
Abstract
Cholangiocytes, the epithelial cells that line the biliary tree, are the target cells for a group of liver diseases known as the cholangiopathies. Even though cholangiocytes constitute only 4–5 % of the liver cell population, their functions are ubiquitous and range from modification of primary bile via secretion and absorption of water, electrolytes, and other molecules to the reaction of the biliary tract to exogenous insults. These cells are equipped with the machinery to recognize and respond to potentially harmful, endogenous, as well as exogenous microbial-derived molecules. Responses to cholangiocyte injury include: (1) proliferation and ductular expansion; (2) release of pro-inflammatory molecules that recruit and activate resident and nonresident immune cells to respond to infectious and inflammatory injury and promote epithelial repair; and (3) the induction of key cellular processes, including apoptosis, autophagy, and senescence. Dysregulation of these responses likely plays an active role in the development of some if not all of the cholangiopathies. In this chapter, we review selected aspects of the normal biology of cholangiocytes and address the plasticity these cells exhibit when alterations in their microenvironment occur.
This work was supported by grants awarded to NFL: NIH NIDDK DK 24031 (R01), Pathobiology of Hepatic Epithelia; NIH NIDDK DK 57993 (R01), Pathophysiology of Biliary Disease; and NIH NIDDK DK 84567 (P30), Mayo Center for Cell Signaling in Gastroenterology
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The biliary system consists of a network of tubular structures, or bile ducts, inside (intrahepatic bile ducts) and outside of the liver (extrahepatic bile ducts). This system facilitates the flow of bile from the liver to the gallbladder for storage before being secreted into the small intestine after meals to aid in the digestion of dietary fats [1]. Bile ducts are lined by epithelial cells, known as cholangiocytes, that vary in morphology and function. Cholangiocytes are best known for their role in bile modification and secretion; however, over the past decades, other functions have been attributed to these cells. For instance, cholangiocytes are key contributors to the function of the innate and adaptive immune systems, as they are “the first line of defense” in the biliary tract against harmful, gut-derived molecules [2]. These cells express receptors on their apical surface that recognize endogenous and exogenous pathogens, chemicals, microbial products, and xenobiotics present in bile. Upon recognition of potentially injurious agents, cholangiocytes may become activated, secreting pro-inflammatory factors necessary for the recruitment of a variety of different cells, including immune cells, to the site of injury [3] (Table 7.1) (Fig. 7.1). Moreover, activated cholangiocytes secrete profibrotic molecules, such as transforming growth factor beta (TGF-β) [4] and platelet-derived growth factor-BB (PDGF-BB), that can activate myofibroblasts, the main mediators of the wound-healing response [5]. Another mechanism of defense for damaged cholangiocytes against stressors, particularly oncogenic agents, is the termination of cellular replication via the process of cellular senescence [6] (Fig. 7.1). Further, when cholangiocytes become senescent, they can transition to a senescence-associated secretory phenotype (i.e., SASP) characterized by the robust secretion of an array of soluble and intravesical factors that affect neighboring cells. For example, the cholangiocyte SASP is characterized by high secretion levels of pro-inflammatory cytokines, such as interleukins 6 (IL-6) and 8 (IL-8) [6] (Table 7.1) (Fig. 7.1). Importantly, IL-6 may play a role in malignant transition of cholangiocytes [7].

Cholangiocyte plasticity model. Schematic representation of the proposed model of the plasticity of cholangiocytes during biliary injury. The solid arrow lines indicate the transition of normal cholangiocytes through the various disease phenotypes. The dashed arrows suggest that activated or senescent cholangiocytes could resolve back to the normal phenotype. The major key molecules and pathways that participate in each stage are also shown [3, 89]
In this chapter we review selected aspects related to cholangiocyte biology with a particular emphasis on cholangiocyte adaptability to changes in their microenvironment as a mechanistic response to injury. The pathways involved in this cholangiocyte plasticity are also reviewed.
Cholangiocyte Biology
Structural Features
Biliary Tree Anatomy
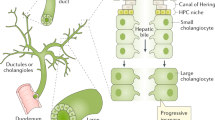
The biliary tree network consists of intrahepatic and extrahepatic ducts [8]. The intrahepatic ducts can be described from four perspectives according to luminal diameter, area, morphology, and physiology [8, 9]. The small ducts (<15 μm) originate from the Canals of Hering and, when combined, give rise to interlobular ducts (10–100 μm) [8]. The merging of two or more septal ducts (100–300 μm) results in the development of large ducts (300–400 μm) [8]. The large ducts combine to form segmental ducts (400–800 μm) and left and right hepatic ducts (>800 μm) from which the extrahepatic ducts emerge. The gallbladder connected to the extrahepatic portion of the biliary tree functions as a storage of bile [8] (Fig. 7.2).

Biliary tract anatomy. The biliary tree is depicted from the finest branches at the Canals of Hering to the small, interlobular, septal, and large ducts. Also shown, the right and left intrahepatic ducts that merge to form the extrahepatic ducts, from where the gallbladder emerges [8]
Cholangiocytes along the biliary tree are morphologically heterogeneous [8]. The small ducts are lined by 4–5 cholangiocytes, termed small cholangiocytes, which exhibit a cuboidal or flattened shape and possess a basement membrane on their basolateral domain. On their apical domain, microvilli and primary cilia face the bile duct lumen. Large bile ducts are lined by columnar-shaped cells known as large cholangiocytes that also express both microvilli and cilia on their apical domain [8]. When compared to small cholangiocytes, large cholangiocytes have a smaller nuclear to cytoplasmic ratio with a higher content of rough endoplasmic reticulum [1]. This feature implies that large cholangiocytes are more differentiated and have less plasticity relative to small cholangiocytes [8].
Cholangiocytes are connected to each other via tight junctions that maintain cholangiocyte polarity through cell-to-cell adhesion [8]. The apical plasma membrane domain faces the ductal lumen, which functions as the secretory pole for ductal bile formation; the basolateral plasma membrane domain faces the extracellular matrix and underlying connective tissue [10], Fig. 7.3.

Cholangiocyte ultrastructure. Transmission electron micrograph of a small mouse cholangiocyte, showing the apical plasma membrane (APM) that faces the ductal lumen. The nucleus, a tight junction (TJ) between two cholangiocytes, and the basolateral plasma membrane (BPM) are also shown
Cholangiocyte Cilia
Each small and large cholangiocyte possesses a primary cilium (~7 μm in length) extending from the apical cholangiocyte membrane into the ductal lumen. Primary cilia are nonmotile, microtubule-based organelles consisting of a membrane-bound axoneme composed of microtubules and a basal body (Fig. 7.4). The axoneme contains a 9 + 0 microtubule arrangement, i.e., nine peripheral microtubule doublets lacking a central pair of microtubules [11, 12]; in contrast, motile cilia have a similar structure but contain two central microtubules (i.e., 9 + 2 structure). The existence of primary cilia was originally reported in various mitotically quiescent mammalian cells by Sergei Sorokin in 1968 [13]. In 2006, primary cilia were described in mouse and rat small and large cholangiocytes [11]. However, their physiological importance was not appreciated until recently when it was demonstrated that primary cilia are involved in mechano-, chemo-, and osmo-sensation [14–18].

Cholangiocyte cilium. Transmission electron micrograph of a small mouse cholangiocyte showing a primary cilium facing the ductal lumen. The basal body of the primary cilium and the apical plasma membrane (APM) are also shown
Functional Features
The main function of intrahepatic cholangiocytes is to modify bile via a series of secretory/absorptive events. These are regulated by several gastrointestinal peptides/hormones including gastrin, endothelin-1, somatostatin, TGR-5, and secretin, which display inhibitory and stimulatory effects on water and bicarbonate (HCO3 −) secretion. These modifications ultimately influence the bile volume, content, tonicity, and alkalinity [8, 14]. While there is considerable species variation, intrahepatic cholangiocytes directly generate up to 40 % of daily bile secretion [2]. Secretory functions are performed mainly by large intrahepatic cholangiocytes via a mechanism dependent on cAMP activation. Large cholangiocytes abundantly express the appropriate ion transport systems and hormone receptors for these functions. For example, cholangiocytes in the large ducts are the major functional anatomic sites for expression of secretin and somatostatin receptors necessary for bile modification and secretion. In contrast, cholangiocytes lining small bile ducts, including the finest branches of the biliary system, do not express the secretin and somatostatin receptors exerting secretory activities independent of cAMP activation [19, 20]. For instance, during injury of large bile ducts, small cholangiocytes, which lack the cystic fibrosis transmembrane conductance regulator (CFTR), activate an alternative pathway for water and electrolyte secretion dependent of Ca+ signaling [21].
Small cholangiocytes, which are mitotically quiescent, proliferate via activation of Ca+2 signaling in response to liver injury and toxins [19, 20]. For instance, small cholangiocytes may replicate upon stimulation with histamine or secretin or injury by α-naphthylisothiocyanate or acute carbon tetrachloride. Le Sage et al. demonstrated that acute administration of carbon tetrachloride to rats induces apoptosis of large cholangiocytes and proliferation of small cholangiocytes. Furthermore, small cholangiocytes acquired de novo expression of secretin receptors. Stimulation of small cholangiocytes with secretin-induced activation of cAMP suggests that upon injury small cholangiocyte may acquire secretory features of large cholangiocytes. This suggested that small cholangiocytes compensated for the functions of the injured large cholangiocytes [22]. Also, after partial hepatectomy, rat small cholangiocytes function as a niche for hepatobiliary progenitor cells. Studies performed in human cholestatic livers and in human-regenerating liver after alcohol-induced injury suggest that human cholangiocytes behave in a similar manner [23]. Thiese et al. reported that acetaminophen-induced hepatic massive necrosis stimulates a niche of stem cells containing small cells positive for the cholangiocyte marker cytokeratin 19 (CK19) within the Canals of Hering [24]. Thus, the main biological properties of small cholangiocytes are their ability to proliferate, to acquire features of large cholangiocytes, to differentiate into hepatocytes, and to be a cell reservoir upon injury [1, 25].
Bile Formation
Ninety-five percent of bile is water with the remaining 5 % consisting of organic solutes such as bile salts, phospholipids, cholesterol, as well as inorganic salts such as Na+, K+, and HCO3 − [26, 27]. Bile is first formed (i.e., primary bile) by hepatocytes and then secreted into the canaliculi via osmotic-dependent excretion of organic solutes across the canalicular membrane drawing water via aquaporin water channels [28]. The principal driver of hepatocyte bile secretion is bile acids (i.e., bile acid-dependent bile flow) [29]. Bile is then modified via absorptive and secretory processes initially by large cholangiocytes via transport of chloride (Cl−), HCO3 −, bile acids (BAs), amino acids, and glucose to modify the water content and alkalinity of bile through a series of hormone-regulated, Ca2+ (calcium)- or cyclic adenosine 3′, 5′-monophosphate (cAMP)-dependent intracellular processes [8, 26].
Moreover, cAMP and/or Ca2+-sensitive basolateral potassium (K+) channels, expressed in cholangiocytes, mediate K+ release which leads to membrane hyperpolarization to maintain the electrical driving force for continued apical Cl− secretion [30]. Under basal conditions, the permeability of the apical membrane is low but can be increased several fold following cAMP stimulation [31, 32]. Furthermore, Cl− secretion and subsequent reuptake is required for HCO3 − secretion by the Cl−/HCO3 − anion exchanger 2 (AE2). Cl− uptake is mediated by the sodium (Na+)/K+/Cl− cotransporter NKCC1, which is localized in the basolateral membrane of rat cholangiocytes. In an electrically neutral manner with stoichiometry of 1Na+:1 K+:2Cl−, a gradient is established, which maintains a high concentration of intracellular Cl− [33]. This is important as HCO3 − is secreted in exchange for luminal Cl−. The movement of ions across the cholangiocyte apical and basolateral membranes promotes osmotic-driven bile secretion [34].
The absorption of ions, BAs, amino acids, and glucose are additional processes that contribute to ductal bile modification [8]. Glucose is removed from bile in a Na+-dependent manner by the Na+-glucose cotransporter, SGLT1, localized in the apical plasma membrane of the bile ducts. Conjugated BAs enter cholangiocytes through the apical Na+ -dependent bile salt uptake transporter (ASBT) [35]. This is a 48 kDa integral membrane protein, localized on the cholangiocyte apical membrane. A truncated form of this transporter (t-ASBT), responsible for the final reabsorption of bile salts from the bile into the blood, is found on the basolateral membrane [36, 37]. To prevent the cytotoxic effects of intracellular BA accumulation, basolateral extrusion of bile salts is mediated by MRP3, a member of the multidrug resistance protein (MRP) subfamily of transporters. MRP3 substrates include the organic anions estradiol-17-glucuronide, bilirubin glucuronide, monovalent bile salts taurocholate and glycocholate, as well as divalent sulfated bile salts [8, 38].
Hepatocytes secrete glutathione into the bile. After glutathione in bile is hydrolyzed, the amino acids, glutamate, cysteine, and glycine are produced and then absorbed by cholangiocytes for the resynthesis of glutathione, which mediates bile salt-independent secretion of canalicular bile. Additionally, taurine and glycine play a key role in the formation of conjugated BAs, preventing the reabsorption of the conjugated BAs as they traffick through the biliary tract [28].
Water Secretion
Water not only plays a major role as the main constituent in bile, but is also involved in the flow of bile and of cholangiocyte signaling pathways via ciliary transduction mechanisms [28]. Osmosis-dependent excretion of ions, organic solutes, and water into the canaliculi establishes osmotic gradients necessary to stimulate bile formation and secretion [8, 26]. Water transport, which is mediated by water channels known as aquaporins (AQPs), plays a key role in ductal bile formation [39]. AQPs are a family of ubiquitously expressed membrane proteins first discovered in the 1980s [35, 36] that form channels allowing the transport of water and small solutes such as glycerol to cross the plasma membrane. The permeability of water across the cell plasma membrane lipid bilayer is increased up to 50 times when AQPs are present relative to plasma membranes lacking AQPs [40]. At least 13 types of AQPs (AQP 0–12) have been described in mammalian cells and have been grouped into three categories according to their functions. Orthodox AQPs (i.e., AQPs 0,1,2,4, and 5) selectively mediate water flow through plasma membranes. Aquaglyceroporins (i.e., AQPs 3,7,9, and 10) allow the passage of water in addition to glycerol and urea. Unorthodox AQPs (i.e., AQPs 6,8,11, and 12) were only recently identified, and their functions remain uncertain [41–43]. AQPs 0, 1, 4, 5, 8, 9, and 11 are all expressed in cholangiocytes [44]. In cholangiocytes, water movement likely occurs principally via a shuttle mechanism involving AQP1, which is localized to both the apical and basolateral domains [39]. Secretin, a gastrointestinal hormone secreted by S cells of the duodenum [45], promotes the movement of intracellular vesicles containing AQP1 to the apical plasma membrane, enhancing osmotic water permeability, a process essential to ductal bile secretion [39]. Furthermore, when vesicles are isolated from the apical and basolateral membranes of bile duct-ligated (BDL) rats treated with secretin, the apical vesicles became enriched in AQP1, while the basolateral vesicles express stable levels of AQP4 [46]. Thus, these observations suggest that AQP1 is regulated and mediates apical water flow, whereas AQP4 is constitutively expressed and mediates the basolateral movement of water [46]. In cholangiocytes isolated from the PCK rat, an animal model of autosomal recessive polycystic kidney disease (ARPKD), AQP1 is overexpressed at the basolateral membrane and may contribute to the expansion of cysts via influx of fluid [47].
Bicarbonate Secretion
Another important function of cholangiocytes is biliary transport of HCO3 −, which maintains bile alkalinity, preventing protonation of bile salts that would otherwise induce bile duct injury. In human and rat cholangiocytes, HCO3 − secretion occurs mainly through the Na+-independent Cl−/HCO3 − exchanger, AE2, and related apical Cl− channels [48]. Biliary secretion of HCO3 − initially requires modulation of intracellular levels of HCO3 − in cholangiocytes. There are two mechanisms by which the intracellular level of HCO3 − is regulated: (i) via direct loading from the basolateral membrane mediated by the Na+/HCO3 − cotransporter or (ii) via carbonic anhydrase-mediated generation of HCO3 − and H+ from hydration of CO2 with water [34]. The basolateral influx of HCO3 − is mediated by the Na+/HCO3 − cotransporter in rats [1] and the Na+-dependent Cl−/HCO3 − anion exchanger in humans [49]. Biliary secretion of HCO3 − also involves the generation in the lumen of a negative potential, requiring activation of Cl− channels and subsequent release of Cl− ions [34]. It is well known that bile ducts express Ca2+-dependent Cl− channels [50]. HCO3 − biliary secretion is influenced by at least three hormones, namely, acetylcholine, somatostatin, and gastrin [51]. Acetylcholine and muscarinic M3 subtype receptor interaction induce an increase in intracellular Ca2+ and activation of the Cl−/HCO3 − ion exchanger AE2. Somatostatin inhibits secretin-stimulated intracellular cAMP synthesis through a somatostatin receptor interaction [52, 53]. In addition, gastrin synthesis, generated by gastric antral G cells, decreases secretin-stimulated cAMP levels through both the downregulation of cyclic adenylate cyclase and decreased expression of secretin receptors [54].
Intracellular Signaling
Cholangiocytes express a number of receptors through which autocrine and paracrine signaling pathways are modulated. Secretin receptors (SR)s are typical G protein-coupled receptors expressed on the basolateral domain of intrahepatic rodent and human large cholangiocytes [55]. In large intrahepatic cholangiocytes, cAMP levels increase upon secretin stimulation [56]. This activation induces phosphorylation of protein kinase A (PKA), which in turn promotes the opening of the apically located Cl− channel (CFTR), resulting in Cl− secretion into bile. This process further activates Cl−/HCO3 − exchange via AE2 resulting in HCO3 − secretion into bile [21, 57]. BDL of rats induces hypercholeresis via secretin-mediated activation of SRs [58] in a mechanism that involves an increased number of secretin receptors per cell [59]. Importantly, studies by Glaser et al. [56] demonstrated that in SR knockout mice, the proliferation of large cholangiocytes is reduced during BDL compared to wild-type BDL mice. In addition, decreased levels of both basal- and secretin-stimulated cAMP as well as reduced phosphorylation of the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) were observed in large cholangiocytes from SR knockout BDL mice compared to large cholangiocytes from wild-type BDL mice. In vitro experiments showed that secretin increased the proliferation of cholangiocytes via cAMP/PKA/ERK1/2 signaling [56].
Cholangiocytes also express the G protein-coupled bile acid receptor, TGR5 (GPBAR-1, M-Bar, or GPR131). TGR5 is a transmembrane receptor linked to cAMP signaling expressed in a variety of human and rodent tissues that is encoded by a gene located on chromosome 1C3 in mouse and 2q35 in humans [60]. In cholangiocytes, TGR5 is found in multiple intracellular locations, including primary cilia on the apical domain, on the non-ciliary portion of the apical membrane, and on the inner and outer membrane of the cholangiocyte nucleus [61]. TGR5 is a major receptor for bile acid signaling in cholangiocytes, and its activation affects intracellular cAMP via coupling to Gαs or Gαi proteins subsequently triggering downstream signaling events [62]. A role for TGR5 in the development of gallstones was proposed by Keitel et al. In vitro experiments from the same study also showed that TGR5 stimulates the CFTR-dependent release of biliary Cl− [63]. As mentioned above, primary cilia are key organelles involved in intracellular signaling and, as such, influence the response to TGR5 cholangiocyte activation. For example, in cholangiocytes, experimentally devoid of primary cilia, stimulation by TGR5 agonists enhanced cAMP activation via Gαi, partially inhibiting ERK signaling, which results in reduced cholangiocyte proliferation [61]. Interestingly, the reverse outcomes were noted when ciliated cholangiocytes were challenged with the same TGR5 agonists [61]. Masyuk et al. demonstrated that primary cilia act as mechanosensors, responding to luminal fluid flow by alterations in intracellular Ca2+ and cAMP. The ciliary proteins involved in this transduction of mechanical stimuli include polycystin-1, a cell surface receptor, and polycystin-2, a Ca2+ channel [14]. Primary cilia also express the transient receptor potential 4 (TRPV4) protein, a Ca2+ permeable, nonselective cation channel, through which they can detect changes in osmolarity [15]. Gradilone et al. demonstrated that hypotonicity induces a rise in intracellular Ca2+ via TRPV4 activation in rat cholangiocytes. Furthermore, in vivo stimulation of cholangiocyte TRPV4 by intrabiliary saline increased adenosine triphosphate (ATP) production, HCO3 − release, and thereby bile movement [15]. The role of cholangiocyte primary cilia as chemosensors has also been demonstrated. Primary cilia on rat cholangiocytes express the purinergic receptors, P2Y12 and P2Y13, that respond to changes in cAPM induced by adenosine diphosphate (ADP) and ATP-γS (nonhydrolyzed analog of ATP), two known agonists of P2Y12 receptors. Moreover, suramin, an inhibitor of P2Y receptors, can prevent the ADP-dependent decrease of cAMP [16].
Cholangiocyte signaling also can occur in response to receptor-mediated recognition of microbial-derived molecules. Receptors involved include Toll-like receptors (TLRs), nucleotide oligomerization domain proteins [3], and purinoceptors [64]. TLRs, a family of conserved receptor proteins critical for pathogen recognition, are present on the apical membranes of cholangiocytes where they are well positioned to detect pathogenic molecules in bile notably pathogen-associated molecular patterns (PAMPs), as well as adaptor proteins such as myeloid differentiation protein 88 (MyD88) and intracellular kinases [65]. Human cholangiocytes express TLRs 1–10, MD-2, MyD88, and downstream effectors of the TLR pathway [66]. The responses triggered by activation of TLRs in cholangiocytes are described later in this chapter.
Communication Between Cells
Cholangiocytes can communicate with each other and with other cells via the release of soluble molecules as well as by secreted extracellular vesicles (ECVs). ECVs are nano-vesicles secreted by various types of benign and malignant cells. Exosomes, a subset of ECVs 30–150 nm in diameter, are membrane-enclosed vesicles present in biological fluids in vivo that shuttle molecules from donor cells to proximal or distant target cells [18]. Exosomes are generated through the invagination of early endosomes, subsequently producing intraluminal vesicles (ILVs) that contain multiple molecules (e.g., proteins, RNA, etc.) that typically result in the formation of multivesicular bodies (MVBs). The dynamic MVB pathways can lead to either: (i) plasma membrane fusion followed by release of ILVs into the extracellular milieu (now termed exosomes) or (ii) lysosomal fusion resulting in degradation [18]. Liver epithelia, including hepatocytes and cholangiocytes, can release exosomes in culture and in vivo, consistent with an important role for exosomes in intercellular communication and signaling. The mechanisms by which exosomes may initiate signaling pathways in target cells remain poorly defined but include: (a) binding to specific membrane receptors to induce intracellular signaling processes; (b) fusion of the exosome with the target cell membrane followed by release of its encapsulated content; and (c) endocytosis of the entire exosome following convex-like membrane bending of the target cell plasma membrane [67].
Cholangiocytes secrete exosomes into the bile duct lumen [18]. Studies using cultured cholangiocytes show that exosomes isolated from bile induce ERK1/2 activation that is dependent on the presence of primary cilia and that can influence ERK1/2-mediated cholangiocyte proliferation and miRNA expression [18]. In addition, it has also been reported that cholangiocytes infected by the protozoan parasite, Cryptosporidium parvum (C. parvum), secrete increased numbers of apically derived exosomes that contain antimicrobial peptides, suggesting a role for cholangiocyte-derived exosomes in response to biliary infection [68].
Basolaterally released exosomes derived from intestinal epithelia as well as from cholangiocytes have also been described; however, their physiological relevance remains unclear [18, 69].
Cholangiocyte Plasticity
Cholangiocytes have the ability to adapt and respond to changes in their microenvironment. For instance, upon injury cholangiocytes become reactive, actively producing and secreting molecules that stimulate immune and wound-healing responses. Furthermore, as a mechanism to prevent malignancy, cholangiocytes undergo a state of senescence in which their proliferative capacity is shut down. Under certain circumstances, however, this mechanism can be bypassed, and cholangiocytes adopt a malignant phenotype characterized by hyperproliferation with a marked production of pro-inflammatory cytokines.
Cholangiocyte Reactivity
Exposure of cholangiocytes to chemicals, microbes, and microbial products can induce cholangiocyte activation [3]. Activated cholangiocytes are characterized by: (i) increased resistance to apoptosis, allowing benign proliferative expansion of cholangiocytes; (ii) increased production and release of cytokines and chemokines that attract immune cells, amplifying the pro-inflammatory response already initiated; (iii) decreased expression of epithelial markers and acquisition of mesenchymal features; and (iv) overproduction and secretion of profibrotic molecules (Table 7.1) (Fig. 7.1) [5]. Reactive cholangiocytes, for example, secrete PDFG-BB, which induces the production of hedgehog (Hh) ligands by myofibroblasts and cholangiocytes. Importantly, activation of the Hh pathway appears necessary for activated cholangiocytes to maintain the reactive phenotype [5].
In an in vitro model of C. parvum infection of cholangiocytes, Chen et al. demonstrated that cholangiocytes respond and defend against this parasite by inducing activation of the nuclear factor kappa B (NF-kB) pathway via TLR-2, TLR-4, and subsequent production of IL-8 and human beta defensin 2 (HBD-2). Both TLRs and HBD-2 are key players of the innate immune system against pathogens [66]. Cholangiocytes can also be exposed to enteric bacterial-derived products via the enterohepatic circulation [70]. For instance, cholangiocytes in patients with primary sclerosing cholangitis (PSC) may be exposed to lipopolysaccharide (LPS), the bioactive part of gram-negative bacteria [71]. Recognition of LPS by cholangiocytes via TLR-4/MyD88 stimulates the NF-kB and N-Ras/ERK pathways. TLR-induced N-Ras activation requires transactivation of the epidermal growth factor receptor (EGFR) and the ADAM metallopeptidase domain 17 (TACE). Stimulation of the N-Ras/ERK pathway and NF-kB activation promote IL-6 expression, a known pro-inflammatory cytokine and mitogen, activating cholangiocyte proliferation [70, 72]. Several other molecules such as hormones, BAs, neuropeptides, and an increase in bile duct pressure are known to induce cholangiocyte proliferation [73].
Bile duct proliferation is another mechanism of response/defense of cholangiocytes upon injury. Acute injury of the biliary tree induces proliferation of large cholangiocytes to maintain internal stability within the bile ducts [3], whereas chronic injury triggers replication of both small and large cholangiocytes [3]. Acute and chronic biliary damage promotes regeneration and repair which modulates bile duct morphogenesis. Fabris et al. demonstrated that in human livers with bile duct injury, reactive bile ducts display features similar to what occurs during the early phase of bile duct morphogenesis [74].
Senescent Cholangiocytes
Cellular senescence is an irreversible state in which cells, arrested in the G1 phase of the cell cycle, can no longer replicate. Cellular senescence is a characteristic of aging and is present in a variety of disorders, e.g., atherosclerosis, osteoarthritis, and chronic obstructive pulmonary disease [75]. It occurs as a result of genotoxic stimulation and constitutes a mechanism to prevent cancer growth as it halts proliferation of injured cells [6]. There are two major, but not mutually exclusive, tumor suppressor pathways that tightly control cellular senescence: the p53 and the p16INK4a/pRB pathways. As mentioned earlier in this chapter, senescent cells may transition to a highly pro-inflammatory phenotype known as the senescence-associated secretory phenotype (SASP) [6]. This term was first proposed by Coppé et al. while studying an array of factors that human pre-senescent and senescent fibroblasts secrete [6]. Fibroblasts undergoing SASP secrete abundant levels of pro-inflammatory cytokines and immuno-attractant chemokines (IL-6, IL-7, IL-8, monocyte chemoattractant protein-2 [MCP-2], and macrophage inflammatory protein-3 alpha), growth regulatory molecules (growth-regulated oncogene, hepatocyte growth factor, and insulin-like growth factor-binding proteins [IGFBPs]), membrane/transmembrane receptors (intracellular adhesion molecules, urokinase receptor, and tumor necrosis factor [TNF] receptors), and survival mediators (osteoprotegerin and fibroblast growth factor) compared to pre-senescent fibroblasts [6].
Senescent cholangiocytes have been reported in different types of liver injury and have been implicated in the pathogenesis of various diseases. Indeed, a positive correlation between cholangiocyte senescence and the degree of rejection in acute liver allograft rejection has been reported [76]. Cholangiocyte senescence has been also associated with the progression of chronic liver diseases such as primary biliary cirrhosis, chronic viral hepatitis, and nonalcoholic steatohepatitis [77]. Further studies from the same group showed an association between fibrosis and inflammation in nonalcoholic fatty liver disease. Furthermore, the number of senescent cholangiocytes increased as the fibrosis progressed. Interestingly, the expression of MCP-1, a SASP secretory factor and chemoattractant of hepatic stellate cells (HSCs) and inflammatory cells, was also upregulated in bile ducts in the late stages of the disease. Coculture experiments also showed increase migration of HSCs toward senescent cholangiocytes. The authors concluded that senescent cholangiocytes most likely produce MCP-1 for the recruitment of HSCs to the sites of injury [78].
Recent studies have demonstrated that cellular senescence may play a key role in the pathogenesis of PSC [75]. Immunofluorescence of human PSC liver sections showed that cholangiocytes are not proliferative and express the senescent markers p16INK4A and γH2A.x, suggesting that cholangiocytes in PSC exhibit increased senescence [75]. Moreover, PSC cholangiocytes produce abundant levels of SASP factors, particularly IL-6, IL-8, plasminogen activator inhibitor-1, and MCP-1 compared to normal and disease control cholangiocytes (Table 7.1) (Fig. 7.1) [75]. As mentioned, SASP components engage in intercellular communication to induce pro-inflammatory and senescent phenotypes in target cells. Importantly, we recapitulated these findings in an in vitro model of stress-induced cholangiocyte senescence. In this same model, we found that cholangiocyte senescence is driven by the N-Ras pathway. Moreover, cholangiocytes isolated from livers of patients with PSC cholangiocytes secrete 23 and 46 times more IL-6 and IL-8, respectively, compared to normal cholangiocytes [79]. At the morphological level, PSC cholangiocytes display an enlarged shape with marked cytoskeletal filamentous proteins [79]. These cells also showed decreased tight junction integrity, evaluated by the low expression level of the tight junction marker ZO-1 [79].
Transformed Cholangiocytes
Neoplastic transformation of cholangiocytes results in the development of cholangiocarcinoma (CCA) [80]. While the molecular mechanisms responsible for the malignant transformation of cholangiocytes and its progression to CCA are still unclear, CCA frequently occurs within bile ducts plagued by chronic inflammation [7].
Aberrant expression of the tyrosine kinase receptor ErbB-2/Neu and prostaglandin endoperoxide synthase cyclooxygenase-2 (COX-2) in biliary epithelia has been implicated in the development and progression of CCA (Table 7.1) (Fig. 7.1). Immunohistochemistry of human bile ducts has demonstrated that both COX-2 and ErbB-2 are several times fold upregulated in PSC and CCA patients compared to normal subjects. There was also a positive correlation between tumor differentiation and the overexpression of COX-2 and ErbB-2 with peak expression of both proteins observed in well-differentiated tumors [81].
In pathological conditions, chronic inflammation promotes oxidative stress via production of abnormal levels of reactive oxygen and nitrogen species. Reactive nitrogen species are generated from nitric oxide (NO). NO is a signaling molecule that at physiological concentrations inhibits inflammation and prevents platelet aggregation and integrin-dependent adhesion. NO is overproduced in a variety of pathological conditions and at high levels can promote carcinogenesis via inhibition of apoptosis, induction of DNA damage, and angiogenesis [82]. Jaiswal et al. demonstrated via immunohistochemistry cholangiocyte DNA damage and de novo production of the inducible nitric oxide synthase (iNOs) in cholangiocytes of patients with PSC [83]. Further studies revealed that iNOs induces malignant transformation of cholangiocytes and CCA progression [84]. The mechanism involves iNOs-dependent production of NO by PSC cholangiocytes. [84]. NO activates the Notch-1 signaling pathway leading to resistance of TRAIL-mediated apoptosis [84].
IL-6 is one of the pro-inflammatory cytokines that is abundantly expressed during chronic bile duct inflammation [7]. It is normally produced by various liver cell types and is particularly secreted at high levels by senescent cholangiocytes in patients with PSC [7, 79]. Several lines of evidence have shown that IL-6 potently stimulates normal cholangiocyte proliferation via autocrine and paracrine mechanisms [72, 73, 85, 86]. The role of IL-6 signaling in liver tumorigenesis and liver cancer progression has also been documented both in vivo and in vitro [87, 88]. Abrogation of the IL-6 signaling pathway by an antihuman IL-6 neutralizing antibody inhibits proliferation of the CCA line KMCH-1 [85]. Furthermore, stimulation of KMCH-1 cells with the pro-inflammatory cytokines IL1-β and TNF-α leads to an upregulation in IL-6 secretion [85]. In vitro and in vivo evidence suggests that IL-6 dysregulation is implicated in cholangiocyte malignant transformation and aggravation of CCA. Meng et al. showed that IL-6 promotes survival of human cell lines from intrahepatic, extrahepatic, and gallbladder tumors via increased expression of the myeloid cell leukemia protein-1, which in turn inhibited apoptosis and decreased sensitivity to chemotherapy [7].
Summary
In this chapter we have selectively summarized the latest literature regarding the biology of normal cholangiocytes. In addition, we present a model of cholangiocyte plasticity that includes the normal functions of cholangiocytes as well as their responses upon injury, focusing on the induction of senescence, the subsequent development of SASP, and ultimately cholangiocyte malignant transformation. The signaling pathways in which injured cholangiocytes communicate are also reviewed. The mechanisms that regulate the responses of cholangiocytes in each stage are not fully understood, and whether cholangiocytes can revert from one stage to another still remains to be elucidated. Understanding what regulates the plasticity of cholangiocytes during disease may lead to finding novel therapeutic targets that could trigger the resolution of the activated, senescent, and SASP phases.
Abbreviations
- ADP:
-
Adenosine diphosphate
- AE2:
-
Anion exchanger 2
- APM:
-
Apical plasma membrane
- AQP:
-
Aquaporin
- ARPKD:
-
Autosomal recessive polycystic kidney disease
- ASBT:
-
Apical Na+-dependent bile salt uptake transporter
- ATP:
-
Adenosine triphosphate
- BA:
-
Bile acids
- BDL:
-
Bile duct ligation
- BPM:
-
Basolateral plasma membrane
- C. parvum :
-
Cryptosporidium parvum
- Ca2+ :
-
Calcium
- cAMP:
-
Cyclic adenosine 3′, 5′- monophosphate
- CCA:
-
Cholangiocarcinoma
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- CK19:
-
Cytokeratin 19
- Cl− :
-
Chloride
- COX-2:
-
Cyclooxygenase-2
- ECVs:
-
Extracellular vesicles
- EGFR:
-
Epidermal growth factor receptor
- ERK1/2:
-
Extracellular signal-regulated protein kinases 1 and 2
- HBD2:
-
Human β-defensin 2
- HCO3 − :
-
Bicarbonate
- Hh:
-
Hedgehog
- HSC:
-
Hepatic stellate cell
- IGFBP:
-
Insulin-like growth factor-binding protein
- IL-6:
-
Interleukin 6
- IL-8:
-
Interleukin 8
- ILV:
-
Intraluminal vesicles
- iNOs:
-
Inducible nitric oxide synthase
- K+ :
-
Potassium
- LPS:
-
Lipopolysaccharide
- MCP:
-
Monocyte chemoattractant protein
- MRP:
-
Multidrug resistance protein
- MVB:
-
Multivesicular bodies
- MyD88:
-
Myeloid differentiation protein 88
- Na+ :
-
Sodium
- SGLT1:
-
Na+-glucose cotransporter
- NF-kB:
-
Nuclear factor kappa B
- NKCC1:
-
Na+/K+/Cl− cotransporter 1
- NO:
-
Nitric oxide
- PDFG-BB:
-
Platelet-derived growth factor-BB
- PKA:
-
Protein kinase A
- PSC:
-
Primary sclerosing cholangitis
- SASP:
-
Senescence-associated secretory phenotype
- SR:
-
Secretin receptor
- t-ASBT:
-
Truncated ASBT
- TGF-β:
-
Transforming growth factor beta
- TJ:
-
Tight junction
- TLR:
-
Toll-like receptors
- TRPV4:
-
Transient receptor potential 4
References
Strazzabosco M, Fabris L. Functional anatomy of normal bile ducts. Anat Rec (Hoboken). 2008;291:653–60.
Syal G, Fausther M, Dranoff JA. Advances in cholangiocyte immunobiology. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1077–86.
O’Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58:1–16.
Milani S, Hermann H, Schuppan D, Stein H, Surrenti C. Transforming growth factors 1 and Are differentially expressed in fibrotic liver disease. Am J Pathol. 1991;139:1221–9.
Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol. 2011;27:268–75.
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of Interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–65.
Tabibian JH, Masyuk AL, Masyuk TV, O’Hara SP, LaRusso NF. Physiology of cholangiocytes. Compr Physiol. 2013;3:1–49.
Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, et al. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal Rat liver. Gastroenterology. 1996;110:1636–43.
Tietz P, Levine S, Holman R, Fretham C, LaRusso NF. Characterization of apical and basolateral plasma membrane domains derived from cultured Rat cholangiocytes. Anal Biochem. 1997;254:192–9.
Huang BQ, Masyuk TV, Muff MA, Tietz PS, Masyuk AI, Larusso NF. Isolation and characterization of cholangiocyte primary cilia. Am J Physiol Gastrointest Liver Physiol. 2006;291:G500–9.
Gradilone SA, Lorenzo Pisarellob MJ, LaRussob NF. Primary Cilia in Tumor Biology: The primary cilium as a therapeutic target in cholangiocarcinoma. Curr Drug Targets. 2015. [Epub ahead of print.]
Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123:499–503.
Masyuk AI, Masyuk TV, LaRusso NF. Physiology of cholangiocytes. In: Johnson LR, editor. Physiology of the gastrointestinal tract. 4th ed. Academic Press; Canada, 2006. p. 1505–33.
Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci U S A. 2007;104:19138–43.
Masyuk AI, Gradilone SA, Banales JM, Huang BQ, Masyuk TV, Lee SO, et al. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol Gastrointest Liver Physiol. 2008;295:G725–34.
Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237:2007–12.
Masyuk AI, Huang BQ, Ward CJ, Gradilone SA, Banales JM, Masyuk TV, et al. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am J Physiol Gastrointest Liver Physiol. 2010;299:G990–9.
Marzioni M, Glaser SS, Francis H, Phinizy JL, LeSage G, Alpini G. Functional heterogeneity of cholangiocytes. Semin Liver Dis. 2002;22:227–40.
LeSage GD, Glaser SS, Marucci L, Benedetti A, Phinizy JL, Rodgers R, et al. Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from BDL rat liver. Am J Physiol. 1999;276:G1289–301.
Afroze S, Meng F, Jensen K, McDaniel K, Rahal K, Onori P, et al. The physiological roles of secretin and its receptor. Ann Transl Med. 2013;1(3):1–14.
LeSage GD, Benedetti A, Glaser S, Marucci L, Tretjak Z, Caligiuri A, et al. Acute carbon tetrachloride feeding selectively damages large, but not small, cholangiocytes from normal rat liver. Hepatology [Research Support, Non-US Gov’t Research Support, US Gov’t, Non-PHS]. 1999;29(2):307–19.
Sell S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology. 1998;27:317–31.
Thiese ND, Saxena R, Portmann BC, Thung SN, Yee H, Chiriboga L, et al. The canals of hering and hepatic stem cells in humans. Hepatology. 1999;30:1425–33.
Sell S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology. 2001;33:738–50.
Boyer JL. Bile formation and secretion. Compr Physiol. 2013;3:1035–78.
Farina A, Dumonceau JM, Lescuyer P. Proteomic analysis of human bile and potential applications for cancer diagnosis. Expert Rev Proteomics. 2009;6:285–301.
Masyuk AI, Masyuk TV, LaRusso NF. Physiology of cholangiocytes. In: Johnson LR, editor. Physiology of the gastrointestinal tract. 5th ed. Elsevier; 2012. p. 1531–57.
Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. Bethesda, MD, 2003;83:633–71.
Feranchak AP, Doctor RB, Troetsch M, Brookman K, Johnson SM, Fitz JG. Calcium-dependent regulation of secretion in biliary epithelial cells: the role of apamin-sensitive SK channels. Gastroenterology. 2004;127:903–13.
Fitz JG, Basavappa S, McGill J, Melhus O, Cohn JA. Regulation of membrane chloride currents in rat bile duct epithelial cells. J Clin Invest. 1993;91:319–28.
McGill JM, Basavappa S, Gettys TW, Fitz JG. Secretin activates Cl- channels in bile duct epithelial cells through a cAMP-dependent mechanism. Am J Physiol. 1994;266:G731–6.
Singh SK, Mennone A, Gigliozzi A, Fraioli F, Boyer JL. Cl(−)-dependent secretory mechanisms in isolated rat bile duct epithelial units. Am J Physiol Gastrointest Liver Physiol. 2001;281:G438–46.
Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–511.
Alpini G, Glaser SS, Rodgers R, Phinizy JL, Robertson WE, Lasater J, et al. Functional expression of the apical Na + −dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology. 1997;113(5):1734–40.
Lazaridis KN, Tietz P, Wu T, Kip S, Dawson PA, LaRusso NF. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc Natl Acad Sci U S A. 2000;97:11092–7.
Alpini G, Glaser S, Alvaro D, Ueno Y, Marzioni M, Francis H, et al. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226–37.
Ballatori N, Truong AT. Glutathione as a primary osmotic driving force in hepatic bile formation. Am J Physiol. 1992;263:G617–24.
Cova E, Gong A, Marinelli RA, LaRusso NF. Water movement across rat bile duct units is transcellular and channel-mediated. Hepatology. 2001;34:456–63.
Day R, Kitchen P, Owen DS, Bland C, Marshall L, Conner AC. Human aquaporins: regulators of transcellular water flow. Biochim Biophys Acta. 1840;2014:1492–506.
Ishibashi K. Aquaporin superfamily with unusual npa boxes: S-aquaporins (superfamily, sip-like and subcellular-aquaporins). Cell Mol Biol (Noisy-le-Grand). 2006;52(7):20–7.
Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y. Aquaporin water channels--from atomic structure to clinical medicine. J Physiol. 2002;542:3–16.
Yeung CH, Callies C, Rojek A, Nielsen S, Cooper TG. Aquaporin isoforms involved in physiological volume regulation of murine spermatozoa. Biol Reprod. 2009;80:350–7.
Masyuk AI, LaRusso NF. Aquaporins in the hepatobiliary system. Hepatology. 2006;43:S75–81.
Gossen D, Poloczek P, Svoboda M, Christophe J. Molecular architecture of secretin receptors: the specific covalent labelling of a 51 kDa peptide after cross-linking of [125I]iodosecretin to intact rat pancreatic acini. FEBS Lett. 1989;243:205–8.
Marinelli RA, Pham LD, Tietz PS, LaRusso NF. Expression of aquaporin-4 water channels in rat cholangiocytes. Hepatology. 2000;31:1313–7.
Banales JM, Masyuk TV, Bogert PS, Huang BQ, Gradilone SA, Lee SO, et al. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173:1637–46.
Beuers U, Maroni L, Elferink RO. The biliary HCO(3)(−) umbrella: experimental evidence revisited. Curr Opin Gastroenterol. 2012;28:253–7.
Strazzabosco M, Joplin R, Zsembery A, Wallace L, Spirli C, Fabris L. Na(+)-dependent and -independent Cl-/HCO-3 exchange mediate cellular HCO3- transport in cultured human intrahepatic bile duct cells. Hepatology. 1997;25:976–85.
Jung J, Lee MG. Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium. 2014;55:376–84.
Hirata K, Nathanson MH. Bile duct epithelia regulate biliary bicarbonate excretion in normal rat liver. Gastroenterology. 2001;121:396–406.
Tietz PS, Alpini G, Pham LD, Larusso NF. Somatostatin inhibits secretin-induced ductal hypercholeresis and exocytosis by cholangiocytes. Am J Physiol. 1995;269:G110–8.
Hogan MC, Masyuk TV, Page L, Holmes 3rd DR, Li X, Bergstralh EJ, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27:3532–9.
Glaser SS, Rodgers RE, Phinizy JL, Robertson WE, Lasater J, Caligiuri A, et al. Gastrin inhibits secretin-induced ductal secretion by interaction with specific receptors on rat cholangiocytes. Am J Physiol. 1997;273:G1061–70.
Glaser SS, Gaudio E, Rao A, Pierce LM, Onori P, Franchitto A, et al. Morphological and functional heterogeneity of the mouse intrahepatic biliary epithelium. Lab Invest. 2009;89:456–69.
Glaser S, Lam IP, Franchitto A, Gaudio E, Onori P, Chow BK, et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology. 2010;52:204–14.
Feranchak AP, Sokol RJ. Cholangiocyte biology and cystic fibrosis liver disease. Semin Liver Dis. 2001;21:471–88.
Alpini G, Ulrich CD, Phillips JO, Pham LD, Miller LJ, LaRusso NF. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am J Physiol. 1994;266:G922–8.
Tietz PS, Hadac EM, Miller LJ, LaRusso NF. Upregulation of secretin receptors on cholangiocytes after bile duct ligation. Regul Pept. 2001;97:1–6.
Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–93.
Masyuk AI, Huang BQ, Radtke BN, Gajdos GB, Splinter PL, Masyuk TV, et al. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1013–24.
Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2015.
Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Häussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50:861–70.
Burnstock G, Vaughn B, Robson SC. Purinergic signalling in the liver in health and disease. Purinergic Signal. 2014;10:51–70.
Rachmilewitz D, Katakura K, Karmeli F, Hayashi T, Reinus C, Rudensky B, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–8.
Chen XM, O’Hara SP, Nelson JB, Splinter PL, Small AJ, Tietz PS. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J Immunol. 2005;175:7447–56.
Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–20.
Hu G, Gong AY, Roth AL, Huang BQ, Ward HD, Zhu G, et al. Release of luminal exosomes contributes to TLR4-mediated epithelial antimicrobial defense. PLoS Pathog. 2013;9(4), e1003261.
Bu HF, Wang X, Tang Y, Koti V, Tan XD. Toll-like receptor 2-mediated peptidoglycan uptake by immature intestinal epithelial cells from apical side and exosome-associated transcellular transcytosis. J Cell Physiol. 2010;222:658–68.
Trussoni CE, Tabibian JH, Splinter PL, O’Hara SP. Lipopolysaccharide (LPS)-induced biliary epithelial cell NRas activation requires Epidermal Growth Factor Receptor (EGFR). PLoS One. 2015;10:1–15.
Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol. 1998;29:409–16.
O’Hara SP, Splinter PL, Trussoni CE, Gajdos GB, Lineswala PN, LaRusso NF. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J Biol Chem. 2011;286:30352–60.
Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–43.
Fabris L, Strazzabosco M, Crosby HA, Ballardini G, Hubscher SG, Kelly DA, et al. Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations. Am J Pathol. 2000;156:1599–611.
Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by Way of N-Ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;6:2263–75.
Brain JG, Robertson H, Thompson E, Humphreys EH, Gardner A, Booth TA, et al. Biliary epithelial senescence and plasticity in acute cellular rejection. Am J Transplant. 2013;13:1688–702.
Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakamuna Y. Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol. 2010;133:212–23.
Chiba M, Sasaki M, Kitamura S, Ikeda H, Sato Y, Nakanuma Y. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J Clin Pathol. 2011;64:564–70.
Tabibian JH, Trussoni CE, O’Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest. 2014;94:1126–33.
Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc. 2015;90:791–800.
Endo K, Yoon BI, Pairojkul C, Demetris AJ, Sirica AE. ERBB-2 overexpression and cyclooxygenase-2 Up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36:439–50.
Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–53.
Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, Gores GJ. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology. 2001;120:190–9.
Ishimura N, Bronk SF, Gores GJ. Inducible nitric oxide synthase Up-regulates notch-1 in mouse cholangiocytes: implications for carcinogenesis. Gastroenterology. 2005;128:1354–68.
Park J, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30:1128–33.
Xiao Y, Wang J, Yan W, Zhou Y, Chen Y, Zhou K, et al. Dysregulated miR-124 and miR-200 expression contribute to cholangiocyte proliferation in the cholestatic liver by targeting IL- 6/STAT3 signalling. J Hepatol. 2015;62:889–96.
Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M, et al. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. EMBO J. 1998;5588–97.
Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;6:1393–404.
Fabris L, Strazzabosco M. Epithelial–mesenchymal interactions in biliary diseases. Semin Liver Dis. 2011;31:11–32.
Li ZR, Wei JL, Li ZZ. Mucins 1-shRNA inhibit the proliferation and HIF-1alpha protein expression on human cholangiocarcinoma cells. Cell Biol Int. 2013;37:121–5.
Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51:146–53.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Loarca, L. et al. (2017). Cholangiocyte Biology. In: Forman, L. (eds) Primary Sclerosing Cholangitis. Springer, Cham. https://doi.org/10.1007/978-3-319-40908-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-40908-5_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40906-1
Online ISBN: 978-3-319-40908-5
eBook Packages: MedicineMedicine (R0)