicells for drug delivery. These minicells are bacterial cells of approximately 400 nm, devoid of a nucleus and produced by mutants in which genes responsible for cell division have been inactivated (MacDiarmid and Brahmbhatt 2011).
2.3.1 Erbitux®EDVsPAC
Targeted minicells for the treatment of solid tumors are under development by EnGeneIc. A wide range of chemotherapeutic drugs can be incorporated in the minicells, including DOX, paclitaxel (PAC) and cisplatin (MacDiarmid et al. 2007). Additionally, minicells can be loaded in a similar fashion with siRNA or with plasmid DNA encoding short hairpin RNA (shRNA) (MacDiarmid et al. 2009). Tumor targeting of minicells is achieved by bispecific antibodies which recognize both the O-polysaccharide component of the lipopolysaccharide present on the minicell surface and a cell surface receptor overexpressed on tumor cells such as EGFR (MacDiarmid et al. 2011). Cell specific association, uptake and toxicity of EGFR-targeted minicells loaded with DOX (EGFRminicellsDOX) was demonstrated in EGFR-expressing MDA-MB-468 human breast cancer cells (MacDiarmid et al. 2007). In several human tumor xenograft models in vivo (breast, lung, ovarian, lung, leukemia), different minicell formulations including EGFRminicellsDOX, EGFR-targeted minicells loaded with PAC (EGFRminicellsPAC) and HER2-targeted minicells loaded with DOX (HER2minicellsDOX) demonstrated strong anti-tumor activity compared to ligand-lacking control formulations (MacDiarmid et al. 2007). For comparison, 100-fold higher doses of Doxil® (100 μg) were needed to achieve similar anti-tumor effects of EGFRminicellsDOX (1 μg) in mice bearing breast cancer xenografts (MacDiarmid et al. 2007). The anti-cancer effect of DOX-loaded minicells was further demonstrated by tumor regression in two dogs with advanced T cell non-Hodgkin’s lymphoma, and safety of minicells was demonstrated by multiple consecutive iv-injections in three healthy pigs (MacDiarmid et al. 2007). Most interestingly, drug-resistance of colon cancer cells could be reversed with sequential treatment of EGFR-targeted minicells loaded with shRNA specific for the MDR P glycoprotein MDR1 (EGFRminicellsshMDR1) followed by targeted minicells loaded with chemotherapeutics. Furthermore, the sequential combination treatment effectively reversed MDR in colon, breast and uterine xenograft models in vivo (MacDiarmid et al. 2009). Intermediate results of a phase I safety and tolerability study were recently presented (Solomon et al. 2012). Multiple doses of intravenously administered EGFRminicellsPAC were generally well tolerated in 28 patients with advanced solid tumors and a dose of 1 × 1010 minicells are recommended for phase II studies (Solomon et al. 2012). A phase I/II study with EGFR-targeted minicells loaded with DOX in patients with glioma is also planned (EnGeneIC Ltd. 2013).
2.4 Retroviral Vectors
The unraveling of the retroviral life cycle basic principles led to the introduction of replication-incompetent retroviruses in the 1980s (Mann et al. 1983). Non-replicating retroviral vectors are able to efficiently integrate their genetic payload in the DNA of the target cell, making them attractive nanocarriers for gene therapy.
2.4.1 Rexin-G
Rexin-G is murine leukemia virus-based nanomedicine for the treatment of osteosarcoma, soft tissue sarcoma and pancreatic cancer developed by Epeius Biotechnologies. The main issue with retroviral vectors has been the lack of tissue specificity (Hall et al. 2000). However, Rexin-G is the first retrovector targeted to tumors and associated neovasculature via a high-affinity collagen-binding motif derived from von Willebrand factor. Rexin-G elicits anti-tumor effects by interfering with cell cycle control with a mutant cyclin G1 gene (Gordon and Hall 2010b). In human tumor xenografts, Rexin-G markedly inhibited tumor growth and increased survival compared to ligand-lacking controls (Gordon et al. 2001). Results from early phase I/II clinical trials in the Philippines for the treatment of metastatic pancreatic cancer and other solid tumors showed that Rexin-G was well tolerated, did not induce organ damage and that there were signs of antitumor activity (Gordon et al. 2004, 2006). In phase I/II clinical trials in the U.S.A., for the treatment of advanced or metastatic pancreatic cancer, Rexin-G was well tolerated in phase I studies but there was no evidence of an anti-tumor response (Galanis et al. 2008). In phase II of these clinical trials which involved higher doses of Rexin-G, no dose-limiting toxicity was found. At none of the doses tested, organ-related toxicity, signs of an antibody response, off-target transfection or presence of replication-competent retrovirus were observed. A correlation between Rexin-G dosage and overall survival was established (Chawla et al. 2010). Similar results were found in phase I/II and phase II trials for the treatment of sarcoma and osteosarcoma (Chawla et al. 2009). Based on these results Rexin-G gained orphan drug status for treatment of soft tissue sarcoma, osteosarcoma and pancreatic cancer in the U.S.A. (Gordon et al. 2006). Of note, during clinical trials Rexin-G treatment was associated with improvement of physiological conditions (liver function, ascites, blood chemistry, wound healing) presumably due to the targeting of exposed collagen by Rexin-G (Gordon and Hall 2009).
3 Discussion
The scope of this review was to provide an overview of ligand-targeted PNMs undergoing clinical evaluation and to reveal the added delivery benefits of the conjugated targeting ligands. Although 13 ligand-targeted PNMs have progressed into clinical trials, the contribution of targeting ligands to therapeutic efficacy of PNMs in humans has not yet been unambiguously proven. Twelve ligand-targeted PNMs are currently under active evaluation while the development of MCC-465 appears to have been discontinued. Limited access to (pre)clinical data for SEL-068 and 2B3-101 prevents detailed discussion of these products.
With the exception of the anti-nicotine vaccine SEL-068, all of the described ligand-targeted PNMs have been developed for the treatment of solid malignant neoplasms. As cancer remains the leading cause of death in the world today, the medical need to design more effective and safer anti-cancer drugs is evident. The anti-tumor effect of ligand-lacking PNMs, largely mediated by the EPR effect, may be further enhanced by the addition of targeting ligands to increase target cell specificity and internalization (reviewed elsewhere (Peer et al. 2007; Davis et al. 2008; Jain and Stylianopoulos 2010)).
The encapsulated therapeutic agent in seven anti-cancer nanomedicines is an established chemotherapeutic compound such as doxorubicin (MM-302, anti-EGFR ILS-DOX, MCC-465, 2B3-101), oxaliplatin (MBP-426), docetaxel (BIND-014) or paclitaxel (Erbitux®EDVsPAC). These compounds have been previously approved by the FDA either as free drug or formulated as ligand-lacking PNMs, thus lowering the development risk and reducing regulatory issues for the new ligand-targeted formulations in development. Four ligand-targeted PNMs contain plasmid DNA or siRNA (SGT-53, SGT-94, CALAA-01, Rexin-G). These molecules are unable to pass cell membranes and are dependent on ligand-induced receptor-mediated internalization for therapeutic activity. The two vaccine formulations targeted to antigen-presenting cells (APC) contain antigen and adjuvants to stimulate the immune system to produce cytotoxic T-cells (Lipovaxin-MM) or neutralizing antibodies (SEL-068).
Of the 13 discussed ligand-targeted PNMs, the exploited particulate nanocarrier of 8 formulations are lipid-based, 3 are based on polymeric NPs, 1 on a retroviral vector and 1 on a bacterial vector. The application of established lipid-based particulate nanocarriers is likely due to the clinical experience gained with these systems as ligand-lacking PNMs, and to reduce development risks and regulatory issues associated with novel nanocarrier systems. For example, MM-302 and anti-EGFR ILs-DOX consist of a similar formulation as Doxil® but targeting ligands are introduced by post-insertion of micelles bearing targeting ligands for tumor targeting (Nellis et al. 2005a, b, Mamot et al. 2012b). The rapid development of ligand-targeted PNMs based on polymers is noteworthy. Such systems are characterized by the production of self-assembling polymeric NP and high-throughput strategies giving advantages in terms of large-scale manufacturing and batch-to-batch variation. CALAA-01 is formulated prior to systemic administration by self-assembly of the (ligand-modfied) polymeric components and siRNA (Davis 2009). BIND-014 and SEL-068, based on the AccurinsTM technology, were developed by the design of pre-functionalized triblock co-polymers to create a library of self-assembling targeted polymeric nanoparticles allowing efficient tailoring of physicochemical characteristics (Gu et al. 2008; Shi et al. 2011). Genetic engineering has led to the development of the replication incompetent retroviral vector Rexin-G, which is generated in human producer cells to generate a targeted biocompatible ligand-targeted PNM with a size of approximately 100 nm (Gordon and Hall 2009). Interestingly, bacterially-derived minicells employed for the generation of Erbitux®EDVsPAC are characterized by a larger size (400 nm) compared to other PNMs (MacDiarmid and Brahmbhatt 2011). It has been shown that the cut-off size of permeable tumor vasculature in the majority of tumors varies between 380 and 780 nm (Yuan et al. 1995; Hobbs et al. 1998). However, the size of synthetic nanocarriers is generally designed to remain below 200 nm to avoid rapid uptake by the MPS and to enhance tumor penetration.
With regard to physicochemical properties of PNMs, the effect of parameters such as total entrapped drug and free drug content, release kinetics and surface characteristics should be acknowledged. Many nanomedicines aim at increasing the MTD due to an improved safety profile. If the overall dose (entrapped + free drug) is significantly increased, the free fraction may become dose-limiting, especially in case of highly potent drugs. Therefore, free drug content and release profile represent important parameters to take into account. Disclosure of release kinetics under physiologically relevant conditions is therefore encouraged.
Regarding the targeting ligand utilized for the discussed ligand-targeted PNMs, 4 nanomedicines target the transferrin receptor (TfR). Specific tumor markers such as EGFR, HER2 and PSMA are targeted by four nanomedicines (the exact target receptor of MCC-465 is not known). In contrast, a tumor stromal target is exploited by one ligand-targeted PNM. Both vaccine formulations target APC and one formulation is designed for crossing the BBB via GSH transporters. The TfR is a well-established target for cancer treatment by virtue of its overexpression on a range of tumors (Daniels et al. 2012). Attachment of transferrin to PNMs for targeting is exploited by MBP-426 and CALAA-01. The lipoplex formulations SGT-53 and SGT-94 also target the TfR but make use of antibody fragments instead of transferrin (Xu et al. 2002). Antibody fragments are smaller than transferrin and recombinant expression allows efficient large scale production and high quality control reducing batch-to-batch variation. When compared to full monoclonal antibodies, the use of antibody fragments for targeting is preferred because they lack the Fc part of the antibody, preventing rapid recognition by cells of the immune system and subsequent clearance of the ligand-targeted PNM. In the case of Erbitux®EDVsPAC minicells, the Fc region is present, but complement-mediated toxicity is inhibited as protein A/G blocks the Fc part of the conjugated monoclonal antibodies (MacDiarmid et al. 2011). However, an antibody response to the O-polysaccharide component of the bispecific antibody was observed in phase I trials (Solomon et al. 2012). Interestingly, while most ligand-targeted PNMs are directed to a single surface receptor overexpressed on tumor cells, Rexin-G is equipped with more promiscuous high-affinity collagen-binding motifs as targeting ligands, resulting in efficient drug delivery to tumor cells, stroma cells, neovasculature and sites of metastasis without apparent significant toxicity towards healthy tissues (Gordon and Hall 2010a). This indicates that proteins overexpressed on tumor cells can be used to discriminate between tumor and healthy cells, but it may be beneficial for robust anti-cancer effects to target the tumor stroma rather than solely tumor cells. The two vaccine products show that ligand-targeted PNMs can also be directed to antigen presenting cells for the generation of targeted vaccines. It is likely that their prolonged circulation time allows the nanomedicines to reach target sites and activate cells of the immune system. Besides general vaccine applications, the vaccine strategy can be applied to design effective anti-cancer nanomedicines that are not hampered by limitations of direct tumor cell targeting (Lammers et al. 2012).
It is generally believed that ligand-lacking and ligand-targeted PNMs have comparable PK parameters, biodistribution and tumor targeting profiles. However, surface characteristics play an important role in interaction with blood components and cell membranes. Modification of PNMs with targeting ligands may therefore alter PK and biodistribution profiles. In addition, in case of ligand-targeted PNMs specific effects of a carefully chosen ligand can be outweighed by aspecific interactions due to charge interactions and adsorbance of proteins to the nanomedicine shell. Studies comparing pharmacokinetics and biodistribution of ligand-lacking versus ligand-modified PNMs are scarce and more importantly: impossible in the clinical setting.
Localization studies in animal models which compared the ligand-targeted formulation to the corresponding ligand-lacking one have only been reported for 7 ligand-targeted PNMs (Table 7.2). Of those 7, 4 reported increased target localization compared to ligand-lacking PNM in vivo, while the other three studies demonstrated comparable target localization values for ligand-lacking and ligand-targeted PNMs. For MM-302, anti-EGFR ILs-DOX and CALAA-01, it was demonstrated that in murine xenograft models overall tumor accumulation was similar for ligand-lacking and ligand-targeted PNMs. However, in studies performed with MBP-462, SGT-53, BIND-014 and Erbitux®EDVsPAC, a higher degree of tumor localization of the ligand-targeted PNM relative to the ligand-lacking PNM was observed. In the case of 5 ligand-targeted PNMs, literature has reported on improved in vitro cellular internalization versus ligand-lacking PNMs. The publications on MM-302 and anti-EGFR ILs-DOX reported results for in vivo cell internalization versus ligand-lacking PNMs, and both showed improvement over ligand-lacking PNMs. Intermediate results from a phase I trial with CALAA-01 reported on target cell internalization in tumor biopsies of three patients (Davis et al. 2010). Dose dependent presence of the transgene was reported in biopsies from metastatic lesions of three patients treated with SGT-53, while no transgene presence was detected in skin biopsies (Senzer et al. 2013). In light of these results it is possible that, while ligand-targeted and ligand-lacking formulations are both dependent on extravasation from the circulation into the tumor, ligand-targeted PNMs are retained longer in the tumor than their ligand-lacking counterparts due to increased cellular internalization or other targeting-ligand mediated interactions within the target. However, the number of ligand-targeted PNMs tested for in vivo target cell internalization is too limited to provide conclusive evidence. Therefore, in tumors where the EPR effect is present, the use of targeting ligands may only be useful to increase cellular internalization or in cases where the targeting ligand itself has intrinsic anti-tumor effects.
Regarding efficacy, all ligand-targeted PNMs included in this overview have shown increased efficacy in vitro and in vivo compared to their ligand-lacking counterparts (with the exception of SEL-068). Although in vitro and in vivo model systems do not provide definite proof of efficacy in humans and no data beyond phase I and II trials have been reported as of yet, these results are encouraging for the concept of ligand-targeted PNMs. In some cases signs of efficacy in phase I and/or II trials were observed, but these studies did not include ligand-lacking PNM controls.
How improved efficacy is related to the presence of a targeting ligand cannot be resolved because the comparison of PK and distribution of ligand-targeted versus ligand-lacking PNMs is often not included. As mentioned previously, such comparisons are scarce and clinical trials are not designed to compare ligand-lacking and ligand-targeted PNMs.
In the majority of the cases described in this review, there is insufficient literature that has reported on cellular and animal studies in which the ligand-targeted PNMs have been compared to ligand-lacking PNMs regarding the parameters in Table 7.2. The only exception is MM-302, which reported superior results to ligand-lacking PNMs in all of these parameters except target localization in animal studies. This indicates that the improved efficacy of MM-302 might be due to improved cellular internalization but since several other ligand-targeted PNMs reported improved target localization in animal studies, this does not necessarily hold true for all ligand-targeted PNMs. Since phase I clinical trials for most of the ligand-targeted PNMs reported in this overview are still ongoing, not all results have been published as of yet. Treatment with ligand-targeted PNMs seemed to be well tolerated in patients in the studies that have been published so far.
In the majority of the discussed ligand-targeted PNMs, the toxicity seems comparable to that of ligand-lacking PNMs in terms of MTD and dose-limiting toxicities (DLT) (Table 7.3). For example, the MTD of MM-302 (40 mg/m2) (Wickham and Futch 2012), anti-EGFR ILs-DOX (50 mg/m2) (Mamot et al. 2012b) and MCC-465 (45.5 mg/m2) (Matsumura et al. 2004) is comparable to that of Doxil® (50 mg/m2) indicating that conjugating targeting ligands to PNMs does not seem to alter the toxicity profile. However, results from a phase I trial with Erbitux®EDVsPAC report a different DLT compared to albumin-bound paclitaxel which may be related to the bacterially-derived particulate nanocarrier or the bispecific antibody (discussed above). It has to be noted that the MTD values for most of the discussed ligand-targeted are based on results obtained from smaller phase I trials and may change after larger phase II/III trials.
4 Future Directions
Ligand-targeted PNMs may prove beneficial in increasing drug exposure due to increased target cell uptake and target tissue retention compared to ligand-lacking PNMs. Additionally, there are several applications where the use of ligand-targeted PNMs may have advantages over ligand-lacking PNMs.
-
1.
Ligand-targeted approaches are crucial for molecules that need to localize intracellularly for therapeutic activity but are not capable of crossing cellular membranes, such as nucleic acids. As a consequence, the development of systemically administered gene (regulating) therapy is evolving concurrently with the development of efficient ligand-targeted particulate nanocarriers (Pecot et al. 2011). The therapeutic potential of RNA interference is illustrated by CALAA-01, which decreased target protein expression in a patient’s tumor in a phase I trial (Davis et al. 2010). The feasibility of therapeutic DNA is demonstrated by Rexin-G, which has shown promising anti-tumor activity in patients including inhibition of metastatic lesions, angiogenesis and intractable or resistant tumors (Gordon and Hall 2009).
-
2.
One common mechanism underlying MDR of tumors is the overexpression of drug-efflux pumps, which actively expel anti-cancer drugs. ligand-targeted PNMs may be able to circumvent MDR by virtue of another cellular fate after receptor-mediated endocytosis rather than passive diffusion over cell membranes of free drug released by ligand-lacking PNMs (Gao et al. 2012). For example, anti-EGFR ILs-DOX showed significantly enhanced antitumor activity in a MDR breast cancer xenograft tumor model compared to free DOX and PLD (Mamot et al. 2012a).
-
3.
Ligand-targeted approaches can also be exploited to generate nanomedicines that exploit two therapeutic strategies simultaneously in order to achieve additive or synergistic anti-tumor effects. For example, DOX-loaded polymeric micelles decorated with intrinsically active anti-EGFR nanobodies significantly reduced tumor growth and prolonged survival of tumor-bearing mice when compared to DOX-loaded micelles without attached targeting ligands (Talelli et al. 2013).
-
4.
An alternative approach to the targeted delivery of anti-cancer drugs to tumor cells is targeting of the tumor blood supply. The endothelial cells of the tumor vasculature are readily accessible to targeted nanomedicines circulating in the bloodstream and more genetically stable than tumor cells limiting the occurrence of drug resistance phenomena. Delivery of DOX by ligand-targeted PNMs targeting ανβ3 integrins overexpressed on tumor neovasculature reduced tumor growth of DOX-insensitive tumors while PLD did not (Schiffelers et al. 2003). In line with these results, it was shown that DOX-loaded ligand-targeted PNMs targeting ανβ3 integrins suppressed metastasis (Murphy et al. 2008).
-
5.
Besides the development of ligand-targeted PNMs for cancer treatment, ligand-targeted approaches can be exploited for the generation of effective vaccines as demonstrated by the clinical evaluation of Lipovaxin-MM and SEL-068.
-
6.
The development of effective nanomedicines for the treatment of CNS remains challenging due to the presence of the BBB. In addition to the physical barrier, metabolic barriers and drug-efflux transporters results in a restriction of drugs that are able to cross the BBB in adequate amounts to reach therapeutic activity (Wong et al. 2012). Ligand-targeted approaches may be more effective compared to unencapsulated drugs or ligand-lacking nanomedicines as they can improve drug delivery to the CNS via receptor-mediated transcytosis (Pinzon-Daza et al. 2013), exemplified by the clinical evaluation of 2B3-101.
To determine the feasibility of clinically relevant ligand-targeted PNMs, further preclinical studies focused on relation between physicochemical properties (nanocarrier type, size and surface characteristics) in combination with targeting ligand properties (type and size) and biodistribution, safety and efficacy are encouraged. Current knowledge of nanotechnology, tumor biology and interactions of nanomedicines in the human body is (too) limited. To advance the applicability of ligand-targeted PNMs, lessons learnt from their bench-to-bedside translation have revealed key issues that need to be addressed including in vitro/in vivo characterization of PNM physicochemical properties (Cho et al. 2013), choice of appropriate animal models (Lammers et al. 2012) and the influence of receptor expression levels on ligand-targeted PNM efficacy (Hendriks et al. 2013). The applicability of ligand-targeted PNMs is ultimately determined by the balance between clinical benefits versus safety and cost-effectiveness of the production process (Cheng et al. 2012).
The efficacy and safety of ligand-targeted PNMs has been shown in animals, but the evidence for the added delivery value of target ligand-coupling to nanomedicines in humans remains to be established. Progress of the ligand-targeted PNMs described in this review through clinical trials will reveal in the upcoming years if ligand-targeted PNMs will represent safe and efficacious drugs in the future.
Abbreviations
- AD-PEG:
-
Adamantane-conjugated polyethylene glycol
- ADC:
-
antibody-drug conjugate
- APC:
-
antigen-presenting cell
- BBB:
-
blood-brain barrier
- CDP:
-
cyclodextrin-containing polymer
- CNS:
-
central nervous system
- CTL:
-
cytotoxic T cells
- DC:
-
dendritic cell
- DC-SIGN:
-
dendritic cell-specific intracellular adhesion molecule 3-grabbing non-integrin
- DLT:
-
dose-limiting toxicity
- DTXL:
-
docetaxel
- DOX:
-
doxorubicin
- EPR:
-
enhanced permeability and retention
- EGFR:
-
epidermal growth factor receptor
- GSH:
-
glutathione
- ILs:
-
immunoliposomes
- IFN-γ:
-
interferon-gamma
- LPS:
-
lipopolysaccharide
- RRM2:
-
M2 subunit of ribonucleotide reductase
- MDR:
-
multi drug resistance
- MPS:
-
mononuclear phagocyte system
- MTD:
-
maximum tolerated dose
- NGPE:
-
N-glutaryl-phosphatidylethanolamine
- L-OHP:
-
oxaliplatin
- PAC:
-
paclitaxel
- PNM:
-
particulate nanomedicine
- PLD:
-
PEGylated liposomal doxorubicin
- PK:
-
pharmacokinetic
- PLA:
-
poly(d,l-lactide)
- PEG:
-
polyethylene glycol
- PLGA:
-
poly(lactic-co-glycolic acid)
- PSMA:
-
prostate-specific membrane antigen
- ACUPA:
-
S,S-2-[3-[5-amino-1-carboxypentyl]-ureido]-pentanedioic acid
- scFv:
-
single-chain antibody fragment
- shRNA:
-
short hairpin RNA
- Tf:
-
transferrin
- Tf-AD-PEG:
-
transferrin-conjugated adamantane-conjugated polyethylene glycol
- TfR:
-
transferrin receptor
References
Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF (1977) Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J Biol Chem 252(11):3582–3586
Adair JR, Howard PW, Hartley JA, Williams DG, Chester KA (2012) Antibody-drug conjugates – a perfect synergy. Expert Opin Biol Ther 12(9):1191–1206
Allen TM, Cullis PR (2013) Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 65(1):36–48
Altin JG, Parish CR (2006) Liposomal vaccines – targeting the delivery of antigen. Methods 40(1):39–52
Altin J, Atmosukarto I, De Wildt RM, Parish C, Price J. Composition for targeting dendritic cells. US 2011/0212168 A1. November 2009 (filing date)
Bae YH, Park K (2011) Targeted drug delivery to tumors: myths, reality and possibility. J Control Release 153(3):198–205
Bangham AD, Standish MM, Watkins JC (1965) Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13(1):238–252
Barenholz Y (2012) Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release 160(2):117–134
Bartlett DW, Davis ME (2007) Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug Chem 18(2):456–468
Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME (2007) Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A 104(39):15549–15554
Bellocq NC, Pun SH, Jensen GS, Davis ME (2003) Transferrin-containing, cyclodextrin polymer-based particles for tumor-targeted gene delivery. Bioconjug Chem 14(6):1122–1132
BIND Biosciences. A phase 1 open label, safety, pharmacokinetic and pharmacodynamic dose escalation study of BIND-014 (Docetaxel Nanoparticles for Injectable Suspension), given by intravenous infusion to patients with advanced or metastatic cancer. In: ClinicalTrials.gov [Internet], Bethesda (MD): National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01300533
BIND Biosciences. A phase 2 study to determine the safety and efficacy of BIND-014 (Docetaxel Nanoparticles for Injectable Suspension) as second-line therapy to patients with non-small cell lung cancer. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01792479
BIND Biosciences. A phase 2 study to determine the safety and efficacy of BIND-014 (Docetaxel Nanoparticles for Injectable Suspension), administered to patients with metastatic castration-resistant prostate cancer. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01812746.
Blume G, Cevc G (1990) Liposomes for the sustained drug release in vivo. Biochim Biophys Acta 1029(1):91–97
Calando Pharmaceuticals. A phase I, dose-escalating study of the safety of intravenous CALAA-01 in adults with solid tumors refractory to standard-of-care therapies. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT00689065
Casi G, Neri D (2012) Antibody-drug conjugates: basic concepts, examples and future perspectives. J Control Release 161(2):422–428
Cerqueira NM, Pereira S, Fernandes PA, Ramos MJ (2005) Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy. Curr Med Chem 12(11):1283–1294
Chang SS, O’Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB (1999) Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin Cancer Res 5(10):2674–2681
Chawla SP, Chua VS, Fernandez L, Quon D, Saralou A, Blackwelder WC, Hall FL, Gordon EM (2009) Phase I/II and phase II studies of targeted gene delivery in vivo: intravenous Rexin-G for chemotherapy-resistant sarcoma and osteosarcoma. Mol Ther 17(9):1651–1657
Chawla SP, Chua VS, Fernandez L, Quon D, Blackwelder WC, Gordon EM, Hall FL (2010) Advanced phase I/II studies of targeted gene delivery in vivo: intravenous Rexin-G for gemcitabine-resistant metastatic pancreatic cancer. Mol Ther 18(2):435–441
Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC (2007) Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 28(5):869–876
Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A (2012) Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science 338(6109):903–910
Cho EJ, Holback H, Liu KC, Abouelmagd SA, Park J, Yeo Y (2013) Nanoparticle characterization: state of the art, challenges, and emerging technologies. Mol Pharm 10(6):2093–2110
Couvreur P, Kante B, Roland M, Speiser P (1979) Adsorption of antineoplastic drugs to polyalkylcyanoacrylate nanoparticles and their release in calf serum. J Pharm Sci 68(12):1521–1524
Crielaard BJ, Lammers T, Schiffelers RM, Storm G (2012) Drug targeting systems for inflammatory disease: one for all, all for one. J Control Release 161(2):225–234
Dams ET, Laverman P, Oyen WJ, Storm G, Scherphof GL, van Der Meer JW, Corstens FH, Boerman OC (2000) Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther 292(3):1071–1079
Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML (2012) The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta 1820(3):291–317
Davis ME (2009) The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm 6(3):659–668
Davis ME, Chen ZG, Shin DM (2008) Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov 7(9):771–782
Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A (2010) Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464(7291):1067–1070
Drexler KE, Peterson C, Pergamit G (1991) Unbounding the future: the nanotechnology revolution. William Morrow/Quill Books, New York
Duncan R, Gaspar R (2011) Nanomedicine(s) under the microscope. Mol Pharm 8(6):2101–2141
EnGeneIC Ltd. A phase 1/2 study to determine the effect of doxorubicin loaded EnGeneIC delivery vehicles on progression free survival in patients with Recurrent Glioblastoma Multiforme (GBM). In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved August 2013. Available from https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=363821
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J (2013) The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine 9(1):1–14
European Medical Research Counsils (2004) European science foundation forward look on Nanomedicine 2005
Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R (2004) Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res 64(21):7668–7672
Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R (2006) Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A 103(16):6315–6320
Gabizon AA (2001) Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Investig 19(4):424–436
Gaillard PJ, Gladdines W, Appeldoorn CCM, Rip J, Boogerd WJ, Beijnen JH, Brandsma D, van Tellingen O (2012a) Development of glutathione pegylated liposomal doxorubicin (2B3-101) for the treatment of brain cancer [abstract]. In: Proceedings of the 103rd annual meeting of the American Association for Cancer Research, Chicago, Illinois. AACR; 2012. Abstract nr 5687
Gaillard PJ, Visser CC, Appeldoorn CC, Rip J (2012b) Targeted blood-to-brain drug delivery –10 key development criteria. Curr Pharm Biotechnol 13(12):2328–2339
Galanis E, Carlson SK, Foster NR, Lowe V, Quevedo F, McWilliams RR, Grothey A, Jatoi A, Alberts SR, Rubin J (2008) Phase I trial of a pathotropic retroviral vector expressing a cytocidal cyclin G1 construct (Rexin-G) in patients with advanced pancreatic cancer. Mol Ther 16(5):979–984
Ganta S, Devalapally H, Shahiwala A, Amiji M (2008) A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release 126(3):187–204
Gao Z, Zhang L, Sun Y (2012) Nanotechnology applied to overcome tumor drug resistance. J Control Release 162(1):45–55
Geretti E, Reynolds J, Hendriks B, Eckelhofer I, Espelin C, Gaddy D, Klinz S, Lee H, Leonard S, Olivier K, Agresta S, Niyikiza C, Nielsen U, Wickham T (2011) HER2-targeted liposomal doxorubicin MM-302 has a favorable cardiosafety profile in preclinical models [abstract]. Molecular Targets and Cancer Therapeutics, San Fransisco, California: AACR-NCI-EORTC; 2011. Abstract nr C90
Goniewicz ML, Delijewski M (2013) Nicotine vaccines to treat tobacco dependence. Hum Vaccin Immunother 9(1):13–25
Gordon EM, Hall FL (2009) The ‘timely’ development of Rexin-G: first targeted injectable gene vector (review). Int J Oncol 35(2):229–238
Gordon EM, Hall FL (2010a) Noteworthy clinical case studies in cancer gene therapy: tumor-targeted Rexin-G advances as an efficacious anti-cancer agent. Int J Oncol 36(6):1341–1353
Gordon EM, Hall FL (2010b) Rexin-G, a targeted genetic medicine for cancer. Expert Opin Biol Ther 10(5):819–832
Gordon EM, Liu PX, Chen ZH, Liu L, Whitley MD, Gee C, Groshen S, Hinton DR, Beart RW, Hall FL (2000) Inhibition of metastatic tumor growth in nude mice by portal vein infusions of matrix-targeted retroviral vectors bearing a cytocidal cyclin G1 construct. Cancer Res 60(13):3343–3347
Gordon EM, Chen ZH, Liu L, Whitley M, Wei D, Groshen S, Hinton DR, Anderson WF, Beart RW Jr, Hall FL (2001) Systemic administration of a matrix-targeted retroviral vector is efficacious for cancer gene therapy in mice. Hum Gene Ther 12(2):193–204
Gordon EM, Cornelio GH, Lorenzo CC 3rd, Levy JP, Reed RA, Liu L, Hall FL (2004) First clinical experience using a ‘pathotropic’ injectable retroviral vector (Rexin-G) as intervention for stage IV pancreatic cancer. Int J Oncol 24(1):177–185
Gordon EM, Lopez FF, Cornelio GH, Lorenzo CC 3rd, Levy JP, Reed RA, Liu L, Bruckner HW, Hall FL (2006) Pathotropic nanoparticles for cancer gene therapy Rexin-G IV: three-year clinical experience. Int J Oncol 29(5):1053–1064
Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E (2000) Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res 6(4):1205–1218
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R (1994) Biodegradable long-circulating polymeric nanospheres. Science 263(5153):1600–1603
Gros L, Ringsdorf H, Schupp H (1981) Polymeric antitumor agents on a molecular and on a cellular level? Angew Chem Int Ed Engl 20(4):305–325
Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC (2008) Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A 105(7):2586–2591
Hall FL, Liu L, Zhu NL, Stapfer M, Anderson WF, Beart RW, Gordon EM (2000) Molecular engineering of matrix-targeted retroviral vectors incorporating a surveillance function inherent in von Willebrand factor. Hum Gene Ther 11(7):983–993
Hamaguchi T, Matsumura Y, Nakanishi Y, Muro K, Yamada Y, Shimada Y, Shirao K, Niki H, Hosokawa S, Tagawa T, Kakizoe T (2004) Antitumor effect of MCC-465, pegylated liposomal doxorubicin tagged with newly developed monoclonal antibody GAH, in colorectal cancer xenografts. Cancer Sci 95(7):608–613
Heidel JD, Liu JY, Yen Y, Zhou B, Heale BS, Rossi JJ, Bartlett DW, Davis ME (2007a) Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin Cancer Res 13(7):2207–2215
Heidel JD, Yu Z, Liu JY, Rele SM, Liang Y, Zeidan RK, Kornbrust DJ, Davis ME (2007b) Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci U S A 104(14):5715–5721
Hendriks BS, Klinz SG, Reynolds JG, Espelin CW, Gaddy DF, Wickham TJ (2013) Impact of tumor HER2/ERBB2 expression level on HER2-targeted liposomal doxorubicin-mediated drug delivery: multiple low-affinity interactions lead to a threshold effect. Mol Cancer Ther 12:1816
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK (1998) Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A 95(8):4607–4612
Hoffman AS (2008) The origins and evolution of “controlled” drug delivery systems. J Control Release 132(3):153–163
Hosokawa S, Tagawa T, Niki H, Hirakawa Y, Nohga K, Nagaike K (2003) Efficacy of immunoliposomes on cancer models in a cell-surface-antigen-density-dependent manner. Br J Cancer 89(8):1545–1551
Hosokawa S, Tagawa T, Niki H, Hirakawa Y, Ito N, Nohga K, Nagaike K (2004) Establishment and evaluation of cancer-specific human monoclonal antibody GAH for targeting chemotherapy using immunoliposomes. Hybrid Hybridomics 23(2):109–120
Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S (2012) Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med 4(128):128ra139
Huh AJ, Kwon YJ (2011) ‘Nanoantibiotics’: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J Control Release 156(2):128–145
Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ (2005) Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res 65(19):8984–8992
Ibrahim NK, Desai N, Legha S, Soon-Shiong P, Theriault RL, Rivera E, Esmaeli B, Ring SE, Bedikian A, Hortobagyi GN, Ellerhorst JA (2002) Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin Cancer Res 8(5):1038–1044
Ishida O, Maruyama K, Tanahashi H, Iwatsuru M, Sasaki K, Eriguchi M, Yanagie H (2001) Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res 18(7):1042–1048
Izbicka E, Diaz A, Campos D, Wick M, Takimoto C, Nagasawa M, Hashimoto C (2007) Evaluation of antitumor activity and biomarkers for MBP-426 in human pancreatic cancer xenograft models in vivo [abstract]. In: Proceedings of the 98th annual meeting of the American Association for Cancer Research, Los Angeles, California (CA): AACR; 2007. Abstract nr 5586
Jain RK, Stylianopoulos T (2010) Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 7(11):653–664
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC (2012) Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev 41(7):2971–3010
Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P, Benz CC, Papahadjopoulos D (1997) Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry 36(1):66–75
Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW (2006) Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 66(13):6732–6740
Kishimoto K, Altreuter D, Johnston L Keller P, Pittet L (2012) SEL-068 A fully synthetic nanoparticle vaccine for smoking cessation and relapse prevention [abstract]. In: Society for research on nicotine and tobacco 18th annual meeting, Houston, Texas: SRNT; 2012. Abstract nr POS1-1
Klibanov AL, Maruyama K, Torchilin VP, Huang L (1990) Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett 268(1):235–237
Klinz S, Eckelhofer I, Gaddy D, Geretti E, Hendriks B, Lee H, Leonard S, Reynolds J, Wickham T (2011) MM-302, a HER2-targeted liposomal doxorubicin, shows binding/uptake and efficacy in HER2 2+ cells and xenograft models [abstract]. In: Proceedings of the 102nd annual meeting of the American Association for Cancer Research, Orlando, Florida: AACR; 2011. Abstract nr 3637
Knop K, Hoogenboom R, Fischer D, Schubert US (2010) Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl 49(36):6288–6308
Kwon IK, Lee SC, Han B, Park K (2012) Analysis on the current status of targeted drug delivery to tumors. J Control Release 164(2):108–114
Lammers T, Kiessling F, Hennink WE, Storm G (2012) Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release 161(2):175–187
Laverman P, Carstens MG, Boerman OC, Dams ET, Oyen WJ, van Rooijen N, Corstens FH, Storm G (2001) Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection. J Pharmacol Exp Ther 298(2):607–612
Lipotek Pty. Ltd. A phase I open-label study of the safety and immunogenicity of escalating doses of Lipovaxin-MM, a novel melanoma immunotherapeutic, in patients with metastatic melanoma. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01052142
Lobatto ME, Fuster V, Fayad ZA, Mulder WJ (2011) Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov 10(11):835–852
Lupold SE, Hicke BJ, Lin Y, Coffey DS (2002) Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62(14):4029–4033
MacDiarmid JA, Brahmbhatt H (2011) Minicells: versatile vectors for targeted drug or si/shRNA cancer therapy. Curr Opin Biotechnol 22(6):909–916
MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, Haasdyk JE, Dickson KA, Brahmbhatt VN, Pattison ST, James AC, Al Bakri G, Straw RC, Stillman B, Graham RM, Brahmbhatt H (2007) Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 11(5):431–445
MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, Sedliarou I, Wetzel S, Kochar K, Brahmbhatt VN, Phillips L, Pattison ST, Petti C, Stillman B, Graham RM, Brahmbhatt H (2009) Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug. Nat Biotechnol 27(7):643–651
Maeda H, Nakamura H, Fang J (2013) The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev 65(1):71–79
Maindrault-Goebel F, de Gramont A, Louvet C, Andre T, Carola E, Mabro M, Artru P, Gilles V, Lotz JP, Izrael V, Krulik M (2001) High-dose intensity oxaliplatin added to the simplified bimonthly leucovorin and 5-fluorouracil regimen as second-line therapy for metastatic colorectal cancer (FOLFOX 7). Eur J Cancer 37(8):1000–1005
Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW (2003) Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res 63(12):3154–3161
Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW (2005) Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 65(24):11631–11638
Mamot C, Ritschard R, Wicki A, Kung W, Schuller J, Herrmann R, Rochlitz C (2012a) Immunoliposomal delivery of doxorubicin can overcome multidrug resistance mechanisms in EGFR-overexpressing tumor cells. J Drug Target 20(5):422–432
Mamot C, Ritschard R, Wicki A, Stehle G, Dieterle T, Bubendorf L, Hilker C, Deuster S, Herrmann R, Rochlitz C (2012b) Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: a phase 1 dose-escalation study. Lancet Oncol 13(12):1234–1241
Mann R, Mulligan RC, Baltimore D (1983) Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 33(1):153–159
Maresca KP, Hillier SM, Femia FJ, Keith D, Barone C, Joyal JL, Zimmerman CN, Kozikowski AP, Barrett JA, Eckelman WC, Babich JW (2009) A series of halogenated heterodimeric inhibitors of prostate specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer. J Med Chem 52(2):347–357
Matsumura Y, Maeda H (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 46(12 Pt 1):6387–6392
Matsumura Y, Gotoh M, Muro K, Yamada Y, Shirao K, Shimada Y, Okuwa M, Matsumoto S, Miyata Y, Ohkura H, Chin K, Baba S, Yamao T, Kannami A, Takamatsu Y, Ito K, Takahashi K (2004) Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 15(3):517–525
Mebiopharm Co. Ltd. A phase I, open label study of MBP-426 given by intravenous infusion in patients with advanced or metastatic solid tumors. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT00355888
Mebiopharm Co. Ltd. A phase Ib/II study of MBP-426 in patients with second line gastric, gastro esophageal, or esophageal adenocarcinoma. In: ClinicalTrials.gov [Internet], Bethesda (MD): National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT00964080
Merrimack Pharmaceuticals Inc. A phase 1, multi-center, open-label, dose-escalation, safety, and pharmacokinetic clinical study of intravenously administered MM-302 in patients with advanced breast cancer. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01304797
Moghimi SM, Andersen AJ, Hashemi SH, Lettiero B, Ahmadvand D, Hunter AC, Andresen TL, Hamad I, Szebeni J (2010) Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: the challenges ahead. J Control Release 146(2):175–181
Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, Wrasidlo W, Cheresh DA (2008) Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A 105(27):9343–9348
Nellis DF, Ekstrom DL, Kirpotin DB, Zhu J, Andersson R, Broadt TL, Ouellette TF, Perkins SC, Roach JM, Drummond DC, Hong K, Marks JD, Park JW, Giardina SL (2005a) Preclinical manufacture of an anti-HER2 scFv-PEG-DSPE, liposome-inserting conjugate. 1. Gram-scale production and purification. Biotechnol Prog 21(1):205–220
Nellis DF, Giardina SL, Janini GM, Shenoy SR, Marks JD, Tsai R, Drummond DC, Hong K, Park JW, Ouellette TF, Perkins SC, Kirpotin DB (2005b) Preclinical manufacture of anti-HER2 liposome-inserting, scFv-PEG-lipid conjugate. 2. Conjugate micelle identity, purity, stability, and potency analysis. Biotechnol Prog 21(1):221–232
Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC (2004) Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets 8(4):335–353
Park JW, Hong K, Carter P, Asgari H, Guo LY, Keller GA, Wirth C, Shalaby R, Kotts C, Wood WI et al (1995) Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A 92(5):1327–1331
Park JW, Kirpotin DB, Hong K, Shalaby R, Shao Y, Nielsen UB, Marks JD, Papahadjopoulos D, Benz CC (2001) Tumor targeting using anti-her2 immunoliposomes. J Control Release 74(1–3):95–113
Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC (2002) Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res 8(4):1172–1181
Park JW, Benz CC, Martin FJ (2004) Future directions of liposome- and immunoliposome-based cancer therapeutics. Semin Oncol 31(6 Suppl 13):196–205
Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK (2011) RNA interference in the clinic: challenges and future directions. Nat Rev Cancer 11(1):59–67
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R (2007) Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2(12):751–760
Petros RA, DeSimone JM (2010) Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov 9(8):615–627
Pinzon-Daza ML, Campia I, Kopecka J, Garzon R, Ghigo D, Rigant C (2013) Nanoparticle- and liposome-carried drugs: new strategies for active targeting and drug delivery across blood–brain barrier. Curr Drug Metab 14(6):625–640
Pirollo KF, Rait A, Zhou Q, Zhang XQ, Zhou J, Kim CS, Benedict WF, Chang EH (2008) Tumor-targeting nanocomplex delivery of novel tumor suppressor RB94 chemosensitizes bladder carcinoma cells in vitro and in vivo. Clin Cancer Res 14(7):2190–2198
Pittet L, Altreuter D, Ilyinskii P, Fraser C, Gao Y, Baldwin S, Keegan M, Johnston L, Kishimoto T (2012) Development and preclinical evaluation of SEL-068, a novel targeted Synthetic Vaccine Particle (tSVPTM) for smoking cessation and relapse prevention that generates high titers of antibodies against nicotine [abstract]. In: Immunology 2012 – 99th meeting of The American Association of Immunologists, Boston, Massachusetts: AAI; 2012. Abstract nr 75.11
Rahman MA, Amin AR, Wang X, Zuckerman JE, Choi CH, Zhou B, Wang D, Nannapaneni S, Koenig L, Chen Z, Chen ZG, Yen Y, Davis ME, Shin DM (2012) Systemic delivery of siRNA nanoparticles targeting RRM2 suppresses head and neck tumor growth. J Control Release 159(3):384–392
Raymond E, Chaney SG, Taamma A, Cvitkovic E (1998) Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol 9(10):1053–1071
Rip J, Appeldoorn C, Manca F, Dorland R, Van Kregten J, Gaillard P (2010) Receptor-mediated delivery of drugs across the blood-brain barrier [abstract]. Pharmacology and toxicology of the blood-brain barrier: state of the art, needs for future research and expected benefits for the EU, Brussel, Belgium: COST; 2010. Abstract nr 5464
Romberg B, Hennink WE, Storm G (2008) Sheddable coatings for long-circulating nanoparticles. Pharm Res 25(1):55–71
Sankhala KK, Mita AC, Adinin R, Wood L, Beeram M, Bullock S, Yamagata N, Matsuno K, Fujisawa T, Phan A (2009) A phase I pharmacokinetic (PK) study of MBP-426, a novel liposome encapsulated oxaliplatin [abstract]. ASCO Annual Meeting, Orlando, Florida: ASCO; 2009. Abstract nr 2535
Schiffelers RM, Koning GA, ten Hagen TL, Fens MH, Schraa AJ, Janssen AP, Kok RJ, Molema G, Storm G (2003) Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 91(1–2):115–122
Selecta Biosciences Inc. A phase 1 randomized, double-blind, placebo-controlled study to assess the safety and pharmacodynamics of increasing subcutaneous doses of SEL-068 in healthy non-smoker and smoker adults. In: ClinicalTrials.gov [Internet], Bethesda (MD): National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01478893
Senzer NN, Matsuno K, Yamagata N, Fujisawa T, Wasserman E, Sutherland W, Sharma S, Phan A (2009) MBP-426, a novel liposome-encapsulated oxaliplatin, in combination with 5-FU/leucovorin (LV): Phase I results of a Phase I/II study in gastro-esophageal adenocarcinoma, with pharmacokinetics [abstract]. Molecular Targets and Cancer Therapeutics, Boston, Massachusetts: AACR-NCI-EORTC; 2009. Abstract nr C36
Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, Nunan R, Pirollo KF, Rait A, Chang EH (2013) Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther 21(5):1096–1103
Shi J, Xiao Z, Kamaly N, Farokhzad OC (2011) Self-assembled targeted nanoparticles: evolution of technologies and bench to bedside translation. Acc Chem Res 44(10):1123–1134
Shimada K, Kasamatsu T, Hasegawa T, Kanai Y, Niki H, Tagawa T, Matsumura Y (2005) The cytotoxicity and intracellular distributions of MCC-465, pegylated liposomal doxorubicin tagged with newly developed monoclonal antibody GAH, in ovarian cancer cell lines [abstract]. In: Proceedings of the 96th Annual Meeting of the American Association for Cancer Research, Anaheim, California: AACR; 2005. Abstract nr 1427
Sievers EL, Senter PD (2013) Antibody-drug conjugates in cancer therapy. Annu Rev Med 64:15–29
Solomon B, Desai J, Scott A, Rosenthal M, McArthur G, Rischin D, Segelov E, MacDiarmid J, Brambhatt H (2012) First-in-man, multicenter, phase I trial evaluating the safety of first-in-class therapeutic, EGFR-targeted, paclitaxel-packaged minicells [abstract]. In: 24th EORTC – NCI – AACR symposium on molecular targets and cancer therapeutics, Dublin, Ireland: EORTC – NCI – AACR; 2012. Abstract nr 585
Srikanth M, Kessler JA (2012) Nanotechnology-novel therapeutics for CNS disorders. Nat Rev Neurol 8(6):307–318
Suzuki R, Takizawa T, Kuwata Y, Mutoh M, Ishiguro N, Utoguchi N, Shinohara A, Eriguchi M, Yanagie H, Maruyama K (2008) Effective anti-tumor activity of oxaliplatin encapsulated in transferrin-PEG-liposome. Int J Pharm 346(1–2):143–150
Svenson S (2012) Clinical translation of nanomedicines. Curr Opinion Solid State Mater Sci 16(6):287–294
Synergene Therapeutics Inc. A phase I open-label safety and pharmacokinetic study of escalating doses of SGT-53 for infusion in subjects with advanced solid tumors. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT00470613
Synergene Therapeutics Inc. A phase I study of systemic gene therapy with SGT-94 in patients with solid tumors. In: ClinicalTrials.gov [Internet], Bethesda (MD), National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01517464
Talelli M, Oliveira S, Rijcken CJ, Pieters EH, Etrych T, Ulbrich K, van Nostrum RC, Storm G, Hennink WE, Lammers T (2013) Intrinsically active nanobody-modified polymeric micelles for tumor-targeted combination therapy. Biomaterials 34(4):1255–1260
to-BBB technologies B.V. An open-label, phase I/IIa, dose escalating study of 2B3-101 in patients with solid tumors and brain metastases or recurrent malignant glioma. In: ClinicalTrials.gov [Internet], Bethesda (MD): National Library of Medicine (US). Retrieved August 2013. Available from http://clinicaltrials.gov/ct2/show/NCT01386580
University Hospital Basel Switzerland. A phase I study of doxorubicin-loaded anti-EGFR immunoliposomes in patients with advanced solid tumors. In: ClinicalTrials.gov [Internet], Bethesda (MD): National Library of Medicine (US). Retrieved July 2013. Available from http://www.clinicaltrials.gov/ct2/show/NCT01702129/
Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, Yehoshua Z, Libson E, Muggia FM, Gabizon A (1995) Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol 13(7):1777–1785
van Broekhoven CL, Parish CR, Demangel C, Britton WJ, Altin JG (2004) Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res 64(12):4357–4365
Von Hoff DD, Mita M, Eisenberg P, LoRusso P, Weiss G, Sachdev J, Mita A, Low S, Hrkach J, Summa J, Berk G, Ramanathan R (2013) A phase I study of BIND-014, a PSMA-targeted nanoparticle containing docetaxel, in patients with refractory solid tumors [abstract]. In: Proceedings of the 104th annual meeting of the American Association for Cancer Research, Washington, DC: AACR; 2013. Abstract nr LB-203
Wickham T, Futch K (2012) A phase I study of MM-302, a HER2-targeted liposomal doxorubicin, in patients with advanced, HER2- positive breast cancer [abstract]. In: 35th annual CTRC-AACR San Antonio breast cancer symposium, San Antonio, Texas: CTRC-AACR; 2012. Abstract nr P5-18-09
Wickham T, Reynolds J, Drummond D, Kirpotin D, Lahdenranta J, Leonard S, Geretti E, Lee H, Klinz S, Hendriks B, Olivier K, Eckelhofer I, Park J, Benz C, Moyo V, Niyikiza C, Nielsen U (2010) Preclinical safety and activity of MM-302, a HER2-targeted liposomal doxorubicin designed to have an improved safety and efficacy profile over approved anthracyclines [abstract]. In: 33rd annual CTRC-AACR San Antonio breast cancer symposium, San Antonio, Texas: CTRC-AACR; 2010. Abstract nr P3-14-09
Wong HL, Wu XY, Bendayan R (2012) Nanotechnological advances for the delivery of CNS therapeutics. Adv Drug Deliv Rev 64(7):686–700
Xu HJ, Xu K, Zhou Y, Li J, Benedict WF, Hu SX (1994) Enhanced tumor cell growth suppression by an N-terminal truncated retinoblastoma protein. Proc Natl Acad Sci U S A 91(21):9837–9841
Xu HJ, Zhou Y, Seigne J, Perng GS, Mixon M, Zhang C, Li J, Benedict WF, Hu SX (1996) Enhanced tumor suppressor gene therapy via replication-deficient adenovirus vectors expressing an N-terminal truncated retinoblastoma protein. Cancer Res 56(10):2245–2249
Xu L, Pirollo KF, Chang EH (1997) Transferrin-liposome-mediated p53 sensitization of squamous cell carcinoma of the head and neck to radiation in vitro. Hum Gene Ther 8(4):467–475
Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, Rait A, Chang EH (2001) Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med 7(10):723–734
Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, Cruz I, Xiang LM, Pirollo KF, Chang EH (2002) Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther 1(5):337–346
Yu W, Pirollo KF, Rait A, Yu B, Xiang LM, Huang WQ, Zhou Q, Ertem G, Chang EH (2004) A sterically stabilized immunolipoplex for systemic administration of a therapeutic gene. Gene Ther 11(19):1434–1440
Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK (1995) Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res 55(17):3752–3756
Zhang X, Multani AS, Zhou JH, Shay JW, McConkey D, Dong L, Kim CS, Rosser CJ, Pathak S, Benedict WF (2003) Adenoviral-mediated retinoblastoma 94 produces rapid telomere erosion, chromosomal crisis, and caspase-dependent apoptosis in bladder cancer and immortalized human urothelial cells but not in normal urothelial cells. Cancer Res 63(4):760–765
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
van der Meel, R., Vehmeijer, L.J.C., Kok, R.J., Storm, G., van Gaal, E.V.B. (2016). Ligand-targeted Particulate Nanomedicines Undergoing Clinical Evaluation: Current Status. In: Prokop, A., Weissig, V. (eds) Intracellular Delivery III. Fundamental Biomedical Technologies. Springer, Cham. https://doi.org/10.1007/978-3-319-43525-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-43525-1_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43523-7
Online ISBN: 978-3-319-43525-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)