
Abstract
Radiotherapy (RT) predominantly is aimed to induce DNA damage in tumour cells that results in reduction of their clonogenicity and finally in tumour cell death. Adaptation of RT with higher single doses has become necessary and led to a more detailed view on what kind of tumour cell death is induced and which immunological consequences result from it. RT is capable of rendering tumour cells immunogenic by modifying the tumour cell phenotype and the microenvironment. Danger signals are released as well as the senescence-associated secretory phenotype. This results in maturation of dendritic cells and priming of cytotoxic T cells as well as in activation of natural killer cells. However, RT on the other hand can also result in immune suppressive events including apoptosis induction and foster tumour cell proliferation. That’s why RT is nowadays increasingly combined with selected immunotherapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Radiotherapy
- DNA damage
- Apoptosis
- Necrosis
- Autophagy
- Danger signals
- Senescence-associated secretory phenotype
- Immunogenic cell death
- Immunotherapy
7.1 Introduction
Two months after the announcement of the discovery of X-rays by Conrad Röntgen on November 30 1895, E. H. Grubbé, a medical student living in Chicago at that time, applied the X-rays therapeutically for the treatment of breast cancer and inflammatory lesions. He was provident and protected the surrounding healthy tissues by a sheet of lead taken from a tea chest. This was the hour of birth of radiotherapy (RT) [1]. The second classical cytotoxic treatment option for cancer disease is chemotherapy . The latter was ultimately discovered by physicians to treat cancer in the First World War. They observed that leukocytes disappeared in humans who survived mustard gas (dichloroethyl sulphide) exposure. They concluded that every poison could be also a potential efficacious remedy [2]. Until today, the three classical columns of cancer therapy are still chemotherapy (CT), RT and, the oldest form of tumour treatment, surgery.
During the last decades, immunotherapy (IT) accrued and multimodal therapies make nowadays more and more their way into clinical practice [3]. These cancer treatment modalities were formerly classified as those acting locally (surgery and RT) and those systemically (CT, IT). However, local modification of tumour cells might also result in secondary systemic responses. The focus of this article is therefore set on the ability of RT to induce distinct forms of tumour cell death and on the subsequent systemic consequences.
7.2 DNA Damage Induction and Repair Capacity as Basis for Local Efficacy of Radiotherapy
The most sensitive cellular structure for radiation is the deoxyribonucleic acid (DNA). X-rays as exogenous DNA damaging source can induce DNA single-strand breaks (SSB), double-strand breaks (DSB), oxidation of DNA bases and non-DSB clustered DNA lesions [4]. The damage is induced either by direct action of radiation on the DNA or mostly secondary by reactive oxygen species (ROS) or reactive nitrogen species (RNS) [5]. Irrespective of the DNA damage sources, the DNA damage response (DDR) is activated consecutively. Several DNA repair pathways have evolved like homologous recombination (HR), non-homologous end-joining (NHEJ), back-up NHEJ (B-NHEJ) nucleotide (NER) and base excision repair (BER) as well as mismatch repair (MMR) dependent on size and modality of the DNA damage [6].
The success or failure of standard clinical radiation treatment has mainly been determined by the four R’s of radiobiology: repair of DNA damage, reoxygenation of hypoxic tumour areas, redistribution of cells in the cell cycle and repopulation [7, 8]. Tumour cells usually less effectively repair sublethal DNA damage compared to healthy tissue cells. This is one reason why repeated irradiation, namely fractionated irradiation, is beneficial since the healthy tissue can regenerate during the radiation break. Furthermore, time is created to allow reoxygenation of hypoxic tumour areas. This highly enhances the radiosensitivity of the tumour cells [9]. The latter also exit the radioresistant S-phase of the cell cycle during radiation breaks and become more sensitive for re-irradiation [10]. However, the breaks should not be too long to avoid repopulation of tumour cells. These are the reasons for delivering radiation in lower doses but repeated fractions.
One has to keep always in mind that the local irradiation of the tumour has to fulfil two main requirements: On the one hand the tumour control probability (TCP) must be as high as possible, but on the other hand the normal tissue complication probability (NTCP) has to be as small as possible [11]. Therefore, the applied dose is finely balanced between minimal, justifiable NTCP matched with a maximal TCP. The linear quadratic model is still the basis for clinicians to estimate the total dose and fractions of irradiation for the respective tumour entities. The dose of irradiation that is necessary to destroy tumour cells and the tolerance dose for healthy tissue is known by clinicians based on long-lasting experience with classical fractionated RT with a single dose of 1.8–2.0 Gy. α/β values were defined long ago for tissues. This was based on observations in mice, namely when and to what extent irradiation causes damage in certain organs [12, 13]. High values characterise early reacting tissue with rare repair and fast repopulation, as e.g. the skin (α/β: 9–19 Gy) and many tumours. Late reacting tissues such as kidney have α/β values <5 Gy and high repair capacity. During fractionated irradiation, the late reacting tissue can regenerate during the radiation breaks and is thereby spared.
Adaption of radiation schemes is necessary for distinct tumour entities since, e.g. prostate cancer has exceptionally low values of α/β. Here, the use of a higher dose per fraction is indicated on this radiobiological basis as it is also currently intensively discussed for breast cancer [14].
It has become feasible to deliver higher single doses due to technical advancements in planning procedures (e.g. intensity-modulated RT), accuracy of dose application (e.g. image-guided RT) and application of protons and heavy ions for RT. How novel techniques in RT change the standards for cancer treatment has recently been comprehensively summarised by Durante et al. and Orth et al. [15, 16].
7.3 Radiotherapy Induces Different Cell Death Modalities
7.3.1 Mitotic Catastrophe
If the DNA damage cannot be properly repaired by the radiation-exposed cells, they execute cell death. Mitotic catastrophe , a type of cell death that occurs during mitosis, was considered for a long time by radiobiologists to be the only way cells die after irradiation. In mammalian cells it is the failure to undergo complete mitosis after DNA damage. This results in multi-ploidy and counting of multinucleated cells is the basis for detection of mitotic catastrophe [17]. The combination of cell cycle checkpoint deficiencies and specific types of DNA damage most likely lead to mitotic catastrophe and cancer cells are especially prone to that [18]. Nevertheless, there is no consensus on the distinctive morphological appearance of mitotic catastrophe as far as the extent of chromatin condensation. The latter is, however, also the morphological hallmark of apoptosis [19].
7.3.2 Senescence
Cells also evolved a bypass to deal with persistent DNA damage, namely senescence. It was first described by Hayflick and colleagues, who demonstrated that as a consequence of telomere shortening with each cycle of DNA replication human fibroblasts do not proliferate until infinity in culture [20]. Senescent cells are characterised by low expression of proteins driving proliferation, morphological changes as increase in volume and, if adherent, flattered morphology. They further highly express senescence-associated acidic lysosomal β-galactosidase. The latter is a manifestation of residual lysosomal activity at a suboptimal pH and it becomes detectable due to the increased lysosomal content in senescent cells [21]. Telomere erosion , DNA damage and oncogenic signalling induce senescence, the so-called replicative, stress and oncogene-induced senescence, respectively. It has always been in the attention of oncologists since it is the basis for prolonged or ideally permanent growth arrest of tumour cells.
However, senescent cells can regain proliferative capacity in a p53-dependent manner after radiation exposure while cells undergoing apoptosis do not. This was especially demonstrated in vitro, as for p53 wild-type MCF-7 compared to MDA-MB231 breast cancer cells with mutant p53 [22]. One should additionally keep in mind that caspase proficiency might be related to it, since MCF-7 cells are deficient for caspase-3 and MDA-MB231 cells not. We recently showed that the in vitro immunogenic potential of caspase-3 proficient breast cancer cells with basal low immunogenicity is increased by hypofractionated irradiation and that of caspase-3 deficient ones not [23].
Since senescent cells remain in a metabolic active state they cannot be defined as dead [24]. They actively shape the microenvironment and the expression and secretion of immune modulating proteins changes during the induction and establishment of senescence [25]. This has been termed as senescence-associated secretory phenotype (SASP) [26]. Senescent cells activate a self-amplifying secretory network. The SASP includes pro-inflammatory cytokines like Interleukin (IL)-1α, IL-1β, IL-6 and IL-8, chemokines and growth factors and thereby connects local senescent cells with systemic inflammatory events [27, 28].
7.3.3 Autophagy
Not only radiation-induced forms of cell demise and inflammation are interconnected, but also additionally the DNA damage response, as demonstrated for autophagy. The latter is a conserved lysosomal pathway for degrading cytoplasmic proteins, macromolecules and organelles. It is kind of a cellular recycling factory unit that also promotes energy efficiency through adenosine triphosphate (ATP) generation. It further mediates damage control by removing non-functional proteins and organelles. A detailed summary on the molecular and cellular mechanisms of autophagy was provided by Glick and colleagues [29]. Autophagy can be monitored by autophagosome formation, but usage of multiple assays is recommended for its detection [30]. We here focus on the impact of autophagy on radiosensitivity, DNA damage response and inflammation.
Cancer cells exploit autophagy to adapt to nutrient limiting, metabolically stressful and hypoxic tumour microenvironment, since the physiological function of autophagy is related to the maintenance of cellular homeostasis under cellular stress [31]. Additionally, a non-protective form of autophagy does exist. Here, the cell is carrying out autophagy-mediated degrading functions, but autophagy inhibition does not lead to sensitisation for radiation or drugs [32]. Furthermore, autophagy can be cytotoxic [33] or cytostatic. The latter one is characterised by prolonged growth inhibition and reduced clonogenic survival without resulting in cell death induction [34]. Because of cytotoxic and cytostatic autophagy, cancer cells most likely often display a reduced autophagy. Overexpression of Beclin 1, a Bcl-2-interacting coiled-coil protein, inhibits cellular proliferation and has autophagy-promoting activity. Beclin-1 expression is absent or frequently low in cancer, e.g. in prostate, breast and ovarian cancer [35].
The relationship between DNA repair and autophagy in cancer cells is just fragmentarily understood. Autophagy has been shown to regulate some of the DNA repair proteins after DNA damage (summarised in [36]). Furthermore, evidence was provided that a mechanistic link between processing of DNA damage and activation of autophagy does exist [37]. In a mouse model of poly-microbial sepsis it was elegantly demonstrated that DNA damaging chemotherapeutics like anthracyclines improved the survival of the septic mice without affecting bacterial burden. This was not a sole effect of suppression of release of inflammatory cytokines like IL-1β and danger signals like high-mobility group box 1 (HMGB1) that could also be achieved by antibiotics, but also of promoting tissue protection from inflammatory damage. This was achieved by autophagy induction in dependence of the activation of the DNA damage response [38, 39]. Recently, hints were identified that defective autophagy in vivo caused an absence or reduction in regulatory proteins critical to both homologous recombination (HR) and non-homologous end joining (NHEJ) DNA damage repair pathways. Further, a failure to induce these proteins in response to radiation was asserted [40]. Cottone and colleagues have identified the activation of autophagy and the release of HMGB1 as key events how colon carcinoma cells recruit leukocytes. Concomitant induction of autophagy to apoptosis by 5-fluorouracil (5-FU) was necessary to induce the leukocytes attraction. They suggest that HMGB1 is translocated to the cytosol and may there promote the activation of autophagy, which in turn fosters further HMGB1 translocation form the nucleus into the cytosol and its consecutive release in the extracellular milieu [41]. Irradiation of tumours with 2 Gy as other DNA-damaging stressor resulted in recruitment of cytotoxic T cells, here in dependence of macrophage differentiation to an iNOS+/M1 phenotype [42]. All these works give on the one hand evidence that after DNA damaging stress not only single cell death forms are induced and that on the other hand interconnections between DNA damage responses, inflammation and systemic immune modulation exist.
7.3.4 Apoptosis
Even though cell death can have many facets the two best known forms are still apoptosis and necrosis. Apoptosis, a form of programmed cell death, is crucial not only during embryonic development, but is present throughout the whole lifetime of multicellular organisms to attain cellular homeostasis. Apoptotic cells are characterised by nuclear and cytoplasmic condensation, nuclear fragmentation and cell shrinkage induced by plasma membrane blebbing [43]. Most importantly and contrary to necrotic cells, apoptotic cells maintain their membrane integrity until late stages of apoptosis execution. Apoptotic cells release and expose a broad range of ‘find me’ and ‘eat me’ signals for phagocytes such as macrophages [44]. The uptake of apoptotic cells occurs in a non- or even anti-inflammatory manner [45]. This immune suppressive effect might contribute to the in part unwanted effects of apoptosis induction by radiotherapy [46].
In response to ionising radiation, apoptosis is predominantly observed in cells of the hematopoietic system [47]. In solid tumours, the multicellular architecture may strongly contribute to render individual tumour cells less susceptible to apoptosis [48]. The TP53 gene provides instructions for making a protein called tumour protein p53 (p53) and is together with the PI3KCA gene that encodes for PI 3-kinases (PI3K) the most mutated gene in all types of cancers [49]. The tumour suppressor p53 primarily functions as a transcription factor, but its binding to the nuclear matrix generally increases after genotoxic stress [50]. p53 is involved in damage recognition, cell-cycle arrest, DNA repair, senescence or apoptosis. Of note is that p53 has roles that do not involve its transactivation functions during DNA repair; it modulates DNA repair processes, except for homologous recombination, by both transactivation-dependent and -independent pathways, as well as damage recognition and apoptosis [51]. It links apoptotic signalling pathways to radiation-induced DNA damage and is capable of directly regulating the Bax-dependent mitochondrial pathway to cell death [52]. In addition to intrinsic apoptosis pathways, extrinsic ones exist based on ligation of death receptors. In response to radiation, proteins of the death receptors are upregulated in a p53 dependent and independent manner [53]. Further, p53 controls signalling-mediated phagocytosis of apoptotic cells through its target Death Domain1α (DD1α) . The latter functions as an engulfment ligand and thereby ensures a proper clearance of cell corpses. This contributes to the maintenance of immune tolerance [54].
Other members of the p53 tumour suppressor family of genes like p73 might compensate the lack of function of p53 and mediate radiation-induced apoptosis [55]. Therefore, the general statement that is mostly based on p53 functionality, that distinct tumours are sensitive for apoptosis after irradiation or not has to be considered critically.
In addition, distinct stimuli can promote an immunogenic variant of apoptosis [24, 56]. Treatment with tumour necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL), e.g. induces membrane calreticulin (CRT) exposure on cancer cells [57]. The pre-apoptotic exposure of the endoplasmic reticulum (ER)-derived CRT together with the late or post-apoptotic release of danger signals like HMGB1 (see below) renders dying tumour cells immunogenic and can be induced by distinct chemotherapeutic agents like anthracyclines and oxaliplatin and by ionising radiation [58]. The exposure pathway of CRT is activated by pre-apoptotic ER stress and mediated via caspase-8-dependent proteolysis of the ER-sessile protein BAP31 and by activation of the pro-apoptotic proteins Bax and Bak [59]. Another scenario where apoptotic cells become immunogenic is that they proceed to secondary necrosis, meaning that they lose their membrane integrity. This happens when the clearance of apoptotic cells is impaired. This clearance defect is present in certain autoimmune diseases or when massive apoptosis occurs, e.g. after multimodal tumour treatments including RT [60, 61]. Secondary necrotic cells are often termed late apoptotic cells. This naming refers to the fact that the cells already underwent the apoptotic programme for a certain time. However, from the immunological point of view, due to the disturbed plasma membrane they behave like necrotic cells (Fig. 7.1).

Ionising radiation induces various tumour cell death modalities. The exposure of tumour cells to ionising radiation results in DNA damage, DNA damage response, ER stress response and in the induction of the displayed cell death forms. Radiation hereby not only impacts on the tumour cell phenotype but also on the tumour cell microenvironment. Of note is that all cell death forms can proceed to necrosis when during time their plasma membrane is disturbed. ATP adenosine triphosphate, CRT calreticulin, ER endoplasmic reticulum, HMGB1 high-mobility group box 1, HSP heat shock protein, LPC lysophosphatidylcholine, ROS reactive oxygen species, SASP senescence-associated secretory phenotype/proteins, 2° secondary
7.3.5 Necrosis
The overall definition of necrosis is that cells have lost their plasma membrane integrity. In Radiation Oncology, the term necrosis was for a long time just linked with radionecrosis, a late side effect of irradiation with high single doses [62]. Soft tissue and bone changes occur and lead in a small percentage of the patients to tissue necrosis.
Beneficial necrosis of tumour cells induced by RT came into the mind of clinicians when data came up that immunogenic cancer cell death has profound clinical and therapeutic implications. Necrotic cells release danger-associated molecular patterns (DAMPs) like HMGB1, heat shock proteins (HSP), nucleotides or uric acid that trigger the activation of both, the innate and the adaptive immune system [63]. Primary necrosis was considered as a non-physiological form of cell death induced by trauma, ROS, pathogens and massive toxicity in general. However, similar to apoptosis, necrosis can also occur in a regulated fashion, meaning that a genetically encoded molecular machinery runs. The so-called necroptosis , which is dependent on the receptor interacting protein (RIP) kinases RIP1 and RIP3 can be induced by factors such as tumour necrosis factor (TNF), Fas Ligand or TRAIL and utilises the same initial signalling cascade as cell-death receptor-induced apoptosis [64]. Necroptosis further requires the substrate of RIP3K, the mixed lineage kinase like (MLKL). Necroptosis can be manipulated by inhibitors such as necrostatin 1, which blocks RIP1 kinase activity [65, 66]. Mounting evidence exists that many of the currently used anticancer agents are capable of engaging necroptotic signalling pathways. This offers the opportunity to reactivate cell death programmes in human malignancies, especially in those being considered as apoptosis resistant [67].
In colorectal cancer cell lines, predominantly necrosis was inducible by RT and/or hyperthermia concomitantly with an increased expression of RIP1 [68]. We recently demonstrated that necroptosis is inducible with the pan caspase inhibitor zVAD-fmk in poorly immunogenic B16 melanoma cells [69]. Combination of RT, CT and immune stimulation by hyperthermia and zVAD-fmk resulted in significant tumour growth retardation compared to treatments without zVAD-fmk. This was dependent on the adaptive immune system, HMGB1 and nucleotides. Therapy-induced immunogenic cancer cell death might therefore be the key event in triggering anti-tumour immune responses.
7.4 Immunogenic Cancer Cell Death
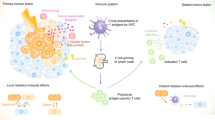
The definition of immunogenic cancer cell death is based on molecular and cellular mechanisms as well as certain in vivo characteristics [70]. Non-immunogenic cell death is characterised by PS exposure and swift clearance of the dying and stressed cells by macrophages. Concomitantly, apoptotic-cell derived blebs [71] and radiation-induced TGF-beta [72] might result in inhibition of anti-tumour immune responses [73] (Fig. 7.2). In contrast, immunogenic cancer cell death is mostly connected with the release of the DAMPs HMGB1 and ATP and with the exposure of CRT. Additionally, further immune activating danger signals like Hsp70 and immunostimulatory cytokines like TNF-α and IL-1β are released [74].

Radiation-induced immunogenic cancer cell death results in activation of the innate and adaptive immune system. Treatment of tumour cells with ionising radiation can induce non-immunogenic cancer cell death, namely apoptotic tumour cells that do expose phosphatidylserine (black dots) on the outer membrane leaflet and secrete TGF-β. They are finally cleared by macrophages in an anti-inflammatory manner. On the other hand, very early apoptotic cells that do expose CRT are immunogenic, as well as senescent cells, cells undergoing autophagy and necrotic cells mainly by release of danger signals, inflammatory cytokines and SASP. Especially a mixture of these cell death modalities is highly immunogenic and results in maturation of DC, consecutive priming of T cells and activation of NK cells. ATP adenosine triphosphate, CRT calreticulin, HMGB1 high-mobility group box 1, HSP heat shock protein, iDC immature dendritic cell, IL interleukin, mDC mature dendritic cell, NK natural killer, SASP senescence-associated secretory phenotype/proteins, TGF-β transforming growth factor beta, TNF tumour necrosis factor
This results in maturation and activation of DCs and ensuing priming of tumour-specific CD8+ T cells. Furthermore, NK cells can be activated by immunogenic cells including their microenvironment [75] (Fig. 7.2). For the in vivo examination of the immunogenic potential of tumour cells, both an immunisation and a therapeutic assay should be used. Both are based on the comparison of tumour growth in wild type compared to immune deficient mice: treatments that do induce immunogenic tumour cell death do result in retarded tumour growth only in wild-type animals [70]. That the tolerance has been actually broken and a memory immune response has been indeed induced should be tested with challenge experiments in animals that were primarily cured. Of note is that antineoplastic regimens that do engage immune effector mechanisms also achieve the same result without inducing immunogenic cancer cell death [76]. Therefore, multiple additional in vitro testing including functional assays with primary immune cells is mandatory to define immunogenic cancer cell death [77].
Besides DAMPs that are associated with immunogenic cell death, the SASP fosters the recruitment of immune cells. Therefore, the SASP is supposed to also act as a danger signal for the immune system aiming to eradicate potentially transformed or damaged cells in a CD4+ T cell and macrophage-dependent manner [78]. Furthermore, radiation-induced senescence in tumours has been shown to lead to an increased adaptive immune response through the recruitment and proliferation of tumour specific cytotoxic CD8+ T-lymphocytes [79].
Besides senescence, activation of autophagy contributes to recruitment of immune cells [41], as necrotic and apoptotic tumour cells, too [80]. High numbers of apoptotic cells, e.g. are sufficient to trigger DC maturation and antigen presentation, even in the absence of released danger signals [81]. This suggests that in vivo, combinations of apoptotic cell death, necrotic cell death, autophagic cell death and senescence trigger the induction of anti-tumour immune responses in a concerted action (Fig. 7.2).
7.5 Systemic Effects of Radiation
The insufficient immunological control of tumours is one hallmark of cancer [82]. Tumours must escape immune surveillance during development and when being established. The cancer immunoediting consists of the elimination, equilibrium and escape phase [83]. In the elimination phase, the immune system is capable of stopping cancer development and destroys tumour cells. In the equilibrium phase a latent state exists, while in the escape phase the immunological defence mechanisms fail and the tumour progresses. The immune system is not only involved in cancer prevention and development but also in cancer therapy [84].
RT might contribute to overcome tumour escape by modifying the phenotype of the tumour cells [85, 86]. In the ideal case, radiation generates an in situ vaccine. However, mostly immune responses to model antigens expressed by tumours have been examined. It remained uncertain whether RT can prime T cells specific for endogenous antigens expressed by poorly immunogenic tumours. Vanpouille-Box and colleagues recently demonstrated that this is also possible, however only when combining RT with blockade of TGF-beta and/or PD-1 [87]. The generated T cells were effective at causing regression of the irradiated tumours but also of non-irradiated metastases.
The so-called out-of-field or abscopal effects of RT are best when RT is combined with further immune activation [88]. To avoid the “mystic” wording abscopal and due to continuously growing numbers of preclinical and clinical studies that immune reactions mediate abscopal responses, they should be better termed RT-induced systemic immune-mediated effects [74]. The key mechanisms involved in ionising radiation-induced systemic effects were recently comprehensively summarised by Mavragani and colleagues [89].
7.6 Immunogenicity of Distinct Doses of RT and of Combination with Immunotherapies
Nowadays, due to technical improvements, RT is delivered in various fractionations. Standard fractionation consists of single doses of 1.8–2.2 Gy (one fraction per day, 5 days a week continuing for 3–7 weeks) and hypofractionation of 3–20 Gy (one fraction a day given for 1–3 days a week) [90]. The available data whether standard fractionation is as immunogenic as fewer applications with higher single doses (hypofractionation) or a very high single dose (radiosurgery) are not conclusive. Irradiation with a high single dose of 10 Gy of glioblastoma mouse tumours induced tumour growth retardation, increased the influx of CD8+ T cells and decreased that of Treg. However, significant improvement of long-term survival was only achieved when combining radiosurgery with blockade of the immune checkpoint molecule programmed cell death protein 1 (PD-1) [91]. While a single high dose of 20 Gy was as effective as 3 × 8 Gy or 5 × 6 Gy in retarding growth of the irradiated tumour, only fractionated irradiation in combination with an antibody against the immune checkpoint protein cytotoxic T-lymphocyte antigen 4 (CTLA-4) induced tumour growth retardation also outside of the irradiation field, as here shown in a mouse breast carcinoma model [92]. In ex vivo assays with human tumour and immune cells, the activation of DCs was similar when getting into contact with norm- or hypofractionated irradiated colorectal cancer cells, but much less after a single irradiation with 15 Gy [93].
Nevertheless, the current hypothesis is that higher doses might impact more strongly on intratumoural induction and production of type I interferon (IFN) with consecutive triggering of innate and adaptive immune mechanisms [94]. Ablative RT dramatically increases T-cell priming in draining lymphoid tissues, leading to both reduction of the primary tumour and of distant metastasis in dependence of CD8+ T cells. These immune responses are greatly amplified by addition of immunotherapy [95]. Lower single doses used in standard fractionation might especially impact on tumour vascularisation and therewith connected infiltration of immune cells [42, 96] (Fig. 7.4). Definite is that combination of RT with further immune activation induces the most striking anti-tumour immune reactions [85]. As already outlined shortly earlier, in response to radiation, tumour cells increase the surface expression of adhesion molecules, death receptors, stress-induced ligands, cryptic antigens and stimulatory molecules, such as MHC I and CD80, thereby becoming more sensitive to T cell-mediated cytotoxicity [86]. In the tumour microenvironment, pro-inflammatory molecules increase and maturation of DCs, antigen presentation and lymph node migration is fostered [97]. On the other hand, the immune cells might also be killed by radiation and pro-tumourigenic factors can be upregulated [98]. Consequently, radiation regimens have to be optimised and adjusted to maximise immunostimulatory functions and for the successful combination with other treatments, including IT [99].
Primarily radiation-induced immune suppression by, e.g. upregulation of PD-L1 on tumour cells has to be exploited for multimodal therapies with checkpoint inhibitors. These are currently the most promising therapies for induction of long-lasting anti-tumour effects as seen by a plateau in the patients’ survival curves [100, 101]. Checkpoint-blockade inhibitors improve adaptive immune responses induced by the RT-mediated increase in tumour antigens and tumours with high somatic mutation prevalence do respond best [102]. Nevertheless, not all of these selected patients respond. Therefore, the most beneficial combination with selected RT schemes and the chronological sequence of application of RT and IT has still to be identified [60]. We just recently summarised preclinical and clinical data on how the immune modulating properties of RT can be exploited for the combined treatment of cancer with immune checkpoint inhibitors [74].
7.7 Immune Suppressive and Proliferation Promoting Effects of Radiotherapy
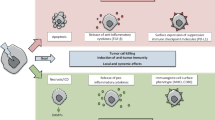
As almost always, two sides of the coin exist. X-rays can also reinforce immunosuppressive pathways (Fig. 7.3).

Radiation-induced immune suppression and tumour cell proliferation. Tumour cells exposed to ionising radiation can acquire an immune suppressive phenotype characterised by the expression of checkpoint inhibitor ligands such as PD-L1, the secretion of TGF-β, the infiltration of regulatory T cells and myeloid-derived suppressor cells into the tumour, and by inducing immune suppressive apoptosis. The latter is connected to reduced infiltration of eosinophils into the tumour, by M2 macrophage polarisation and by caspase-3, fractalkine and EGF-dependent increased tumour cell proliferation. Further, during and after RT, radioresistant cancer stem cells could be selected as well as Langerhans cells that generate in turn immune suppressive Treg. CSF-1 macrophage colony-stimulating factor 1, EGF epidermal growth factor, LC Langerhans cell, MDSC myeloid-derived suppressor cell, PD-L1 programmed cell death protein 1 ligand, PGE2 prostaglandin E2, TGF-β transforming growth factor beta, Treg regulatory T cell
Treg are intrinsically radioresistant which might lead to their intratumoural enrichment during RT. In the tumour microenvironment, Treg acquire a highly suppressive phenotype which is further increased by RT [103]. This is one rationale for combination of RT with further IT, as already mentioned earlier for checkpoint inhibitors. Short-term ablation of Treg in advanced spontaneous tumours induces both high numbers of dead tumour cells and in combination with RT significantly reduced metastatic tumour progression concomitant with prolonged survival [104].
As Treg, Langerhans cells (LC) are quite resistant immune cells [105]. Recently, it was found that LC resisted damage by irradiation because of their intrinsic expression of the cyclin-dependent kinase inhibitor CDKN1A (p21). Further, the LC-mediated generation of Treg was enhanced by radiation and directly correlated with the growth of the skin tumour [106].
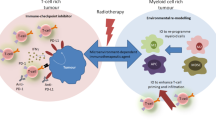
RT might further induce the macrophage colony-stimulating factor CSF1 in tumours and myeloid-derived suppressor cells accumulate in the tumour as well as in spleen, lung, lymph nodes and peripheral blood in a prostate cancer model [107]. This is again a convincing fact why especially combination of RT with immune modulation with CSF1 inhibitors in this case triggers beneficial anti-tumour responses (Fig. 7.4). Therapies have to be optimised in a way that the positive immunological impact of RT on anti-cancer responses outweighs the negative ones [108, 109]. Recently, it was demonstrated that granulocyte-macrophage colony-stimulating factor as a potent stimulator of DC maturation in combination with local RT generates abscopal responses in patients with metastatic solid tumours such as non-small cell lung and breast cancer [110].

Therapeutic exploitation of norm- and hypofractionated radiotherapy in combination with immune therapies for cancer treatment. For the induction of local and systemic tumour control, norm- and/or hypofractionated radiotherapy has to be combined with selected immune therapies to overcome the immune suppressive pathways depicted in Fig. 7.3. The most prominent events related to norm-fractionated radiotherapy, hypofractionated radiotherapy and immunotherapy are depicted in blue, red and green, respectively. CSF-1 macrophage colony-stimulating factor 1, CXCR4 chemokine (C-X-C Motif) receptor 4, EGFR epidermal growth factor receptor, LC Langerhans cell, PD-1 programmed cell death protein 1, TGF-β transforming growth factor beta, Treg regulatory T cell
Again, we should also have in mind the local as well as systemic consequences of RT. Apoptosis induction by RT is beneficial with regard to local tumour cell killing, but not inevitably from the immunological point of view [111]. Ford et al. recently demonstrated for B cell lymphomas that apoptotic tumour cells promote tumour growth, angiogenesis and accumulation of tumour-associated macrophages (TAM) resulting from in situ macrophage proliferation [112]. TAM are one of the major inflammatory cells that infiltrate tumours and epidemiological studies depict a correlation between TAM density and poor cancer prognosis [113]. Tissue destruction, even a small one occurring when taking a biopsy, may result in polarisation of macrophages to an M2 phenotype that could foster accelerated tumour progression [114].
Tumour cell apoptosis does thus not only impact on the immune system but also on proliferation of surrounding cells. Already in 1956 it was described that tumours killed by X-rays stimulate the proliferation of viable tumour cells [115]. It has been suggested that this is dependent on trophic substances derived from the tumour cells but also of the tumour bed, the microenvironment [116]. Recently, Chaurio et al. demonstrated that in an allogenic situation UV-B-irradiated apoptotic cells stimulate the growth of co-implanted viable tumour cells. These experiments were conducted in immune competent mice [117]. Since UV-B induces a mixture of apoptotic and necrotic cells it would be worth to examine in the future how distinct forms of tumour cells death impact on the proliferation of viable tumour cells and what mixture of cell death forms results predominantly in fostering of tumour cell proliferation and/or induction of anti-tumour immunity, respectively.
But what are the radiation-induced trophic substances that stimulate tumour cell proliferation? Apoptotic cells release a variety of “find-me” signalling factors, including nucleotides, the lipid lysophosphatidylcholine (LPC) and proteins such as fractalkine (summarised in [118]). The latter mediates the chemotaxis of macrophages to apoptotic lymphocytes [119]. Therefore, it might indirectly induce viable tumour cell proliferation by attracting macrophages into the tumour that are there polarised to M2 macrophages and directly by transactivation of the epidermal growth factor (EGF) pathway in the tumour cells [120]. This might be a further reason why combined treatments of tumours with RT and EGF receptor tyrosine kinase inhibitors are efficient [121]. Huang et al. demonstrated that caspase-3 is central in regulating the growth-promoting properties of dying cells by inducing the release of arachidonic acid and the production of prostaglandin E2 (PGE2) being a key regulator of tumour growth. Of special note is that caspase-3 was activated during RT [122]. RT-induced apoptosis may indeed lead to caspase 3-dependent tumour cell repopulation [46], but on the other hand caspase-3 is important to trigger immunogenic cancer cell death after hypofractionated irradiation [23]. Since TAM interacting with apoptotic tumour cells are central to activating multiple oncogenic pathways, to promote tumour cell growth and survival, angiogenesis, remodelling and metastasis [118] the aim should be to predominately induce necroptotic cancer cell death by RT [67, 68] and to concurrently target TAM, e.g. by pharmacologic blockade of chemokine (C-X-C Motif) receptor 4 (CXCR4) [123]. Massive necrosis should be induced to counteract the reduction of the immunogenicity of the necrotic cells by lactoferrin [124].
Interestingly, lactoferrin also functions as a “keep-out” signal to granulocytes. Since activated eosinophils were recently demonstrated to be essential for tumour rejection in the presence of tumour-specific CD8+ T cells and for an M1-like phenotype of macrophages [125], tumour promoting effects of apoptotic cells might also be connected to this. To summarise, apoptosis is central in conditioning the tumour microenvironment [126] (Figs. 7.1 and 7.3). This almost mandatorily demands that RT is combined with selected immune therapies to counteract the in part non-beneficial pro-tumourigenic effects of RT (Fig. 7.4). The same applies for possible selection of radioresistant cancer stem cells during and after RT [8, 127]. Mesenchymal stem cells are highly sensitive to small molecule receptor kinase inhibitors and combination treatments incorporating RT [128].
7.8 Conclusions
Even though approximately 60 % of patients with solid tumours are treated with RT, much fewer studies evaluating local therapies are published in high-impact oncology and medicine literature compared to systemic and targeted therapies [129]. Fortunately, a paradigm shift has been implemented during the last years: besides the local effects of RT on the DNA, also non-DNA targeted effects, the so-called systemic ones, do exist [130]. In former times it was predominantly publicised that only immune suppressive effects of RT exist. This has been questioned by many studies and it has become clear that a timely restricted radiation-induced decrease of immune cells does not automatically indicate that the immune system is functionally impaired [131]. The growing knowledge on the various forms of tumour cell death that can be induced by RT and/or CT has paved the way for combination of RT with IT [70, 132]. As it is common for the immune system that nearly every mechanism has wanted and unwanted effects independent of the existing state, also tumour cell death induction by RT can be beneficial for local and systemic tumour control (Fig. 7.2) and on the other hand even promote tumour cell proliferation and repopulation (Fig. 7.3). This highlights that a very sophisticated view on cell death induction by RT including the triggered cell death pathways and resulting cell death forms is mandatory [80, 133]. This becomes particularly important when further improving combination therapies consisting of RT, targeted therapies and immunotherapy (Fig. 7.4). Radiation-induced cell death is the mediator that broadens the modes of action of RT from a local level to a systemic one.
References
Lederman M. The early history of radiotherapy: 1895-1939. Int J Radiat Oncol Biol Phys. 1981;7:639–48.
Goodman LS, Wintrobe MM, Dameshek W, et al. Landmark article Sept. 21, 1946: nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. By Louis S. Goodman, Maxwell M. Wintrobe, William Dameshek, Morton J. Goodman, Alfred Gilman and Margaret T. McLennan. JAMA. 1984;251:2255–61.
Kalbasi A, June CH, Haas N, et al. Radiation and immunotherapy: a synergistic combination. J Clin Invest. 2013;123:2756–63.
Aziz K, Nowsheen S, Pantelias G, et al. Targeting DNA damage and repair: embracing the pharmacological era for successful cancer therapy. Pharmacol Ther. 2012;133:334–50.
Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125.
Iliakis G. Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother Oncol. 2009;92:310–5.
Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nat Rev Cancer. 2004;4:737–47.
Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells. 2010;28:639–48.
Helbig L, Koi L, Bruchner K, et al. Hypoxia-inducible factor pathway inhibition resolves tumor hypoxia and improves local tumor control after single-dose irradiation. Int J Radiat Oncol Biol Phys. 2014;88:159–66.
Murata T, Akagi K, Kimura H, et al. Analysis of cell kinetics after irradiation by flow cytometry: proliferative ability of G2-blocked cells after cell sorting. Oncol Rep. 1998;5:385–8.
Maier P, Wenz F, Herskind C. Radioprotection of normal tissue cells. Strahlenther Onkol. 2014;190:745–52.
Travis EL, Parkins CS, Down JD, et al. Repair in mouse lung between multiple small doses of X rays. Radiat Res. 1983;94:326–39.
Williams MV, Denekamp J, Fowler JF. A review of alpha/beta ratios for experimental tumors: implications for clinical studies of altered fractionation. Int J Radiat Oncol Biol Phys. 1985;11:87–96.
Qi XS, White J, Li XA. Is alpha/beta for breast cancer really low? Radiother Oncol. 2011;100:282–8.
Durante M, Reppingen N, Held KD. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol Med. 2013;19:565–82.
Orth M, Lauber K, Niyazi M, et al. Current concepts in clinical radiation oncology. Radiat Environ Biophys. 2014;53:1–29.
Amornwichet N, Oike T, Shibata A, et al. Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One. 2014;9, e115121.
Fragkos M, Beard P. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS One. 2011;6, e22946.
Castedo M, Perfettini JL, Roumier T, et al. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37.
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36.
Kurz DJ, Decary S, Hong Y, et al. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–22.
Jones KR, Elmore LW, Jackson-Cook C, et al. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol. 2005;81:445–58.
Kotter B, Frey B, Winderl M, et al. The in vitro immunogenic potential of caspase-3 proficient breast cancer cells with basal low immunogenicity is increased by hypofractionated irradiation. Radiat Oncol. 2015;10:197.
Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11.
Shelton DN, Chang E, Whittier PS, et al. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–45.
Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18.
Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544.
Dalby KN, Tekedereli I, Lopez-Berestein G, et al. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–9.
Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74:647–51.
Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8.
Yang Y, Yang Y, Yang X, et al. Autophagy and its function in radiosensitivity. Tumour Biol. 2015;36:4079–87.
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6.
Zhang D, Tang B, Xie X, et al. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol Ther. 2015;16:1005–13.
SenGupta T, Torgersen ML, Kassahun H, et al. Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat Commun. 2013;4:2674.
Figueiredo N, Chora A, Raquel H, et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39:874–84.
Medzhitov R. Septic shock: on the importance of being tolerant. Immunity. 2013;39:799–800.
Lin W, Yuan N, Wang Z, et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci Rep. 2015;5:12362.
Cottone L, Capobianco A, Gualteroni C, et al. 5-Fluorouracil causes leukocytes attraction in the peritoneal cavity by activating autophagy and HMGB1 release in colon carcinoma cells. Int J Cancer. 2015;136:1381–9.
Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Lauber K, Blumenthal SG, Waibel M, et al. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–87.
Voll RE, Herrmann M, Roth EA, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1.
Lauber K, Munoz LE, Berens C, et al. Apoptosis induction and tumor cell repopulation: the yin and yang of radiotherapy. Radiat Oncol. 2011;6:176.
Belka C, Heinrich V, Marini P, et al. Ionizing radiation and the activation of caspase-8 in highly apoptosis-sensitive lymphoma cells. Int J Radiat Biol. 1999;75:1257–64.
Kerbel RS. Impact of multicellular resistance on the survival of solid tumors, including micrometastases. Invasion Metastasis. 1994;14:50–60.
Rafii A, Touboul C, Al Thani H, et al. Where cancer genomics should go next: a clinician’s perspective. Hum Mol Genet. 2014;23:R69–75.
Jiang M, Axe T, Holgate R, et al. p53 binds the nuclear matrix in normal cells: binding involves the proline-rich domain of p53 and increases following genotoxic stress. Oncogene. 2001;20:5449–58.
Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6:44–55.
Baptiste N, Prives C. p53 in the cytoplasm: a question of overkill? Cell. 2004;116:487–9.
Sheikh MS, Burns TF, Huang Y, et al. p53-dependent and -independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Res. 1998;58:1593–8.
Yoon KW, Byun S, Kwon E, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015;349:1261669.
Wakatsuki M, Ohno T, Iwakawa M, et al. p73 protein expression correlates with radiation-induced apoptosis in the lack of p53 response to radiation therapy for cervical cancer. Int J Radiat Oncol Biol Phys. 2008;70:1189–94.
Kroemer G, Galluzzi L, Kepp O, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
de Bruyn M, Wiersma VR, Helfrich W, et al. The ever-expanding immunomodulatory role of calreticulin in cancer immunity. Front Oncol. 2015;5:35.
Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91.
Zitvogel L, Kepp O, Senovilla L, et al. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res. 2010;16:3100–4.
Frey B, Rubner Y, Wunderlich R, et al. Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation—implications for cancer therapies. Curr Med Chem. 2012;19:1751–64.
Gaipl US, Sheriff A, Franz S, et al. Inefficient clearance of dying cells and autoreactivity. Curr Top Microbiol Immunol. 2006;305:161–76.
Kocher M, Wittig A, Piroth MD, et al. Stereotactic radiosurgery for treatment of brain metastases. A report of the DEGRO Working Group on Stereotactic Radiotherapy. Strahlenther Onkol. 2014;190:521–32.
Krysko O, Love Aaes T, Bachert C, et al. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4, e631.
Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95.
Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21.
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20.
Fulda S. Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol. 2014;35:51–6.
Mantel F, Frey B, Haslinger S, et al. Combination of ionising irradiation and hyperthermia activates programmed apoptotic and necrotic cell death pathways in human colorectal carcinoma cells. Strahlenther Onkol. 2010;186:587–99.
Werthmoller N, Frey B, Wunderlich R, et al. Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death Dis. 2015;6, e1761.
Kepp O, Senovilla L, Vitale I, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3, e955691.
Stadler K, Frey B, Munoz LE, et al. Photopheresis with UV-A light and 8-methoxypsoralen leads to cell death and to release of blebs with anti-inflammatory phenotype in activated and non-activated lymphocytes. Biochem Biophys Res Commun. 2009;386:71–6.
Pineda JR, Daynac M, Chicheportiche A, et al. Vascular-derived TGF-beta increases in the stem cell niche and perturbs neurogenesis during aging and following irradiation in the adult mouse brain. EMBO Mol Med. 2013;5:548–62.
Xie Y, Bai O, Yuan J, et al. Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-beta1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res. 2009;69:7756–66.
Derer A, Deloch L, Rubner Y, et al. Radio-immunotherapy-induced immunogenic cancer cells as basis for induction of systemic anti-tumor immune responses—pre-clinical evidence and ongoing clinical applications. Front Immunol. 2015;6:1–19.
Multhoff G, Pockley AG, Streffer C, et al. Dual role of heat shock proteins (HSPs) in anti-tumor immunity. Curr Mol Med. 2012;12:1174–82.
Galluzzi L, Senovilla L, Zitvogel L, et al. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33.
Fucikova J, Moserova I, Truxova I, et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer. 2014;135:1165–77.
Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51.
Meng Y, Efimova EV, Hamzeh KW, et al. Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther. 2012;20:1046–55.
Lauber K, Ernst A, Orth M, et al. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front Oncol. 2012;2:116.
Rovere P, Vallinoto C, Bondanza A, et al. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol. 1998;161:4467–71.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8.
Candeias SM, Gaipl US. The immune system in cancer prevention, development and therapy. Anticancer Agents Med Chem. 2015;16:101–7.
Frey B, Rubner Y, Kulzer L, et al. Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol Immunother. 2014;63:29–36.
Hodge JW, Ardiani A, Farsaci B, et al. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol. 2012;39:323–39.
Vanpouille-Box C, Diamond JM, Pilones KA, et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75:2232–42.
Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58:862–70.
Mavragani IV, Laskaratou DA, Frey B, et al. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review. Toxicol Res. 2016;5:12–33.
Prasanna A, Ahmed MM, Mohiuddin M, et al. Exploiting sensitization windows of opportunity in hyper and hypo-fractionated radiation therapy. J Thorac Dis. 2014;6:287–302.
Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9.
Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88.
Kulzer L, Rubner Y, Deloch L, et al. Norm- and hypo-fractionated radiotherapy is capable of activating human dendritic cells. J Immunotoxicol. 2014;11:328–36.
Burnette BC, Liang H, Lee Y, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–96.
Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–95.
Ganss R, Ryschich E, Klar E, et al. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–70.
Hatfield P, Merrick A, Harrington K, et al. Radiation-induced cell death and dendritic cells: potential for cancer immunotherapy? Clin Oncol (R Coll Radiol). 2005;17:1–11.
Barcellos-Hoff MH, Derynck R, Tsang ML, et al. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest. 1994;93:892–9.
Shahabi V, Postow MA, Tuck D, et al. Immune-priming of the tumor microenvironment by radiotherapy: rationale for combination with immunotherapy to improve anticancer efficacy. Am J Clin Oncol. 2015;38:90–7.
Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300–9.
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Persa E, Balogh A, Safrany G, et al. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. 2015;368:252–61.
Bos PD, Plitas G, Rudra D, et al. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–66.
Durakovic N, Bezak KB, Skarica M, et al. Host-derived Langerhans cells persist after MHC-matched allografting independent of donor T cells and critically influence the alloresponses mediated by donor lymphocyte infusions. J Immunol. 2006;177:4414–25.
Price JG, Idoyaga J, Salmon H, et al. CDKN1A regulates Langerhans cell survival and promotes Treg cell generation upon exposure to ionizing irradiation. Nat Immunol. 2015;16:1060–8.
Xu J, Escamilla J, Mok S, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–94.
Frey B, Gaipl US. Radio-immunotherapy: the focused beam expands. Lancet Oncol. 2015;16:742–3.
Zitvogel L, Kroemer G. Subversion of anticancer immunosurveillance by radiotherapy. Nat Immunol. 2015;16:1005–7.
Golden EB, Chhabra A, Chachoua A, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015;16:795–803.
Lauber K, Herrmann M. Tumor biology: with a little help from my dying friends. Curr Biol. 2015;25:R198–201.
Ford CA, Petrova S, Pound JD, et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr Biol. 2015;25:577–88.
Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. 2013;35:585–600.
Weber M, Moebius P, Buttner-Herold M, et al. Macrophage polarisation changes within the time between diagnostic biopsy and tumour resection in oral squamous cell carcinomas—an immunohistochemical study. Br J Cancer. 2015;113:510–9.
Revesz L. Effect of tumour cells killed by x-rays upon the growth of admixed viable cells. Nature. 1956;178:1391–2.
van den Brenk HA, Crowe MC, Stone MG. Reactions of the tumour bed to lethally irradiated tumour cells, and the Revesz effect. Br J Cancer. 1977;36:94–104.
Chaurio R, Janko C, Schorn C, et al. UVB-irradiated apoptotic cells induce accelerated growth of co-implanted viable tumor cells in immune competent mice. Autoimmunity. 2013;46:317–22.
Willems JJ, Arnold BP, Gregory CD. Sinister self-sacrifice: the contribution of apoptosis to malignancy. Front Immunol. 2014;5:299.
Truman LA, Ford CA, Pasikowska M, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–36.
Tardaguila M, Mira E, Garcia-Cabezas MA, et al. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013;73:4461–73.
Moschini I, Dell’Anna C, Losardo PL, et al. Radiotherapy of non-small-cell lung cancer in the era of EGFR gene mutations and EGF receptor tyrosine kinase inhibitors. Future Oncol. 2015;11:2329–42.
Huang Q, Li F, Liu X, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–6.
Hughes R, Qian BZ, Rowan C, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75:3479–91.
Miles K, Clarke DJ, Lu W, et al. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol. 2009;183:2122–32.
Carretero R, Sektioglu IM, Garbi N, et al. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol. 2015;16:609–17.
Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94.
Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Biol Sci. 2011;7:517–35.
Nicolay NH, Sommer E, Perez RL, et al. Mesenchymal stem cells are sensitive to treatment with kinase inhibitors and ionizing radiation. Strahlenther Onkol. 2014;190:1037–45.
Holliday EB, Ahmed AA, Yoo SK, et al. Does cancer literature reflect multidisciplinary practice? A systematic review of oncology studies in the medical literature over a 20-year period. Int J Radiat Oncol Biol Phys. 2015;92:721–31.
Kadhim M, Salomaa S, Wright E, et al. Non-targeted effects of ionising radiation—implications for low dose risk. Mutat Res. 2013;752:84–98.
Belka C, Ottinger H, Kreuzfelder E, et al. Impact of localized radiotherapy on blood immune cells counts and function in humans. Radiother Oncol. 1999;50:199–204.
Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015;1(9):1325–32.
Frey B, Hehlgans S, Rodel F, et al. Modulation of inflammation by low and high doses of ionizing radiation: implications for benign and malign diseases. Cancer Lett. 2015;368(2):230–7.
Acknowledgments
This work is in part funded by the German Federal Ministry of Education and Research (BMBF; m4 Cluster, 16EX1021R and GREWIS, 02NUK017G) and the European Commission (DoReMi, European Atomic Energy Community’s Seventh Framework Programme (FP7/2007–2011) under grant agreement no. 249689).
Conflict of Interest Statement
All authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Frey, B., Derer, A., Scheithauer, H., Wunderlich, R., Fietkau, R., Gaipl, U.S. (2016). Cancer Cell Death-Inducing Radiotherapy: Impact on Local Tumour Control, Tumour Cell Proliferation and Induction of Systemic Anti-tumour Immunity. In: Gregory, C. (eds) Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy. Advances in Experimental Medicine and Biology, vol 930. Springer, Cham. https://doi.org/10.1007/978-3-319-39406-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-39406-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39404-6
Online ISBN: 978-3-319-39406-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)