
Abstract
A wide range of congenital anomalies may affect the whole gastrointestinal tract, from the esophagus to the rectum. High intestinal obstructions are those occurring proximal to the ileum, whereas low obstructions involve the distal ileum and colon and typically result in dilation of several bowel loops. These abnormalities are a significant cause of morbidity in children and, less frequently, in adults. Most of these congenital anomalies, especially those involving the small intestine, present with obstructive symptoms, while some of them with abdominal pain, vomiting, and gastrointestinal bleeding. Definitive therapy is surgical, and in some cases, it has to be performed urgently. Their diagnosis may require several imaging modalities in order to tailor the most appropriate treatment. Some of them, such as gastric or high intestinal obstruction, do not usually require further investigation; on the other hand, low obstructions require a barium enema study. US has become important for the evaluation of some of these abnormalities; it can help to differentiate a small bowel obstruction from a colonic obstruction. In addition, it may help to suggest possible anorectal malformations through dynamic procedures. A detailed anatomical investigation is crucial in tailoring the most appropriate surgical approach for each patient. For this reason MRI is essential because it well depicts the pelvic and perineal anatomy in order to evaluate the location and morphology of the anorectal malformation and to study the grade of development of the puborectalis and sphincteric musculature before the surgery.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Esophageal Atresia and Tracheoesophageal Fistula
Esophageal atresia (EA), isolated or associated with tracheoesophageal fistula (TEF), was first described by Thomas Gibson in 1697, but it was only in 1941 that Cameron Haight performed the first successful surgical repair of this anomaly (Mortell and Azizkhan 2009; Pinheiro et al. 2012).
The overall incidence is approximately 1:3–5000 live-born infants (de Jong et al. 2010). In the majority of cases, it is sporadic, but the incidence is higher in twins, 2.56 higher than in singletons (Seo et al. 2010; Holland and Fitzgerald 2010).
These pathological entities represent a complex of congenital anomalies characterized by an incomplete formation of the primitive esophagus or by an abnormal communication between the trachea and esophagus.
The pathologic mechanism behind this malformation is still unknown, although it is thought to be a developmental disorder involving the division of the primitive foregut into trachea and esophagus occurring since the fourth week of gestation (Cumming 1975). There are three main theories explaining this phenomenon: the first theory states that the evagination of the tracheal diverticulum begins with the primitive digestive tube, which, growing in the caudal direction very quickly, results into a separation of trachea from esophagus, which is still part of the digestive tube. According to this mechanism, the tracheoesophageal malformation results from a tracheal growth failure.
The second theory postulates the formation of a septum in the coronal plane of the primitive digestive tube, which separates the trachea ventrally and the esophagus dorsally; a failure in this process might result into a tracheoesophageal malformation. The third theory combines the previous two, suggesting that the growth of the tracheal diverticulum occurs together with the separation of the digestive tube, resulting in the final separation of the trachea from the esophagus. The loss of a portion of the previously formed tube due to a regression toward the main part of the embryo may result in the EA (Ioannides and Copp 2009).
Some reports described this entity occurring as a multifactorial complex, involving both genetic and environmental factors; 6–10 % of patients have a defined genetic syndrome (Genevieve et al. 2007). There are also molecular and morphogenic factors related to EA, such as apoptosis, the Sox2, Shh, Gli-2, Gli-3, and FOX genes, and the transcription factors Nkx2.1 and Tbx4. A failure in the expression of one of these genes or in their apoptotic programs could be responsible for EA (El-Gohary et al. 2010; Shaw-Smith 2010). Furthermore, environmental factors increasing the risk to develop EA have been described; for example, maternal alcohol and smoking, maternal exposure to methimazole and diethylstilbestrol (DES), exogenous sex hormones, infectious disease, and maternal diabetes are severe factor risks. A role for vitamin A deficiency has been suggested in the development of EA/TEF, since it caused severe congenital anomalies in the offspring of pregnant rats, including agenesis of the lung and TEF (Di Gianantonio et al. 2001; Nora et al. 1976; Wong-Gibbons et al. 2008; Felix et al. 2007).
As evident, no specific environmental risk factors has been identified.
In 25 % of cases, this anomaly is associated with other gastrointestinal malformations, such as pyloric stenosis, annular pancreas, imperforate anus, and the VACTERL complex (vertebral, anal, cardiac, tracheal, esophageal, renal, and limb anomalies), which is the best known group of anomalies associated with EEA/TEF.
Other recognized associations are trisomy 18 and 21, CHARGE syndrome, and Potter syndrome.
2 Classification
The classification of EA anomalies is based on the location of atresia and the presence of any associated tracheoesophageal fistula.
The first classification was described by Vogt in 1929 and then modified by Gross in 1953. According to the last one, five types of congenital EA are possible (Fig. 1.1):

These drawings show the five types of EA/TEF. In (a) pure esophageal atresia; in (b) and (c), respectively, it is associated with a proximal or distal fistula with the trachea or main bronchus. In (d) there are both the fistulas and in (e) you can find a tracheoesophageal fistula without esophageal atresia
-
Type A: pure esophageal atresia without fistula.
-
Type B: esophageal atresia with a fistula between the trachea and the proximal esophageal pouch.
-
Type C: esophageal atresia with fistula between the trachea or the main bronchus and the distal esophageal segment.
-
Type D: esophageal atresia with both proximal and distal fistulas.
-
Type E: tracheoesophageal fistula “H or N” shaped without atresia. It is called N-type fistula because it courses obliquely from the esophagus upward to the trachea. Most of them are at thoracic inlet (T2–T3).
Type C is by far the most common, occurring in 82–86 % of cases. Then we have type A atresia (8–9 % of cases), followed by type E (6 % of cases). The less common are type B and C occurring in about 1 % of cases (Spitz 2007).
3 Diagnosis
3.1 Prenatal Diagnosis
US, performed between the 16th and 20th week of gestation, can demonstrate the presence of polyhydramnios, reduced intraluminal liquid, and the absence of gastric bubble, signs suggesting EA, although they are not specific criteria, reporting a PPV of 44 % and 56 %, respectively.
The so-called upper pouch sign, the dilatation of the upper blind fundus of the atresic esophagus, may be observed during fetal deglutition at the 32nd gestational week (Holland and Fitzgerald 2010; Houben and Curry 2008); nevertheless this sign could not be observed also in experienced hands.
In the presence of a high US suspicion of EA/TEF, MR can be performed, having a sensitivity of 100 % to diagnose these anomalies: the diagnostic criterion is represented by the lack of visualization of the intrathoracic region of the esophagus (Pinheiro et al. 2012; Houben and Curry 2008).
3.2 Neonatal Diagnosis
3.2.1 Clinical Findings
The impossibility to pass a catheter beyond 11 or 12 cm is the primary sign of EA in the delivery room (Lahdes-Vasama et al. 2009).
In the nursery room, the inability to swallow saliva or milk, aspiration during early feeding, or cyanosis can make us to suspect the presence of EA. In type E atresia, the clinical diagnosis may be performed later in years because of delayed symptoms such as cough while swallowing, distended abdomen, and recurrent pneumonia.
3.2.2 Diagnostic Imaging
3.2.2.1 Plain X-ray
The confirmation requires an anteroposterior and lateral chest radiography revealing the proximal blind pouch distended with air (Fig. 1.2). The abdomen must always be included in the radiograph to find out air in the gastrointestinal tract, in cases of distal fistula. In fact, in types A and B, there is no any sign of gas in stomach and intestinal tract (Fig. 1.3), whereas in types C and D, we can observe gas distending the bowel loops (Fig. 1.4) (Berrocal et al. 1999a).

Esophageal atresia: abdomen x-ray shows air-filled dilated proximal esophageal pouch. The presence of air in the bowel suggests a distal tracheoesophageal fistula

Esophageal atresia with upper fistula. The nasogastric tube stops in the upper esophagus. Associated findings are right pneumothorax and left pneumonia. No gas in the abdomen

Esophageal atresia with lower fistula along with pneumonia of the upper left lobe. Esophagogram shows a distended proximal pouch
A paramount detail to convey to the surgeon is the position of the aortic arch, because it helps to choose the best surgical approach; when chest x-ray does not recognize the side of the arch, it is indicated to perform an echocardiography to plan the surgical method and to prognosticate the outcome; in fact a right-sided aortic arch (2.5 % of cases) implies a very challenging repair requiring a left thoracotomy.
Radiographically, it can be possible to recognize the type of EA by advancing a radiopaque feeding tube through the nose to the level of atresia (Fig. 1.5); the tube will stop and curl coming in front of a blind end. The proximal pouch may be distended by a gentle injection of air or a little amount (1–2 ml) of water-soluble contrast medium via the tube (Fig. 1.5).

Esophageal atresia with lower fistula. (a) Frontal radiograph shows the nasogastric tube arresting within the upper esophageal pouch. It is clearly depicted in air distention of the stomach and jejunum. (b, c) Esophagogram shows a complete upper esophageal obstruction
Isolated long-gap EA (>2 vertebral bodies or a gap >3 cm) is associated with 13 pairs of ribs.
When a fistula occurs, atelectasis and pneumonia of the upper right lobe are detected in 50 % of cases (see Figs. 1.3 and 1.4).
When type E is suspected, the best way to demonstrate the fistula is to perform a tube esophagogram under fluoroscopic guidance. It is always preferable to use an iodinate nonionic water-soluble contrast material, diluted with water at 50 %. It is administered both directly and through a nasogastric tube located at the gastroesophageal junction. The patient is in right lateral position, with the lower limb slightly raised with respect to the upper limb, in order to promote the opacification of the tracheoesophageal fistula, running upward and rightward (Fig. 1.6). The contrast media must be aspirated back immediately at the end of the examination.

Tracheoesophageal fistula without atresia. Esophagogram shows a communication between the thoracic esophagus and trachea (white arrow). Contrast media fill the whole tracheobronchial tree
Iodinated water-soluble contrast media is usually preferred to the diluted barium because the latter one could cause a severe respiratory distress if aspirated, being a quickly solidifying substance.
During the preoperative period, it has been proposed to perform tracheobronchoscopy to vcarina, to recognize other anomalies, and to occlude the TEF with a balloon, in order to improve the mechanical ventilation and to avoid gastric distension and gastroesophageal reflux.
4 Treatment
In the absence of severe malformations, the primary correction of EA and TEF is the best treatment option. In some cases the primary anastomosis cannot be performed because of a “long gap” separating the two ends of the esophagus; to overcome these difficult cases, many options have been suggested, such as the Livaditis myotomy, the Foker technique, and the extensive mobilization of the distal segment through the diaphragmatic hiatus. Nevertheless, some complications may occur if the anastomosis is tailored under tension, such as gastroesophageal reflux, esophageal stricture, and a high leak rate (Seitz et al. 2006; Sharma et al. 2000; Alabbad et al. 2009).
In type A the surgical approach consists of tailoring of a gastrostomy for feeding and a suctioning of the blind pouch to protect the airways. The following options for reconstructions involve or a delayed primary repair using the native esophagus or its replacement using colon or stomach.
In type E the operation requires a neck dissection to expose the anomaly, then the division and the repair of the fistula. This technique is characterized by a quite high risk of recurrent laryngeal nerve injury; in the recent years, an Nd:YAG laser has been used for this purpose, although new data are necessary because experience is limited and it is not widely recommended.
During the postoperative period, if the esophageal anastomosis has been performed under tension, the patient is electively paralyzed and ventilated for 5–7 days. If a transanastomotic feeding tube has been inserted, the feeding usually starts 48 h after the surgery and proceeds very slowly (Alabbad et al. 2009). Only when the child starts to swallow, the oral feeding may start. The primary complications after surgery are represented by the leak and stenosis of anastomosis (Fig. 1.7), esophageal dysmotility, gastroesophageal reflux, fistula recurrence, scoliosis, and respiratory disorders.

Esophageal atresia: esophagogram shows a postsurgery stenosis
Some patients may have an uneventful postoperative period, whereas others may come across some respiratory or esophageal disorders that can affect their ability to develop adaptive behaviors.
5 Esophageal Stenosis and Webs
They are considered as a variant of esophageal atresia and are often associated with TEF.
Congenital stenosis often appears as a 2–3 mm constriction in the middle or distal esophagus. Their differential diagnosis must include gastroesophageal reflux, surgery-related strictures, and corrosive ingestion.
Congenital webs are identified on barium enema studies as smooth and thin defect in the esophagus and can be located at the same level of the fistula.
6 Stomach
A complete gastric agenesis never occurs. Microgastria, a congenitally small stomach, is a very rare condition occurring both isolated and in association with other anomalies, especially the heterotaxia syndrome with asplenia. At imaging the typical finding is represented by an enlarged esophagus with a small midline stomach (Applegate et al. 1999).
A complete obstruction involving the gastric outlet is very rare, except when caused by extrinsic conditions, such as congenital peritoneal bands exercising pressure on gastric walls or by pancreatic tissue within gastric wall (Berrocal et al. 1999a).
Gastric atresia is also a rare condition, accounting for <1 % of all congenital intestinal obstructions, and it is typically localized to the antrum or pyloric region. It is thought to be a consequence of localized vascular occlusion in fetal life rather than to failed bowel canalization, mainly because of the absence of epithelial perforation in the stomach as in the esophagus and duodenum.
Pyloric atresia is divided into three types: (1) complete atresia with no connection between the stomach and duodenum, (2) complete atresia with a fibrous band connecting the stomach and duodenum, and (3) gastric membrane or a diaphragm.
Clinical presentation is characterized by non-bilious vomiting within the first few hours after delivery and abdominal distention (Moore 1989).
It can be familial or associated with epidermolysis bullosa.
Its diagnosis requires an abdomen x-ray showing the so-called single bubble sign, caused by the distention of the stomach close to the obstruction with the absence of gas in bowel lumen. Contrast media is not needed.
The partial gastric outlet obstruction is commonly caused by incomplete prepyloric diaphragm, antral stenosis, aberrant pancreatic tissue in gastric antrum, and antral duplication cysts.
At abdomen x-rays, it is possible to detect gastric distention with small amount of gas within the small bowel, depending on the site of the obstruction.
Barium enema and US can help to identify the defect; in fact barium enhances the presence of membranes as thin (2–3 mm) and linear filling defects interrupting the barium column, resulting in “pseudo double bulb” appearance, because the inflowing barium outlines at first the space between the antrum and the pylorus and then the duodenal bulb (Clements et al. 1979).
When the stomach is filled with clear fluid, US can detect the membrane as an echogenic band extending centrally from the lesser to greater curvatures in the prepyloric region (Chew et al. 1992; Van Winckel et al. 1994).
The aberrant pancreatic tissue can be found more commonly in the gastric antrum and may cause intermittent obstruction because it prolapses into the pylorus. Contrast media is able to highlight a smooth, dome-shaped filling defect of 1–3 cm with a central umbilication on the larger curvature (Lai and Tompkins 1986).
Gastric duplication accounts for about 7 % of gastrointestinal tract duplications. Most of them are located on the greater curvature and are noncommunicating. The clinical symptoms depend on their size, their location, and the presence of any communication with any tract of digestive tube.
They are usually found out during childhood with vomiting and abdominal pain, although most of them are asymptomatic.
Abdomen x-ray and contrast enema can display a paragastric mass displacing the stomach, but US, CT, or MRI can better detect a well-defined cystic mass close to the greater curvature, appearing at sonogram having an echogenic inner rim with a hypoechoic outer muscle layers.
7 Duodenum
7.1 Complete Duodenal Obstruction
Complete duodenal obstruction is generally caused by duodenal atresia, resulting from a defect of recanalization during the 9th–11th week of gestation; it does not seem to be related to intrauterine vascular insults, as occurs in lower small bowel atresia.
It is characterized by a reported incidence of about 1:5000–10,000 live births, with most recent data indicating 0.9 per 10,000 (Best et al. 2012).
Unlike other intestinal atresia, it is often associated with other congenital anomalies, such as congenital heart disease, additional intestinal atresia, VACTERL syndrome, malrotation, annular pancreas, biliary tract and mandibulofacial anomalies, and trisomy 21, which is present in almost 30 % of cases (Hertzberg and Bowie 1990).
Four main types of duodenal atresia have been described (Fig. 1.8): type I is characterized by a complete mucosal membrane or a diaphragm within the muscularis with an intact serosa, thus maintaining the continuity of the bowel. In type 2 there is a fibrous cord linking the two duodenum segments that are discontinuous. Type 3 does not consider any connection between the proximal and distal segments of the duodenum. In type IV we have multiple atretic segments such as a “string of sausages” (Grosfeld et al. 1979).

These drawings show the main types of duodenal atresia. In (a–b) there is a complete mucosal membrane or a diaphragm within the muscularis with an intact serosa, thus maintaining the continuity of the bowel. In (c) there is a fibrous cord linking the two duodenal segments. In (d) does not consider any connection between the proximal and distal segments of the duodenum
Other causes of duodenal obstruction are annular pancreas, midgut volvulus, duodenal web, Ladd bands, and pre-duodenal portal vein.
Annular pancreas is an abnormal portion of pancreatic tissue arising from the head and encircling the second part of duodenum forming a ring; when a complete ring is formed, a total and severe duodenal obstruction occurs, while if the ring is incomplete, obstructive symptoms may occur later in life (Norton et al. 1992).
Midgut volvulus is the most dramatic result of intestinal malrotation, and, when present, its radiographic findings are indistinguishable from duodenal atresia in such cases. As a consequence, any duodenal obstruction presenting at birth must be considered as a midgut volvulus until proved otherwise (Berrocal et al. 1999a).
8 Diagnosis
8.1 Clinical Manifestations
The initial manifestation is often specific, usually polyhydramnios due to the infant’s inability to swallow and to absorb the amniotic fluid. After birth, the typical manifestation is vomiting, which is delayed until after the first feeding, and bilious, because the obstruction is located beyond the ampulla of Vater.
The vomiting is going to increase dramatically thereafter. Abdominal distention and feeding difficulties are often associated (Brantberg et al. 2002).
8.2 Diagnostic Imaging
Perinatal US can detect the presence of polyhydramnios, which is an early clue to the possibility of duodenal atresia since about 15 % of neonates showing this finding will have a gastrointestinal anomaly and about 80 % of cases with duodenal atresia have polyhydramnios. An important US finding, detected both ante- and postnatally, is the “double bubble” sign indicating the distention of proximal duodenum and stomach associated with lack of gas in the lower bowel (Correia-Pinto and Ribeiro 2014).
In cases of complete duodenal atresia, no gas will be detected distal to the proximal duodenum. This is the classic finding discovered on abdomen x-ray also (Fig. 1.9), and when it is present, no other radiographic studies are required.

Frontal x-ray depicts a markedly distended stomach and duodenum (double bubble sign), without evidence of gas in the rest of bowel tract, typical of duodenal atresia
Contrast media diluted with water at 50 % and administered via a nasogastric tube shows gastric and duodenal distention without any sign of contrast media in the distal duodenum (Fig. 1.10). Nevertheless, the upper gastrointestinal study is not indicated when the abdomen x-ray does not show any certain sign of duodenal obstruction, because the administration of contrast media may lead to aspiration.

(a) Plain film shows gas distention of the stomach and proximal duodenum; no gas is seenthe rest of the abdomen. (b) Barium study confirms the duodenal atresia with the double bubble sign
It is important to notice that this sign is also present in other conditions, such as intestinal malrotation; for this reason, some authors suggested performing contrast enema to make sure of the right diagnosis, since in intestinal atresia a microcolon of disuse would be found, while a malpositioned colon and cecum would suggest a malrotation (Laya et al. 2015; Strouse 2008).
9 Treatment
A naso- or orogastric tube is placed in order to decompress the stomach and to reduce the risk of aspiration; intravenous fluids are given to support vital functions.
Once obtained the hemodynamical stability, the little patient can undergo the surgical repair, via laparotomy or laparoscopy, performing a duodenoduodenostomy or a duodenojejunostomy. While operating, it is always fundamental to rule out other gastrointestinal anomalies, such as malrotation, bowel atresia, or annular pancreas (Son et al. 2015; van der Zee 2011).
Long-term outcome is very favorable with survival rates of about 90 %. Among the causes of morbidity and mortality, associated anomalies and ultrashort bowel syndrome have to be mentioned, requiring long-term parenteral nutrition.
10 Partial Duodenal Obstruction
10.1 Duodenal Stenosis and Duodenal Web
Partial duodenal obstruction can be caused by duodenal stenosis and web, Ladd bands, annular pancreas, midgut volvulus, pre-duodenal portal vein, and duplication cyst.
As seen in patients with duodenal atresia, also in duodenal stenosis, the radiologic sign of “double bubble” is present, but in this case, a small amount of gas in the lower gastrointestinal tract is visible (Fig. 1.11).

(a) Radiograph performed before shows gas distention of the stomach and of the proximal aspect ofthe duodenum; note the presence of little amount of air in the intestinal loops (b) Barium study depicts a severe duodenal stenosis with the double bubble sign
Performing UGI studies is useful and is performed with an iodinated water-soluble contrast media diluted with water at 50 %, administered via a nasogastric tube. It allows to visualize a slow progression of contrast media through the stenotic segment of the duodenum to the distal bowel; in addition, it is helpful to differentiate it from a midgut volvulus in which the small intestine twists like a corkscrew around the superior mesenteric artery. This finding can be visualized by performing color Doppler US also, recognizing the so-called whirlpool sign of a midgut volvulus, characterized by the clockwise wrapping of the superior mesenteric vein and the mesentery around the superior mesenteric artery (Materne 2001).
Duodenal webs are small congenital obstructive membranes of the mucosa and submucosa layers, with a central pinhole opening representing the functional web.
They occur because of an incomplete bowel lumen recanalization during the eighth to tenth week of gestation. They are typically located in the second portion of the duodenum (85–90 % of cases), less frequently in the third and fourth portion.
Their incidence is approximately 1:10,000–40,000 live births. In the majority of cases, they are congenital, although some reports suggest they can be a complication of a long-term therapy with nonsteroidal anti-inflammatories. They are often associated with other congenital conditions, such as malrotation, trisomy 21, annular pancreas, and congenital heart disease.
Prenatal US can show polyhydramnios and growth failure, associated with a dilated stomach.
After birth, clinical presentations usually consist of vomiting, food refusal, and failure to thrive; in some reports gastrointestinal bleeding has been described (Nagpal et al. 1993; Al Shahwani et al. 2013).
Abdominal x-rays may demonstrate a double bubble sign or be normal, while upper GI series may reveal the “wind-sock sign,” which is the result of the long-term pressure of peristalsis against the stenotic portion of the duodenum, leading to distal stretching of the web, creating an intraluminal pseudodiverticulum. The finding is characterized by a thin radiolucent membrane representing barium filling the lumen and around the diaphragm.
Because of the partial obstruction, these congenital anomalies may be diagnosed later in life with respect to those with duodenal atresia, or they are diagnosed incidentally because the patient undergoes examinations for other reasons.
10.2 Treatment
Surgical options are duodenoduodenostomy or duodenotomy with web lysis, via laparotomy or laparoscopy. When proximal duodenal lumen is equal or exceeds 5 cm, it can be possible to choose for imbrications or tapering duodenoplasty (Sarin et al. 2012). Possible complications after surgery are bleeding and pancreatitis, whereas the endoscopic approach may lead to incomplete repair of the web. In the presence of malrotation, Ladd procedures are to be performed.
The general outcome is good and, as for other gastrointestinal malformations, depends mainly on associated congenital anomalies (Escobar et al. 2004).
11 Jejunum and Ileum
Jejunoileal atresias are commonly caused by an intrauterine ischemic insult, related to a vascular cause or secondary to a volvulus, intussusception, malrotation, or intestinal strangulation via a hernia. Also maternal smoking and cocaine abuse have been indicated as possible causes of intestinal atresia.
Their incidence is about 1:50,000–10,000 live births, affecting both sexes equally.
They can affect the small bowel everywhere, although the distal portion of the ileum is the most involved (Baglaj et al. 2008).
The association with other congenital malformations is less common, and, when present, cystic fibrosis and malrotation are the most commonly associated conditions.
A form of hereditary intestinal atresia is the multiple intestinal atresia (HMIA), a severe variant firstly described in 1970 by Mishalany and Der Kaloustian and Guttman et al. This is an autosomal recessive disorder affecting multiple bowel segments from the pylorus to the rectum, with long intestinal segments frequently occluded. It occurs commonly in French Canadians and can be associated with combined immunodeficiency. Studies reported a linkage with the mutation of tetratricopeptide repeat domain-7A (TTC7A) gene, fundamental for the development of the thymus and intestinal epithelium. This condition is characterized by a poor outcome since after surgical repair, the patients keep on having low gastrointestinal functions although preserving the intestinal length to avoid the short gut syndrome (Ali et al. 2011).
Intestinal stenoses mainly result from three causes: external compression, intramural narrowing from the rest of heterotypic tissue, and incompletely perforated intraluminal web. Extrinsic compressions are most commonly confined to the duodenum, resulting from peritoneal bands or annular pancreas (Free and Gerald 1968).
11.1 Classification
The classification of jejunoileal atresia was firstly provided by Louw and then refined by Martin and Zerella and by Grosfield et al. The classification considers four types of intestinal atresia based on anatomic features. Type I is characterized by a mucosa and submucosa web with continuity of the proximal and distal muscular layers with an intact mesentery. Type II consists of a fibrous cord connecting the atretic bowel ends. Type III has two subtypes: in the IIIa form, there is an atretic segment with a mesenteric defect “V shaped” also and IIIb is the so-called apple-peel deformity, in which you have a proximal jejunal atresia with an extensive mesenteric defect with the bowel wrapped around a single artery. Type IV describes multiple atresias appearing like a “string of sausage” (Grosfeld et al. 1968) (Fig. 1.12).

These drawings show the four types of jejunoileal atresia, note the lost of mesenteric integrity and the presence of multiple stenoses
11.1.1 Clinical Presentation
The typical presentation is a newborn in the first 1–2 days of life with bilious vomiting and abdominal distention which depends on the location of atresia, with more distal lesions having more distentions. Infants may experience hyperbilirubinemia and feeding difficulties also. A history of maternal polyhydramnios is often associated. In more distal lesions, meconium may not pass; in more proximal lesions meconium may pass because of generation of success entericus (Frischer and Azizkhan 2012).
11.1.2 Diagnosis
Prenatal ultrasound may show dilated, echogenic bowel and maternal polyhydramnios in about one-third of cases. In ultrasound uncertain cases, MRI can be performed.
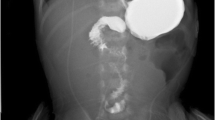
Postnatally, the first step to perform is an abdomen x-ray which reveals several dilated bowel loops, more than the double bubble of duodenal atresia but less than in the lower obstructions (Fig. 1.13). Typical is the so-called triple bubble sign, indicating the dilatation of stomach, duodenum, and proximal jejunum; the disproportionate dilatation of the bowel proximal to the atresia results in a bulbous contour suggestive of congenital small bowel obstruction (Fig. 1.14).

Jejunal atresia. Air distention of the stomach and first jejunal loops, with signs of obstruction

(a–b) Frontal plain films show jejunal atresia along with intestinal occlusion. “Triple bubble” sign (b)
In cases of partial obstruction, a small amount of air can be observed distally; in some cases, a little amount of air can be injected through a nasogastric tube to discriminate between a partial and a total obstruction. In isolated duodenum of jejunal atresia, the colon is normal in size because of intestinal secretions by the distal bowel loops making a normal caliber colon; instead, in ileal atresia the colon is diffusely reduced in caliber (functional microcolon), well demonstrable by performing a contrast enema (Morris et al. 2016; Berrocal et al. 1999b).
The fluoroscopic gastrointestinal study is performed as already described earlier; the enema can show a distal unused colon (microcolon) which is not in its right position in the abdomen, leading to some grades of malrotation (Fig. 1.15). It is often easy to observe the retrograde flow of enema in the distal ileum until the site of ill or jejunal atresia.

Multiple intestinal atresias. (a, b) Frontal and lateral radiographs show intestinal obstruction with air-fluid levels. (c) Barium enema study demonstrates an unused microcolon occupying the left abdomen
In most instances an upper gastrointestinal study is not required before surgical repair; but when performed it may help to exclude other obstructive disorders (i.e., meconium ileus or Hirschsprung’s disease), especially when dilated small bowel or the colon cannot be differentiated on plain radiography. If meconium ileus is present, Gastrografin can be used as enema which can help to evacuate the meconium, being a hypertonic solution, as well as make a diagnosis (Di Giacomo et al. 2015).
Moreover, contrast media can be performed in cases of partial obstruction, demonstrating the presence and the grade of stenosis (Figs. 1.16 and 1.17) or a mucosal web with a small opening. It can also help to identify the location of the cecum to rule out anomalies of rotation and fixation.

(a) Radiograph shows dilatation of the stomach and duodenal loop. Gas is seen in jejunal loops. (b) Barium study demonstrated a jejunal stenosis at the level of Treitz ligament

(a) Abdominal plain radiogram shows a dilated stomach and irregular distribution of the air in the abdomen; (b) after a barium enema study one can observe a dilated stomach and proximal duodenum, with air in the distal bowel loops, suggesting a duodenal stenosis
Also sonography can be used to differentiate meconium ileus and ill atresia, because the former is characterized by dilated loops filled with echogenic material, while in ill atresia, the bowel content is echo-poor.
The presence of intraperitoneal calcifications suggests a meconium peritonitis as a consequence of intrauterine intestinal perforation and can be seen in 12 % of cases (Frischer and Azizkhan 2012); intestinal perforation can occur also after birth (Fig. 1.18).

(a) The fluoroscopic gastrointestinal study shows a duodenal stenosis because of dilatation of the stomach and proximal duodenum. (b, c) The same patient in AP and LL projections: intestinal perforation is evident also
The “apple-peel” syndrome is characterized by proximal jejunal atresia with the absence of mid-small intestine and the dorsal mesentery as a consequence of intrauterine occlusion of the distal superior mesenteric artery. Therefore, the distal bowel derives its bloody supply from the proximal superior mesenteric artery and the distal small bowel spirals around its single vascular supply resembling an apple peel.
11.1.3 Treatment
At first, infant needs to be stabilized and needs of a nasogastric tube to decompress the stomach and to minimize the aspiration. Broad-spectrum antibiotics must be administered. Surgical repair can be performed both laparoscopically and via laparotomy. The choices of operation depend on the pathological findings, associated alterations (malrotation, volvulus, gastroschisis, meconium peritonitis), and the length of the remaining bowel. In patients with a normal length remaining bowel, a resection of the atretic loop can be performed; in case with a short bowel left, a proximal tapering enteroplasty or intestinal plication has been proposed to preserve bowel length (Juang and Snyder 2012).
For these patients, it is also of utmost importance to document a complete distal patency; therefore, an intraluminal catheter and a saline solution flush can often identify additional distal atresias. In fact, while a primary resection or a tapering enteroplasty is needed for the initial atresia, in cases of multiple atresias, the distal ones are not distended; a direct end-to-end anastomosis without resection for type II or IIIA distal atresia separated by a cord or a gap or an enterotomy and web excision with traverse closure of the enterotomy for distal type I web are acceptable.
Postoperative complications are anastomotic leak, infection, and short gut syndrome. These patients may suffer from feeding intolerance also, especially in the presence of meconium peritonitis, short bowel syndrome, and luminal discrepancy (Wang et al. 2014).
The prognosis is good with >90 % of survival; the prognosis for those suffering of short gut syndrome depends on the length of the remaining bowel, the dependence on parenteral nutrition, and the presence of the ileocecal valve (Calisti et al. 2012).
12 Colon
Colonic atresia is relatively less common compared to ileal atresia and is one of the rarest causes of neonatal intestinal obstructions, having an incidence of 5–15 % of all intestinal atresias.
It is usually caused by a fetal extrinsic mesenteric vascular obstruction associated with internal hernia, volvulus, and intussusception or strangulation.
An intraluminal vascular obstruction has also been advocated as a possible cause of CA. Some authors proposed the theory of emboli that, originating from the placenta and reaching the fetal mesenteric circulation, may cause intrinsic vascular obstruction bypassing the lung circulation (Etensel et al. 2005; Erskine 1970).
Puri and Fujimoto stated that multiple intestinal atresias may derive from a malformative process of the gastrointestinal tract (Puri and Fujimoto 1988).
Varicella has been claimed to play a role in the pathogenesis of CA; in fact it seems that it might cause an injury to the enteric plexus leading to poor blood vessel development and ischemic condition, resulting in intestinal atresia (Sauve and Leung 2003).
In about two thirds of cases, it has been reported as an isolated anomaly, although it can be associated with other congenital anomalies, in particular fixation and malrotation. In fact it seems that, causing problems related with location and formation of proximal and distal segments, they both cause CA or are caused by colonic atresia (Landes et al. 1994; Sarin 2000; Benawra et al. 1981).
It commonly affects the colon proximal to the splenic flexure. Atresias affecting the ascending colon are often indistinguishable from obstructions of the distal ileum.
Affected neonates complain with abnormal abdominal distention, vomiting, and failure to pass meconium. The differential diagnosis includes ileal atresia, meconium ileus or peritonitis, functional immaturity of the colon, and Hirschsprung’s disease (Berrocal et al. 1999b).
12.1 Classification
The classification of CA was firstly provided by Louw and then refined by Marin and Zerella and Grosfeld (Grosfeld et al. 1979; Louw 1967; Martin and Zerella 1976). It consists of four types of CA: type I represents a mucosal defect with an intact mesentery. Type II is characterized by a fibrous cord connecting the atretic bowel ends. Type IIIa consists of an atretic segment with a “V-shaped” mesenteric defect; type IIIb is the so-called apple-peel deformity in which we have a proximal colonic atresia, and the distal bowel is supplied by a single retrograde blood vessel. In type IV there are multiple atretic segments.
12.2 Diagnosis
Prenatal US shows a characteristic finding of a colon with apparent haustra and showing an intestinal coursing at the periphery of the abdomen (Anderson et al. 1993).
Plain radiography shows features of low intestinal obstruction with air-fluid levels due to retained meconium, multiple dilated bowel loops, and the absence of air in the rectum. The colon proximal to the point of atresia is disproportionately dilated, and a mottled pattern of gas and feces can be identified.
Colonic enema study has been already described: enema typically shows a distal unused colon (microcolon) (Fig. 1.15c) with obstruction to the retrograde flow of contrast medium at the site of atresia with the more proximal dilated colon ending in a blind pouch. Nevertheless, it cannot define the length of bowel involved nor whether a proximal lesion exists (Winters et al. 1992).
Performing enema, different radiological signs have been described; that is, in type I defect, it can be observed the “wind-sock sign” because the contrast media pushes against the membrane. In type III there is the “hook” sign described on the microcolon sign (Azzie et al. 2002; Blair and Jamieson 2001).
US findings of atretic colon include dilation of the distal small bowel and proximal colon whose content appears markedly echogenic due to the retained meconium. The differential diagnosis between small bowel and colonic obstruction can be performed only if the distal portions of the colon are visualized and appear collapsed (Pasto et al. 1984).
12.3 Treatment
Previous studies recommended resection with primary anastomosis for lesions located close to the splenic flexure and colostomy with delayed anastomosis for atresia distal to this point (Coran and Eraklis 1969; Schiller et al. 1979).
In the recent years, it has been proposed to perform resection and primary anastomosis, regardless of the location of the atresia, when the newborn’s condition permits: in fact the distal and proximal ends close to the atresia are different for innervation, vascularity, and size (Pohlson et al. 1988).
Cox et al. proposed that this approach could be safely performed with a diameter variance of 3:1 (proximal:distal) and without other distal mechanical obstructions (Cox et al. 2005).
In the absence of small bowel atresias, the normal length of small intestine can guarantee a normal bowel function.
When the ileocecal valve is intact, colonic atresia may appear as a closed loop obstruction with a high risk of perforation (Watts et al. 2003).
Regarding survival and postoperative complications, CA is the most favorable type of intestinal atresias. In these cases the overall mortality is 10 % or less; nevertheless, delayed diagnosis (>4 days) may result in a very high rate of mortality, about 100 % (Karnak et al. 2001; Tao et al. 1987).
13 Anorectal Malformations
Anorectal malformations (ARMs) comprise a wide group of congenital anomalies involving the distal anus, the rectum, and the genitourinary tract. The estimated incidence is about 1:5000 live births, affecting males and females with a similar frequency. Most of them are represented by imperforate anus with the distal enteric component ending blindly (atresia) or through a fistula into the genital or urinary tract or into the perineum. They are usually associated with other congenital anomalies in up to 70 % of cases, such as cardiac, vertebral (i.e., genesis and atresia of the sacrum, vertebral dysplasia, and tethered cord syndrome), renal, and limb anomalies.
In particular, urogenital anomalies are the most common encountered in up to 60 % of patients complaining with hydronephrosis and vesicoureteral reflux (Berrocal et al. 1999b; Levitt and Peña 2007).
13.1 Embryology
In the early embryonic like the development of the anorectum, the urogenital sinus and the caudal neural tube are strictly related. In fact, between the fourth and sixth week of gestation, the primitive hindgut and the allantois (primitive urogenital sinus) enter into the cloaca, and the urorectal septum develops some infoldings of the lateral cloacal walls. At the same time, the developing neural tube and the mesodermal compartment, growing longitudinally, are responsible for the initial curvature of the embryo; therefore, the distance between the cloacal membrane and the tip of the urorectal septum is reduced. At the end of week 7, the urorectal septum and the cloacal membrane are located at the same level. The cloaca is then divided into a ventral part (the urogenital sinus) and a dorsal part (the rectum and proximal anal canal). Between them, the tip of the urorectal septum becomes the perineal area. At this time, the cloacal membrane ruptures and opens two orifices in the perineum: one ventral or urogenital and one dorsal or anal. Simultaneously, a secondary occlusion of the anorectal canal takes place, which then will rupture and recanalize by apoptosis at the end of week 8 (Nievelstein et al. 1993, 1998, 2002).
Anomalies in the development of the anorectal septum are the most involved in the ARMs.
They are divided into two main groups depending on the period in which the anomalies occur; the anomalies characterized by an early abnormal development of the dorsal part of the cloaca and the cloacal membrane typically manifest as an ectopic anal orifice or fistula. Those anomalies due to a later abnormal recanalization of the secondary occluded anal orifice manifest as abnormal anus in a normal position.
13.2 Classification
The best known classification of ARMs is that provided by Wingspread in 1984, which divides these anomalies into three groups, low, intermediate, or high, depending on the location of the rectal pouch with respect to the puborectal sling (Stephens et al. 1988).
A low-type ARM is defined as a rectal pouch located below the level of the puborectal muscle.
An intermediate- or high-type ARM is characterized by a rectal pouch located at or above the level of the puborectal sling.
In 2005 the Krickenbeck Conference established a new classification depending on the presence or absence of fistulas and their location, as well as the position of the rectal pouch. This classification provides five types of fistulas: rectoperineal, rectovestibular, rectourethral bulbar, rectourethral prostatic, and rectovesical. The rectovaginal fistula is a variant of cloacal anomaly (Holschneider et al. 2005).
While the Wingspread classification indicates the location of the rectal pouch, the Krickenbeck’s one gives anatomic evaluation about the rectal pouch but also indications about any fistulas. This is an important finding for the surgeon who can anticipate the extent of mobilization of the atretic rectal segment and define the most appropriate surgical approach.
Finally there is the one provided by Gans, which is the simplest and most used. This classification comprised three types of ARMs: rectal atresia, in which the anus is open but the rectum above the anus is atretic and no fistula is present. Then we have the ectopic anus, occurring when the terminal bowel fails to descend causing lack of communication with the anus. As a consequence, the rectum opens via a fistula at an abnormal location, such as the vestibule, vagina, urethra, bladder, and cloaca. In imperforate anus the distal bowel ends blindly without any fistula (Gans 1970) (Figs. 1.19 and 1.20).

These drawings show the different types of male ARMs with or without rectal atresia and with or whithout the presence of a rectal fistula with the urinary tract. The anus can be ectopic or imperforate

These drawings show the different types of female ARMs with anorectal agenesia or rectal atresia with or without fistula. In the most complex malformation there is a common outlet: the cloaca
13.3 Diagnosis
A prenatal diagnosis is very difficult to reach, occurring in about 16 % of cases. Associated findings are oligohydramnios, abdominal or pelvic cystic masses, fetal ascites, hydronephrosis, and intestinal distention. In the presence of a distended bladder with oligohydramnios, a possible ARM should be considered.
When multiple pelvic cystic structures are observed in a female fetus, one may take in mind it could correspond to a distended bladder or a unique or septate fluid-filled vagina; therefore a cloacal anomaly should be suspected. The differential diagnosis includes hydrometrocolpos related to imperforate hymen (Bischoff et al. 2010; Calvo-Garcia et al. 2011).
Fetal MR imaging in the third trimester of gestation can help confirm ARMs. In a normal fetus, urine has signal intensity similar to that of fluid, whereas normal meconium appears hyperintense on T1-weighted images and hypointense on T2-weighted images. In some fetuses with ARMs, mainly long common-channel cloacal anomalies in females and some rectourinary fistulas in males, increased signal intensity in the rectum, and decreased signal intensity in the bladder can be observed on T2-weighted images as a result of the mixing of urine and meconium.
Nevertheless, in the majority of cases, ARMs are diagnosed at birth. All diagnostic techniques can be used (Miele et al. 2006; Miele and Di Giampietro 2014). In infants with a rectoperineal or a rectovestibular fistula (external fistulas), the diagnosis of a low type of ARM is evident, and a perineal surgical procedure can be performed early. If no external fistula is evident, the passage of meconium through the vagina or with the urine may become evident after 24–48 h of life, delaying the diagnosis of an intermediate or high type of ARM (Alamo et al. 2010).
There are different opinions about the most useful imaging studies to perform in neonates with ARMs. Imaging studies in the first 2 days of life include radiography of the thorax, spine, and pelvis with cardiac, perineal, abdominal, pelvic, and spine US to detect possible associated anomalies.
In the past the invertography was performed to determine the level of atresia (Fig. 1.21); nowadays its use is to be discouraged as it causes unuseful stress. In fact, during invertogram, the baby keeps on crying causing obliteration of the lower rectum because of the contraction of puborectalis sling, and the rectum, being pushed into a cephalic direction, causes an error in ARM classification (Niedzielski 2005).

Anorectal atresia. Invertogram. A small metal object is fixed to the anal dimple
In the presence of fistula, in invertogram this one becomes the highest point of the rectum, and some gas may escape causing less distention of the rectum.
In a prone cross-table lateral view with babies heat in genupectoral position, the fistula becomes the lowest point and the rectum is better distended with a better delineation of rectal gas (Fig. 1.22).

Anorectal atresia. Lateral radiograph shows a distended rectal pouch. A small metal object is fixed to the anal dimple
The “M line” runs horizontally through the junction of the lower third and upper two thirds of the ischium; it describes the level of the puborectal sling, and it is used to classify lesions as high, intermediate, or low on lateral radiographs.
Plain radiography of the thorax, spine, and pelvis in the anteroposterior and lateral views allow identification of associated cardiac, costal, and vertebral anomalies (Narasimha Rao et al. 1983).
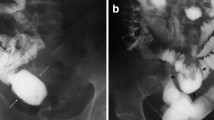
Perineal US is an excellent technique for evaluating the distance between the rectum distal pouch and the perineum and even the location of any rectourogenital fistulas (Figs. 1.23 and 1.24) (Niedzielski 2005; Oppenheimer et al. 1983). It is usually performed with a 10–12-MHz high-resolution linear array transducer by using the transperineal approach, with the child in a supine position and the pelvis and legs elevated. The distance between the rectal pouch and the anus is measured in the midline sagittal plane through the perineum. A distance of greater than 15 mm indicates a high type of ARM, whereas a distance of less than 15 mm suggests a low type of malformation (Haber et al. 2007; Haber 2009). Nevertheless, differentiation between a low and a high lesion in a relatively low position may be difficult, and any crying by the patient can increase the intra-abdominal pressure, displacing the distal rectal pouch to the perineum and shortening this distance. Fistulas may be identified as linear tracts connecting the rectal pouch to the bladder, urethra, or posterior wall of the vagina.

Anorectal atresia with bladder fistula. (a) US shows a fluid-filled urinary bladder and a not-distended rectal pouch behind. (b) During the urinary bladder voiding phase, you can observe the filling of rectal pouch, indicating a communication between the rectum and bladder

Anorectal atresia with bladder fistula. (a) US shows a fluid-filled urinary bladder and a rectal pouch distended by corpuscolated material backward (b) During the urinary bladder voiding phase, you can observe the additional filling of rectal pouch, indicating a communication between the rectum and bladder
Abdominal US evaluation of the urinary tract is limited in the first 24 h after birth, because the physiologic dehydration and the reduced urinary output cause the lack of upper tract dilatation. However, detection of any genitourinary anomalies requires a voiding cystourethrography (Boemers et al. 1999).
Spinal US provides accurate information about the morphology and integrity of the sacrum and distal vertebral column, allowing identification of the level of the medullary cone and demonstration of any presacral mass. In these cases complementary spinal MR imaging is mandatory.
Cystography and/or colostography are routinely performed to delineate associated fistulas between rectum and urinary tract as well as vesicoureteral reflux (Figs. 1.23 and 1.24).
CT and MR are the techniques of choice to delineate pelvic anatomy, in particular the puborectalis sling and the external sphincter. In neonates with ARMs, a combined protocol of pelvic and spinal MR would be acceptable (Figs. 1.25 and 1.26).

Anorectal malformation. Image from colostography depicts a rectourethral fistula and anal atresia

Colostography shows an anorectal atresia with perineal fistula
Boys presenting with ARM usually show other genital anomalies like hypospadias, micropenis, and ambiguous genitalia. MR imaging typically displays normal sphincteric development with the anterior part of the rectum anteriorly dislocated and a rectoperineal fistula.
Male patients commonly suffer from the high or intermediate form of ARM, with a rectourethral fistula and almost normal perineal and gluteal anatomy with a well-developed levator ani and sphincteric muscle at MR imaging (Ratan et al. 2004; Peña and Hong 2000).
In girls, any perineal or vestibular fistula can be well diagnosed at clinical examination. This is a low type of ARM, in which rectum and vagina are well separated in most of cases.
The most common anomaly depicted at MR imaging is the rectovestibular fistula, in which both levator ani and external sphincter are well developed.
Cloacal anomaly is well diagnosed at clinical inspection, where a unique opening is seen for rectum, vagina, and urethra, but it is more difficult to delineate at MR because of the association of other genital anomalies, such as hydrocolpos, and a septum in uterus and vagina. However, MR can show an underdeveloped levator ani muscle which appears even atrophic along with a nonvisible external anal sphincter (Nievelstein et al. 1993; Peña and Hong 2000).
The most frequent clinical syndromes associated with ARMs are the VACTERL syndrome, the caudal regression syndrome, and the Currarino syndrome.
The prevalence of VACTERL is about 1.10,000–40,000 live births, and in most of cases, it is sporadic.
Its diagnosis can be suspected in the presence of US findings of a single umbilical artery and polyhydramnios along with vertebral, renal, limb, and cardiac malformations. These patients have a high prevalence of spinal dysraphism, tethered cord syndrome, and genitourinary conditions. In these cases MR imaging is mandatory.
Caudal regression syndrome is a neural tube defect presenting with developmental anomalies of the distal spine along with complete or partial agenesis of the os sacrum and pelvic deformity and anomalies affecting the neural tube, gastrointestinal and genitourinary tract, limbs, and heart. Clinically patients suffer from a complete neurological deficit as well as no control of the bladder and bowel.
Prenatal US findings show interruption of the distal spine because of the absence of thoracic and lumbar vertebrae or the sacrum with fusion of pelvic bones and typical frog-like position of lower limbs (Solomon 2011; Solomon et al. 2011; Kuo et al. 2007). Also in this case, MR imaging is required after birth to find out the level of spinal agenesis.
Currarino syndrome was first described in 1981 to describe the association of ARM, sacrococcygeal defect, and a presacral mass (teratoma, dermoid cyst, meningocele, neuroenteric cyst) responsible for a tethered cord syndrome.
This is an autosomal dominant disorder with incomplete penetrance. Patients usually present with intractable constipation (Currarino et al. 1981; Alamo et al. 2013).
Lateral x-ray depicts the sacral anomaly with preservation of the first sacral vertebra and dysplasia of the last sacral vertebrae, with a sickle or scimitar appearance. MR imaging well depicts the presacral mass, the tethered cord, and the high atrophy of the sphincteric muscle.
13.4 Treatment
The mainstay of therapy is to obtain a good quality of life with acceptable levels of bowel control and normal sexual and reproductive abilities.
The low-type ARM can be managed early with opening of the rectal pouch and ligature of the fistula, when present.
The intermediate and high-type ARMs are treated with colostomy at first and then with a definitive repair at a later age, by performing a posterior sagittal anorectoplasty alone or along with laparoscopic rectoplasty in a second intervention (deVries and Peña 1982; Georgeson et al. 2000). Prior the definitive repair, a high-pressure distal colostogram is required to confirm the level of rectal atresia and to identify any fistulous tract. Through a Foley catheter, a water-soluble contrast medium is injected under mild pressure to inflate its balloon and occlude the stoma. The injection should be continued until the patient starts voiding to increase the possibility to visualize the fistula (Gross et al. 1991; Gupta and Guglani 2005).
High fistulas, mainly the rectourethral prostatic or the rectovesical in boys, are very difficult to visualize on a sagittal approach, so that a laparotomy or laparoscopy may be performed.
References
Al Shahwani N, Mandhan P, Elkadhi A, Ali MJ, Latif A (2013) Congenital duodenal obstruction associated with Down’s syndrome presenting with hematemesis. J Surg Case Rep 2013(12). pii: rjt108. doi:10.1093/jscr/rjt108
Alabbad SI, Ryckman J, Puligandla PS, Shaw K, Nguyen LT, Laberge JM (2009) Use of transanastomotic feeding tubes during esophageal atresia repair. J Pediatr Surg 44:902–905
Alamo L, Laswad T, Schnyder P et al (2010) Fetal MRI as complement to US in the diagnosis and characterization of anomalies of the genito-urinary tract. Eur J Radiol 76(2):258–264
Alamo L, Meyrat BF, Meuwly JY, Meuli RA, Gudinchet F (2013) Anorectal malformations: finding the pathway out of the labyrinth. Radiographic 33:491–512
Ali YA, Rahman S, Bhat V, Al Thani S, Ismail A, Bassiouny I (2011) Hereditary multiple intestinal atresia (HMIA) with severe combined immunodeficiency (SCID): a case report of two siblings and review of the literature on MIA, HMIA, and HMIA with immunodeficiency over the last 50 years. BMJ Case Rep. doi:10.1136/bcr.05.2010.3031. pii: bcr0520103031
Anderson N, Malpas T, Robertson R (1993) Prenatal diagnosis of colon atresia. Pediatr Radiol 23:63–64
Applegate KE, Goske MJ, Pierce G, Murphy D (1999) Situs revisited: imaging of the heterotaxy syndrome. RadioGraphics 19:837–852; discussion, 853–854
Azzie G, Craw S, Beasley SW (2002) Colonic atresia: from suspicion to confirmation on pre-operative radiology. J Paediatr Child Health 38:518–520
Baglaj M, Carachi R, Lawther S (2008) Multiple atresia of the small intestine: a 20-year review. Eur J Pediatr Surg 18:13–18
Benawra R, Puppala BL, Mangurten HH, Booth C, Bassuk A (1981) Familial occurrence of congenital colonic atresia. J Pediatr 99:435–436
Berrocal T, Torres I, Gutierrez J, Prieto C, del Hoyo ML, Lamas M (1999a) Congenital anomalies of the upper gastro-intestinal tract. Radiographics 19:855–872
Berrocal T, Lamas M, Gutieerrez J, Torres I, Prieto C, del Hoyo ML (1999b) Congenital anomalies of the small intestine, colon, and rectum. Radiographics 19(5):1219–1236
Best KE, Tennant PW, Addor MC, Bianchi F, Boyd P, Calzolari E et al (2012) Epidemiology of small intestinal atresia in Europe: a register based study. Arch Dis Child Fetal Neonatal Ed 97(5):F353–F358
Bischoff A, Levitt MA, Lim FY, Guimarães C, Peña A (2010) Prenatal diagnosis of cloacal malformations. Pediatr Surg Int 26(11):1071–1075
Blair GK, Jamieson DH (2001) Colon atresia-type III. J Pediatr Surg 36:530–531
Boemers TM, Beek FJ, Bax NM (1999) Guidelines for the urological screening and initial management of lower urinary tract dysfunction in children with anorectal malformations: the ARGUS protocol. BJU Int 83(6):662–671
Brantberg A, Blaas HG, Salvesen KA, Haugen SE, Mollerlokken G, Eik-Nes SH (2002) Fetal duodenal obstructions: increased risk of prenatal sudden death. Ultrasound Obstet Gynecol 20(5):439–446
Calisti A, Olivieri C, Coletta R, Briganti V, Oriolo L, Giannino G (2012) Jejunoileal atresia: factors affecting the outcome and long-term sequelae. J Clin Neonatol 1(1):38–41
Calvo-Garcia MA, Kline-Fath BM, Levitt MA et al (2011) Fetal MRI clues to diagnose cloacal malformations. Pediatr Radiol 41(9):1117–1128
Chew AL, Friedwald JP, Donovan C (1992) Diagnosis of congenital astral web by ultrasound. Pediatr Radiol 22:342–343
Clements JL, Jinkins JR, Torres WE et al (1979) Antral mucosal diaphragm in adults. AJR Am J Roentgenol 133:1105–1109
Coran AG, Eraklis AJ (1969) Atresia of the colon. Surgery 65:828–831
Correia-Pinto J, Ribeiro A (2014) Congenital duodenal obstruction and double-bubble sign. N Engl J Med 371(11):e16
Cox SG, Numaouglu A, Millar AJW, Rode H (2005) Colonic atresia: spectrum of presentations and pitfalls in management. A review of 14 cases. Pediatr Surg Int 21:813–818
Cumming WA (1975) Esophageal atresia and tracheoesophageal fistula. Radiol Clin N Am 13:277–285
Currarino G, Coln D, Votteler T (1981) Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol 137(2):395–398
de Jong EM, Felix JF, de Klein A, Tibboel D (2010) Etiology of esophageal atresia and tracheoesophageal fistula: “mind the gap”. Curr Gastroenterol Rep 12:215–222
deVries PA, Peña A (1982) Posterior sagittal anorectoplasty. J Pediatr Surg 17(5):638–643
Di Giacomo V, Trinci M, Van der Byl G, Catania VD, Calisti A, Miele V (2015) Ultrasound in newborns and children suffering from nontraumatic acute abdominal pain: imaging with clinical and surgical correlation. J Ultrasound 18:385–393. doi:10.1007/s40477-014-0087-4. Epub 2014 Apr 9
Di Gianantonio E, Schaefer C, Matroiacovo PP et al (2001) Adverse effects of prenatal methimazole exposure. Teratology 64:262–266
El-Gohary Y, Gittes GK, Tovar JA (2010) Congenital anomalies of the esophagus. Semin Pediatr Surg 19:186–193
Erskine JM (1970) Colonic stenosis in the newborn: the possible thromboembolic etiology of intestinal stenosis and atresia. J Pediatr Surg 5:321–333
Escobar MA, Ladd AP, Grosfeld JL, West KW, Rescorla FJ, Scherer LR 3rd et al (2004) Duodenal atresia and stenosis: long-term follow-up over 30 years. J Pediatr Surg 39(6):867–871
Etensel B, Temir G, Karkiner A et al (2005) Atresia of the colon. J Pediatr Surg 40:1258–1268
Felix JF, Steegers-Theunissen RP, de Walle HE, de Klein A, Torfs CP, Tibboel D (2007) Esophageal atresia and tracheo-esophageal fistula in children of women exposed to diethylstilbestrol in utero. Am J Obstet Gynecol 197:e31–e35
Free EA, Gerald B (1968) Duodenal obstruction in the newborn due to annular pancreas. Am J Roentgenol 103(2):321–325
Frischer JS, Azizkhan RG (2012) Jejunoileal atresia and stenosis. In: Coran AG (ed) Pediatric surgery, 7th edn. Elsevier Saunders, Philadelphia
Gans SL (1970) Classification of anorectal anomalies: a critical analysis. J Pediatr Surg 5:511–513
Genevieve D, de Pontual L, Amiel J, Sarnacki S, Lyonnet S (2007) An overview of isolated and syndromic esophageal atresia. Clin Genet 71:392–399
Georgeson KE, Inge TH, Albanese CT (2000) Laparoscopically assisted anorectal pull-through for high imperforate anus: a new technique. J Pediatr Surg 35(6):927–930, discussion 930–931
Grosfeld JL, Ballantine TVN, Shoemaker R (1968) Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg 14:368–375
Grosfeld JL, Ballantine TV, Shoemaker R (1979) Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg 14(3):368–375
Gross GW, Wolfson PJ, Pena (1991) An augmented-pressure colostogram in imperforate anus with fistula. Pediatr Radiol 21(8):560–562
Gupta AK, Guglani B (2005) Imaging of congenital anomalies of the gastrointestinal tract. Indian J Pediatr 72:403–414
Haber HP (2009) Ultrasonography of imperforate anus in neonate: an approach correlated with current surgical concepts. Ultraschall Med 30(2):189–195
Haber HP, Seitz G, Warmann SW, Fuchs J (2007) Transperineal sonography for determination of the type of imperforate anus. AJR Am J Roentgenol 189(6):1525–1529
Hertzberg BS, Bowie JD (1990) Fetal gastrointestinal abnormalities. Radiol Clin N Am 28:101–114
Holland AJ, Fitzgerald DA (2010) Oesophageal atresia and trachea-esophageal fistula: current management strategies and complications. Pediatr Respir Rev 11:100–106, quiz 106–107
Holschneider A, Hutson J, Peña A et al (2005) Preliminary report on the international conference for the development of standards for the treatment of anorectal malformations. J Pediatr Surg 40(10):1521–1526
Houben CH, Curry JI (2008) Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula. Prenat Diagn 28:667–675
Ioannides AS, Copp AJ (2009) Embryology of esophageal atresia. Semin Pediatr Surg 18:2–11
Juang D, Snyder CL (2012) Neonatal bowel obstruction. Surg Clin North Am 92(3):685–711
Karnak I, Ciftci AO, Senocak ME, Tanyel FC, Buyukpamukcu N (2001) Colonic atresia: surgical management and outcome. Pediatr Surg Int 17:631–635
Kuo MF, Tsai Y, Hsu WM, Chen RS, Tu YK, Wang HS (2007) Tethered spinal cord and VACTERL association. J Neurosurg 106(3 Suppl):201–204
Lahdes-Vasama TT, Sihvonen R, Iber T (2009) Perforation of the upper and lower segments of atretic esophagus (type C) secondary to nasogastric tube insertion. Pediatr Surg Int 25:537–538
Lai ECS, Tompkins RK (1986) Heterotopic pancreas: review of 26-year experience. Am J Surg 151:697–700
Landes A, Shuckett B, Skarsgard E (1994) Non-fixation of the colon in colonic atresia: a new finding. Pediatr Radiol 24:167–169
Laya BF, Andres MM, Conception NDP, Dizon RH (2015) Patterns of microcolon: imaging strategies for diagnosis of lower intestinal obstruction in neonates. J Am Osteopat Coll Radiol 4(1):1–11
Levitt MA, Peña A (2007) Anorectal malformations. Orphanet J Rare Dis 2:33
Louw JH (1967) Resection and end-to-end anastomosis in the management of atresia and stenosis of the small bowel. Surgery 62:940–950
Martin LW, Zerella JT (1976) Jejunoileal atresia: a proposed classification. J Pediatr Surg 11:399–403
Materne R (2001) The duodenal wind sock sign. Radiology 218(3):749–750
Miele V, Di Giampietro I (2014) Diagnostic imaging in emergency. Salute Soc (2EN): 127–138. doi:10.3280/SES2014-002010EN
Miele V, Andreoli C, Grassi R (2006) The management of emergency radiology: key facts. Eur J Radiol 59:311–314. Epub 2006 Jun 27
Moore CCM (1989) Congenital gastric outlet obstruction. J Pediatr Surg 24:1241–1246
Morris G, Kennedy A Jr, Cochran W (2016) Small bowel congenital anomalies: a review and update. Curr Gastroenterol Rep 18:16. doi:10.1007/s11894-016-0490-4
Mortell AE, Azizkhan RG (2009) Esophageal atresia repair with thoracotomy: the Cincinnati contemporary experience. Semin Pediatr Surg 18:12–19
Nagpal R, Schnaufer L, Altschuler SM (1993) Duodenal web presenting with gastrointestinal bleeding in a seven-month-old infant. J Pediatr Gastroenterol Nutr 16(1):90–92
Narasimha Rao KL, Prasad GR, Katariya S, Yadav K, Mitra SK, Patak IC (1983) Prone cross table lateral view: an alternative to the invertogram in imperforate anus. AJR Am J Roentgenol 140:227–229
Niedzielski JK (2005) Invertography versus ultrasonography and distal colostography for the determination of bowel skin distance in children with anorectal malformations. Eur J Pediatr Surg 15(4):262–267
Nievelstein RA, Hartwig NG, Vermeij-Keers C, Valk J (1993) Embryonic development of the mammalian caudal neural tube. Teratology 48(1):21–31
Nievelstein RA, van der Werff JF, Verbeek FJ, Valk J, Vermeij-Keers C (1998) Normal and abnormal embryonic development of the anorectum in human embryos. Teratology 57(2):70–78
Nievelstein RA, Vos A, Valk J, Vermeij-Keers C (2002) Magnetic resonance imaging in children with anorectal malformations: embryologic implications. J Pediatr Surg 37(8):1138–1145
Nora JJ, Nora AH, Pervinche AG, Ingram JV, Fountain AK, Peterson MJ (1976) Congenital abnormalities and first trimester exposure to progestagen/oestrogen (letter). Lancet 1:313–314
Norton KI, Tenreiro R, Rabinowitz JG (1992) Sonographic demonstration of annular pancreas and a distal duodenal diaphragm in a newborn. Pediatr Radiol 22:66–67
Oppenheimer DA, Carroll BA, Shochat SJ (1983) Sonography of imperforate anus. Radiology 148:127–128
Pasto ME, Deiling JM, O’Hara AE, Rifkin MD, Goldberg BB (1984) Neonatal colonic atresia: ultrasound findings. Pediatr Radiol 14:346–348
Peña A, Hong A (2000) Advances in the management of anorectal malformations. Am J Surg 180(5):370–376
Pinheiro PFM, Simoes AC, Pereira RM (2012) Current knowledge on esophageal atresia. World J Gastroenterol 18(28):3662–3672
Pohlson EC, Hatch EI Jr, Glick PL, Tapper D (1988) Individualized management of colonic atresia. Am J Surg 155:690–692
Puri P, Fujimoto T (1988) New observations on the pathogenesis of multiple intestinal atresias. J Pediatr Surg 23:221–225
Ratan SK, Rattan KN, Pandey RM, Mittal A, Magu S, Sodhi PK (2004) Associated congenital anomalies in patients with anorectal malformations: a need for developing a uniform practical approach. J Pediatr Surg 39(11):1706–1711
Sarin YK (2000) Pyloric atresia associated with intestinal atresias. Indian Pediatr 37:205–207
Sarin YK, Sharma A, Sinha S, Deshpande VP (2012) Duodenal webs: an experience with 18 patients. J Neonatal Surg 1(2):20
Sauve RS, Leung AK (2003) Congenital varicella syndrome with colonic atresias. Clin Pediatr (Phila) 42:451
Schiller M, Aviad I, Freund H (1979) Congenital colonic atresia and stenosis. Am J Surg 138:721–724
Seitz G, Warmann SW, Schaefer J, Poets CF, Fuchs J (2006) Primary repair of esophageal atresia in extremely low birth weight infants: a single-center experience and review of the literature. Biol Neonate 90:247–251
Seo J, do Kim Y, Kim AR, Kim DY, Kim SC, Kim IK et al (2010) An 18-year experience of tracheoesophageal fistula and esophageal atresia. Korean J Pediatr 53:705–710
Sharma AK, Shekhawat NS, Agrawal LD, Chaturvedi V, Kothari SK, Goel D (2000) Esophageal atresia and tracheoesophageal fistula: a review of 25 years’ experience. Pediatr Surg Int 16:478–482
Shaw-Smith C (2010) Genetic factors in esophageal atresia, trachea-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet 53:6–13
Solomon BD (2011) VACTERL/VATER association. Orphanet J Rare Dis 6:56–67
Solomon BD, Raam MS, Pineda-Alvarez DE (2011) Analysis of genitourinary anomalies in patients with VACTERL (vertebral anomalies, anal atresia, cardiac malformations, tracheo-esophageal fistula, renal anomalies, limb abnormalities) association. Congenit Anom (Kyoto) 51(2):87–91
Son TN, Liem NT, Kien HH (2015) Laparoscopic simple oblique duodenoduodenostomy in management of congenital duodenal obstruction in children. J Laparoendosc Adv Surg Tech A 25(2):163–166
Spitz L (2007) Oesophageal atresia. Orphanet J Rare Dis 2:24
Stephens FD, Smith ED, Paoul NW (1988) Anorectal malformations in children: update 1988. Liss, New York
Strouse PJ (2008) Malrotation. Semin Roentgenol 43(1):7–14
Tao HA, Lin CC, Huang FY, Yeh ML, Shih SL (1987) Congenital atresia of the colon: a case report. Acta Pediatr Sin 28:120–122
van der Zee DC (2011) Laparoscopic repair of duodenal atresia: revisited. World J Surg 35(8):1781–1784
Van Winckel MAJM, Afschrift MB, Vande Walle JGJ (1994) Ultrasound diagnosis of a prepyloric diaphragm. J Clin Ultrasound 22:141–143
Wang J, Du L, Cai W, Pan W, Yan W (2014) Prolonged feeding difficulties after surgical correction on intestinal atresia: a 13-year experience. J Pediatr Surg 49(11):1593–1597
Watts AC, Sabharwal AJ, MacKinlay GA, Munro FD (2003) Congenital colonic atresia: should primary anastomosis always be the goal? Pediatr Surg Int 19:14–17
Winters WD, Weinberger E, Hatch EI (1992) Atresia of the colon in neonates: radiographic findings. AJR 159:1273–1276
Wong-Gibbons DL, Romitti PA, Sun L, Moore CA, Reefhuis J, Bell EM, Olshan AF (2008) Maternal periconceptional exposure to cigarette smoking and alcohol and esophageal atresia +/− trachea-esophageal fistula. Birth Defects Res A Clin Mol Teratol 82:776–784
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Miele, V., Piccolo, C.L., Saracco, V., Napoletano, M., Trinci, M., Brunese, L. (2016). Intestinal Stenosis and Atresia. In: Miele, V., Trinci, M. (eds) Imaging Non-traumatic Abdominal Emergencies in Pediatric Patients. Springer, Cham. https://doi.org/10.1007/978-3-319-41866-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-41866-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41865-0
Online ISBN: 978-3-319-41866-7
eBook Packages: MedicineMedicine (R0)