
Abstract
Primary sclerosing cholangitis (PSC) is a chronic, idiopathic liver disease which, despite extensive laboratory-based investigation, translational studies, and clinical trials, lacks established medical treatment. In this chapter, we provide a succinct, comprehensive review of research regarding the potential role for ursodeoxycholic acid as a pharmacotherapy for PSC.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Cholestatic liver disease
- Biliary tract diseases
- Cholestasis
- Pharmacotherapy
- Bile acids
- Medical management
- Algorithm
- Pruritus
- Quality of life
- Outcomes
Overview and Clinical Epidemiology
Primary sclerosing cholangitis (PSC) is a chronic, cholestatic disorder of the liver characterized by three major features: biliary inflammation and periductal fibrosis on liver histology, multifocal biliary strictures alternating with segmental ductal dilatation on cholangiography, and a cholestatic serum biochemical profile [1, 2]. Unlike most other cholangiopathies, i.e., disorders primarily of or affecting the biliary tract [3, 4], PSC can affect individuals of essentially all ages and racial backgrounds, remains etiopathogenically perplexing, and lacks established medical therapy despite decades of laboratory-based investigation, translational studies, and clinical trials [1, 5]. It is because of these factors that PSC has, unfortunately, been regarded as the “black box” of liver disease [6].
Although the fundamental underpinnings and optimal management approaches for PSC remain uncertain, it is clear that, as a result of these uncertainties and the generally progressive nature of PSC, there is substantial public health and patient-level burden due to this disorder. Indeed, PSC represents a major risk factor for cholangiocarcinoma (CCA) [7], carries a median liver transplantation (LT)-free survival of 15 years [8], and (despite its rarity) is a leading indication for LT in countries around the world [9]. Although LT can be curative for PSC and PSC-associated CCA, it is only performed in highly selected patients and centers, and even suitable candidates may experience recurrent disease (≈3–4 % per year) [10]. Lastly, quality of life (QOL) is also significantly impaired in patients with PSC, both pre- and post-LT, and is related to debilitating symptoms such as pruritus and fatigue as well as the unpredictable disease course and complications related to coexisting inflammatory bowel disease (IBD) [11–13].
Proposed Etiopathogenesis of and the Basis of Bile Acid Therapy in PSC
Although PSC remains an idiopathic disorder, prevailing hypotheses regarding its etiopathogenesis suggest that a disruption of gut-liver axis signaling at various levels may play a fundamental role [6]. These hypotheses are largely based on the premise that enterohepatic generation and/or circulation of microbial metabolites, derivatives, or other molecules can initiate and perpetuate aberrant or exaggerated cellular responses and subsequent biliary injury. This has been the subject of ongoing investigation over the last several decades, with the goal being to identify potentially causal molecules and pathways and develop targeted therapies accordingly.
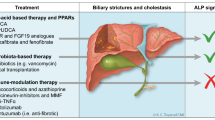
Representing perhaps the most widely investigated molecule and certainly the most extensively studied pharmacotherapy in PSC is ursodeoxycholic acid (UDCA) [14]. First isolated over a century ago from Thalarctos maritimus (now known as Ursus maritimus), i.e., the polar bear, UDCA is a hydrophilic, 3,7-dihydroxy bile acid (BA). In most vertebrates, including Homo sapiens, UDCA is a secondary BA and only a minor component (<5 %) of the BA pool; the major known exception among vertebrates is the Ursidae family, particularly Ursus americanus (the American black bear), wherein UDCA is typically a relatively major component (>5–30 %) of the BA pool [6, 15]. BA physiology and the potential therapeutic applications of BA therapies are shown in Fig. 11.1 and discussed in greater detail in recent review articles [16, 17].

Bile acid physiology and circulation: an avenue for therapeutic applications. Bile acids (BAs) are synthesized by hepatocytes and subsequently secreted into canalicular bile by means of specialized hepatocyte canalicular membrane transporters. Canalicular bile drains into the biliary tree and is modified by the epithelial cells lining it, that is, cholangiocytes. Bile then drains into the proximal small bowel, that is, duodenum, and is metabolized by enteric bacteria. Approximately 95 % of BAs are reabsorbed in the terminal ileum and enter the portal vein to be recycled back to the liver via the enterohepatic circulation. Once in the sinusoids of the liver, BAs can be taken up by hepatocytes and secreted back into bile. A fraction of (unconjugated) BAs in the biliary tree is taken up by cholangiocytes at the apical membrane (i.e., prior to reaching the small intestine) and returned to the liver sinusoids via the cholehepatic shunt. Some endogenous and synthetic BAs as well as BA analogs have considerably distinct pharmacologic properties, including but not limited to the degree to which they are cholehepatically shunted (e.g., nor-UDCA being a potent stimulator of cholehepatic shunting) or their potency for agonizing receptors such as the farnesoid X receptor (e.g., obeticholic acid being a potent FXR agonist). The unique properties of some BAs and BA analogs can be harnessed for therapeutic purposes in hepatobiliary diseases including PSC; indeed, this represents an area of ongoing biomedical research. Key: AE2 anion exchange protein 2, ASBT apical sodium-dependent bile acid transporter, BSEP bile salt export pump, MRP multidrug resistance protein, NTCP Na+ (sodium)-taurocholate cotransporting polypeptide, OATP organic anion-transporting polypeptide, OST organic solute transporter, t-ASBT truncated apical sodium-dependent bile acid transporter, TGR5 G protein-coupled bile acid receptor 1 (Adapted with permission from the Mayo Foundation for Medical Education and Research. All rights reserved)
Based on studies in patients as well as various lines of experimental (e.g., model system) data, the mechanisms through which UDCA is believed to exert therapeutic effects in cholestatic disorders include dilution of hydrophobic (or otherwise “toxic”) BAs, promotion of their excretion, upregulation of the biliary bicarbonate umbrella [18, 19], immunomodulation, and anti-inflammatory actions [2, 12, 15, 20–22. In addition, recent data suggest that UDCA may have anti-senescent properties [23]; while the liver has traditionally been regarded as an organ resistant to aging [24], recent studies have shown cellular senescence (in particular cholangiocyte senescence) to be increased in PSC [5], and this finding has been regarded as a marker and driver of biliary injury [23, 25].
Perhaps somewhat surprisingly, evidence supporting a therapeutic role for UDCA in PSC (or animal models thereof) has been inconsistent, with some studies even suggesting detrimental effects at high doses (discussed further below) [19, 26, 27]. As a result, because of the lack of consistently perceived benefits, in their respective practice guidelines, the American Association for the Study of Liver Diseases (AASLD) [21] and European Association for the Study of the Liver (EASL) [20] advise against and provide no specific recommendation, respectively, regarding the use of UDCA in patients with PSC.
Clinical Trials of UDCA in PSC
The earliest clinical studies of UDCA were published in the late 1980s [21, 28, 29] and, albeit uncontrolled, demonstrated promising symptomatic and objective improvements among patients with PSC [30]. These studies soon led to the first randomized controlled trial (RCT) of UDCA, which demonstrated significant improvements in multiple biochemical end points as well as in liver histology [31]. Since then, seven other RCTs have been conducted, initially with low (10–15 mg per kg body weight per day [mg/kg/d])-, then intermediate (17–23 mg/kg/day)-, and most recently high-dose (28–30 mg/kg/day) UDCA (Table 11.1) [14]. While low-dose UDCA was repeatedly shown to yield biochemical improvements, it has not been convincingly shown to improve outcomes, and thus its routine use in PSC is not recommended [21].
High-dose UDCA has been studied in PSC and shown to be associated with an increase in serious adverse outcomes. Specifically, treatment with 28–30 mg/kg/day was found to be associated with a significantly increased risk of major adverse events in a recent RCT of 150 patients with PSC, which was stopped early [19]. At the time of study termination (6 years’ post-study initiation), 30 patients in the UDCA group (39 %) versus 19 patients in the placebo group (26 %) had reached one of the preestablished clinical end points, namely, development of cirrhosis, varices, CCA, LT, or death. After adjustment for baseline characteristics, the risk of a primary end point was 2.3 times greater for patients receiving UDCA compared to the placebo group (p < 0.01) and 2.1 times greater for death, LT, or LT listing criteria (p = 0.038). In addition, serious adverse events were more common in the UDCA group compared to the placebo group (63 % versus 37 %, p < 0.01). While the mechanisms of these inferior outcomes with high-dose UDCA remain uncertain, they may ostensibly be due to toxic metabolites of supratherapeutic UDCA administration and seem to be particularly affect patients with early-stage disease [27]. Based on these results, high-dose UDCA is not recommended in PSC and should not be prescribed.
To date, the most intriguing and favorable RCT-derived data supporting the role of UDCA in PSC have been with use of intermediate-dose UDCA. For example, Mitchell et al. [32] found significant improvements in serum biochemistries, hepatic fibrosis stage, and cholangiographic appearance among patients treated with intermediate-dose UDCA (Table 11.1). Subsequently, and in the largest RCT of UDCA to date, Olsson et al. [33] reported a 34 % relative reduction in need for LT, 31 % relative reduction in mortality, and 22 % relative reduction in diagnosis of CCA. These results did not reach statistical significance, perhaps due to the low incidence of these “hard end points” as well as inability to enroll the planned number of study participants; however, they have been regarded as showing a trend toward such by various expert investigators, many of whom continue to offer intermediate-dose UDCA to select patients with PSC (discussed in the subsequent section) [1, 6, 34]. This practice is supported by several long-term-outcome studies by our group and others from within the last several years which have shown that patients with persistently elevated ALP who achieve clinically significant improvement or normalization of ALP with UDCA therapy have decreased risk of major adverse events (e.g., CCA, need for LT, or liver-related death) [30, 35–37].
Of interest is a recent prospective European study evaluating the effects of 3 months of UDCA withdrawal on serum biochemical tests as well as QOL and symptoms among 26 patients with PSC who were receiving UDCA at a dose of 10–15 mg/kg/day [34]. At the end of UDCA withdrawal, there was a significant (76 %) increase in ALP as well as ALT, AST, bilirubin, and Mayo PSC risk score. Changes in QOL were variable across specific parameters as well as within individual patients, and the majority did not change significantly; there was, however, near doubling in pruritus rating, and this coincided with worsened fatigue in 42 % and deterioration in overall general health (a domain of the short form-36 quality of life instrument) in 60 % of patients. This study represents the largest prospective evaluation of UDCA withdrawal in PSC, and despite several limitations [6], it suggests therapeutic benefit in at least a subset of patients with PSC.
Potential Chemopreventive Properties of UDCA Against Colorectal Cancer
A small body of data suggests that UDCA may play a chemopreventive role against colorectal cancer (CRC) in individuals with PSC-IBD. For example, in a cross-sectional study of 59 patients with PSC-UC undergoing colonoscopic surveillance, UDCA use was associated with decreased prevalence of colonic dysplasia [38]. In another randomized, placebo-controlled trial of 52 patients with PSC-UC, UDCA use was associated with a relative risk of 0.26 for developing colorectal dysplasia or CRC [39]. While specific recommendations have been made regarding CRC prevention in PSC-IBD [40], routine use of UDCA for this indication has not been recommended as additional studies remain needed to confirm its putative chemopreventive properties [21].
UDCA in Clinical Practice
Use of UDCA in routine clinical practice is highly varied among gastroenterologists and even among subspecialized hepatologists within individual referral centers. This is likely a result of mixed views as to the potential benefits of UDCA therapy and the paucity of consistent, high-quality data to suggest a definite therapeutic impact. It is interesting to note that although it is well described that >20 % of patients with another cholestatic liver disease, primary biliary cirrhosis, are nonresponders to UDCA, this drug is still widely recommended as primary therapy; even in patients who are unlikely to respond (e.g., established cirrhosis) or seem to have no or minimal response to UDCA, societal guidelines do not recommend withholding it, perhaps with the hope being that some degree of benefit might still be achieved. Nevertheless, and for reasons that have not been well studied, there appears to be more reticence toward UDCA in PSC as compared to primary biliary cirrhosis, although many clinicians continue to use UDCA in patients with PSC.
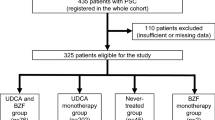
Until safer and more effective pharmacotherapies become available, our current practice is to offer a trial of intermediate-dose UDCA (17–23 mg/kg/day) to patients with compensated PSC whose serum ALP remains >1.5× the upper limit of normal after 1 year since the time of diagnosis [45] or who have troublesome symptoms of cholestasis (e.g., pruritus), as shown in Fig. 11.2. If UDCA is not symptomatically well tolerated or if clinically significant improvement in ALP is not achieved, we discontinue UDCA treatment. These decisions are made with patients’ direct involvement and input and based on careful interpretation of the available biomedical literature [6, 30–36, Ref Annals of Hep [DOI pending]]. Implementation of UDCA in this manner (1) offers patients with PSC the opportunity to potentially benefit from UDCA, (2) lends itself to prospective study in order to help expedite evidence-based treatment recommendations, and (3) can be implemented alongside novel experimental pharmacotherapies (e.g., nor-UDCA, the preclinical data for which indicate that it may well be more effective when used in combination with UDCA).

Proposed algorithm for UDCA use in clinical practice and trials in PSC. *Surveillance and management options reviewed elsewhere [4]. **Consider referral to subspecialist in cholestatic liver disease and/or to tertiary care center. †Also consider decreasing UDCA dose to the lowest dose which maintains biochemical and/or symptomatic response on an individualized basis. Key: ALP serum alkaline phosphatase; CA 19-9 carbohydrate antigen 19-9, MRCP magnetic resonance cholangiopancreatography, PSC primary sclerosing cholangitis, UDCA ursodeoxycholic acid, ULN, upper limit of normal
UDCA in PSC: Conclusions
Although many questions remain unanswered, given the morbidity and mortality of PSC, we believe that the existing evidence supports a role for judicious use of UDCA in patients with PSC, particularly in the absence of safer and more effective therapeutic options. Treatment with UDCA can be implemented in unison with ongoing efforts to develop and rigorously test-emerging therapies through basic, translational, and clinical research endeavors.
The study of PSC pharmacotherapeutics appears to now be better positioned than ever, and with continued innovation, collaboration, and investigation, an even more broadly therapeutic treatment seems likely in the near future.
References
Tabibian JH, Lindor KD. Primary sclerosing cholangitis: a review and update on therapeutic developments. Expert Rev Gastroenterol Hepatol. 2013;7(2):103–14.
Pollheimer MJ, Halilbasic E, Fickert P, Trauner M. Pathogenesis of primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2011;25(6):727–39.
Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc. 2015;90(6):791–800.
O’Hara SP, Gradilone SA, Masyuk TV, Tabibian JH, LaRusso NF. MicroRNAs in cholangiopathies. Curr Pathobiol Rep. 2014;2(3):133–42.
Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-Ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;59(6):2263–75.
Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?). Hepatology. 2014;60(3):785–8.
Tabibian JH, Lindor K. Challenges of cholangiocarcinoma detection in patients with primary sclerosing cholangitis. J Anal Oncol. 2012;1:50–5.
Wiesner RH, Grambsch PM, Dickson ER, et al. Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology. 1989;10(4):430–6.
Bjoro K, Brandsaeter B, Foss A, Schrumpf E. Liver transplantation in primary sclerosing cholangitis. Semin Liver Dis. 2006;26(1):69–79.
Alabraba E, Nightingale P, Gunson B, et al. A re-evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allografts. Liver Transpl. 2009;15(3):330–40.
Aberg F, Hockerstedt K, Roine RP, Sintonen H, Isoniemi H. Influence of liver-disease etiology on long-term quality of life and employment after liver transplantation. Clin Transplant. 2012;26(5):729–35.
Benito de Valle M, Rahman M, Lindkvist B, Bjornsson E, Chapman R, Kalaitzakis E. Factors that reduce health-related quality of life in patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2012;10(7):769–775.e2.
Tabibian A, Tabibian JH, Beckman LJ, Raffals LL, Papadakis KA, Kane SV. Predictors of health-related quality of life and adherence in Crohn’s disease and ulcerative colitis: implications for clinical management. Dig Dis Sci. 2015;60(5):1366–74.
Triantos CK, Koukias NM, Nikolopoulou VN, Burroughs AK. Meta-analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011;34(8):901–10.
Hagey LR, Crombie DL, Espinosa E, Carey MC, Igimi H, Hofmann AF. Ursodeoxycholic acid in the Ursidae: biliary bile acids of bears, pandas, and related carnivores. J Lipid Res. 1993;34(11):1911–7.
Tabibian JH, Masyuk AI, Masyuk TV, O’Hara SP, LaRusso NF. Physiology of cholangiocytes. Compr Physiol. 2013;3(1):541–65.
Maillette de Buy Wenniger LJ, Oude Elferink RP, Beuers U. Molecular targets for the treatment of fibrosing cholangiopathies. Clin Pharmacol Ther. 2012;92(3):381–7.
Fickert P, Fuchsbichler A, Wagner M, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127(1):261–74.
Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50(3):808–14.
European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237–67.
Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51(2):660–78.
Hofmann AF. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology. 2009;49(5):1403–18.
Tabibian JH, O’Hara SP, Trussoni CE, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. 2016;63(1):185–96.
Verma S, Tachtatzis P, Penrhyn-Lowe S, et al. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology. 2012;56(4):1510–20.
O’Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58(3):575–82.
Fickert P, Zollner G, Fuchsbichler A, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123(4):1238–51.
Imam MH, Sinakos E, Gossard AA, Kowdley KV, Luketic VA, Edwyn Harrison M, McCashland T, et al. High-dose ursodeoxycholic acid increases risk of adverse outcomes in patients with early stage primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011;34(10):1185–92.
Chazouilleres O, Poupon R, Capron JP, et al. Ursodeoxycholic acid for primary sclerosing cholangitis. J Hepatol. 1990;11(1):120–3.
O’Brien CB, Senior JR, Arora-Mirchandani R, Batta AK, Salen G. Ursodeoxycholic acid for the treatment of primary sclerosing cholangitis: a 30-month pilot study. Hepatology. 1991;14(5):838–47.
Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43(4):309–13.
Beuers U, Spengler U, Kruis W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology. 1992;16(3):707–14.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121(4):900–7.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129(5):1464–72.
Wunsch E, Trottier J, Milkiewicz M, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology. 2014;60(3):931–40.
Lindstrom L, Hultcrantz R, Boberg KM, Friis-Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2013;11(7):841–6.
Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2013;58(2):329–34.
Rupp C, Rossler A, Halibasic E, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther. 2014;40(11–12):1292–301.
Tung BY, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89–95.
Pardi DS, et al. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–93.
Tabibian JH, Moradkhani A, Topazian MD. Colorectal cancer surveillance in primary sclerosing cholangitis and inflammatory bowel disease. Ann Hepatol. 2015;14(4):564–6.
Lo SK, Herrmann R, Chapman RW, et al. Ursodeoxycholic acid in primary sclerosing cholangitis: a double-blind placebo controlled trial. Hepatology. 1992;16.92A.
Stiehl A, Walker S, Stiehl L, et al. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3-year pilot study with a placebo-controlled study period. J Hepatol. 1994;20(1):57–64.
De Maria N, Colantoni A, Rosenbloom, Van Thiel DH. Ursodeoxycholic acid does not improve the clinical course of primary sclerosing cholangitis over a 2-year period. Hepatogastroenterology. 1996;43(12):1472–9.
Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med. 1997;336(10):691–5.
Hilscher M, Enders FB, Carey EJ, et al. Alkaline phosphatase normalization is a biomarker of improved survival in primary sclerosing cholangitis. Ann Hepatol. 2016;15(2):246–53.
Conflicts of Interest, Disclosures
James H. Tabibian – none
Keith D. Lindor – unpaid consultant for Shire and Intercept
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tabibian, J.H., Lindor, K.D. (2017). Ursodeoxycholic Acid Treatment in Primary Sclerosing Cholangitis. In: Forman, L. (eds) Primary Sclerosing Cholangitis. Springer, Cham. https://doi.org/10.1007/978-3-319-40908-5_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-40908-5_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40906-1
Online ISBN: 978-3-319-40908-5
eBook Packages: MedicineMedicine (R0)