
Abstract
Worldwide, the incidences of cancer are rising. Various environmental and genetic factors are predisposing individuals to cancer. Majority of these factors result in upregulation of pro-survival pathways, downregulation of tumor suppressors, and chronic inflammation. The ratio of w-6/w-3 polyunsaturated fatty acids (PUFAs) plays very crucial role in the initiation and progression of cancer. Low w-6/w-3 PUFA ratio has been shown beneficial in managing the hallmarks of cancer cell. Enormous data from cancer cell line and in vivo cancer models have given insight into the mechanisms underlying the anticancer effects of w-3 PUFAs. Here, we discussed major possible mechanisms for beneficial effects of w-3 PUFAs as evidenced by the preclinical in vitro cancer cell line models and in vivo models.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Anticancer
- Lipid peroxidation
- Lipid rafts
- Eicosanoids
- Mitochondrial hyperpolarization
- Inflammation
- Apoptosis
- Metastasis
- Immunomodulation
- Fat-1 transgenic mouse model
Introduction
Cell–cell interaction, cell–extracellular matrix interaction, and proliferative and anti-proliferative signals such as tumor suppressor proteins are critical in tissue homeostasis and cellular quiescence. For normal cell to proliferate, stimulatory signals are essential which are activated by growth factors, extracellular matrix components, and cell–cell interaction molecules [1]. In addition to this, a mammalian cell has limited capacity to proliferate with some exceptions such as stem cells and germ cells which are known as Hayflick limit. When cells grow old or insulted by physicochemical stimuli, they enter senescence phase, dead cells are removed, and new cells take their place. When this regulated cascade is disturbed, uncontrolled cell growth leading to cancer may arise. Majority of the cancers are linked with somatic mutations in proto-oncogenes and/or tumor suppressor genes and/or DNA repair genes. Cancers are also linked with environmental factors. Cancer-causing environmental exposures include substances, such as the chemicals in tobacco smoke, and radiation, such as ultraviolet rays from the sun. Among various cancer-causing factors, chronic inflammation has been shown to play major role [1–3]. Besides being pro-tumorigenic, inflammatory factors also influence immune response toward tumors.
Cellular functions such as membrane structure and fluidity, cellular metabolism and energy production, cell signaling, and cellular interactions are attributed to fatty acids [1, 4]. Dietary inclusion of w-3 FA from marine and/or plant origin has beneficial effect on various health conditions. Omega-3 fatty acids (w-3 FAs) are known to help reduce the risk of heart disease, lower triglycerides, and relieve inflammatory conditions such as rheumatoid arthritis and inflammatory bowel disease. Emerging evidence from both experimental and epidemiological studies suggests w-3 FAs may play a role in cancer prevention [5]. Several clinical and preclinical studies have pointed out that w-3 FA consumption is associated with decreased cancer risk of the breast, prostate, colon, and kidneys [6]. The main FAs in this group are α-linolenic acid (ALA), and the long-chain FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Like linoleic acid (LA, parent FA from w-6 PUFA family), ALA is an essential FA for humans, as it is required for normal function and body is unable to make it. EPA and DHA can be synthesized in the body from ALA. LA and ALA are converted to w-6 (AA) and w-3 (EPA and DHA) LC-PUFAs, respectively, by series of reactions conducted by desaturases and elongases in endoplasmic reticulum. Both these pathways share same set of enzymes for the conversion to respective PUFA leading to competition between w-6 and w-3 PUFAs for their metabolic conversion [1]. Synthesis of DHA takes place in peroxisomes by β-oxidation of 24:6w-3.
Experimental data show that w-3 PUFAs may act on the hallmarks of cancer cells, including the newly added criteria by Hanahan and Weinberg [7]. Evidence accumulated from numerous experimental systems indicates that w-3 FAs may exert an antitumor action by altering the cell membrane phospholipid composition and, consequently, affecting the expression and function of numerous receptors, proteins, and lipid-derived signaling molecules.
Various mechanisms have been explained for the effect of w-3 PUFA’s beneficial effect in cancer [8]. Here we will look into major mechanisms involved in anticancer effect of w-3 PUFA in cancer.
Role in Lipid Peroxidation
Among many anti-cancer mechanisms studied for w-3 PUFA, major one is lipid peroxidation [9–11]. w-3 PUFAs get incorporated into the tumor cell membranes, which results in the increased susceptibility of these cells for lipid peroxidation and accumulation of lipid peroxidation products (reactive lipid hydroperoxides) to cytostatic or cytotoxic levels.
Oxidative damage is key mechanism for many known chemotherapeutic agents such as doxorubicin and mitomycin. Supplementing the diet of tumor xenograft-bearing mice with w-3 PUFAs results in increased efficacy of these chemotherapeutic agents. The increased efficacy of these drugs has been assigned to increase the susceptibility of tumor cell membranes to lipid peroxidation [12].
w-3 PUFA-promoted cell death is mainly through ROS-induced apoptosis as shown in various cell lines where intracellular ROS levels were elevated and treatment with antioxidants abrogates effect of w-3 PUFA-induced cell death. Cell death is mainly via apoptosis where caspases 8, 3, and 9 are shown to execute the death process in human pancreatic cancer cells [13].
In vivo xenograft data support the conclusion that EPA-induced cell death is mainly mediated through the formation of intracellular ROS where animals were fed with fish oil diet and 3-nitroso-cysteine was used as a marker for protein nitration and cellular oxidative stress in vivo [13].
Role in Mitochondrial Membrane Lipid Peroxidation
Epidemiological data suggest that while high levels of saturated fatty acids are positively associated with cancer risk, w-3 FAs seem to prevent cancer [14–17].
Protective action of w-3 FAs against cancer has also been supported by in vitro and in vivo studies. Consumption of dietary fiber enhances butyrate production resulting in enhanced protective action [18, 19]. Due to enhancement of unsaturation of mitochondrial phospholipids, especially cardiolipin, w-3 FAs prime butyrate to initiate apoptotic cascade. So there is a synergy between w-3 FAs and butyrate.
The role of DHA in mediating mitochondrial membrane lipid oxidation causing apoptosis in colonocytes has been elucidated [20]. It has been observed that (i) DHA and butyrate synergistically enhance lipid oxidation, (ii) mitochondrial-targeted antioxidant protects cells from oxidative stress, (iii) DHA enhances mitochondrial membrane potential, and (iv) mitochondrial-targeted antioxidant suppresses butyrate-induced apoptosis.
DHA, via mitochondrial ROS over production, simultaneously induces autophagy and apoptosis in cancer cells [21]. Autophagy and apoptosis are self-destructive processes that share many key regulators. Cytotoxicity is restricted to w-3 PUFAs as arachidonic acid had no effect. DHA incorporation into mitochondrial membrane phospholipids and enhanced mitochondrial lipid oxidation and ROS generation seems to be contributing to the antitumor activity of w-3 PUFAs. Mitochondrial ROS generation induced by w-3 PUFAs that inhibit PI3K/mTOR may be responsible for the autophagy and apoptosis.
There is evidence that dietary fiber that increases butyrate levels in colon is protective against colorectal cancer [22]. It has also been shown that butyrate and omega-3 fatty acids work coordinately and protect against colon tumorigenesis which is primarily by increasing apoptosis [23]. Interestingly, it has been shown that DHA alters colonocyte mitochondrial membrane composition, thereby creating permissive environment for apoptosis [19]. Further, it has also been shown that mitochondrial lipid oxidation products, membrane phospholipid-derived hydroperoxides (LOOH), play an important role in DHA and butyrate-induced apoptosis [24].
It is known that finely tuned intracellular homeostasis and compartmentalization of Ca2+ can lead to cell death. It is now recognized that mitochondria play a key role in both apoptosis and necrosis by regulating intracellular Ca2+ homeostasis, activation of caspases, and the release of ROS [25]. Mitochondria can be regarded as critical checkpoint in Ca2+ signaling, and in certain situations mitochondrial Ca2+ accumulation is a trigger for cytochrome C release and the induction of apoptosis [26]. DHA and butyrate synergistically induce colonocyte apoptosis by enhancing mitochondrial Ca2+ accumulation [23]. DHA along with butyrate enhances ROS and LOOH production and causes a change in mitochondrial transition permeability. DHA amplifies the butyrate action up to 43 %. DHA alone, however, has no significant action. The synergistic action of enhancement of apoptosis is possibly due to Ca2+-mediated intrinsic mitochondrial pathway [27].
Cancer cell mitochondria are hyperpolarized [28], along with decreased levels of mitochondria-derived ROS and also decreased expression of Kv channels. The decreased ROS production can close redox-sensitive MTP. Increased hexokinase levels support the compensatory aerobic glycolysis in cancer cells. In summary, hyperpolarization of mitochondria, blocking entry of pyruvate into mitochondria, increased glucose uptake, and aerobic glycolysis are some of the measures taken by the cancer cells to avoid apoptosis. Targeting mitochondria by modifying mitochondrial membrane composition by omega-3 fatty acids, depolarization of the mitochondrial membrane to allow entry of pyruvate into mitochondria, restoring normal TCA cycle and apoptotic pathways, can be a very effective strategy to treat all types of cancer. In this respect, there exists a lot of excitement about dichloroacetate (DCA). DCA reverses cancer cell abnormal metabolism from aerobic glycolysis to glucose oxidation by reducing the activity of mitochondrial pyruvate dehydrogenase kinase 1 (PDK1). DCA reduces high mitochondrial membrane potential and increases mitochondrial reactive oxygen species (ROS) in malignant, but not in normal cells.
Figure 12.1 represents various key modifications or adaptations cancer possesses to avoid apoptosis.

Illustration depicts various strategies of cancer cells to avoid apoptosis. Mitochondrial hyperpolarization, aerobic glycolysis in association with upregulation of hexokinase, and inhibition of PDH. Various agents that target these modifications alone or in combination resulting in cancer cell apoptosis are marked in bold letters
Role in Inflammation
Inflammation has been shown to play very crucial role in tumorigenesis [29] and is involved in the every single step of tumorigenesis, right from tumor initiation, tumor promotion to metastatic progression. Various types of immune and inflammatory cells are frequently present within tumors. Microenvironment of the tumor is influenced by these cells through the production of cytokines, chemokines (such as TNF α, IL-1, and IL-6), growth factors, prostaglandins, and reactive oxygen and nitrogen species. Most of the tumors trigger inflammatory cascade that in turn modulates tumor microenvironment in favor of cancer cells [29]. Signaling pathways involved in the pro-tumorigenic effects of inflammation are often subjected to a feed-forward loop in tumor cells. For example, activation of NF-kB in immune cells induces production of cytokines that activate NF-kB in cancer cells to induce chemokines that attract more inflammatory cells into the tumor [30]. The cytokine IL-6 promotes oncogenesis by stimulation of NF-kB and STAT-3 signaling [31].
Transcription factors, NF-kB and STAT3, are at the center stage in various inflammatory processes as they control various cytokines involved in the inflammatory process [32]. Both these transcription factors are shown to be involved in the tumorigenesis and are considered cancer therapy targets [2]. Figure 12.2 represents the impact of inflammation on tumor initiation and progression.

Tumor cell mass and tumor-associated various cells such as stromal cells, tumor-associated macrophages show feed-forward association resulting in the microenvironment that will allow sustained survival, continuous proliferation, angiogenesis, and metastasis
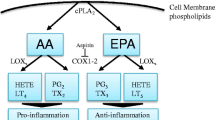
w-6 and w-3 PUFAs are stored in the phospholipids of the cytosolic leaflet of cell and organelle membrane in esterified form and can be mobilized by phospholipase A2 [33]. Free or membrane-released FAs are acted upon by cyclooxygenases (COX), lipoxygenases (LOX), and cytochrome p450 (CYP450). Resulting metabolites are known as eicosanoids, which act as short-lived hormone-like lipids. These eicosanoids are involved in the various processes such as platelet aggregation, cellular growth, and cell differentiation [6]. As mentioned earlier, there exists competition between w-3 and w-6 PUFAs for desaturases and elongases [34]. The eicosanoids from w-6 PUFA (AA) are pro-inflammatory (series 2 prostaglandins and thromboxanes, and series 4 leukotrienes and lipoxins) while those derived from w-3 PUFA and CYP450 metabolites of w-6 PUFA (series 3 prostaglandins and thromboxanes, E-series and D-series resolvins, series 5 leukotrienes, EEQs, HEPEs, merlines, EDPs, EETs, nHETEs, and DiHETEs) are considered anti-inflammatory or immunologically less reactive. That is why, a higher level of ALA results in reduced synthesis of AA from LA, in turn resulting into low levels of pro-inflammatory AA eicosanoids [35].
NF-kB mediates inflammatory process by controlling cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) [32]. COX2 [36–40] and/or iNOS levels are elevated in many cancer types [41]. PGE2 (AA metabolite of COX) is shown to induce tumor cell proliferation. PGE2 stimulates the expression and activation of aromatase enzyme which coverts androgens to estrogens. A significant correlation has been established between aromatase and cyclooxygenase enzyme systems at gene and protein levels indicating autocrine and paracrine mechanisms may be involved in hormone-dependent cancer development via growth stimulation from local estrogen biosynthesis [42, 43]. A strong epidemiological evidence associates the estrogen to breast, endometrial, and uterine cancers [44]. 3-series and 5-series prostaglandins and leukotrienes derived from EPA and DHA are shown to have anti-inflammatory activity. These are shown to reduce PGE2 production by 60 % and LTB4 by 75 % in human peripheral blood mononuclear cells [45]. Thus, increasing w-3 PUFA levels with lowering w-6 levels may play important role in managing tumor-associated inflammation.
Apart from inhibiting expression of COX-2 and iNOS target genes, DHA treatment in Caco-2 cell line results in simultaneous reprogramming of genes involved in differentiation such as p21(Waf1/Cip1), p27, and apoptosis by activating caspases such as caspase-3 [46].
Supplementation of w-3 PUFA to neuroblastoma cells reduced the AA-derived eicosanoids [8, 47]. In these cells, DHA and its metabolites induced apoptosis by intrinsic pathway. 17-hydroperoxy-DHA showed the highest cytotoxic potency. Additive or synergistic effect has been seen with DHA and COX-2 inhibitor celecoxib in neuroblastoma cells [48] and other human cancer cell lines [49]. Mechanism seems to be COX2-dependent (inhibiting AA metabolism by blocking COX-2) and COX-2-independent (modulation of heat shock proteins and NF-kB activity and steroid receptors) [1, 49].
In cultured pancreatic cells and human monocytes, w-3 PUFAs inhibit NF-kB activity directly. Inhibitory effect is mediated by decreased degradation of IkB, inhibitory subunit of NF-kB [30, 33]. w-3 FAs are converted by COX-2 into OH derivatives, which are then metabolized to eFoX. eFoX inhibits pro-inflammatory responses and induces nrf2-dependent gene expression (nrf2 regulates major antioxidant pathways including glutathione) [50] and PPARγ activation and adduct to protein and glutathione [51].
In colon xenograft model, COX-2 has been shown to decrease apoptosis by modulating expression of bcl-2 gene expression. In this model, tumor growth was restricted by w-3 PUFA supplementation through downregulation of COX-2. In prostate tumor model, beneficial effect is mediated by downregulating bad phosphorylation resulting in the upregulation of tumor cell apoptosis [1].
Tissue-associated macrophages play important role in the inflammatory cascade. Macrophages express GPR120, an orphan G protein-coupled receptor which works as a w-3 FA receptor or sensor. In the Toll-like receptor signaling pathway and TNFα inflammatory pathway, TAK1 phosphorylation is essential for downstream JNK and NF-kB pathway activation as TAK1 is converging point. Binding of DHA to GPR120 results in the inhibition of TAK1 phosphorylation and inhibition of downstream NF-kB and JNK activation in β-arrestins-dependent manner [52]. Both these w-3 PUFAs inhibit NF-kB activation and TNF-α secretion in macrophages.
An opposing effect of EPA was reported by Wu et al. In contrast to the inhibitory effect of w-3 PUFA in prostate cancer, FFA4 (GPR120) and its agonist EPA have oncogenic role in colon cancer. In Caco-2 colon cancer cell line, EPA activates p70S6K through FFA4-mediated signaling [53].
Enzymes 5-LOX, 15-LOX, or acetylated COX act on the EPA and/or DHA to produce E-series and D-series resolvins. Protectins from DHA are also formed by these enzymes. Discovery of resolvins and protectins in 2002 helped to understand the mechanism underlying the w-3 PUFA beneficial effects [9–11].
Lipoxins, resolvins, and protectins also increase the expression of CCR5 receptors on T cells and aging PMN, which help clearing local chemokine depots from the inflammatory site. Apoptotic neutrophils are then phagocytized by macrophages, leading to neutrophil clearance and release of anti-inflammatory and inflammation resolution cytokines such as transforming growth factor-beta1 [54, 55].
Best studied eicosanoid from w-3 PUFAs is EPA-derived resolvin E1 (RvE1). In colorectal cancer cell lines, RvE1 shows anticancer activity by inhibiting NF-kB inflammatory pathway through G protein-coupled receptors ChemR23 and BLT1 [56, 57].
Role in Physicochemical Properties of Lipid Rafts and Signal Transduction
Lipid rafts are important membrane domains for cell signaling and drug accumulation inside the cell as they are enriched with many regulatory proteins, some growth factor receptors, and drug efflux pumps [58]. Lipid rafts are highly ordered and less fluid than the surrounding membrane due to presence of cholesterol and sphingolipids mainly sphingomyelin (containing saturated fatty acids) [58, 59]. Cholesterol maintains the microdomains of the lipid rafts. Supplementing growth medium with EPA or DHA results in the alteration in the structure and composition of the lipid rafts as cholesterol and sphingomyelin levels go down. Due to long carbon chain and unsaturated nature, incorporation of EPA or DHA results in disturbance in lipid raft organization and structure resulting in suppression of the raft-associated cell signaling [46, 60–64].
Wu et al. have shown that EPA and DHA activate neutral sphingomyelinase that hydrolyzes sphingomyelin to ceramide which has apoptotic function [65].
PUFAs act on membrane-associated signal transduction. Upregulation of pro-survival pathways such as EGFR, Ras/Raf, and PI3K/mTOR pathway is seen in many malignancies. Unsaturated FAs particularly w-3 PUFA [66] (mainly DHA) are able to modulate the activity of various intracellular signaling pathways mediated by calcium, protein kinase C (PKC), mitogen-activated protein kinases (MAPKs), epithelial growth factor receptor (EGFR), and Ras [1, 4].
Schley et al. have shown in the MDA MB231 cells that the incorporation of EPA or DHA in the lipid rafts results in the depletion of cholesterol followed by expulsion of EGFR from the microdomains and its phosphorylation. p38MAPK (downstream of EGFR) gets phosphorylated after EGFR phosphorylation. p38MAPK phosphorylation is associated with apoptosis in MDA MB231 cells [67].
Activation and overexpression of EGFR have been seen in many malignancies including breast cancer. In ER-ve breast cancer cell lines, DHA treatment results in inhibition of EGFR activation and slight decrease in the expression of EGFR. Inhibitory effect is mediated through caspase-3 or caspase-8 pathways [36, 60, 68, 69].
In MCF7 cell line, ALA alone was able to downregulate HER-2 expression by 79 % which was further confirmed by downregulation of expression of Ki-67 and PCNA [45].
Nuclear receptors, peroxisome proliferator-activated receptors (PPARs), function as ligand-activated transcription factors and regulate PPAR response elements (PPREs). These PPREs are found in regulatory regions of variety of genes involved in lipid metabolism and homeostasis, cell proliferation, cell differentiation, and inflammatory responses. PUFA and eicosanoids are naturally occurring PPAR ligands. In in vitro studies on breast cancer cell lines, PPARγ activation induces apoptosis via syndecan-1 [1, 70, 71]. In rats with induced mammary carcinogenesis, dietary supplementation with a low w-6/w-3PUFA ratio (1:14.6) was shown to increase PPARγ protein content, which was paralleled with a reduction of tumor burden. All these evidences indicate PPARγ activation is beneficial for controlling breast cancer and a potential role for PPARγ ligands (w-3 PUFA) in the treatment of breast cancer [45].
Ca+2 is a major intracellular factor playing role in signaling pathways that are involved in various cellular processes such as growth, proliferation, gene expression, contraction, secretion, and metabolism. Majority of these processes require sustained increase in the intracellular-free calcium. Store-operated calcium influx (SOCI) pathway governs the influx of calcium from extracellular matrix into the cells. In an aggressive thymoma rat models, w-3 PUFA-containing diet resulted in low levels of SOCI and reduced tumor growth [72].
Once taken up by cells or released from the cell membrane, DHA acts as a ligand to different nuclear receptors, such as the PPARγ and the RXR receptor. In conjunction with these receptors that act as transcription factors, DHA helps regulate various biological functions ranging from homeostasis and lipid metabolism to cell differentiation and cell death [6, 56, 73].
Figure 12.3 represents major cellular processes modulated by w-3 PUFA.

w-3 PUFA metabolites, i.e., eicosanoids, modulate cellular processes by interacting with various receptors or altering signaling pathways. Overall, modulation of signaling pathways results in downregulating inflammatory cascade, enhanced fatty acid degradation in association with lowered fatty acid synthesis, and low expression of EMT markers ultimately resulting in the cell death
Role in Limitless Cell Proliferation
Limitless proliferation is one of the hallmarks of cancer cell. w-3 PUFAs, EPA, and DHA may inhibit mitosis by at least three different mechanisms such as both these w-3 PUFAs inhibit AA-induced activation of PKC and further mitosis, by downregulating Ras and AP1 oncoproteins which are associated with many cancers and are known to stimulate mitosis and by inhibiting synthesis of pro-mitosis eicosanoids derived from AA. These mechanisms have been demonstrated in the breast and colon cancer cell lines. In colon cancer animal models, w-3 PUFA inclusion in the diet resulted into prevention of K-Ras mutation and Ras membrane expression [1, 74].
Role in Apoptosis
bcl2 family proteins regulate important mechanism of cell death, i.e., programmed cell death by apoptosis. Vast amount of clinical and preclinical data indicates that, in majority of cancers, expression of pro- or anti-apoptotic bcl2 family proteins is altered. These proteins promote or inhibit apoptosis by the release of effectors and executors from mitochondria.
In in vitro studies in breast cancer, colon cancer cell lines highlight that treatment with EPA and/or DHA shifts the balance between pro- and anti-apoptotic bcl2 proteins in favor of cancer cell death. EPA and DHA may downregulate anti-apoptotic bcl2 and Bcl-xL proteins and upregulate pro-apoptotic Bak and Bcl-xS protein level [1]. Both these FAs are shown to initiate apoptosis process by cytochrome C release from mitochondrial membrane and direct binding with PPARγ. Similar mechanisms were illustrated in the rat models of various cancers such as in colon cancer rat model, feeding animals with fish oil resulted into lower levels of bcl2 and apoptosis of cancer cells [75, 76].
Role in Adhesion and Angiogenesis
Adhesion and angiogenesis are first two critical steps in the tumor establishment. Cytoskeletal rearrangement is essential for cellular adhesion which is controlled by Rho GTPase. DHA has also been shown to decrease TNFα-induced monocyte rolling, adhesion, and transmigration. Probably by down regulating Rho GTPase, ICAM-1, and VCAM-1, DHA can inhibit adhesion [6].
Experimentally, it has been shown that, once the tumor is established, for a tumor to grow beyond the size 1–2 mm3, substantial new blood vessel development is essential to cater the increasing nutrient demand [77]. As the tumor grows, core of the tumor becomes necrotic resulting into cell death and release of pro-inflammatory factors such as IL-1 and high-mobility group box-1 (HMGB1) which in turn trigger neo-angiogenesis [78, 79].
It is known that inflammation, hypoxia and mechanical stress may result in the endothelial cell activation or release of cytokines involved in abluminal sprouting. Basement membrane proteolysis for capillary sprouting is mediated by MMPs. EPA has been shown to downregulate MMP2 and 9. In in vivo experiments, similar findings were observed where mouse xenograft models for colon and breast cancer showed low tumor microvessel density and VEGF levels in EPA-fed mice against control group [77]
Tumor-associated macrophages secrete pro-inflammatory and pro-angiogenic factors which are directly regulated by NF-kB, STAT3, and AP1 [30]. Inhibition of NF-kB, STAT3, CCL2, CXCL12, and depletion of TAM result in reduced angiogenesis and tumor growth [80].
NO and COX-2 also regulate VEGF-mediated angiogenesis. Nitric oxide (NO) promotes tumor growth (by enhancing invasiveness), endothelial cell survival, and proliferation, and inhibits apoptosis. Inducible nitric oxide synthase (iNOS) stimulates NO production which in turn increases PGE2 production, which is implicated in tumor growth and progression. EPA and DHA have been demonstrated to inhibit NO production and iNOS expression in murine macrophages and downregulate NO and nuclear factor-kappaB (NF-kB) in human colon cancer cell lines.
DHA inhibits angiogenesis probably by decreasing levels of VEGF, PDGF, and platelet-derived endothelial cell growth factor [6].
In CYP450-mediated metabolism, epoxyeicosatrienoic acids (EETs) are derived from w-6 AA and epoxydocosapentaenoic acids (EDPs) from w-3 DHA. In terms of angiogenesis and cancer, EETs are pro-angiogenic and have been shown to accelerate tumor growth and metastasis by stimulation of tumor angiogenesis. In angiogenesis model, Matrigel plug assay where inducers were VEGF and bFGF, EDP inhibited vascularization. One of these EDP metabolites, 19,20-EDP dramatically inhibited endothelial tube formation after a 6-h treatment in human umbilical vein endothelial cells (HUVECs) by inhibiting migration of the HUVEC cells. 19,20-EDP directly targets endothelial cells to suppress angiogenesis, primarily via suppression of endothelial cell migration. The 19,20-EDP also inhibited the activity of matrix metalloproteinase 2 (MMP-2) but with a weak activity. 19,20-EDP inhibited angiogenesis via blocking VEGF–VEGFR2 signaling. Overexpression of CYP epoxygenases in cancer cells or endothelial cells accelerates tumor growth and metastasis, which are largely attributed to AA-derived EETs [81, 82]. Thus, lowering the ratio of w-6/w-3 FA may result in the lowered synthesis of pro-inflammatory and pro-angiogenic EETs and higher levels of anti-inflammatory and anti-angiogenic EDPs.
VEGFR2 is the therapeutic target of numerous angiogenesis inhibitors. But, VEGF-VEFGR2 inhibitors are routinely associated with induction of hypertension. EDPs are extremely potent vasodilators which give them unique advantage in antiangiogenic agent that can be used in cancer therapy avoiding hypertension [81].
Role in Metastasis
AA-derived eicosanoids are involved in the every step of metastatic cascade such as intravascular tumor cell–tumor cell interaction, interactions with platelets and forming aggregates, and secretion of type IV collagenase essential for invasion [83]. As mentioned earlier, eicosanoids derived from EPA and in particular DHA [72] are biologically less active and inhibit the actions of AA-derived eicosanoids [81]. CD44 is pro-metastasis molecule, expressed by aggressive cancer cells, and is essential for the EMT. DHA may downregulate the expression of CD44. Anti-metastatic properties of DHA are highlighted in many animal models such as breast cancer xenograft model and colon cancer models. Preoperative and/or postoperative DHA dietary supplementation in these models results in low bone and lung metastasis, respectively [84].
Role in Immunomodulation
Among the multiple subtypes present in macrophages, M2-type macrophages have tumor growth-promoting properties, higher expression of immunosuppressive cytokines such as TGF-β, induce invasion and angiogenesis, and promote metastasis. TAMs are induced to M2 polarization by cytokines such as IL-4 and IL-13(TH-2 cytokines), which assists in tumor promotion and development. Macrophage recruitment and polarization to M2 type are regulated by growth factors such as M-CSF and chemokines such as MCP-1. Both M-CSF and MCP-1 expressions are upregulated in many cancer types such as prostate cancer. In vitro studies using PC3 prostate cancer cell line and THP1 cells (induced to show M2-type functional properties) gave similar findings. Supplementation of EPA/DHA resulted in lowered macrophage recruitment by downregulating M-CSF and MCP-1. Data from similar experimental system suggest involvement of PPARγ and NF-kB [85]. w-3 PUFA from fish oil is shown to decrease TH-1 and TH-2 cytokine responses at mucosal level [86]. Tumor promotion functions of TAM include extracellular matrix remolding, promotion of tumor cell invasion, angiogenesis, lympho-angiogenesis, metastasis, and immunosuppression [87].
Similar to TAM, tumor-associated neutrophils (TANs) show tremendous plasticity. The N2-type TAN phenotype driven by TGF-β is negatively correlated with clinical outcome. Tumor microenvironment influences the pro-tumorigenic functional properties of TANs. These properties include release of elastases, MMPs, oncostatin M, etc., and enzymes or growth factors that ultimately result in tumor cell proliferation, invasion, and angiogenesis [87, 88].
Figure 12.4 represents functional molecules from TAM and TAN involved in tumor progression

TAM and TAN modulate tumor survival, angiogenesis, and metastasis by virtue of various factors such as interleukins, growth factors, and proteases
Role in Combination with Chemotherapeutic Agents and Drug Resistance
Majority of current chemotherapeutic treatments are associated with adverse side effects and resistance development against single targeted therapeutic drug. This unwanted chemotherapy-related toxicity is mainly due to requirement of high dose to achieve clinically significant efficacy.
In vitro combination studies with w-3 PUFA and various standard chemotherapeutic agents show additive or synergistic effects. In the colon cancer cell lines, synergistic combination effect of DHA with 5FU was independent of p53 status and was executed by intrinsic apoptotic pathway [89–94]. Apart from involvement of mitochondrial apoptotic pathway, distinct gene expression profile suggests role played by some other pathway [95]. Among various mechanisms elucidated for the beneficial combination effects, lipid peroxidation and ROS generation have been major players along with modulation of lipid rafts resulting into drug efflux [96–98]. It has been shown that lipid peroxidation initiated by DHA is required for this beneficial combination effects. The preclinical cell line-based data indicate DHA pretreatment reverts chemo-resistance for doxorubicin, taxol, docetaxel, arsenic trioxide, vincristine, and cisplatin. It has been shown in various resistant cell lines that degree of resistance is lowered after DHA treatment [99].
Drug resistance is influencing the mortality rates in various cancers. ABC transporters (P-glycoprotein (Pgp/ABCB1), multidrug resistance-related proteins (MRPs/ABCCs), and breast cancer resistance protein (BCRP/ABCG2)) limit the intracellular accumulation of several anticancer agents by inducing the efflux of chemotherapeutic drugs. High level of cholesterol synthesis has been linked with multidrug resistance especially in colon cancer. The activity of Pgp and BCRP is directly related to the amount of cholesterol in the plasma membrane. Significant amount of these drug efflux pumps is embedded in cholesterol-rich domains of plasma membranes especially detergent-resistant membranes (DRMs). DHA acts as a strong DRM-interrupting agent. Due to long carbon chain and high level of unsaturation, DHA poorly fits in the cholesterol-rich DRM increasing degree of lipid unsaturation and altering the physicochemical properties of these compartments [100].
3-hydroxy-3methylglutaryl-coenzyme A reductase (HMGCoAR) is a crucial enzyme in the biosynthesis of cholesterol. EPA and/or DHA in cell line-specific manner has been shown to reduce HMGCoAR activity and restore ubiquitination level in drug-resistant cell lines. It has been shown that the inhibitory effect of EPA and DHA is cell line specific and controversial, for example, in SW480 cells, DHA has very little effect on de novo cholesterol synthesis and increases SREBP-2 which induces several genes involved in cholesterol synthesis.
Maheo et al. have reported contrasting data. Accordingly, irrespective of the breast cancer cell line studied, DHA incorporation in the cellular membrane did not result into intracellular doxorubicin accumulation [101].
High levels of prostaglandins and COX2 are associated with many human cancers such as breast, cervix, lung, skin, colon, and prostate. Celecoxib (COX-2 inhibitor) shows potential in cancer treatment, but use is limited because of dose-induced toxicity. In colon cancer cell line, combination of celecoxib and DHA resulted in induction of apoptosis at much lower doses compared to individual treatment has been shown that combination with DHA reduces side effects (toxicity) allowing increase in the dose [46, 61–64, 102–104]. Underlying mechanism is associated with NF-kB p65 translocation, and PPARc and RXRa are involved in the mechanism [48]. Similar synergistic effect was seen in the combination of curcumin and DHA. The combinations were also found to suppress iNOS, COX-2, 5-lipoxygenase (5-LOX), and cPLA2 activities [105]. The anticancer effect of 1a,25(OH)2D3 is well documented [47], but the doses required to achieve clinically significant effect may result in the hypercalcemia. Combination of 1a,25(OH)2D3 with DHA has synergistic effect in glioblastoma and lung cancer cell line [99].
Experimental Evidence from Animal Studies
Various xenografts models have been studied to explore effect of dietary PUFA on tumor growth or tumor burden. The inhibitory effects of n-3 PUFA on tumor growth have been well documented for breast cancer in rodent xenograft models such as MDA Mb 435, MDA MB231, MCF7, and R3230AC [106]. When these xenograft-bearing mice were fed with fish oil or menhaden oil showed slow tumor growth and low tumor volume compared to control group, xenograft models fed with w6-rich diet. Modification of cell membrane structure and composition, high incorporation of w-3 PUFA into the tumor phospholipids, which further reduced pro-inflammatory eicosanoid synthesis from AA, and lipid peroxidation in the tumor cells are underlying this beneficial effect of w-3 against tumor progression. The antitumor property of w-3 PUFA was supported by Ki-67 immunohistochemical staining (absent in the resting cell). Similar observations were seen in case of prostate cancer fed with ALA-rich flaxseed oil.
The fat-1 transgenic mouse model provides evidence that DHA and DHA-derived compounds may have significance in cancer development. These mice carry a gene that encodes a desaturase that catalyzes conversion of w-6/w-3 FAs, a feature that is lacking in most mammals, including humans. In this mouse model, melanoma formation and growth, colitis-associated colon cancer growth, and prostate cancer growth were all inhibited compared to tumor growth in non-transgenic animals [48].
In another study, aggressive HER-2-positive BC model (MMTV-neu-YD5) was crossed with fat-1 mice yielding mice that were able to convert w-6 PUFA to w-3 PUFA and susceptible to mammary tumor growth were fed with w-3 FAs-containing or w-6 FA-containing feed. In both these approaches, tumor incidence was dramatically reduced along with prolonged tumor latency. Effect of the w-3 PUFA was dose-dependent [107].
In line with the previous study, the observed dose-dependent effects of w-3 PUFA were associated with a dose-dependent change in the fatty acid profile, reflecting a decreased w-6/w-3 ratio in the mammary glands and an increased EPA and DHA in tumor phospholipid classes [45].
Various chemical carcinogen [e.g., 7,12-dimethylbenz (α) anthracene]-induced tumor models were used to determine effect of w-3 FAs or ration of w-6/w-3 FAs on tumor growth, tumor burden, and apoptotic index. As seen in these models, tumor burden and tumor multiplicity went down simultaneously increasing apoptotic index after w3-containing feed. Mechanistically, oncogenic pathway proteins such as HER2, pAkt, and bcl2 were downregulated, and pro-apoptotic proteins such as Bax and p53 were upregulated [45].
Conclusion
Effects of w-3 PUFA are mediated by displacing AA in the cell membrane phospholipids and inhibiting pro-inflammatory, pro-angiogenic, and pro-metastatic AA eicosanoids. Lipid peroxidation and oxidative stress are the possible major mechanisms for w-3 PUFA-induced cell death. EPA and DHA are shown to attenuate pro-survival and anti-apoptotic pathways which results in inhibition of cancer cell proliferation and induction of apoptosis. EPA and DHA both act as immune modulators controlling activity of tumor-associated immune cells and microenvironment.
Figure 12.5 summarizes various signaling pathways mitigated by high w-3/w-6 PUFA ratio.

High w-3/w-6 FA ratio downregulates cell survival and proliferation by inducing apoptosis or autophagy, and alters tumor cell metabolism and tumor microenvironment by immunomodulation. It restricts neo-angiogenesis and metastasis. Downregulation of inflammatory mediators in favor of tumor mass reduction and induction of apoptosis in cancer cells seems to be important mechanism
Abbreviations
- ALX/FPR2:
-
Lipoxin A4 receptor/formyl peptide receptor 2 (ALX/FPR2)
- AP1:
-
Activator protein 1
- Bak:
-
Bcl-2 homologous antagonist killer
- BAX:
-
Bcl-2-associated X protein
- bcl-2:
-
B-cell lymphoma 2
- BCRP:
-
Breast cancer resistance protein
- bFGF:
-
Basic fibroblast growth factor
- BLT1:
-
Leukotriene B4 receptor 1
- Bv8:
-
Prokineticin-2
- CCL2:
-
Chemokine (C–C motif) ligand 2
- CCR5:
-
C–C chemokine receptor type 5
- ChemR23:
-
Chemerin Receptor 23
- COX:
-
Cyclooxygenase
- cPLA2:
-
Cytosolic phospholipases A2
- CXCL12:
-
C–X–C motif chemokine 12
- CYP450:
-
Cytochrome P450
- DCA:
-
Dichloroacetate
- DHA:
-
Docosahexaenoic acid
- DRM:
-
Detergent-resistant membranes
- EDPs:
-
Epoxydocosapentaenoic acids
- EETs:
-
Epoxyeicosatrienoic acids
- EEQs:
-
Epoxyeicosatetraenoic acids
- eFoX:
-
Electrophile oxo-derivative
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- EGFR:
-
Epithelial growth factor receptor
- EPA:
-
Eicosapentaenoic acid
- EMT:
-
Epithelial–mesenchymal transition
- ER:
-
Estrogen receptor
- ETC:
-
Electron transport chain
- FA:
-
Fatty acids
- FASN:
-
Fatty acid synthase
- GPR120:
-
G protein-coupled receptor 120
- GPR32:
-
G protein-coupled receptor 32
- HEPEs:
-
Hydroxyeicosapentaenoic acids
- HER-2:
-
Human epidermal growth factor receptor 2
- HETEs:
-
Hydroxyeicosatetraenoic acids
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia-inducible factor
- HMGB1:
-
High-mobility group box-1
- HMGCoAR:
-
3-hydroxy-3methylglutaryl-coenzyme A reductase
- HUVECs:
-
Human umbilical vein endothelial cells
- HEPE:
-
Hydroxyeicosapentaenoic acid
- ICAM-1:
-
Intercellular adhesion molecule 1
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- IL-1:
-
Interleukin 1
- IL-10:
-
Interleukin 10
- IL-6:
-
Interleukin 6
- iNOS:
-
Inducible nitric oxide synthase
- JNK:
-
c-Jun N-terminal kinas
- LA:
-
Linoleic acid
- LC-PUFA:
-
Long-chain polyunsaturated fatty acid
- LOX:
-
Lipoxygenase
- LTB4:
-
Leukotriene B4
- MAPKs:
-
Mitogen-activated protein kinases
- MCP-1:
-
Monocyte chemotactic protein 1
- M-CSF:
-
Macrophage colony-stimulating factor
- MMPs:
-
Matrix metalloproteases
- MRPs:
-
Multidrug resistance-related proteins
- MTP:
-
Mitochondrial permeability transition
- w-3:
-
Omega-3
- w-6:
-
Omega-6
- NF-kB:
-
Nuclear factor-kappaB
- NO:
-
Nitric oxide
- nrf2:
-
Nuclear factor (erythroid-derived 2)-like 2
- OSM:
-
Oncostatin M
- PCNA:
-
Proliferating cell nuclear antigen
- PDGF:
-
Platelet-derived growth factor
- PDH:
-
Pyruvate dehydrogenase
- PDK:
-
Pyruvate dehydrogenase kinase
- Pgp/ABCB1:
-
P-glycoprotein/ATP-binding cassette subfamily B member
- PI3K/mTOR:
-
Phosphoinositide 3-kinase/mammalian target for rapamycin
- PKC:
-
Protein kinase C
- PMN:
-
Polymorphonuclear
- PPARγ:
-
Peroxisome proliferator-activated receptors
- PPREs:
-
PPAR response elements
- PUFA:
-
Polyunsaturated fatty acids
- ROS:
-
Reactive oxygen species
- RXR:
-
Retinoid X receptor
- SOCI:
-
Store-operated calcium influx
- SREBP1:
-
Sterol regulatory element-binding protein-1
- STAT-3:
-
Signal transducer and activator of transcription 3
- TAK1:
-
Transforming growth factor β-activated kinase-1
- TAM:
-
Tumor-associated macrophages
- TAN:
-
Tumor-associated neutrophils
- TCA cycle:
-
Tricarboxylic acid cycle
- TGF-β:
-
Transforming growth factor-beta
- TNF α:
-
Tumor necrosis factor-alpha
- VCAM-1:
-
Vascular cell adhesion protein 1
- VEGF:
-
Vascular endothelial growth factor
- ALA:
-
α-linolenic acid
References
Stephenson JA, Al-Taan O, Arshad A, Morgan B, Metcalfe MS, Dennison AR. The multifaceted effects of omega-3 polyunsaturated fatty acids on the hallmarks of cancer. J Lipids. 2013;2013 (Article ID 261247).
Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15:425.
Anand P, Kunnumakkara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25(9):2097–116.
Nathalie V, Lajoie-Mazenc I, Auge N, Suc I, Frisach MF, et al. Activation of epithelial growth factor receptor pathway by unsaturated fatty acids. Circ Res. 1999;85:892–9.
Ries A, Trottenberg P, Elsner F. A systematic review on the role of fish oil for the treatment of cachexia in advanced cancer: an EPCRC cachexia guidelines project. Palliat Med. 2012;26:294–304.
Giessman H, Johnson JI, Kogner P. Omega-3 fatty acids in cancer, the protectors of good and the killers of evil? Exp Cell Res. 2010;316:1365–73.
Hanahan D, Weinberg RA. The hallmarks of cancer review. Cell. 2000;100:57–70.
Signori C, El-Bayoumy K, Russo J, Thompson HJ, Richie JP, Hartman TJ, et al. Chemoprevention of breast cancer by fish oil in preclinical models: trials and tribulations. Cancer Res. 2011;71(19):1–6.
Erickson KL, Hubbard NE. Fatty acids and breast cancer: the role of stem cells. Prostaglandins LeukotEssent Fatty Acids. 2010;82:237–41.
Pauwels EK, Kairemo K. Fatty acid facts, part II: role in the prevention of carcinogenesis, or, more fish on the dish? Drug News Perspect. 2008;21:504–10.
Tapiero H, Ba GN, Couvreur P, Tew KD. Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed Pharmacother. 2002;56(5):215–22.
Elaine HW, Munoz J Jr, Cameron I. Role of lipid peroxidation and antioxidant enzymes in omega 3 fatty acids induced suppression of breast cancer xenograft growth in mice. Cancer Cell Int. 2002;2:10.
Fukui M, Kang KS, Okada K, Zhu BT. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: role of ros accumulation, caspase-8 activation, and autophagy induction. J Cell Biochem. 2012;114(1):192–203.
Epstein MM, Kasperzyk JL, Mucci LA, Giovannucci E, Price A, Wolk A, et al. Dietary fatty acid intake and prostate cancer survival in Örebro County, Sweden. Am J Epidemiol. 2012;176(3):240–52.
Fernandez E, Chatenoud L, La Vecchia C, Negri E, Franceschi S. Fish consumption and cancer risk. Am J Clin Nutr. 1999;70(1):85–90.
Caygill CPJ, Hill MJ. Fish n-3 fatty acids and human colorectal and breast cancer. Eur J Cancer Prev. 1995;4:329–32.
Courtney ED, et al. Eicosapentaenoic acid (EPA) reduces crypt cell proliferation and increases apoptosis in normal colonic mucosa in subjects with a history of colorectal adenomas. Int J Colorectal Dis. 2007;22(7):765–76.
Chang WC, Chapkin RS, Lupton JR. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18(4):721–30.
Hong MY, Chapkin RS, Barhoumi R, et al. Fish oil increases mitochondrial phospholipid unsaturation, upregulating reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis. 2002;23:1919–25.
Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, et al. The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis. 2005;26(11):1914–21.
Shin S, Jing K, Jeong S, Kim N, Song K-S, Heo J-Y, et al. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-Mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p 53. BioMed Res Int. 2013;2013 (Article ID 568671).
Bingham SA, Day NE, Luben R, et al. Dietary fibre in food and protection against colorectal cancer in European prospective investigation into cancer and nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496–501.
Kolar SSN, Barhoumi R, Lupton JR, Chapkin RS. Docosahexaenoic acid and butyrate synergistically induce colonocyte apoptosis by enhancing mitochondrial Ca2+ accumulation. Cancer Res. 2007;67:5561–8.
Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, et al. The role of docosahexaenoic acid mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis. 2005;26:1914–21.
Nutt LK, Chandra J, Pataer A, et al. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem. 2002;277:20301–8.
Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999;18(22):6349–61.
Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86(1):369–408.
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer related inflammation. Nature. 2008;454:436–44.
Griennikov SI, Greten FR, Karin M. immunity inflammation and cancer. Cell. 2010;140(6):883–99.
Fiala M. Curcumin and omega-3 fatty acids enhance NK cell-induced apoptosis of pancreatic cancer cells but curcumin inhibits interferon-γ production: benefits of omega-3 with curcumin against cancer. Molecules. 2015;20(2):3020–6.
Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11.
Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res. 2008;47(2):147–55.
Abedi E, Sahari MA. Long-chain polyunsaturated fatty acid sources and evaluation of their nutritional and functional properties. Food Sci Nutr. 2014;2(5):443–63.
Wahli W, et al. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23(7):351–63.
Rogers KR, Kikawa KD, Mouradian M, Hernandez K, McKinnon KM, Ahwah SM. Docosahexaenoic acid alters epidermal growth factor receptor related signaling by disrupting its lipid raft association. Carcinogenesis. 2010;31(9):1523–30.
Karmali RA, Reichel P, Cohen LA, Terano T, Hirai A, Tamura Y, et al. The effects of dietary omega-3 fatty acids on the DU-145 transplantable human prostatic tumor. Anticancer Res. 1987;7:1173–80.
Galli C, Calder PC. Effects of fat and fatty acids intake on inflammatory and immune responses. A critical review. Ann Nutr Metab. 2009;55:123–39.
Fradet V, Cheng I, Casey G, Witte JS. Dietary omega3 fatty acids, cyclooxygenase-2 genetic variation, and aggressive prostate cancer risk. Clin Cancer Res. 2009;15(7):2559–66.
Wang D, DuBois RN. The role of the PGE2–aromatase pathway in obesity-associated breast inflammation. Cancer Discov. 2012;2(4):308–10.
Surh Y-J, Chun K-S, Cha H-H, Han SS, Keum Y-S, Park K-K, Lee SS. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat Res Fundam Mol Mech Mutagen. 2001;480–481:243–68.
Brodie MH, Lu Q, Long BJ, Fulton A, Chen T, Macpherson N, et al. Aromatase and COX-2 expression in human breast cancers. J Steroid Biochem Mol Biol. 2001;79(1–5):41–7.
Brueggemeier RW, Quinn AL, Parrett ML, Joarder FS, Harris RE, Robertson FM. Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999;140(1–2):27–35.
Bhat H. Estrogen’s role in cancer. Columbia Univ Health Sci. 2003;2(10).
Liu J, Ma DWL. The Role of n-3 polyunsaturated fatty acids in the prevention and treatment of breast cancer. Nutrients. 2014;6(11):5184–223.
Narayanan BA, Narayanan NK, Simi B, Reddy BS. Modulation of inducible nitric oxide synthase and related proinflammatory genes by the omega-3 fatty acid docosahexaenoic acid in human colon cancer cells. Cancer Res. 2003;63:972–9.
Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin. 2010;39:401–18.
Gleissman H, Yang R, Martinod K, Lindskog M, Serhan CN, Johnsen JI, et al. Docosahexaenoic acid metabolome in neural tumors: identification of cytotoxic intermediates. FASEB J. 2010;24(3):906–15.
Narayanan NK, Narayanan BA, Reddy BS. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-kappaB, and steroid hormone receptors. Int J Oncol. 2005;26(3):785–92.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47.
Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, et al. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–41.
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin sensitizing effect. Cell. 2010;142(5):687–98.
Liu Z, Hopkins MM, Zhang Z, Quisenberry CB, Fix LC, Galvan BM, et al. Omega-3 fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J Pharmacol Exp Ther. 2015;352:380–94.
Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and pro-resolving lipid mediators. Annu Rev Pathol. 2008;3:279–312.
Lee HJ, Park MK, Lee EJ, Lee CH. Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int J Biochem Cell Biol. 2013;45(12):2801–7.
Hutchinson JM, Volpato M, Loadman P, Nicolaou A, Hull M. Neoplasia and cancer pathogenesis PWE-163 Chemr23 and BLT1 receptor expression in colorectal cancer. Gut. 2013;62:A196–7.
Cockbain AJ, Toogood GJ, Hull MA. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2012;61(1):135–49.
Calder PC, Yaqoob P. Lipid rafts—composition, characterization, and controversies. J Nutr. 2007;137(3):545–7 (American Society for Nutrition).
Anchisi L, Dessi S, Pani A, Mandas A. Cholesterol homeostasis: a key to prevent or slow down neurodegeneration. Frontiers. 2012;3 (Article 486).
Blanckaert V, Ulmann L, Mimouni V, Antol J, Brancquart L, Chénais B. Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int J Oncol. 2010;36:737–42.
Hawk ET, Viner JL, Dannenberg A, DuBois RN. COX-2 in cancer—a player that’s defining the rules. J Natl Cancer Inst. 2002;94:545–6.
McCormick DL, Rao KV, Steele VE, Lubet RA, Kelloff GJ, Bosland MC. Chemoprevention of rat prostate carcinogenesis by 9-cis-retinoic acid. Cancer Res. 1999;59:521–4.
Rao KV, Johnson WD, Bosland MC, Lubet RA, Steele VE, Kelloff GJ, et al. Chemoprevention of rat prostate carcinogenesis by early and delayed administration of dehydroepiandrosterone. Cancer Res. 1999;59:3084–9 (1999).
Swamy MV, Cooma I, Patlolla JM, Simi B, Reddy BS, Rao CV. Modulation of cyclooxygenase-2 activities by the combined action of celecoxib and decosahexaenoic acid: novel strategies for colon cancer prevention and treatment. Mol Cancer Ther. 2004;3:215–21.
Wu M, Harvey KA, Ruzmetov N, Welch ZR, Sech L, Jackson K, et al. Omega-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int J Cancer. 2005;117:340–8.
Flock MR, Harris WS, Kris-Etherton PM. Long-chain omega-3 fatty acids: time to establish a dietary reference intake. Nutr Rev. 2013;71(10):692–707.
Schley PD, Brindley DN, Field CJ. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J Nutr. 2007;548–53.
Corsetto PA, GigliolaMontorfano SZ, Jovenitti IE, Cremona A, Berra B, et al. Effects of n-3 PUFAs on breast cancer cells through their incorporation in plasma membrane. Lipids Health Dis. 2011;10:73.
Kang KS, Wang P, Yamabe N, Fukui M, Jay T, Zhu BT. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PLoS One. 2010;5(4):e10296.
Mobraten K, Haug TM, Kleiveland CR, Lea T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis. 2013;12:101.
Rahman MM, Veigas M, Williams PJ, Fernandes G. DHA is a more potent inhibitor of breast cancer metastasis to bone and related osteolysis than EPA. Breast Cancer Res Treat. 2013;141(3). doi:10.1007/s10549-013-2703-y.
Calviello G, Palozza P, Di Nicuolo F, Maggiano N, Bartoli GM. n–3 PUFA dietary supplementation inhibits proliferation and store-operated calcium influx in thymoma cells growing in Balb/c mice. J Lipid Res. 2000;41:182–8.
Menendez JA, Ropero S, Mehmi I, Atlas E, Colomer R, Lupu R. Overexpression and hyperactivity of breast cancer-associated fatty acid synthase (oncogenic antigen-519) is insensitive to normal arachidonic fatty acid-induced suppression in lipogenic tissues but it is selectively inhibited by tumoricidal alpha-linolenic and gamma-linolenic fatty acids: a novel mechanism by which dietary fat can alter mammary tumorigenesis. Int J Oncol. 2004;24(6):1369–83.
Elaine Hardman W. Omega-3 fatty acids to augment cancer therapy. american society for nutritional sciences. Int Res Conf Food Nutr Cancer. 2002;132:3508S–12S.
Baracos VE, Mazurak VC, Ma DWL. n-3 polyunsaturated fatty acids throughout the cancer trajectory: influence on disease incidence, progression, response to therapy and cancer-associated cachexia. Nutr Res Rev. 2004;17:177–92.
Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27(50):6398–406.
Spencer L, Mann C, Metcalfe M, Webb MB, Pollard C, Spencer D, et al. The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur J Cancer. 2009;45:2077–86.
Vakkila L. Inflammation and necrosis promote tumor growth. Nat Rev Immunol. 2004;4:641–8.
Tang D, Kang R, Zeh HJ III, Lotze MT. High-mobility Group box 1 [HMGB1] and cancer. Biochim Biophys Acta. 2010;1799(1–2):131.
Joyce JA, Pollard JW, et al. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52.
Zhang G, Panigrahy D, Mahakiane LM, Yang J, Liu J-Y, Leea KSS, et al. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. PNAS. 2013;110(16):6530–35.
Calviello G, Serini S. Dietary omega-3 polyunsaturated fatty acids and cancer. Diet Cancer Ser. 2010;1.
Rose DP, Connolly JM, Coleman M. Effect of omega-3 fatty acids on the progression of metastases after the surgical excision of human breast cancer cell solid tumors growing in nude mice. Clin Cancer Res. 1996;2:1751–6.
Merendino N, Costantini L, Manzi L, Molinari R, D’Eliseo D, Velotti F. Dietary ω-3 polyunsaturated fatty acid DHA: a potential adjuvant in the treatment of cancer. BioMed Res Int. 2013;11 pages (ArticleID310186).
Li CC, Hou YC, Yeh CL, Yeh SL. Effects of eicosapentaenoic acid and docosahexaenoic acid on prostate cancer cell migration and invasion induced by tumor-associated macrophages. PLoS ONE. 2014;9(6):e99630.
Cunningham-Rundles S. Is the fatty acid composition of immune cells the key to normal variations in human immune response? Am J Clin Nutr. 2003;77(5):1096–7.
Mantovani A. Macrophages, neutrophils, and cancer: a double edged sword. New J Sci. 2014;2014:14 pages (Article ID 271940).
Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology; 2013.
Menendez JA, Lupu R, Colomer R. Exogenous supplementation with omega-3 polyunsaturated fatty acid docosahexaenoic acid (DHA; 22, 6n-3) synergistically enhances taxane cytotoxicity and downregulates Her-2/neu (c-erbB-2) oncogene expression in human breast cancer cells. Eur J Cancer Prev. 2005;14:263–70.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9.
Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood. 1999;94:2102–11.
Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88:1052–61.
Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–53.
Calviello G, Di Nicuolo F, Serini S, Piccioni E, Boninsegna A, Maggiano N. Docosahexaenoic acid enhances the susceptibility of human colorectal cancer cells to 5-fluorouracil. Cancer Chemother Pharmacol. 2005;55:12–20.
Zhou Z, Zhang L, Mu Q, Lou Y, Gong Z, Shi Y, et al. The effect of combination treatment with docosahexaenoic acid and 5-fluorouracil on the mRNA expression of apoptosisrelated genes, including the novel gene BCL2L12, in gastric cancer cells. In Vitro Cell Dev Biol Anim. 2009;45:69–74.
Guffy MM, North JA, Burns CP. Effect of cellular fatty acid alteration on adriamycin sensitivity in cultured L1210 murine leukemia cells. Cancer Res. 1984;44:1863–6.
Zijlstra JG, de Vries EG, Muskiet FA, Martini IA, Timmer-Bosscha H, Mulder NH. Influence of docosahexaenoic acid in vitro on intracellular adriamycin concentration in lymphocytes and human adriamycin-sensitive and -resistant small-cell lung cancer cell lines, and on cytotoxicity in the tumor cell lines. Int J Cancer. 1987;40:850–6.
Das UN, Madhavi N, Sravan Kumar G, Padma M, Sangeetha P. Can tumour cell drug resistance be reversed by essential fatty acids and their metabolites? Prostaglandins Leukot Essent Fatty Acids. 1998;58:39–54.
Vibet S, Goupille C, Bougnoux P, Steghens JP, Gore J, Maheo K. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free Radic Biol Med. 2008;44(7):1483–91.
Gelsomino G, et al. Omega 3 fatty acids chemosensitize multidrug resistant colon cancer cells by down-regulating cholesterol synthesis and altering detergent resistant membranes composition. Mol Cancer. 2013;12:137.
Maheo K, Vibet S, Steghens JP, Dartigeas C, Lehman M, Bougnoux P, et al. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free Radic Biol Med. 2005;39:742–51.
Tjandrawinata RR, Dahiya R, Hughes-Fulford M. Induction of cyclo-oxygenase-2 mRNA by prostaglandin E2 in human prostatic carcinoma cells. Br J Cancer. 1997;75:1111–8.
Taketo MM. Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst. 1998;90:1529–36.
Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, et al. Cyclo-oxygenase 2, a pharmacological target for the prevention of cancer. Lancet Oncol. 2001;2:544–51.
Saw CL, Huang Y, Kong AN. Synergistic antiinflammatory effects of low doses of curcumin in combination with polyunsaturated fatty acids: docosahexaenoic acid or eicosapentaenoic acid. Biochem Pharmacol. 2010;79:421–30.
Elaine Hardman W, Reddy Avula CP, Fernandes G, Cameron IL. Three percent dietary fish oil concentrate increased efficacy of doxorubicin against MDA-MB 231 breast cancer xenografts. Clin Cancer Res. 2001;7:2041.
MacLennan MB, Clarke SE, Perez K, Wood GA, Muller WJ, Kang JX, et al. Mammary tumor development is directly inhibited by lifelong n-3 polyunsaturated fatty acids. J Nutr Biochem. 2013;24(1):388–95.
Acknowledgments
Authors would like to thank Mr. Aniket Mali for the illustration in Fig. 12.1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Joshi, A.A., Hegde, M.V., Adekar, S.P. (2016). Omega-3 Fatty Acids in Cancer: Insight into the Mechanism of Actions in Preclinical Cancer Models. In: Hegde, M., Zanwar, A., Adekar, S. (eds) Omega-3 Fatty Acids. Springer, Cham. https://doi.org/10.1007/978-3-319-40458-5_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-40458-5_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40456-1
Online ISBN: 978-3-319-40458-5
eBook Packages: MedicineMedicine (R0)