
Abstract
The highly adaptive opportunistic human pathogen, Staphylococcus aureus, contains a large number of integral membrane transport proteins important in homeostasis and pathogenesis. Included in this group are multidrug efflux pumps which play a role in antimicrobial resistance by reducing the intracellular concentration of drug compounds through active extrusion from the cell. S. aureus encodes many such drug efflux proteins, which fall into four out of the six currently recognized drug transporter families. These efflux pumps can confer host resistance against a vast number of clinically relevant antimicrobials, including macrolides, quinolones, streptogramins, and tetracyclines, as well as biocides such as biguanidines, diamidines, and quaternary ammonium compounds. The prevalence of these drug efflux determinants, either chromosomally or plasmid encoded, has been established worldwide, with clinical isolates expressing numerous multidrug transport proteins being continually identified. In addition to the characterized drug transporters, the recent surge in sequencing has also revealed a number of putative drug efflux proteins; some of these may share characteristics of known pumps, but, as seen with the recent addition of the proteobacterial antimicrobial compound efflux family, may represent proteins which are unique.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Staphylococcus aureus
- Multidrug resistance
- Drug efflux pump
- Sav1866
- NorA
- NorB
- NorC
- MgrA
- MepA
- MepR
- QacA
- QacB
- QacC
- QacR
1 Introduction
Identified to be a major cause of both nosocomial- and community-acquired infections, Staphylococcus aureus is a highly adaptive opportunistic bacterium that displays a wide spectrum of pathogenicity, largely owed to its ability to acquire mobile genetic elements encoding virulence and resistance determinants [1, 2]. This adaptive power has given rise to the emergence of methicillin-resistant S. aureus (MRSA) and continues to contribute to the development of new strains resistant against a number of antimicrobial drugs, including one of the last resort drugs, vancomycin [3, 4]. Resistance genes carried on plasmids, transposons, and other mobile elements can be acquired by S. aureus from other Gram-positive bacteria through the process of horizontal gene transfer and genetic recombination, arming it with determinants such as vanA, which originated in enterococci, but now contributes to vancomycin resistance in S. aureus [5, 6].
History has shown that staphylococci are able to act against each new antimicrobial by employing at least one resistance mechanism. These mechanisms include enzymatic modification or destruction of the drug, alteration of the drug target by mutation, enzymatic inactivation resulting in reduced affinity for the drug, and extruding the drug from the cell via an efflux protein [7, 8]. Of these, drug efflux appears to be one of the most widespread resistance mechanisms. In Gram-positive bacteria, such as S. aureus which lack an outer membrane, efflux pumps are vital in limiting the accumulation of toxic compounds within the cell. Found in all living organisms, these proteins fulfill numerous physiological functions including the expulsion and subsequent elimination of endogenous metabolites, bile salts, and host-defense molecules as well as the uptake of essential nutrients [9–12]. Their abundance and the variety of roles they fulfill suggest that drug efflux is a fortuitous event, which has helped to arm bacteria against most, if not eventually all, antimicrobials they come across.
Initially detected on plasmids in staphylococcal strains found to be highly resistant to antimicrobials [13], whole genome sequencing has allowed for the detection of a multitude of putative drug-specific and multidrug transporters. Recent studies analyzing chromosomally encoded multidrug efflux-like proteins from the genomes of three S. aureus strains, 8325, COL, and N315, identified 21 open reading frames encoding putative membrane efflux proteins [14]. Although experimental analysis revealed that overexpression of these did not produce resistance to common multidrug efflux substrates, it is still possible that one or more of these can extrude a yet untested compound and that other genes encoding drug efflux-like proteins, not identified by the method employed in the study, exist. To date there are close to 70 whole S. aureus genomes sequenced (the National Center for Biotechnology Information database: http://ncbi.nlm.nih.gov), but not all have been analyzed, leaving the possibility that there are a number of genes encoding novel drug efflux pumps which are yet to be assessed.
Although the number of identified multidrug transporters continues to increase, experimental structural analysis of these hydrophobic membrane-bound proteins such as X-ray crystallography and nuclear magnetic (NMR) spectroscopy is limited by numerous factors and hampered by the experimental challenges which are inherent to such analyses [15, 16]. As a result of these, biochemical analysis such as site-directed mutagenesis, where residues are substituted with other amino acids carrying a different charge or side-chain volume, is carried out in order to assess their importance in function and/or structure. Such analyses, coupled with computational determination of the three-dimensional structure of a protein, have become essential for both functional and structural analysis of membrane transporters.
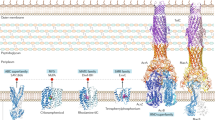
Falling into six different transport protein superfamilies/families (Fig. 7.1), and based on their energy requirements, drug transport proteins can be termed either as primary or secondary transporters. Primary transporters include the ATP-binding cassette (ABC) superfamily, while secondary transporters include the major facilitator superfamily (MFS), the resistance-nodulation-cell division (RND) superfamily, the multidrug and toxic compound extrusion (MATE) family, the small multidrug resistance (SMR) family, and the newly discovered proteobacterial antimicrobial compound efflux (PACE) protein family [17–21]. Although some can be specific for a single drug, numerous membrane transporters can extrude a broad range of structurally diverse antimicrobial compounds and as such are designated multidrug efflux pumps. Both specific and multidrug transport proteins have been identified in S. aureus and at a protein level are indistinguishable (Table 7.1). Of these, characterized membrane transporters within S. aureus include the chromosomally encoded LmrS [35], NorA [38, 39], NorB [42], NorC [43], SdrM [49], MdeA [36, 37], MepA [55], Sav1866 [28], and Tet38 [42], as well as the plasmid-encoded TetA(K) [50], TetA(L) [53], QacA [67], QacB [68], QacC [58], QacJ [61], and QacG transporters [69]. Assessment of these transporters has shown that they are capable of transporting a wide range of substrates and in most cases show an overlap in their substrate profiles.

Diagrammatic representation of transporters from the six major drug transporter superfamilies or families. The representative transport systems are shown along with their energy-coupling mechanisms (i.e., ATP hydrolysis for the ABC superfamily export system and the use of electrochemical gradients for the remaining systems). The export systems classified within the ABC, MFS, SMR, MATE, and PACE families typically transport their substrates (drug, green oval) across the cytoplasmic membrane. However, RND transporters as well as some MFS and ABC systems assemble with periplasmic- and outer membrane-bound proteins to form tripartite systems (in Gram-negative bacteria) that are able to expel substrates across both the cytoplasmic and outer membranes
This chapter summarizes the most recent data on the best characterized multidrug transporter that is a representative from within each transport family identified within S. aureus. These multidrug efflux pumps are the ABC superfamily Sav1866 protein, the MFS QacA/QacB proteins, the MATE family MepA protein, and the SMR QacC protein. Although RND membrane transporters have been identified in S. aureus, including a Mmpl homologue transporter [70] and the SecDF accessory factor to the Sec protein translocation machinery [71], none have been shown to be directly involved in multidrug efflux and as such will not be included in this review. By focusing on a representative of each family/superfamily, the mechanism of transport, substrate profile, structure, and regulation will be discussed.
2 Primary Active Transporters
2.1 The ATP-Binding Cassette Superfamily
Consisting of both uptake and efflux transport systems, ABC transporters represent one of the largest superfamilies of proteins known in both prokaryotic and eukaryotic organisms [72, 73]. Proteins within this family have been shown to import or export a wide range of substrates, including, among others, amino acids, sugars, and lipids [74, 75]. This substrate diversity is mirrored by the myriad physiological roles that ABC transporters play in the cell. These include nutrient uptake, elimination of waste products from the cell, and export of cellular components such as cell wall polysaccharides [76, 77]. Found in all species, their importance is exemplified by the number of proteins certain species express. For example, in Escherichia coli, almost 5 % of its genomic coding capacity is composed of genes encoding ABC transporters [78]. Analysis of ABC transporters has also shown some of them to be medically important, including those involved in cystic fibrosis, various eye diseases, liver disease, and multidrug resistance in cancer cells, among others [79].
Analyses of crystal structures of ABC transporters such as BtuCD, an E. coli vitamin B12 importer [76, 80]; HI1470/1, a metal-chelate transporter from Haemophilus influenzae [81]; and the high-resolution structure of the S. aureus Sav1866 multidrug transporter [82] show that they follow a basic organization which includes two transmembrane domains (TMDs) which act as the substrate-translocation pathway and the nucleotide-binding domains (NBDs), which bind and hydrolyze ATP [83, 84]. Organized as either heterodimers or homodimers, the NBD and TMD subunits can be encoded separately, or as a single polypeptide, and can be arranged with a carboxyl-terminal NBD and an amino-terminal TMD or vice versa [83]. In contrast to the TMDs, NBDs are homologous throughout the ABC superfamily and contain several motifs which are characteristic of this family. These include the Walker A and B motifs, which are present in many nucleotide-binding proteins, motif C which is specific to the ABC superfamily [74] and the stacking aromatic D, H, and Q loops [77].
Structural studies have revealed that the TMDs, containing the substrate-binding sites, are composed of multiple α-helical transmembrane segments (TMSs) that form the transmembrane channel. The TMSs extend, in many cases, into the cytosol where they fold and form a physical interface with the NBDs [85]. The majority of ABC membrane transporters are predicted to have 6 TMSs per domain and function as homodimers, with each transport complex containing 12 TMSs in total. Although exceptions to this rule exist, most ABC transporters conform to this two-times-six TMS paradigm [86].
Despite representing the largest of the six drug transporter families, the number of ABC drug membrane proteins in S. aureus which have been characterized remains low. As mentioned above, the Sav1866 membrane transporter, representing the first staphylococcal drug transporter for which a structure has been determined [82], remains the best characterized of all the currently identified drug membrane transporters and is discussed in more detail below. Msr(A), initially identified on a macrolide resistance plasmid from Staphylococcus epidermidis, is a 488-amino acid protein which confers resistance to 14- and 15-membered ring macrolides, type B streptogramins, and telithromycin, among others [87]. Msr(A) shares high homology with Msr(C) and Msr(D), which are characterized by two fused NBDs but lack identifiable TMDs [88]. Substrate efflux is thought to occur from an interaction of its NBDs with TMDs of other chromosomally encoded ABC transporters [25]. It has also been proposed that Msr(A) does not function as a transport protein but confers resistance though a different mechanism. One possibility is that Msr(A) disassociates erythromycin, from the ribosome by inducing a conformational change which would allow the antibiotic to passively diffuse out of the cell [89].
Displaying a low amino acid sequence identity of 35 % to Msr(A), the Vga(A) protein is also plasmid encoded and, along with the variant Vga(A), and the Vga(B) and Vga(C) proteins, mediates resistance against type A streptogramins, lincosamides, and pleuromutilins [30, 31, 33]. Like Msr(A), the Vga proteins have two NBDs but lack the TMDs, which suggests that these proteins utilize a similar mechanism of transport [13].
Finally, representing the newest addition to the S. aureus ABC transporters is the AbcA multidrug resistance protein [22, 64]. Displaying high similarity to ABC multidrug transporters such as the Lactococcus lactis LmrA, E. coli MsbA, and the S. aureus Sav1866 protein, overexpression of this transporter results in resistance against β-lactams, daptomycin, moenomycin, and dyes such as ethidium [64].
2.2 The ABC Transporter: Sav1866
The S. aureus Sav1866 multidrug transporter was the first ABC membrane transporter structure to be determined at a high resolution of 3.0 Å [28] and is used as a model for homologous human and bacterial ABC multidrug transporters [29, 90]. Although crystallized, little was known about Sav1866 and its role in transport. Functional studies have now revealed that Sav1866 can transport structurally unrelated substrates such as ethidium, tetraphenylphosphonium, and Hoechst 33342 in intact cells as well as from plasma membrane vesicles and proteoliposomes containing purified and functionally reconstituted protein [29].
Following the two-times-six TMS paradigm mentioned above, the solved crystal structure of Sav1866 revealed that this protein consisted of a dimer comprised of two elongated subunits, with each subunit containing an amino-terminal TMD and a carboxyl-terminal NBD [28]. Sav1866 is referred to as a homodimer of half transporters [91], as one TMD is fused to a NBD, which then dimerizes to form a full membrane transporter [28]. These two subunits appear to twist and embrace each other, with the NBD and TMD interacting tightly (Fig. 7.2) [28]. Adopting an outward-facing conformation, the TMD face outward, toward the exterior of the cell, while the NBD subunits are shown to be closely associated and pack around the nucleotide [84]. The two TMDs provide a substrate-translocation pathway, while the two NBDs are the involved in ATP binding and hydrolysis [82].

The Sav1866 structure. The Sav1866 structure (PDB code 2HYD) is shown in an outward-facing orientation from the front and back. The two monomers of the half-transporter are colored blue and green and bound nucleotide is shown in a purple ball and stick representation
The exact mechanisms by which multidrug ABC transporters like Sav1866 export their substrates are still unclear, although analysis of crystal structures from members in both inward and outward conformations that are available for this superfamily has presented a possible mechanism for coupling ATP hydrolysis to transport [92]. Occurring in an outward-open conformation, the hydrolysis of ATP results in the TMDs reorienting to produce an inward-open conformation of the protein, which allows substrate and ATP binding to occur. Substrate extrusion then is mediated by the interaction of substrate and TMDs, which initiates conformational changes in both the TMDs and the NBDs, leading to an outward-open conformation. The substrate is then expelled and the transporter is reset for further transport [93, 94]. More structural, biochemical, and biophysical studies are required to elucidate the nature of these conformational changes employed by ABC transporters.
3 Secondary Active Transporters
3.1 The Major Facilitator Superfamily
The MFS is the largest family of secondary transporters, catalyzing the transport of a diverse range of substrates [95, 96]. As found for ABC transporters, proteins belonging to this superfamily are ubiquitously found in the membranes of all living cells [97, 98], making up ~25 % of all known transport membrane proteins in prokaryotes [96]. These proteins can act as uniporters, symporters, or antiporters, harnessing the power in the electrochemical gradient of H+ or Na+ ions to transport amino acids, peptides, sugars, vitamins, and drugs, among others [99]. This continually expanding superfamily is currently composed of 82 recognized subfamilies, although most of these have been classified, 17 MFS families are composed of as of yet functionally uncharacterized members and are referred to as unknown major facilitators [98].
Well-defined multidrug transporters belong primarily to three different drug:H+ antiporter (DHA) subfamilies; DHA1, DHA2, and DHA3. Proteins belonging to the DHA1 and DHA2 subfamilies are known to efflux a wide range of structurally dissimilar compounds and have been demonstrated to play a major role in bacterial multidrug resistance [100], while members of the DHA3 subfamily, found only in prokaryotes, are shown to efflux antibiotics such as macrolides and tetracyclines [9]. Consisting of between 400 and 600 amino acids, allocation of drug transporters to these subfamilies is primarily based on the number of TMSs. Proteins within the DHA1 and DHA3 subfamily have their amino acids arranged into 12 TMSs, while those belonging to DHA2 are comprised of 14 TMSs [98, 101]. These TMSs are connected by hydrophilic loops, with both the N- and C-termini facing the cytoplasm [102]. Members with 12 TMSs are thought to have evolved from a single 2 TMS hairpin that triplicated, giving rise to a protein with 6 TMSs which through a duplication event resulted in the formation of 12 TMSs. MFS transporters with 14 TMSs are believed to have obtained two centrally localized TMSs as a result of an intragenic duplication event of an adjacent hairpin [98].
Like members of the ABC superfamily, sequence analysis of MFS transporters has identified a number of superfamily- and family-specific sequence motifs [17, 50, 97]. High conservation of these motifs within members implies that they play a role in structure and/or function. Among these motifs, motif C, specific to antiporters, is highly conserved between efflux proteins. Positioned in TMS 5 of all antiporters, it is believed that this motif may influence or specify the direction of substrate transport [103]. Analysis of this motif in the MFS S. aureus tetracycline resistance efflux protein TetA(K) revealed that a number of conserved residues within this motif are important for tetracycline transport [50], with glycine residues identified as conferring conformational plasticity required for drug efflux. However, the best characterized motif is motif A, also known as the MFS-specific motif [104]. Recent structural analysis of the MFS transporter YjaR has demonstrated that residues contained in motif A sense and respond to protonation inside the central cavity [104].
Although the structures of 23 different MFS transporters belonging to a variety of MFS subfamilies have been so far elucidated (http://blanco.biomol.uci.edu/mpstruc/), only two of these are of multidrug transporters. These are the E. coli EmrD multidrug transporter [105] and, solved more recently, the E. coli MdfA drug efflux protein [106]. The MdfA structure (2.0–2.4 Å) is also the first MFS multidrug transporter to be solved in complex with its substrates, chloramphenicol, deoxycholate and N-dodecyl-N,N-dimethylamine-N-oxide. The structure shows approximately 30 residues forming the substrate-binding cavity, of which the majority are shown to be hydrophobic. This is in line with the EmrD structure which also possesses an internal cavity of hydrophobic residues [105]. The crystal structures of these are reminiscent of other MFS-solved structures, such as those for the E. coli lactose permease (LacY) [107, 108] and glycerol-3-phosphate transporter (GlpT) [109], however, differ in the internal cavity, with LacY and GlpT maintaining a hydrophilic cavity [105].
Despite MFS proteins displaying low sequence similarity, different substrate specificity, and transport coupling mechanisms, MFS protein structures described to date exhibit some generalities [100]. Combining data achieved from the analysis of structures of MFS proteins in different states of the transport process, i.e., outward-facing, occluded, and inward-facing states, the mechanism for drug translocation has been postulated [105]. According to this model, substrate translocation is assumed to be facilitated by the interconversion of the inward-and an outward-facing alternating conformation of MFS transporters, using a “rocker-like” mechanism to alternately generate a pathway of access to either surface [105, 110, 111].
Representing the largest group of identified multidrug transporters within S. aureus, MFS members are also among the best characterized bacterial drug transporters [13]. Included, as shown in Table 7.1, is the NorA multidrug transporter, which was one of the first chromosomally identified and characterized staphylococcal MFS proteins [38, 112]. Conferring resistance to a wide range of chemically diverse compounds such as hydrophilic fluoroquinolones, dyes like ethidium bromide and biocides, this 388-amino acid protein is a 12-TMS member of the DHA1 subfamily [113]. In addition to the NorA protein, S. aureus contains MFS-family drug transporters such as the NorB, NorC, LmrS, QacA, SdrM, and MdeA multidrug resistance proteins and Tet38, a tetracycline-specific efflux pump (Table 7.1).
Sharing around 30 % similarity with NorA, the NorB efflux pump confers resistance to a number of compounds, some of which overlap with the NorA substrate profile, while others are NorB specific, such as moxifloxacin and sparfloxacin [42, 114]. Additionally, NorB has been shown to influence bacterial fitness and survival in abscesses alluding to the possibility that this pump may transport other as yet unidentified natural substrates [115]. Predicted to be comprised of 14 TMSs, this protein, along with NorC and Tet38, was identified in a S. aureus mgrA knockout mutant [42, 116]. MgrA is a global regulator of a number of proteins including the Nor multidrug efflux pumps and is discussed in detail below. Analysis of this mgrA knockout mutant revealed resistance to quinolones, tetracyclines, and antimicrobial compounds, which was not conferred by the NorA multidrug efflux protein. In addition to identifying NorB, this study identified the NorC multidrug efflux pump (61 % similarity with NorB), shown to contribute to quinolone resistance [43] and the 14-TMS Tet38 transporter (26 % similarity to the tetracycline exporter TetA(K)), that exports tetracycline, in addition to certain unsaturated fatty acids [65]. The Tet38 protein has also been shown to contribute to S. aureus colonization of mouse skin and aid in the ability of S. aureus to invade and survive within epithelial cells [65, 117].
Sharing the predicted 14-TMS topology identified for the above membrane transporters, the S. aureus MdeA, SdrM, and LmrS multidrug efflux pumps are all chromosomally encoded and display varying degrees of similarity to other MFS proteins. The MdeA protein, initially identified from the N315 clinical strain [36], when overexpressed confers resistance to quaternary ammonium compounds (QACs) and antibiotics. Sequence analysis shows that it is most closely related (62 % similarity) to the LmrB efflux protein from Bacillus subtilis [36]. The SdrM membrane transporter shares 61 % sequence similarity with QacA and has been shown to confer low-level resistance to dyes such as acriflavine and ethidium as well as to the fluoroquinolone, norfloxacin [49]. Finally, representing one of the more recent additions to the S. aureus MFS group of multidrug efflux pumps, the LmrS protein is involved in the increased resistance to chloramphenicol, linezolid, tetraphenylphosphonium, and trimethoprim [35].
3.2 The MFS transporters: QacA and QacB
The most widely studied multidrug efflux pumps in S. aureus are the chromosomally encoded NorA and the plasmid-encoded QacA and QacB membrane transporters [118]. The QacA multidrug efflux pump was the first bacterial multidrug efflux system to be discovered [67] and since has been extensively analyzed [13, 44, 68, 119, 120]. Initially identified in Australian multidrug-resistant strains of S. aureus, the qacA determinant was found to be carried on the pSK1 large multidrug resistance plasmid [67]. Classified within the DHA2 subfamily of the MFS, QacA consists of 514 amino acids that have been demonstrated to be organized into 14 TMSs (Fig. 7.3) [68]. Assessment of resistance has revealed that QacA confers resistance to a broad range of more than 30 cationic and lipophilic antimicrobials, including monovalent cationic dyes such as ethidium and pyronin Y, QACs such as benzalkonium and cetylpyridinium, and bivalent cations such as chlorhexidine and pentamidine [44–46]. QacA has been identified in clinically relevant S. aureus isolates across the world and has been shown to contribute to resistance to disinfectants that are widely used in medical environments and to a number of antiseptics in eye drop formulations [121, 122].

Schematic topological representation of the S. aureus QacA multidrug transporter. Topology of the QacA protein is based on hydropathy analysis. The 14 TMSs are indicated in gray, with residues found to be functionally important highlighted in red
QacA-mediated resistance has been extensively assessed, with results showing that QacA uses a single antiport mechanism for the expulsion of these structurally different compounds [45, 119, 121, 123, 124]. QacA-mediated efflux is dependent on both the Δψ and on the ΔpH components of the proton motive force [46] and is initiated within the inner leaflet of the cytoplasmic membrane, thus preventing the drugs from entering the cytoplasm [46]. Analyses of the kinetic parameters K m and V max have revealed that QacA-mediated transport of substrates adheres to Michaelis-Menten kinetics, with low K m values in the micromolar range for a range of QacA substrates indicating high binding affinity [46].
Closely related to the QacA membrane drug transporter and possibly a progenitor of QacA, the QacB plasmid-encoded protein was initially identified in clinical strains isolated in the early 1950s [125]. Differing by only seven base pairs, the qacB determinant primarily confers resistance against monovalent organic cations, thus exhibiting a more restricted substrate profile than QacA [46, 47, 125]. This difference in substrate specificity stems from an acidic residue at position 323 (Asp323) in TMS 10, which is an uncharged alanine in QacB, that affords the QacA membrane transporter the capacity to transport a wider range of substrates including bivalent cations, which QacB can only poorly transport [45, 46]. This acidic residue is proposed to play a role in the processive-like transport relay recently identified in MFS proteins for bivalent cations [126]. Not only has the QacA TMS 10 region been shown to be involved in the binding of bivalent cations [46, 120] but additionally Gly377, located in TMS 12, may also facilitate the transport of bivalent cationic substrates. Mutagenesis studies moving an acidic residue from TMS 10 to TMS 12 revealed that QacA double mutants Asp323Cys-Gly377Glu and Asp323Cys-Gly377Asp retained an overall capacity to confer resistance to bivalent cations such as chlorhexidine and dequalinium [123]. Other residues also located in TMS 10 have been found to be functionally important, as Met319 along with Asp323 forms the bivalent substrate-binding site, while Gly313 appears to be important for the extrusion of both bivalent and monovalent substrates [120].
Although residues such as Gly313 may play a role in the efflux of both monovalent and bivalent compounds, competition analyses have demonstrated that QacA possesses distinct binding sites for these compounds [46]. Fluorimetric transport assays measuring the efflux of ethidium in the presence of various nonfluorescent monovalent or bivalents substrates showed that monovalent cations such as benzalkonium competitively inhibited QacA-mediated ethidium efflux, while the addition of bivalent cations such as chlorhexidine resulted in noncompetitive inhibition. These results suggested that monovalent compounds may share the same or have overlapping binding sites, whereas bivalent compounds bind at a distinct site or sites other than those occupied by ethidium, a monovalent cation [46]. This is similar to what has been observed in the regulator of QacA, the QacR multidrug-binding repressor protein, as discussed in more detail below.
The differences between QacA and QacB, and their natural variants, have allowed for the assessment of their evolution and their ability to recognize different subsets of substrates. Initially, beginning with the discovery of the prototypical QacA protein, carried on the pSK1 plasmid [67] and the QacB protein, carried on pSK23 [125], these two proteins are an excellent example of fortuitous mutations which can extend the substrate profile of membrane transport proteins. As such, although most likely initially being able to expel only one class of antimicrobials, a single mutation, such as exemplified by the change to a charged residue in QacA at residue position 323, is able to render membrane proteins capable of transporting new classes of compounds.
3.3 The Multidrug and Toxic Compound Extrusion Family
Exhibiting a membrane topology similar to the MFS, members of the MATE family are ubiquitous to all kingdoms [18]. Well-characterized MATE family multidrug efflux proteins include MepA from S. aureus [55, 56] and NorM from Neisseria gonorrhoeae (NorM-NG) [127–129] and Vibrio parahaemolyticus [130, 131]. Phylogenetic analysis has revealed that the MATE family is composed of 3 large families with 14 small subfamilies [132]. In addition to drug efflux of compounds including cationic dyes such as ethidium, aminoglycosides, and fluoroquinolones [129, 133], MATE family proteins are involved in basic mechanisms which maintain homeostasis, by extruding metabolic waste products and xenobiotics in nature [132, 134]. This is achieved using energy stored in either the Na+ or H+ electrochemical gradients.
Membrane transporters of the MATE family have between 400 and 500 amino acids and form 12 TMSs. Although no apparent consensus is observed between MATE proteins, they exhibit around 40 % similarity [134] with multiple sequence alignments revealing slightly conserved regions located in TMS 1 and TMS 7 and within six loops, two extracellular and four cytoplasmic loops. Given this symmetrical repetition of conserved regions, which is distributed between the amino and carboxyl halves of the MATE pumps, it is hypothesized that these transporters evolved from a common ancestral gene that underwent genetic duplication [132], similar to that proposed for MFS proteins.
Although determined to be clinically significant, the transport mechanism of MATE proteins is poorly understood. However, analysis of solved structures of four MATE proteins (http://blanco.biomol.uci.edu/mpstruc/), including structures of the NorM-NG transporter in apo- and substrate-bound forms [129, 131, 135], has revealed a multidrug-binding cavity which is composed largely of negatively charged amino acids, with a limited number of hydrophobic residues. At least three negatively charged amino acids of the NorM-NG transporter were determined to be involved in substrate charge neutralization, as they were essential for precluding electroneutral or negatively charged compounds from being bound and transported, thereby ensuring specificity [129].
Based on the above structural and functional assessment of the NorM-NG transporter, it was suggested that Na+ triggers multidrug extrusion by inducing conformational changes within the protein. The binding of Na+ to a drug-bound transporter promotes the movement of TMS 7 and TMS 8 which causes the drug to disassociate from the binding site. The Na+ bound, drug-free transporter then switches to an inward-facing conformation, where it can then bind to another drug. It is believed that drug binding and the subsequent movement of TMS 7 and TMS 8 weaken Na+ binding. As a result of this change, Na+ is released into the cytoplasm and the transporter takes on an inward-facing conformation, and then the drug-bound protein returns to the outward-facing conformation where the transport cycle is completed [129].
Although Na+-driven, the mechanism of transport is comparable to the mechanism described for H+-coupled transporters. In fact, structural analysis and functional assessment of DinF, a H+-coupled MATE transporter from Bacillus halodurans, also identified a membrane-embedded substrate-binding site. It was suggested that drug-mediated DinF transport resulted from the direct competition between H+ and the drug and occurred through conformational inward- and outward-facing changes [136], which, as discussed above, has also been described for proteins belonging to the MFS.
3.4 The MATE Transporter: MepA
The chromosomally encoded MepA protein was the first and is the only MATE family multidrug transporter to be discovered in S. aureus [55]. Functional analysis of this protein revealed that when overexpressed, it had a broad substrate profile (Table 7.1) that included both monovalent and bivalent biocides such as ethidium and chlorhexidine, respectively, as well as fluoroquinolone agents such as norfloxacin and ciprofloxacin [55]. The expression of mepA is controlled by the transcriptional repressor MepR, a MarR-family member (see below).
Analysis of MepA residues critical for substrate binding and/or translation revealed that like many drug efflux pumps whose substrates are cationic, negatively charged residues play a key role in the substrate-translocation pathway, which in MepA are Glu156 and Asp183, located in TMS 4 and TMS 5, respectively. However, the exact nature of their participation in this process is still to be confirmed [57, 137]. In addition to these, within the substrate-translocation pathway, which in silico modeling has revealed to be formed from TMSs 1, 2, 4, 7, 8, and 10 coming together and forming a central cavity, residues Ser81, Ala161, Met291, and Ala302 have been identified as playing a role in substrate interaction. Further examination of this model also showed a residue unique to the MepA protein, Glu295 located in TMS 8, whose side-chain projected into the putative central cavity. Site-directed mutagenesis studies assessing the possible function of the residue revealed that the size and not the charge of the residue played an important role in the function of MepA, as a substitution with residues smaller than Glu resulted in a reduction in function [137]. Other functionally important residues, which are located outside of the putative central cavity and lying within TMSs 7–11 and the intervening cytoplasmic loops, include Lys242, Asn308, Met312, Asn369, Phe375, Met391, Ala392, and Ala397.
3.5 The Small Multidrug Resistance Family
Prior to the discovery of the PACE family, the SMR family represented the smallest proteins of the membrane transport system. The SMR family can be separated into three subfamilies: the small multidrug pumps (SMP), paired small multidrug resistance pumps (PSMR), and suppressors of groEL mutations (SUG); however, phylogenetic analysis of SMR proteins shows that they are subdivided into two phylogenetic clusters, with only one of these being able to catalyze drug export [138, 139]. As drug transport has not been demonstrated for a number of proteins within the PSMR subfamily, members of this subfamily are spread throughout the two clusters mentioned above. Thus, of the two identified clusters, the largest one is composed of SMR proteins including the multidrug exporters QacC from S. aureus and EmrE from E. coli, which are grouped with QacE, QacEΔ1, QacF, QacH, and QacJ.
By assessing the diversity and evolution of the SMR family, in search for a common SMR progenitor, it was revealed that this family underwent a high frequency of lateral gene transfer and rapid sequence divergence, giving rise to the current variety of SMR proteins [138]. The emergence of PSMR proteins is thought to have occurred through gene duplication events, explaining the shared relationship that some of these proteins exhibit with both the SMR and SUG subfamilies. Additionally SMR-family proteins display regions of conservation with other larger metabolite and multidrug membrane transporters. Fusions of TMSs of SMR proteins and sequence rearrangements are believed to have contributed to the formation of the bacterial/archaeal transporter family containing proteins with five TMSs, which later led to the formation of the drug/metabolite efflux family [102, 140].
Frequently identified on mobile elements, such as integrons and plasmids, these proteins are typically composed of only 100–150 amino acids and form four TMSs [140]. As found for all other membrane transport families, multiple sequence alignments of SMP-subfamily proteins can reveal a number of signature motifs [138, 139]. These motifs lie in each of the predicted TMSs with the highest residue conservation observed in TMS 1 [139]. Assessment of this TMS revealed that a single negatively charged residue, Glu14, is highly conserved in SMR members, and is essential for drug transport [141–144].
Members of the SMP subfamily confer low-level resistance to a variety of antimicrobial agents, including a number of QACs such as benzalkonium and tetraphenylphosphonium in addition to toxic lipophilic cations such as DNA intercalating agents. They have also demonstrated to have the ability to extrude potentially toxic metabolites such as nicotine intermediates out of the cell [139, 140].
Members of the SMR family are known as proton-coupled transporters and are proposed to function as dimers [139, 145, 146]. Currently the only solved structure of a SMR-family protein is that of the E. coli EmrE multidrug transporter, which has been determined at a resolution of 7.0 Å [147] and 3.8 Å [148]. According to the earlier structure, EmrE is an asymmetric homodimer that consists of a bundle of eight TMSs with one substrate molecule that is bound near the center. Such asymmetry was also seen in the crystal structure of EmrE in complex with tetraphenylphosphonium, which showed the asymmetrical arrangement appeared to stem from an antiparallel topology within the homodimer [148]. This antiparallel orientation proposed for EmrE was surprising, as membrane proteins generally insert in one particular orientation and follow the “positive-inside rule” [149]. However, SMR proteins like EmrE are proposed to form dual-topology dimers with the subunits having an inverted topology. This could be due to the fact that EmrE appears to have a weak charge-bias and as such may not conform to this rule [150]. It is possible through genetic manipulation to force EmrE into one or the other orientation, although this does limit the formation of an active transport protein [150].
In addition to QacC, proteins such as QacEΔ1, QacG, QacH, and QacJ have been identified on plasmids within a number of antimicrobial resistant bacteria, including various S. aureus strains isolated from humans [59] and animals [61, 151] and in the food industry [62, 63, 152]. The Qac nomenclature given to these proteins is based on their ability to confer host resistance against QACs [153]. Assessment of resistance has shown that despite conferring resistance to a wide range of QACs and cationic dyes, Qac proteins display their own unique resistance profiles for specific compounds. For example, high levels of resistance to ethidium have been determined for QacH and QacEΔ1, but not for QacG. However, QacG has been shown to confer higher levels of resistance to cetyltrimethylammonium bromide than QacH [139]. Displaying distinct substrate ranges, analysis of human staphylococcal clinical isolates has shown that several qac genes coexist in some isolates and appear to act synergistically to remove different compounds from the bacterial cells [154].
3.6 The SMR Transporter: QacC
The QacC multidrug transporter was the first SMR-family member identified. It was originally cloned from S. aureus strains resistant to disinfectants such as ethidium and benzalkonium chloride [155]. The qacC determinant was found to be located on a small 2.4 kb plasmid identified as pSK89, isolated from an Australian hospital [156]. Homology with pSK89 was also identified with the larger pJE1 and pSK41 plasmids, suggesting that these too carried resistance determinants which were similar or identical to the determinant responsible for the resistance attributed to QacC; as such this determinant was referred to as qacD. Although initially assumed that the qacC gene stemmed from qacD, similar to that seen with the QacA/B relationship, it was later shown that that these genes were identical and thus encoded one protein only, QacC [157]. In addition to QacD, the QacC membrane protein has also been referred to as Smr (staphylococcal multidrug resistance) [157] and Ebr (ethidium bromide resistance protein) [158, 159].
Comparative analysis of the QacC amino acid sequence with other drug efflux proteins such as QacA, the B. subtilis Bmr, and E. coli TetA proteins revealed low sequence identity implying an independent origin for the qacC gene [157]. Further analysis revealed that QacC shared high sequence similarity with proteins belonging to a family of small hydrophobic proteins, which were all encoded by short open reading frames (~330 bp). Of these, QacC was shown to be most closely related to the E. coli MvrC protein, now known as EmrE, showing a 42 % sequence identity [157, 160]. In addition to displaying high homology with EmrE, QacC was also shown to contain a number of highly conserved residues identified in other SMR proteins, including Glu13. Mutagenesis of this residue revealed that a substitution of Glu13 with aspartic acid or glutamine resulted in host cell susceptibility to benzalkonium [141]. This highly conserved carboxylic residue, found to be putatively located within the first transmembrane domain of many SMR proteins, has since been extensively analyzed and shown to be essential for drug/proton binding and translocation in SMR-family proteins [139, 161].
Composed of 107 amino acids, topological assessment of QacC, initially investigated by the construction and analysis of a series of qacC-phoA and qacC-lacZ fusions, supported the proposed four TMS topology seen for SMR proteins (Fig. 7.4) [58]. In addition, the orientation of the N-terminus was shown to be cytoplasmic; however, the location of the C-terminus could not be unequivocally determined. Further analysis, using NMR confirmed that QacC is composed of four TMSs, with residues 6–23 contained in TMS 1, 32–40 TMS 2, 60–71 TMS 3, and 94–101 TMS 4 [162]. As with other members of the SMP subfamily, the QacC functional unit is postulated to be dimeric.

Schematic topological representation of the S. aureus QacC multidrug transporter. Secondary structure of the QacC membrane transport protein is based on hydropathy analysis. Residues shaded in red have been found to be essential for proper QacC function, as analyzed by site-directed mutagenesis [58, 157]
Functional analysis of the QacC multidrug transporter revealed that although possessing a more limited substrate range than QacA, it can confer host resistance to a broad spectrum of antimicrobials including QACs such as cetyltrimethylammonium bromide, cetylpyridinium, benzalkonium, and dequalinium, as well as dyes such as crystal violet, ethidium, proflavine, pyronin Y, and rhodamine 6G [58]. In addition to the essential Glu13 residue, site-directed mutagenesis revealed that two other highly conserved residues, Tyr59 and Trp62, were essential for function [58]. Furthermore, the conserved proline at position 31 was found to be essential for full function among members of the SMR family, as substitutions of this residue with either glycine or alanine residues, which carry similar side-chain volumes resulted in a reduction in resistance to both crystal violet and ethidium, suggesting that Pro31 may reside in an region of the protein that is integral in determining QacC substrate specificity [58]. Finally, Glu24, located in a loop region, was shown to be involved in substrate specificity [157], while Cys42, located within TMS 3, was shown to play a role in substrate recognition and could only be replaced with threonine to maintain wild-type function [58].
4 Regulation of Staphylococcal Drug Efflux Pumps
The regulatory mechanisms used for the expression of multidrug transporters are complex, intertwined, and not completely understood. Regulation of multidrug efflux pumps can occur at global and specific levels, or not at all, as some pumps appear to not be under the control of regulatory proteins. Regulation is complex, as global regulatory proteins can themselves be modulated by other proteins and/or small regulatory RNAs. Global regulatory proteins are defined by their ability to modulate operons belonging to different metabolic pathways [163, 164], while specific regulators, also known as substrate-responsive regulators as they commonly bind to the substrates of the multidrug efflux pumps, can act as transcriptional activators or repressors for individual pumps [118]. All of these regulatory mechanisms are at play in S. aureus providing a means that enable the bacteria to adapt to changing environments, including the presence of antimicrobial agents.
Within S. aureus, the global regulator MgrA, also known as Rat or NorR, regulates the expression of a number of genes in the mgrA regulon including those encoding virulence factors and multidrug resistance efflux pumps such as NorA, NorB, and NorC [42, 43, 165]. MgrA binding itself is regulated by phosphorylation/dephosphorylation by Stk1 and RsbU, respectively [165]. Recently, the importance of MgrA has been extended, as it was shown that small RNAs, namely, RNAIII, stabilize mgrA mRNA by interacting with it, leading to its increased production. RNAIII is integral to the regulation of virulence genes affected by the agr quorum-sensing system in S. aureus [166]. Possessing a helix-turn-helix motif involved in DNA binding, the MgrA protein is a small transcriptional regulator that functions by directly binding to the promoter region of its target [167, 168]. In S. aureus, it can function both as an activator and a repressor, as it can downregulate the expression of NorA and upregulate the expression of NorB [168].
Regulation of efflux pumps can also be carried out at a local level. In addition to the MgrA global regulator, NorA, NorB, and NorC are also regulated by a specific regulator, NorG, which also controls the ATP-dependent membrane protein, AbcA [169]. Among the S. aureus-specific regulators, and one of the best characterized, is the QacR multidrug-binding protein that represses the transcription of the qacA/qacB multidrug transporter genes [170, 171]. QacR functions as a pair of dimers by binding in the absence of QacA substrates to an inverted repeat (IR1) DNA sequence which overlaps the qacA/B transcriptional initiation sites, resulting in repression of qacA/B transcription [172]. Assessment of the substrate recognition profile of QacR has revealed that it binds a number of structurally dissimilar cationic lipophilic antiseptics, disinfectants, and cytotoxins including crystal violet, ethidium, and rhodamine [173, 174], all of which are also QacA substrates. Binding of these substrates induces a coil-to-helix conformational change in QacR which renders the protein unable to bind IR1, triggering its release from the qacA/B promoter region and allowing transcription to occur [172, 175].
Much of what is known pertaining to the structure and function of QacR comes from high-resolution crystal structures of QacR bound to ten structurally diverse compounds [171, 172, 176], including one structure of QacR bound simultaneously to two different drugs [177]. These structures revealed that QacR contains two separate but overlapping drug-binding pockets, which exist in one extended binding site. The orientation of these two pockets, referred to as the “ethidium” and “rhodamine 6G” pockets, shows that the ethidium pocket partially overlaps the rhodamine 6G pocket and lies closer to the proposed drug-binding “portal” entrance than the rhodamine 6G pocket [172]. The volume of the extended binding pocket, when drug-free, is under 400 Å3; however, this expands to 1,100 Å3 when bound to substrate [172], exhibiting immense flexibility. The binding pocket itself is rich in aromatic residues and contains acidic residues which play a role in substrate discrimination by affecting the positioning of the drugs within this pocket [172, 178]. The aromatic residues also play a vital role in the QacR-induction mechanism, whereby drug binding triggers a conformational change of QacR by initiating a coil-to-helix transition of residues Thr89 through to Tyr93, resulting in the elongation of the C-terminus of helix 5 by a turn. This results in the expulsion of Tyr92 and Tyr93 from the interior of the protein leading to the formation of the large multidrug-binding pocket [171, 172, 178]. Upon this conformational change, the relocation of the DNA-binding domain is instigated leading to the QacR protein no longer being able to bind to the IR1-operator site, resulting in derepression of qacA [178]. The presence of these distinct pockets provides an explanation as to why QacR is able to bind both charged and neutral compounds as well as two different drugs simultaneously [172]. However, the exact mechanism by which QacR is able to recognize such a wide variety of compounds is still unknown [173]. This is reminiscent of what is seen in the structure and functional analyses of multidrug transporters to date, as it appears that proteins able to bind multiple compounds generally have a pocket where subsets of residues, commonly charged or aromatic in nature, interplay and interact with each individual substrate.
Regulation of the MepA pump by the MepR repressor protein provides the only known example of how a MATE pump is regulated in bacteria. Like the above discussed global regulator MgrA, MepR is a MarR-family repressor that is encoded upstream of mepA [55, 56, 179, 180]. MepR binds to a seven-base-pair signature sequence contained within operator regions upstream of both mepR and mepA where the mepR operator contains one repeat and that for mepA two, resulting in MepR displaying a higher affinity for the mepA-operator site. Mutations in the mepA-operator site can impede MepR binding and thus produce an elevated expression of MepA [181]. MepR repression can be relieved by the interaction of MepR with cationic, hydrophobic agents resulting in dissociation of MepR from the DNA. Although the majority of MepR ligands are substrates of MepA [179, 181], some selectivity is seen with bis-indole compounds [182]. MepR-inactivating substitution mutants have been reported among bloodstream isolates with reduced susceptibilities to biocides and fluoroquinolones [183]. Analysis of these variants located these mutations to a linker region between the dimerization and DNA-binding domains rather than in the DNA-binding domain per se; three of these have been crystallized and examined in detail [184]. These structures also show MepR possesses a very open conformation that can accommodate multiple ligands likely to influence the structural and allosteric changes of MepR [185].
Analysis of regulators such as those mentioned above is vital, as they provide valuable insights into the substrate range of the transport proteins that they regulate. Additionally, a detailed understanding of the complex nature of how multidrug exporters are controlled and respond to the changing environment that bacteria inhabit may allow for new strategies to be devised to overcome these survival mechanisms and lead to new treatments.
5 Concluding Remarks
The importance of multidrug transporters in antimicrobial resistance has clearly been established. With their wide substrate specificity and ability to produce elevated levels of resistance when overexpressed, they contribute to the high level of resistance exhibited by S. aureus. For bacteria such as S. aureus lacking an outer membrane, drug efflux pumps may also serve as a first line of defense before other more established mechanisms can be established, allowing for survival in a hostile environment [118]. Given its classification as an opportunistic nosocomial pathogen [186–188], the long-term exposure of S. aureus to hospital biocides may also have significantly contributed to its acquisition of numerous membrane drug transport proteins. Such advantageous acquisition can be seen in the prevalence of the qacA, qacB, and qacC determinants, identified in isolates from environments where decontamination with biocides is in use [189, 190], particularly as these genes are commonly carried on mobile genetic elements.
Their prevalence in S. aureus and contribution to resistance has made multidrug efflux pumps, such as those discussed in this review, targets for the development of inhibitors which, when successful, would restore the activity of the antimicrobial agent [133]. Although inhibition studies have led to the development of compounds capable of inhibiting such pumps, for example, inhibition of the Tet efflux pumps [191–193], their development is challenging and complicated by the need to combine them with antimicrobial agents that show similar pharmacokinetic profiles [133]. Thus, the key to the development of inhibitors lies in the understanding of the molecular mechanism of multidrug transporters and highlights the importance of structural and functional analysis as it is through the detailed understanding of their mechanism that effective inhibitors can be designed.
References
Lindsay JA, Holden MT (2004) Staphylococcus aureus: superbug, super genome? Trends Microbiol 12:378–385. doi:10.1016/j.tim.2004.06.004
Alibayov B, Baba-Moussa L, Sina H, Zdenkova K, Demnerova K (2014) Staphylococcus aureus mobile genetic elements. Mol Biol Rep 41:5005–5018. doi:10.1007/s11033-014-3367-3
Hiramatsu K, Aritaka N, Hanaki H, Kawasaki S, Hosoda Y, Hori S, Fukuchi Y, Kobayashi I (1997) Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350:1670–1673. doi:10.1016/S0140-6736(97)07324-8
Limbago BM, Kallen AJ, Zhu W, Eggers P, McDougal LK, Albrecht VS (2014) Report of the 13th vancomycin-resistant Staphylococcus aureus isolate from the United States. J Clin Microbiol 52:998–1002. doi:10.1128/JCM.02187-13
Bozdogan B, Esel D, Whitener C, Browne FA, Appelbaum PC (2003) Antibacterial susceptibility of a vancomycin-resistant Staphylococcus aureus strain isolated at the Hershey Medical Center. J Antimicrob Chemother 52:864–868. doi:10.1093/jac/dkg457
Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT et al (2003) Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med 348:1342–1347. doi:10.1056/NEJMoa025025
Poole K (2002) Mechanisms of bacterial biocide and antibiotic resistance. J Appl Microbiol 92(Suppl):55S–64S. doi:10.1046/j.1365-2672.92.5s1.8.x
Pantosti A, Sanchini A, Monaco M (2007) Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol 2:323–334. doi:10.2217/17460913.2.3.323
Kumar A, Schweizer HP (2005) Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv Drug Deliv Rev 57:1486–1513. doi:10.1016/j.addr.2005.04.004
Piddock LJ (2006) Multidrug-resistance efflux pumps – not just for resistance. Nat Rev Microbiol 4:629–636. doi:10.1038/nrmicro1464
Joo HS, Otto M (2015) Mechanisms of resistance to antimicrobial peptides in staphylococci. Biochim Biophys Acta 1848:3055–3061. doi:10.1016/j.bbamem.2015.02.009
Sannasiddappa TH, Hood GA, Hanson KJ, Costabile A, Gibson GR, Clarke SR (2015) Staphylococcus aureus MnhF mediates cholate efflux and facilitates survival under human colonic conditions. Infect Immun 83:2350–2357. doi:10.1128/IAI.00238-15
Hassan KA, Skurray RA, Brown MH (2007) Active export proteins mediating drug resistance in staphylococci. J Mol Microbiol Biotechnol 12:180–196. doi:10.1159/000099640
Schindler BD, Frempong-Manso E, DeMarco CE, Kosmidis C, Matta V, Seo SM, Kaatz GW (2015) Analyses of multidrug efflux pump-like proteins encoded on the Staphylococcus aureus chromosome. Antimicrob Agents Chemother 59:747–748. doi:10.1128/AAC.04678-14
Cozzetto D, Tramontano A (2008) Advances and pitfalls in protein structure prediction. Curr Protein Peptide Sci 9:567–577. doi:10.2174/138920308786733958
Eswar N, Eramian D, Webb B, Shen MY, Sali A (2008) Protein structure modeling with MODELLER. Methods Mol Biol 426:145–159. doi:10.1007/978-1-60327-058-8_8
Paulsen IT, Brown MH, Skurray RA (1996) Proton-dependent multidrug efflux systems. Microbiol Rev 60:575–608
Brown MH, Paulsen IT, Skurray RA (1999) The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 31:394–395. doi:10.1046/j.1365-2958.1999.01162.x
Collu F, Cascella M (2013) Multidrug resistance and efflux pumps: insights from molecular dynamics simulations. Curr Top Med Chem 13:3165–3183. doi:10.2174/15680266113136660224
Hassan KA, Jackson SM, Penesyan A, Patching SG, Tetu SG, Eijkelkamp BA, Brown MH, Henderson PJ et al (2013) Transcriptomic and biochemical analyses identify a family of chlorhexidine efflux proteins. Proc Natl Acad Sci U S A 110:20254–20259. doi:10.1073/pnas.1317052110
Hassan KA, Liu Q, Henderson PJ, Paulsen IT (2015) Homologs of the Acinetobacter baumannii AceI transporter represent a new family of bacterial multidrug efflux systems. mBio 6:e01982–14. doi:10.1128/mBio.01982-14
Schrader-Fischer G, Berger-Bachi B (2001) The AbcA transporter of Staphylococcus aureus affects cell autolysis. Antimicrob Agents Chemother 45:407–412. doi:10.1128/AAC.45.2.407-412.2001
Truong-Bolduc QC, Hooper DC (2007) The transcriptional regulators NorG and MgrA modulate resistance to both quinolones and β-lactams in Staphylococcus aureus. J Bacteriol 189:2996–3005. doi:10.1128/JB.01819-06
Villet RA, Truong-Bolduc QC, Wang Y, Estabrooks Z, Medeiros H, Hooper DC (2014) Regulation of expression of abcA and its response to environmental conditions. J Bacteriol 196:1532–1539. doi:10.1128/JB.01406-13
Ross JI, Eady EA, Cove JH, Baumberg S (1996) Minimal functional system required for expression of erythromycin resistance by msrA in Staphylococcus aureus RN4220. Gene 183:143–148. doi:10.1016/S0378-1119(96)00541-0
Ross JI, Eady EA, Cove JH, Baumberg S (1995) Identification of a chromosomally encoded ABC-transport system with which the staphylococcal erythromycin exporter MsrA may interact. Gene 153:93–98. doi:10.1016/0378-1119(94)00833-E
Reynolds E, Cove JH (2005) Enhanced resistance to erythromycin is conferred by the enterococcal msrC determinant in Staphylococcus aureus. J Antimicrob Chemother 55:260–264. doi:10.1093/jac/dkh541
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185. doi:10.1038/nature05155
Velamakanni S, Yao Y, Gutmann DA, van Veen HW (2008) Multidrug transport by the ABC transporter Sav 1866 from Staphylococcus aureus. Biochemistry 47:9300–9308. doi:10.1021/bi8006737
Chesneau O, Ligeret H, Hosan-Aghaie N, Morvan A, Dassa E (2005) Molecular analysis of resistance to streptogramin A compounds conferred by the Vga proteins of staphylococci. Antimicrob Agents Chemother 49:973–980. doi:10.1128/AAC.49.3.973-980.2005
Gentry DR, McCloskey L, Gwynn MN, Rittenhouse SF, Scangarella N, Shawar R, Holmes DJ (2008) Genetic characterization of Vga ABC proteins conferring reduced susceptibility to pleuromutilins in Staphylococcus aureus. Antimicrob Agents Chemother 52:4507–4509. doi:10.1128/AAC.00915-08
Allignet J, Loncle V, el Sohl N (1992) Sequence of a staphylococcal plasmid gene, vga, encoding a putative ATP-binding protein involved in resistance to virginiamycin A-like antibiotics. Gene 117:45–51. doi:10.1016/0378-1119(92)90488-B
Kadlec K, Schwarz S (2009) Novel ABC transporter gene, vga(C), located on a multiresistance plasmid from a porcine methicillin-resistant Staphylococcus aureus ST398 strain. Antimicrob Agents Chemother 53:3589–3591. doi:10.1128/AAC.00570-09
Kehrenberg C, Schwarz S (2006) Distribution of florfenicol resistance genes fexA and cfr among chloramphenicol-resistant Staphylococcus isolates. Antimicrob Agents Chemother 50:1156–1163. doi:10.1128/AAC.50.4.1156-1163.2006
Floyd JL, Smith KP, Kumar SH, Floyd JT, Varela MF (2010) LmrS is a multidrug efflux pump of the major facilitator superfamily from Staphylococcus aureus. Antimicrob Agents Chemother 54:5406–5412. doi:10.1128/AAC.00580-10
Huang J, O'Toole PW, Shen W, Amrine-Madsen H, Jiang X, Lobo N, Palmer LM, Voelker L et al (2004) Novel chromosomally encoded multidrug efflux transporter MdeA in Staphylococcus aureus. Antimicrob Agents Chemother 48:909–917. doi:10.1128/AAC.48.3.909-917.2004
Yamada Y, Shiota S, Mizushima T, Kuroda T, Tsuchiya T (2006) Functional gene cloning and characterization of MdeA, a multidrug efflux pump from Staphylococcus aureus. Biol Pharm Bull 29:801–804. doi:10.1248/bpb.29.801
Yoshida H, Bogaki M, Nakamura S, Ubukata K, Konno M (1990) Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J Bacteriol 172:6942–6949
Neyfakh AA, Borsch CM, Kaatz GW (1993) Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob Agents Chemother 37:128–129. doi:10.1128/AAC.37.1.128
Kaatz GW, Seo SM, Ruble CA (1993) Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother 37:1086–1094. doi:10.1128/AAC.37.5.1086
Kaatz GW, Thyagarajan RV, Seo SM (2005) Effect of promoter region mutations and mgrA overexpression on transcription of norA, which encodes a Staphylococcus aureus multidrug efflux transporter. Antimicrob Agents Chemother 49:161–169. doi:10.1128/AAC.49.1.161-169.2005
Truong-Bolduc QC, Dunman PM, Strahilevitz J, Projan SJ, Hooper DC (2005) MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J Bacteriol 187:2395–2405. doi:10.1128/JB.187.7.2395-2405.2005
Truong-Bolduc QC, Strahilevitz J, Hooper DC (2006) NorC, a new efflux pump regulated by MgrA of Staphylococcus aureus. Antimicrob Agents Chemother 50:1104–1107. doi:10.1128/AAC.50.3.1104-1107.2006
Brown MH, Skurray RA (2001) Staphylococcal multidrug efflux protein QacA. J Mol Microbiol Biotechnol 3:163–170
Mitchell BA, Brown MH, Skurray RA (1998) QacA multidrug efflux pump from Staphylococcus aureus: comparative analysis of resistance to diamidines, biguanidines, and guanylhydrazones. Antimicrob Agents Chemother 42:475–477
Mitchell BA, Paulsen IT, Brown MH, Skurray RA (1999) Bioenergetics of the staphylococcal multidrug export protein QacA. Identification of distinct binding sites for monovalent and divalent cations. J Biol Chem 274:3541–3548. doi:10.1074/jbc.274.6.3541
Littlejohn TG, Paulsen IT, Gillespie MT, Tennent JM, Midgley M, Jones IG, Purewal AS, Skurray RA (1992) Substrate specificity and energetics of antiseptic and disinfectant resistance in Staphylococcus aureus. FEMS Microbiol Lett 95:259–265. doi:10.1016/0378-1097(92)90439-U
Leelaporn A, Firth N, Paulsen IT, Hettiaratchi A, Skurray RA (1995) Multidrug resistance plasmid pSK108 from coagulase-negative staphylococci; relationships to Staphylococcus aureus qacC plasmids. Plasmid 34:62–67. doi:10.1006/plas.1995.1034
Yamada Y, Hideka K, Shiota S, Kuroda T, Tsuchiya T (2006) Gene cloning and characterization of SdrM, a chromosomally-encoded multidrug efflux pump, from Staphylococcus aureus. Biol Pharm Bull 29:554–556. doi:10.1248/bpb.29.554
Ginn SL, Brown MH, Skurray RA (2000) The TetA(K) tetracycline/H+ antiporter from Staphylococcus aureus: mutagenesis and functional analysis of motif C. J Bacteriol 182:1492–1498. doi:10.1128/JB.182.6.1492-1498.2000
Guay GG, Khan SA, Rothstein DM (1993) The tet(K) gene of plasmid pT181 of Staphylococcus aureus encodes an efflux protein that contains 14 transmembrane helices. Plasmid 30:163–166. doi:10.1006/plas.1993.1045
Hirata T, Saito A, Nishino K, Tamura N, Yamaguchi A (2004) Effects of efflux transporter genes on susceptibility of Escherichia coli to tigecycline (GAR-936). Antimicrob Agents Chemother 48:2179–2184. doi:10.1128/AAC.48.6.2179-2184.2004
Schwarz S, Cardoso M, Wegener HC (1992) Nucleotide sequence and phylogeny of the tet(L) tetracycline resistance determinant encoded by plasmid pSTE1 from Staphylococcus hyicus. Antimicrob Agents Chemother 36:580–588. doi:10.1128/AAC.36.3.580
Kadlec K, Schwarz S (2010) Identification of a plasmid-borne resistance gene cluster comprising the resistance genes erm(T), dfrK, and tet(L) in a porcine methicillin-resistant Staphylococcus aureus ST398 strain. Antimicrob Agents Chemother 54:915–918. doi:10.1128/AAC.01091-09
Kaatz GW, McAleese F, Seo SM (2005) Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob Agents Chemother 49:1857–1864. doi:10.1128/AAC.49.5.1857-1864.2005
McAleese F, Petersen P, Ruzin A, Dunman PM, Murphy E, Projan SJ, Bradford PA (2005) A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob Agents Chemother 49:1865–1871. doi:10.1128/AAC.49.5.1865-1871.2005
Banchs C, Poulos S, Nimjareansuk WS, Joo YE, Faham S (2014) Substrate binding to the multidrug transporter MepA. Biochim Biophys Acta 1838:2539–2546. doi:10.1016/j.bbamem.2014.06.013
Paulsen IT, Brown MH, Dunstan SJ, Skurray RA (1995) Molecular characterization of the staphylococcal multidrug resistance export protein QacC. J Bacteriol 177:2827–2833
Kazama H, Hamashima H, Sasatsu M, Arai T (1998) Distribution of the antiseptic-resistance gene qacEΔ1 in Gram-positive bacteria. FEMS Microbiol Lett 165:295–299. doi:10.1111/j.1574-6968.1998.tb13160.x
Paulsen IT, Littlejohn TG, Radstrom P, Sundstrom L, Skold O, Swedberg G, Skurray RA (1993) The 3′ conserved segment of integrons contains a gene associated with multidrug resistance to antiseptics and disinfectants. Antimicrob Agents Chemother 37:761–768. doi:10.1128/AAC.37.4.761
Bjorland J, Steinum T, Sunde M, Waage S, Heir E (2003) Novel plasmid-borne gene qacJ mediates resistance to quaternary ammonium compounds in equine Staphylococcus aureus, Staphylococcus simulans, and Staphylococcus intermedius. Antimicrob Agents Chemother 47:3046–3052. doi:10.1128/AAC.47.10.3046-3052.2003
Heir E, Sundheim G, Holck AL (1999) The qacG gene on plasmid pST94 confers resistance to quaternary ammonium compounds in staphylococci isolated from the food industry. J Appl Microbiol 86:378–388. doi:10.1046/j.1365-2672.1999.00672.x
Heir E, Sundheim G, Holck AL (1998) The Staphylococcus qacH gene product: a new member of the SMR family encoding multidrug resistance. FEMS Microbiol Lett 163:49–56. doi:10.1111/j.1574-6968.1998.tb13025.x
Yoshikai H, Kizaki H, Saito Y, Omae Y, Sekimizu K, Kaito C (2016) Multidrug resistance transporter AbcA secretes Staphylococcus aureus cytolytic toxins. J Infect Dis 213:295–304. doi:10.1093/infdis/jiv376
Truong-Bolduc QC, Bolduc GR, Medeiros H, Vyas JM, Wang Y, Hooper DC (2015) Role of the Tet38 efflux pump in Staphylococcus aureus internalization and survival in epithelial cells. Infect Immun 83:4362–4372. doi:10.1128/IAI.00723-15
Guffanti AA, Krulwich TA (1995) Tetracycline/H+ antiport and Na+/H+ antiport catalyzed by the Bacillus subtilis TetA(L) transporter expressed in Escherichia coli. J Bacteriol 177:4557–4561
Tennent JM, Lyon BR, Gillespie MT, May JW, Skurray RA (1985) Cloning and expression of Staphylococcus aureus plasmid-mediated quaternary ammonium resistance in Escherichia coli. Antimicrob Agents Chemother 27:79–83. doi:10.1128/AAC.27.1.79
Paulsen IT, Brown MH, Littlejohn TG, Mitchell BA, Skurray RA (1996) Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc Natl Acad Sci U S A 93:3630–3635
Wong TZ, Zhang M, O’Donoghue M, Boost M (2013) Presence of antiseptic resistance genes in porcine methicillin-resistant Staphylococcus aureus. Vet Microbiol 162:977–979. doi:10.1016/j.vetmic.2012.10.017
Cuaron JA, Dulal S, Song Y, Singh AK, Montelongo CE, Yu W, Nagarajan V, Jayaswal RK et al (2013) Tea tree oil-induced transcriptional alterations in Staphylococcus aureus. Phytother Res 27:390–396. doi:10.1002/ptr.4738
Quiblier C, Zinkernagel AS, Schuepbach RA, Berger-Bachi B, Senn MM (2011) Contribution of SecDF to Staphylococcus aureus resistance and expression of virulence factors. BMC Microbiol 11:72. doi:10.1186/1471-2180-11-72
Rees DC, Johnson E, Lewinson O (2009) ABC transporters: the power to change. Nat Rev Mol Cell Biol 10:218–227. doi:10.1038/nrm2646
Vetrivel U, Subramanian G (2014) Importance of ABC transporters in different tissues. Drug Metabol Drug Interact 29:65–66. doi:10.1515/dmdi-2014-0016
Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113. doi:10.1146/annurev.cb.08.110192.000435
Chang G (2003) Multidrug resistance ABC transporters. FEBS Lett 555:102–105. doi:10.1016/S0014-5793(03)01085-8
Locher KP, Lee AT, Rees DC (2002) The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296:1091–1098. doi:10.1126/science.1071142
Linton KJ (2007) Structure and function of ABC transporters. Phys Chem Chem Phys 22:122–130. doi:10.1152/physiol.00046.2006
Linton KJ, Higgins CF (1998) The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol 28:5–13. doi:10.1046/j.1365-2958.1998.00764.x
Jones PM, O'Mara ML, George AM (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem Sci 34:520–531. doi:10.1016/j.tibs.2009.06.004
Korkhov VM, Mireku SA, Locher KP (2012) Structure of AMP-PNP-bound vitamin B12 transporter BtuCD-F. Nature 490:367–372. doi:10.1038/nature11442
Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC (2007) An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science 315:373–377. doi:10.1126/science.1133488
Dawson RJ, Locher KP (2007) Structure of the multidrug ABC transporter Sav 1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett 581:935–938. doi:10.1016/j.febslet.2007.01.073
Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72:317–364. doi:10.1128/MMBR.00031-07
Du D, van Veen HW, Murakami S, Pos KM, Luisi BF (2015) Structure, mechanism and cooperation of bacterial multidrug transporters. Curr Opin Struct Biol 33:76–91. doi:10.1016/j.sbi.2015.07.015
George AM, Jones PM (2013) An asymmetric post-hydrolysis state of the ABC transporter ATPase dimer. PLoS One 8:e59854. doi:10.1371/journal.pone.0059854
Biemans-Oldehinkel E, Doeven MK, Poolman B (2006) ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett 580:1023–1035. doi:10.1016/j.febslet.2005.11.079
Reynolds ED, Cove JH (2005) Resistance to telithromycin is conferred by msr(A), msrC and msr(D) in Staphylococcus aureus. J Antimicrob Chemother 56:1179–1180. doi:10.1093/jac/dki378
Ross JI, Eady EA, Cove JH, Cunliffe WJ, Baumberg S, Wootton JC (1990) Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol Microbiol 4:1207–1214. doi:10.1111/j.1365-2958.1990.tb00696.x
Reynolds E, Ross JI, Cove JH (2003) Msr(A) and related macrolide/streptogramin resistance determinants: incomplete transporters? Int J Antimicrob Agents 22:228–236. doi:10.1016/S0924-8579(03)00218-8
Beck A, Aanismaa P, Li-Blatter X, Dawson R, Locher K, Seelig A (2013) Sav 1866 from Staphylococcus aureus and P-glycoprotein: similarities and differences in ATPase activity assessed with detergents as allocrites. Biochemistry 52:3297–3309. doi:10.1021/bi400203d
Oldham ML, Davidson AL, Chen J (2008) Structural insights into ABC transporter mechanism. Curr Opin Struct Biol 18:726–733. doi:10.1016/j.sbi.2008.09.007
Locher KP (2009) Structure and mechanism of ATP-binding cassette transporters. Philos Trans R Soc Lond B Biol Sci 364:239–245. doi:10.1098/rstb.2008.0125
Higgins CF (2001) ABC transporters: physiology, structure and mechanism–an overview. Res Microbiol 152:205–210. doi:10.1016/S0923-2508(01)01193-7
ter Beek J, Guskov A, Slotboom DJ (2014) Structural diversity of ABC transporters. J Gen Physiol 143:419–435. doi:10.1085/jgp.201411164
Law CJ, Maloney PC, Wang DN (2008) Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62:289–305. doi:10.1146/annurev.micro.61.080706.093329
Madej MG, Dang S, Yan N, Kaback HR (2013) Evolutionary mix-and-match with MFS transporters. Proc Natl Acad Sci U S A 110:5870–5874. doi:10.1073/pnas.1303538110
Saier MH Jr, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jahn PS et al (1999) The major facilitator superfamily. J Mol Microbiol Biotechnol 1:257–279
Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH Jr (2012) The major facilitator superfamily (MFS) revisited. FEBS J 279:2022–2035. doi:10.1111/j.1742-4658.2012.08588.x
Saier MH Jr, Tran CV, Barabote RD (2006) TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res 34:D181–D186. doi:10.1093/nar/gkj001
Yan N (2013) Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci 38:151–159. doi:10.1016/j.tibs.2013.01.003
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Saier MH Jr (2003) Tracing pathways of transport protein evolution. Mol Microbiol 48:1145–1156. doi:10.1046/j.1365-2958.2003.03499.x
Varela MF, Sansom CE, Griffith JK (1995) Mutational analysis and molecular modelling of an amino acid sequence motif conserved in antiporters but not symporters in a transporter superfamily. Mol Membr Biol 12:313–319
Jiang D, Zhao Y, Wang X, Fan J, Heng J, Liu X, Feng W, Kang X et al (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc Natl Acad Sci U S A 110:14664–14669. doi:10.1073/pnas.1308127110
Yin Y, He X, Szewczyk P, Nguyen T, Chang G (2006) Structure of the multidrug transporter EmrD from Escherichia coli. Science 312:741–744. doi:10.1126/science.1125629
Heng J, Zhao Y, Liu M, Liu Y, Fan J, Wang X, Zhao Y, Zhang XC (2015) Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res 25:1060–1073. doi:10.1038/cr.2015.94
Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301:610–615. doi:10.1126/science.1088196
Guan L, Mirza O, Verner G, Iwata S, Kaback HR (2007) Structural determination of wild-type lactose permease. Proc Natl Acad Sci U S A 104:15294–15298. doi:10.1073/pnas.0707688104
Huang Y, Lemieux MJ, Song J, Auer M, Wang DN (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301:616–620. doi:10.1126/science.1087619
Karpowich NK, Wang DN (2008) Symmetric transporters for asymmetric transport. Science 321:781–782. doi:10.1126/science.1161495
Huang YW, Hu RM, Chu FY, Lin HR, Yang TC (2013) Characterization of a major facilitator superfamily (MFS) tripartite efflux pump EmrCABsm from Stenotrophomonas maltophilia. J Antimicrob Chemother 68:2498–2505. doi:10.1093/jac/dkt250
Ubukata K, Itoh-Yamashita N, Konno M (1989) Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother 33:1535–1539. doi:10.1128/JB.01819-06
Yu JL, Grinius L, Hooper DC (2002) NorA functions as a multidrug efflux protein in both cytoplasmic membrane vesicles and reconstituted proteoliposomes. J Bacteriol 184:1370–1377. doi:10.1128/JB.184.5.1370-1377.2002
Truong-Bolduc QC, Bolduc GR, Okumura R, Celino B, Bevis J, Liao CH, Hooper DC (2011) Implication of the NorB efflux pump in the adaptation of Staphylococcus aureus to growth at acid pH and in resistance to moxifloxacin. Antimicrob Agents Chemother 55:3214–3219. doi:10.1128/AAC.00289-11
Ding Y, Onodera Y, Lee JC, Hooper DC (2008) NorB, an efflux pump in Staphylococcus aureus strain MW2, contributes to bacterial fitness in abscesses. J Bacteriol 190:7123–7129. doi:10.1128/jb.00655-08
Truong-Bolduc QC, Zhang X, Hooper DC (2003) Characterization of NorR protein, a multifunctional regulator of norA expression in Staphylococcus aureus. J Bacteriol 185:3127–3138. doi:10.1128/JB.185.10.3127-3138.2003
Truong-Bolduc QC, Villet RA, Estabrooks ZA, Hooper DC (2014) Native efflux pumps contribute resistance to antimicrobials of skin and the ability of Staphylococcus aureus to colonize skin. J Infect Dis 209:1485–1493. doi:10.1093/infdis/jit660
Costa SS, Viveiros M, Amaral L, Couto I (2013) Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol J 7:59–71. doi:10.2174/1874285801307010059
Hassan KA, Souhani T, Skurray RA, Brown MH (2008) Analysis of tryptophan residues in the staphylococcal multidrug transporter QacA reveals long-distance functional associations of residues on opposite sides of the membrane. J Bacteriol 190:2441–2449. doi:10.1128/JB.01864-07
Xu Z, O’Rourke BA, Skurray RA, Brown MH (2006) Role of transmembrane segment 10 in efflux mediated by the staphylococcal multidrug transport protein QacA. J Biol Chem 281:792–799. doi:10.1074/jbc.M508676200
Cervinkova D, Babak V, Marosevic D, Kubikova I, Jaglic Z (2013) The role of the qacA gene in mediating resistance to quaternary ammonium compounds. Microb Drug Resist 19:160–167. doi:10.1089/mdr.2012.0154
Shi GS, Boost M, Cho P (2015) Prevalence of antiseptic-resistance genes in staphylococci isolated from orthokeratology lens and spectacle wearers in Hong Kong. Invest Ophthalmol Vis Sci 56:3069–3074. doi:10.1167/iovs.15-16550
Hassan KA, Skurray RA, Brown MH (2007) Transmembrane helix 12 of the Staphylococcus aureus multidrug transporter QacA lines the bivalent cationic drug binding pocket. J Bacteriol 189:9131–9134. doi:10.1128/JB.01492-07
Hassan KA, Xu Z, Watkins RE, Brennan RG, Skurray RA, Brown MH (2009) Optimized production and analysis of the staphylococcal multidrug efflux protein QacA. Protein Expr Purif 64:118–124. doi:10.1016/j.pep.2008.11.009
Paulsen IT, Brown MH, Skurray RA (1998) Characterization of the earliest known Staphylococcus aureus plasmid encoding a multidrug efflux system. J Bacteriol 180:3477–3479
Fluman N, Adler J, Rotenberg SA, Brown MH, Bibi E (2014) Export of a single drug molecule in two transport cycles by a multidrug efflux pump. Nat Commun 5:4615. doi:10.1038/ncomms5615
Rouquette-Loughlin C, Dunham SA, Kuhn M, Balthazar JT, Shafer WM (2003) The NorM efflux pump of Neisseria gonorrhoeae and Neisseria meningitidis recognizes antimicrobial cationic compounds. J Bacteriol 185:1101–1106. doi:10.1128/JB.185.3.1101-1106.2003
Long F, Rouquette-Loughlin C, Shafer WM, Yu EW (2008) Functional cloning and characterization of the multidrug efflux pumps NorM from Neisseria gonorrhoeae and YdhE from Escherichia coli. Antimicrob Agents Chemother 52:3052–3060. doi:10.1128/AAC.00475-08
Lu M, Symersky J, Radchenko M, Koide A, Guo Y, Nie R, Koide S (2013) Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci U S A 110:2099–2104. doi:10.1073/pnas.1219901110
Morita Y, Kodama K, Shiota S, Mine T, Kataoka A, Mizushima T, Tsuchiya T (1998) NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob Agents Chemother 42:1778–1782
He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, Chang G (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467:991–994. doi:10.1038/nature09408
Moriyama Y, Hiasa M, Matsumoto T, Omote H (2008) Multidrug and toxic compound extrusion (MATE)-type proteins as anchor transporters for the excretion of metabolic waste products and xenobiotics. Xenobiotica 38:1107–1118. doi:10.1080/00498250701883753
Li X-Z, Nikaido H (2004) Efflux-mediated drug resistance in bacteria. Drugs 64:159–204. doi:10.2165/00003495-200464020-00004
Omote H, HiasaI M, Matsumoto T, Otsuka M, Moriyama Y (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci 27:587–593. doi:10.1016/j.tips.2006.09.001
Tanaka Y, Hipolito CJ, Maturana AD, Ito K, Kuroda T, Higuchi T, Katoh T, Kato HE et al (2013) Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496:247–251. doi:10.1038/nature12014
Lu M, Radchenko M, Symersky J, Nie R, Guo Y (2013) Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat Struct Mol Biol 20:1310–1317. doi:10.1038/nsmb.2687
Schindler BD, Patel D, Seo SM, Kaatz GW (2013) Mutagenesis and modeling to predict structural and functional characteristics of the Staphylococcus aureus MepA multidrug efflux pump. J Bacteriol 195:523–533. doi:10.1128/JB.01679-12
Paulsen IT, Skurray RA, Tam R, Saier MH Jr, Turner RJ, Weiner JH, Goldberg EB, Grinius LL (1996) The SMR family: a novel family of multidrug efflux proteins involved with the efflux of lipophilic drugs. Mol Microbiol 19:1167–1175. doi:10.1111/j.1365-2958.1996.tb02462.x
Bay DC, Rommens KL, Turner RJ (2008) Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim Biophys Acta 1778:1814–1838. doi:10.1016/j.bbamem.2007.08.015
Bay DC, Turner RJ (2009) Diversity and evolution of the small multidrug resistance protein family. BMC Evol Biol 9:140. doi:10.1186/1471-2148-9-140
Grinius LL, Goldberg EB (1994) Bacterial multidrug resistance is due to a single membrane protein which functions as a drug pump. J Biol Chem 269:29998–30004
Muth TR, Schuldiner S (2000) A membrane-embedded glutamate is required for ligand binding to the multidrug transporter EmrE. EMBO J 19:234–240. doi:10.1093/emboj/19.2.234
Yerushalmi H, Schuldiner S (2000) An essential glutamyl residue in EmrE, a multidrug antiporter from Escherichia coli. J Biol Chem 275:5264–5269. doi:10.1074/jbc.275.8.5264
Gutman N, Steiner-Mordoch S, Schuldiner S (2003) An amino acid cluster around the essential Glu-14 is part of the substrate- and proton-binding domain of EmrE, a multidrug transporter from Escherichia coli. J Biol Chem 278:16082–16087. doi:10.1074/jbc.M213120200
Korkhov VM, Tate CG (2008) Electron crystallography reveals plasticity within the drug binding site of the small multidrug transporter EmrE. J Mol Biol 377:1094–1103. doi:10.1016/j.jmb.2008.01.056
Schuldiner S (2009) EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim Biophys Acta 1794:748–762. doi:10.1016/j.bbapap.2008.12.018
Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG (2003) Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J 22:6175–6181. doi:10.1093/emboj/cdg611
Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G (2007) X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A 104:18999–19004. doi:10.1073/pnas.0709387104
von Heijne G (1986) The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J 5:3021–3027
Rapp M, Seppala S, Granseth E, von Heijne G (2007) Emulating membrane protein evolution by rational design. Science 315:1282–1284. doi:10.1126/science.1135406
Bjorland J, Steinum T, Kvitle B, Waage S, Sunde M, Heir E (2005) Widespread distribution of disinfectant resistance genes among staphylococci of bovine and caprine origin in Norway. J Clin Microbiol 43:4363–4368. doi:10.1128/JCM.43.9.4363-4368.2005
Heir E, Sundheim G, Holck AL (1999) Identification and characterization of quaternary ammonium compound resistant staphylococci from the food industry. Int J Food Microbiol 48:211–219. doi:10.1016/S0168-1605(99)00044-6
Wassenaar TM, Ussery D, Nielsen LN, Ingmer H (2015) Review and phylogenetic analysis of qac genes that reduce susceptibility to quaternary ammonium compounds in Staphylococcus species. Eur J Microbiol Immunol 5:44–61. doi:10.1556/EUJMI-D-14-00038
Correa JE, De Paulis A, Predari S, Sordelli DO, Jeric PE (2008) First report of qacG, qacH and qacJ genes in Staphylococcus haemolyticus human clinical isolates. J Antimicrob Chemother 62:956–960. doi:10.1093/jac/dkn327
Littlejohn TG, DiBerardino D, Messerotti LJ, Spiers SJ, Skurray RA (1991) Structure and evolution of a family of genes encoding antiseptic and disinfectant resistance in Staphylococcus aureus. Gene 101:59–66. doi:10.1016/0378-1119(91)90224-Y
Lyon BR, Skurray R (1987) Antimicrobial resistance of Staphylococcus aureus: genetic basis. Microbiol Rev 51:88–134
Grinius L, Dreguniene G, Goldberg EB, Liao CH, Projan SJ (1992) A staphylococcal multidrug resistance gene product is a member of a new protein family. Plasmid 27:119–129. doi:10.1016/0147-619X(92)90012-Y
Sasatsu M, Shima K, Shibata Y, Kono M (1989) Nucleotide sequence of a gene that encodes resistance to ethidium bromide from a transferable plasmid in Staphylococcus aureus. Nucleic Acids Res 17:10103. doi:10.1093/nar/17.23.10103
Sasatsu M, Shirai Y, Hase M, Noguchi N, Kono M, Behr H, Freney J, Arai T (1995) The origin of the antiseptic-resistance gene ebr in Staphylococcus aureus. Microbios 84:161–169
Yerushalmi H, Lebendiker M, Schuldiner S (1995) EmrE, an Escherichia coli 12-kDa multidrug transporter, exchanges toxic cations and H+ and is soluble in organic solvents. J Biol Chem 270:6856–6863
Yerushalmi H, Mordoch SS, Schuldiner S (2001) A single carboxyl mutant of the multidrug transporter EmrE is fully functional. J Biol Chem 276:12744–12748. doi:10.1074/jbc.M010979200
Poget SF, Harris R, Cahill SM, Girvin ME (2010) 1H, 13C, 15N backbone NMR assignments of the Staphylococcus aureus small multidrug-resistance pump (Smr) in a functionally active conformation. Biomol NMR Assign 4:139–142. doi:10.1007/s12104-010-9228-7
Gottesman S (1984) Bacterial regulation: global regulatory networks. Ann Rev Genet 18:415–441. doi:10.1146/annurev.ge.18.120184.002215
Martinez-Antonio A, Collado-Vides J (2003) Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol 6:482–489. doi:10.1016/j.mib.2003.09.002
Truong-Bolduc QC, Hooper DC (2010) Phosphorylation of MgrA and its effect on expression of the NorA and NorB efflux pumps of Staphylococcus aureus. J Bacteriol 192:2525–2534. doi:10.1128/JB.00018-10
Gupta RK, Luong TT, Lee CY (2015) RNAIII of the Staphylococcus aureus agr system activates global regulator MgrA by stabilizing mRNA. Proc Natl Acad Sci U S A 112:14036–14041. doi:10.1073/pnas.1509251112
Ingavale S, van Wamel W, Luong TT, Lee CY, Cheung AL (2005) Rat/MgrA, a regulator of autolysis, is a regulator of virulence genes in Staphylococcus aureus. Infect Immun 73:1423–1431. doi:10.1128/IAI.73.3.1423-1431.2005
Luong TT, Dunman PM, Murphy E, Projan SJ, Lee CY (2006) Transcription profiling of the mgrA regulon in Staphylococcus aureus. J Bacteriol 188:1899–1910. doi:10.1128/JB.188.5.1899-1910.2006
Truong-Bolduc QC, Dunman PM, Eidem T, Hooper DC (2011) Transcriptional profiling analysis of the global regulator NorG, a GntR-like protein of Staphylococcus aureus. J Bacteriol 193:6207–6214. doi:10.1128/JB.05847-11
Grkovic S, Brown MH, Roberts NJ, Paulsen IT, Skurray RA (1998) QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J Biol Chem 273:18665–18673. doi:10.1074/jbc.273.29.18665
Brooks BE, Piro KM, Brennan RG (2007) Multidrug-binding transcription factor QacR binds the bivalent aromatic diamidines DB75 and DB359 in multiple positions. J Am Chem Soc 129:8389–8395. doi:10.1021/ja072576v
Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG (2001) Structural mechanisms of QacR induction and multidrug recognition. Science 294:2158–2163. doi:10.1126/science.1066020
Schumacher MA, Brennan RG (2003) Deciphering the molecular basis of multidrug recognition: crystal structures of the Staphylococcus aureus multidrug binding transcription regulator QacR. Res Microbiol 154:69–77. doi:10.1016/S0923-2508(02)00013-X
Theis T, Skurray RA, Brown MH (2007) Identification of suitable internal controls to study expression of a Staphylococcus aureus multidrug resistance system by quantitative real-time PCR. J Microbiol Methods 70:355–362. doi:10.1016/j.mimet.2007.05.011
Grkovic S, Hardie KM, Brown MH, Skurray RA (2003) Interactions of the QacR multidrug-binding protein with structurally diverse ligands: implications for the evolution of the binding pocket. Biochemistry 42:15226–15236. doi:10.1021/bi035447+
Murray DS, Schumacher MA, Brennan RG (2004) Crystal structures of QacR-diamidine complexes reveal additional multidrug-binding modes and a novel mechanism of drug charge neutralization. J Biol Chem 279:14365–14371. doi:10.1074/jbc.M313870200
Schumacher MA, Miller MC, Brennan RG (2004) Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J 23:2923–2930. doi:10.1038/sj.emboj.7600288
Peters KM, Brooks BE, Schumacher MA, Skurray RA, Brennan RG, Brown MH (2011) A single acidic residue can guide binding site selection but does not govern QacR cationic-drug affinity. PLoS One 6:e15974. doi:10.1371/journal.pone.0015974
Kaatz GW, DeMarco CE, Seo SM (2006) MepR, a repressor of the Staphylococcus aureus MATE family multidrug efflux pump MepA, is a substrate-responsive regulatory protein. Antimicrob Agents Chemother 50:1276–1281. doi:10.1128/AAC.50.4.1276-1281.2006
Luong TT, Newell SW, Lee CY (2003) Mgr, a novel global regulator in Staphylococcus aureus. J Bacteriol 185:3703–3710. doi:10.1128/JB.185.13.3703-3710.2003
Schindler BD, Seo SM, Birukou I, Brennan RG, Kaatz GW (2015) Mutations within the mepA operator affect binding of the MepR regulatory protein and its induction by MepA substrates in Staphylococcus aureus. J Bacteriol 197:1104–1114. doi:10.1128/JB.02558-14
Opperman TJ, Williams JD, Houseweart C, Panchal RG, Bavari S, Peet NP, Moir DT, Bowlin TL (2010) Efflux-mediated bis-indole resistance in Staphylococcus aureus reveals differential substrate specificities for MepA and MepR. Bioorg Med Chem 18:2123–2130. doi:10.1016/j.bmc.2010.02.005
DeMarco CE, Cushing LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW (2007) Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob Agents Chemother 51:3235–3239. doi:10.1128/AAC.00430-07
Birukou I, Tonthat NK, Seo SM, Schindler BD, Kaatz GW, Brennan RG (2013) The molecular mechanisms of allosteric mutations impairing MepR repressor function in multidrug-resistant strains of Staphylococcus aureus. mBio 4:e00528–13. doi:10.1128/mBio.00528-13
Kumaraswami M, Schuman JT, Seo SM, Kaatz GW, Brennan RG (2009) Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res 37:1211–1224. doi:10.1093/nar/gkn1046
Archer GL (1998) Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis 26:1179–1181. doi:10.1086/520289
Tong SY, Holden MT, Nickerson EK, Cooper BS, Koser CU, Cori A, Jombart T, Cauchemez S et al (2015) Genome sequencing defines phylogeny and spread of methicillin-resistant Staphylococcus aureus in a high transmission setting. Genome Res 25:111–118. doi:10.1101/gr.174730.114
Conceicao T, Coelho C, de Lencastre H, Aires-de-Sousa M (2015) High prevalence of biocide resistance determinants in Staphylococcus aureus isolates from three African countries. Antimicrob Agents Chemother 60:678–681. doi:10.1128/AAC.02140-15
Fraise AP (2002) Biocide abuse and antimicrobial resistance – a cause for concern? J Antimicrob Chemother 49:11–12. doi:10.1093/jac/49.1.11
Vali L, Davies SE, Lai LL, Dave J, Amyes SG (2008) Frequency of biocide resistance genes, antibiotic resistance and the effect of chlorhexidine exposure on clinical methicillin-resistant Staphylococcus aureus isolates. J Antimicrob Chemother 61:524–532. doi:10.1093/jac/dkm520
Hirata T, Wakatabe R, Nielsen J, Someya Y, Fujihira E, Kimura T, Yamaguchi A (1997) A novel compound, 1,1-dimethyl-5(1-hydroxypropyl)-4,6,7-trimethylindan, is an effective inhibitor of the tet(K) gene-encoded metal-tetracycline/H+ antiporter of Staphylococcus aureus. FEBS Lett 412:337–340. doi:10.1016/S0014-5793(97)00796-5
Nelson ML, Levy SB (1999) Reversal of tetracycline resistance mediated by different bacterial tetracycline resistance determinants by an inhibitor of the Tet(B) antiport protein. Antimicrob Agents Chemother 43:1719–1724
Gibbons S, Udo EE (2000) The effect of reserpine, a modulator of multidrug efflux pumps, on the in vitro activity of tetracycline against clinical isolates of methicillin resistant Staphylococcus aureus (MRSA) possessing the tet(K) determinant. Phytother Res 14:139–140. doi:10.1002/(SICI)1099-1573(200003)
Acknowledgments
This work was financially supported by the National Health and Medical Research Council Australia Project Grant 1002670 and a Weizmann-Australia Joint Research Program. S.A.S. is the recipient of an Australian Postgraduate Award.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Sapula, S.A., Brown, M.H. (2016). Antimicrobial Drug Efflux Pumps in Staphylococcus aureus . In: Li, XZ., Elkins, C., Zgurskaya, H. (eds) Efflux-Mediated Antimicrobial Resistance in Bacteria. Adis, Cham. https://doi.org/10.1007/978-3-319-39658-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-39658-3_7
Published:
Publisher Name: Adis, Cham
Print ISBN: 978-3-319-39656-9
Online ISBN: 978-3-319-39658-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)