
Abstract
In 2002, a syndrome of tremor, ataxia, cognitive decline, and the presence of unique ubiquitin staining intranuclear inclusions in the brain was discovered in premutation males carrying an expansion of between 55 and 200 CGG trinucleotide repeats on the FMR1 gene. This clinical syndrome is now known as fragile X-associated tremor/ataxia (FXTAS) and has been found in both male and female carriers of the expanded premutation allele. The goal of this chapter is to summarize what is known about the anatomical pathology associated with the fragile X premutation and particularly in those individuals with FXTAS. Neuropathology in FXTAS was initially found in the central nervous system, but recent evidence has demonstrated pathological features, including intranuclear inclusions, in the peripheral nervous system, the enteric nervous system, and the neuroendocrine system. The precise cellular dysfunctions that underlie these pathologic features are currently under intense investigation with the goal of prevention and treatment of this devastating disorder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset, progressive neurodegenerative disorder that affects many carriers of an FMR1 premutation, an expanded trinucleotide repeat sequence (CGG) in the 5′ untranslated region (5′ UTR) of the FMR1 gene. This gene is polymorphic, and in unaffected individuals there are roughly 5–45 CGG repeats, while individuals with a premutation carry an allele of 55–200 CGG repeats and show a two-to-eightfold increase in levels of the FMR1 mRNA (see Chap. 6).
Patients with FXTAS typically show cerebellar ataxia, tremor, cognitive deficits, peripheral neuropathy, autonomic dysfunction, and psychiatric involvement (see Chaps. 1 and 3). The disorder is thought to arise from a toxic RNA gain of function that is caused by overexpression of the expanded CGG FMR1 mRNA in premutation carriers (Allen et al. 2005; Kenneson et al. 2001; Tassone et al. 2000). Magnetic resonance imaging (MRI) also shows that patients with FXTAS have mild to moderate brain atrophy in both cerebrum and cerebellum and white matter changes in cerebrum and cerebellum. Increased T2 signal intensity in the middle cerebellar peduncles (MCP ) is commonly found in subjects affected by FXTAS (see Chap. 4) (Brunberg et al. 2002). Finally, the neuropathological hallmark of FXTAS is the presence of eosinophilic intranuclear inclusions in both neurons and astrocytes. These inclusions are found throughout the brain and in the autonomic nervous system as well as in non-nervous system tissues (e.g., pancreas). In light of these unique findings, the proposed diagnostic criteria for FXTAS include the presence of intranuclear inclusions as a major criterion (Hagerman and Hagerman 2004; Jacquemont et al. 2004).
Intranuclear Inclusions
Intranuclear inclusions are the distinctive pathological finding among premutation carriers affected by FXTAS. They have also been observed in a knock-in mouse models of the FMR1 premutation (see Chap. 8). Observed initially in human brain tissues (Greco et al. 2002), eosinophilic intranuclear inclusions are widely distributed in both neurons and astrocytes, being found in many different regions throughout the brain, including the frontal cortex, hippocampus, ependymal cells, choroid plexus, brainstem nuclei, and cerebellum (Greco et al. 2002, 2006). Given the significance of clinical symptomology related to the limbic system, it is important to note that the highest percentage of inclusions in FXTAS cases is in the hippocampus . Immunohistochemically, these inclusions stain positive for ubiquitin, lamin A/C, and a number of heat-shock proteins (Iwahashi et al. 2006). They stain negative for tau isoforms, α-synuclein, and polyglutamine peptides and appear to reflect a new class of nuclear inclusion disorder as compared to other triplet repeat disorders, such as Huntington’s disease and some of the spinocerebellar atrophies (SCA). FXTAS is also distinct from neuronal intranuclear inclusion disorder (NIID) (Hagerman and Hagerman 2004). Furthermore, these inclusions do not contain any single predominant protein species; the most prominent protein accounts for only roughly 7 % of the total protein mass (Iwahashi et al. 2006). Also noteworthy is that in patients with FXTAS, and in contrast to patients with CAG repeat degenerative disorders and inclusions, the protein product of the FMR1 gene, FMRP, is structurally normal and present at relatively normal or only slightly reduced expression levels, due to the fact that the expanded CGG repeat occurs in a non-coding portion (5′ UTR) of the gene.
A highly efficient, flow-based isolation and purification of inclusions from postmortem FXTAS brain tissues has allowed for mass spectrometric analysis of the entire protein complement of the isolated inclusions as well as follow-up immunohistochemical analysis to conclusively identify more than 20 inclusion-associated proteins. Several proteins appear to be ubiquinated and/or polyubiquinated in these purified inclusions, but ubiquinated proteins are the minority (Iwahashi et al. 2006). Ubiquitin is present within intracellular aggregates of a wide range of neurological disorders, not just FXTAS (Woulfe 2008). In the case of FXTAS, ubiquitin, a proteosomal degradation product , is utilized as a marker for isolation or detection of intranuclear inclusions by immunostaining (Greco et al. 2002, 2006; Iwahashi et al. 2006). Among the proteins identified within the inclusions are the RNA-binding protein, hnRNP A2, several intermediate filament (IF) proteins, including lamin A/C, the small heat-shock protein αB-crystallin, α-internexin, and other neurofilament (NF) proteins. The FMR1 mRNA is present, but only as a minor component within the inclusions (Tassone et al. 2004b), and FMRP has not yet been found to be present in the inclusions. All of the proteins found in the inclusions are potential candidates for involvement in the RNA gain of function that may underlie FXTAS pathology (Hagerman and Hagerman 2004; Iwahashi et al. 2006).
The time course of inclusion formation relative to clinical onset of disease is not yet known, nor is it understood whether the intranuclear inclusions are directly causative of FXTAS pathophysiology and symptomology, or simply a reflection of the progression of the disorder. If the inclusion materials are active or neurotoxic, they may contribute directly to damage to the nervous system. However, it is also possible that the intranuclear inclusions may represent a protective mechanism, serving as a repository for disabled enzymes and their products as has been proposed for Huntington’s disease (Bowman et al. 2005). Intranuclear inclusions have also been reported in a fragile X premutation female carrier that showed no clinical symptoms associated with FXTAS (Tassone et al. 2012). These data suggest that the presence of intranuclear inclusions may be a consequence of, but not sufficient to lead to an FXTAS diagnosis (Tassone et al. 2012). Inclusions have been also described in a fragile X mosaic male (Pretto et al. 2013). A very low number of inclusions have been also detected in three older adults with FXS. These low levels suggest that the clinical course in these three subjects would not have been influenced by contributions from RNA toxicity (Hunsaker et al. 2011). The youngest deceased case of FXTAS (stage 2) described to date is a 36-year-old male that presented with both neuronal and astrocytic inclusions. The number of cells with inclusions and its size was comparable to that in older cases. These data indicate that even FXTAS is thought to be a disorder of aging in carriers of FMR1 premutation, it can occur earlier in adult life, particularly if another disease process is occurring, such as substance abuse (as in this case) that may exacerbate the pathological process of FXTAS (Martinez-Cerdeno et al. 2015).
Recently, the presence of inclusions positive for a polyglycine containing FMRP protein (FMRpolyG) have been described in the CNS and non-CNS organs in FXTAS. Todd and colleagues (Todd et al. 2013) demonstrated that through initiation at a near-ATG codon located in the 5′UTR of the FMR1 gene a polyglycine-containing protein, FMRpolyG, is expressed. This protein accumulates in brain of FXTAS patients. It is also present in ubiquitin-positive inclusions in Drosophila, cell culture, and mouse disease models. Using two novel antibodies, they confirmed the existence of FMRpolyG-positive intranuclear inclusions in postmortem non-CNS material of a premutation carrier with FXTAS. Furthermore, the described colocalization of FMRpolyG and ubiquitin is found in the vast majority of inclusions. The presence of FMRpolyG-positive intranuclear inclusions in heart, kidney, adrenal gland, and thyroid is consistent with the unexplained medical comorbidities reported in some patients with FXTAS, including thyroid disease, cardiac arrhythmias, hypertension, migraine, impotence, and neuropathy (Buijsen et al. 2015). FMRpolyG have been also described in ovarian stromal cells of a woman with FXPOI but not in the ovaries of control subjects. The FMRpolyG-positive inclusions colocalized with ubiquitin-positive inclusions. Similar inclusions were also observed in the pituitary of a man with FXTAS but not in control subjects. Similarly, ovaries of exCGG-KI mice, but not wild-type mice, contained numerous inclusions in the stromal cells that stained for both FMRpolyG and ubiquitin (Buijsen et al. 2015).
It is unclear whether the FMR1 premutation predisposes individuals to or accelerates the course of other degenerative diseases of the central nervous system (or vice versa), being this a topic of active investigation . A number of FXTAS cases that have come to autopsy showed Lewy body formation in the substantia nigra, whether or not Parkinson’s disease (PD) was clinically identified (Greco et al. 2002). An additional report described an elderly woman with cognitive impairment that was attributed to early AD that had both manifestations of FXTAS and premature ovarian failure (Al-Hinti et al. 2007). In addition, two females carrying the FXTAS premutation who suffered early onset Alzheimer’s disease (AD) symptomology along with tremor and ataxia showed histopathological features of both AD and FXTAS (Greco et al., unpublished data). The superior and middle temporal gyri in these two women showed as high a percentage of intranuclear inclusions as seen in the hippocampus (unpublished data). In a reported case of concurrent FXTAS and multiple sclerosis (MS), the patient showed patchy and diffuse signal intensity alterations in white matter on T2-weighted MRI scans. Histologically, there were numerous regions of demyelination as well as the presence of intranuclear inclusions (Fig. 5.1). Recently, the first case of case of FMR1 premutation with Prader-Willi Phenotype (PWP) and FXTAS has been described. This patient presented with eosinophilic and ubiquitin-positive intranuclear inclusions throughout his brain, as is common in FXTAS and suffered multiple architectural cortical abnormalities that may be related to neuroblast migration problems during cerebral cortex prenatal development, including abnormal gyration, mild ventricle enlargement, thin cortex, thin corpus callosum, and a left small inferior olive (Martínez-Cerdeño et al., unpublished data).

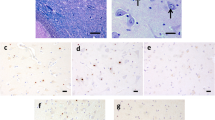
(a) Severe subcortical white matter degeneration , as seen in some cases of FXTAS autopsy brains; (b) Corresponding patchy white matter loss affecting cortical U-fibers (×40, LFB–PAS stain); (c) Occasional swollen axons can be identified in cerebral and cerebellar white matter and middle cerebellar peduncles
Brain Pathology
Gross Brain Pathology
In FXTAS, white matter disease is broad within the cerebrum and cerebellum, and is accompanied by a mild to moderate cortical atrophy and ventriculomegaly. The brain also presents with brainstem atrophy, especially of the pons. There are no notable gross structural changes in the spinal cord. When Parkinson’s disease is concurrent, the substantia nigra is pale. When Alzheimer’s disease is concurrent, cortical atrophy is often more prominent than that usually seen in FXTAS alone (Greco et al. 2002). In a reported case of concurrent FXTAS and multiple sclerosis, the patient showed patchy and diffuse signal intensity alterations in white matter on T2-weighted MRI scans. Histologically, there were numerous regions of demyelination as well as the presence of intranuclear inclusions (Fig. 5.1), (Greco et al. 2008). In a high percentage of FXTAS cases, increased T2 signal intensity is present in the middle cerebellar peduncles and can also be seen in the deep cerebellar white matter and brainstem (Brunberg et al. 2002), (See Chap. 4).
Microscopic Brain Pathology
Inclusions on H&E stains are discrete, hyaline-appearing, eosinophilic, round to slightly ovoid bodies (Fig. 5.2). They typically measure 2–5 μm in diameter and are single within a nucleus, except for those in Purkinje cells that are often twin inclusions (Ariza et al., 2016). They are periodic acid-Schiff (PAS), silver, tau, and neurofilament (NF) negative but stain positively for ubiquitin. Although inclusions have been identified in neurons throughout the brain, they have not been seen in Betz cells. Purkinje cell loss beyond that expected with otherwise normal aging and axonal swellings/torpedoes are commonly seen in FXTAS. No correlation have been found between the loss of Purkinje cells and the presence of single or twin inclusions within Purkinje cells (Ariza et al., 2016). Bergmann gliosis accompanies Purkinje cell loss in the cerebellum (Greco et al. 2002, 2006). Inclusions are also present in neurons of the dentate nucleus, and neurons and astrocytes throughout the cerebellum. When clinical PD has been diagnosed, cytoplasmic Lewy bodies are seen in pigmented neurons of the substantia nigra, and when FXTAS coexists the intranuclear inclusions can be seen in the pigmented neurons of the substantia nigra, whether or not cytoplasmic Lewy bodies are present. Two cases of PD and the characteristic radiological (MCP) sign demonstrated widespread p62-positive and ubiquitin-positive and 1C2-negative neuronal and glial intranuclear inclusions with mild Purkinje cell depletion consistent with FXTAS. There was also loss of pigmented neurons in the substantia nigra with α-synuclein-positive Lewy bodies (De Pablo-Fernandez et al. 2015). In both symptomatic male and female premutation carriers who also carried a diagnosis of AD, the intranuclear inclusions of FXTAS could be seen in pyramidal neurons of the hippocampus that also contained neurofibrillary tangles. Tree women with FXTAS plus dementia showed the presence of intranuclear inclusions and sufficient number of cortical amyloid plaques and neurofibrillary tangles to make AD a highly likely cause of dementia. A fourth case had dementia with cortical Lewy bodies and inclusions (Tassone et al. 2012).

(a) Neuronal and astrocytic intranuclear inclusions, CA4 (×400, H&E stain). (b) Ubiquitin immunoperoxidase stain showing numerous intranuclear inclusions, CA4, of hippocampus (×200). (c) Intranuclear inclusions, similar to those seen in the nervous system, are identified in Leydig cells (testosterone producing) of the testicles (×200, H&E). (d) Epicardial fat pad autonomic ganglion cell harboring an intranuclear inclusion (×200, H&E)
In astrocytes, intranuclear inclusions are usually surrounded by a clear halo, although this may be an artifact of tissue preparation. They are present diffusely in both protoplasmic astrocytes of gray matter and fibrillary astrocytes of white matter of the brain and spinal cord. They are also seen in pituicytes, the modified astrocytes of the posterior pituitary gland (Louis et al. 2006). Inclusions are also present in cells of the choroid plexus and ependyma, both of which have astrocyte lineages. In contrast, they are rarely present in microglia and have not been identified in oligodendrocyte nuclei or endothelial cells of the brain. The appearance of intranuclear inclusions is similar between males and females and between brain and spinal cord. While the spinal cord is otherwise grossly unremarkable, intranuclear inclusions have been identified in astrocytes and autonomic neurons of the intermediolateral column of the spinal cord but not in anterior horn cells (Gokden et al. 2008; Greco et al. 2002, 2006). Gokden et al. (2008) have also observed intranuclear inclusions in paraspinal sympathetic ganglia.
The appearance of intranuclear inclusions by electron microscopy is similar in neurons and astrocytes and appears as non-membrane-bound collections of granulofilamentous material (Greco et al. 2002). The filaments appear as straight rod-like proteins arranged in a haphazard manner (Gokden et al. 2008; Greco et al. 2002). The ultrastructural appearance of these inclusions is, however, otherwise not particularly informative.
Morphometric Analysis: Neuronal and Inclusion Counts
Percentages of neurons and astrocytes containing intranuclear inclusions were originally determined only in one study, for frontal cortex, hippocampus, and the ventral pontine region of 8 male FXTAS patients and 10 normal (no neurological disease), age-matched control subjects. Quantification of inclusions was carried out using a computer-based imaging and cell-counting system (StereoInvestigator, MBI, Inc., Williston, VT) on H&E-stained slides. The number of neurons and astrocytes with intranuclear inclusions (actual counts and percentages) is presented in Table 5.1 (summary of Tables 3–5 from (Greco et al. 2006)). No inclusions were seen in control cases. Inclusions were later described in Pukinje neurons in 65% of the FXTAS cases analyzed (26 cases). Of those, more than half of the cases (9/17) possessed twin adjacent nuclear inclusions. For the 17 cases with inclusions, Purkinje cells containing inclusions accounted for 9.1± 0.9 % of the total Purkinje cells. Those Purkinje cells with twin inclusions comprised 16.7 ± 2.7 of all Purkinje cells with inclusions (Ariza et al, 2016). The presence of inclusions in Purkinje cells was correlated with age). Other quantification studies include that of the youngest deceased case ever diagnosed with FXTAS, a man in his 30s, that contained 9.5 % of cells in the prefrontal cortex (14.2 % astrocytes; 4.8 % neurons), 6.4 % in CA1 (18 % astrocytes; 9.6 % neurons), 5.7 % in caudatum (20 % astrocytes; 6.4 % neurons), and 5.3 % in cerebellum (3.2 % astrocytes; 6.0 % neurons) (Martinez-Cerdeno et al. 2015). The case with FXTAS and PWP presented inclusions within the cerebral and cerebellar cortices in 14 % of cells, while the hippocampus had 3 % in the molecular layer, 14 % in the pyramidal layer, and 11 % in the granular layer. Intranuclear inclusions in astrocytes were massive, occupying up to 80 % of the nucleus, while in neurons the inclusions were smaller and about the size of the nucleolus (Martínez-Cerdeño et al., unpublished data).
In general, more and bigger intranuclear inclusions were observed in astrocytes than in neurons (the hippocampal CA1 subregion was an exception, but the pyramidal cell layer was counted, which contains relatively few astrocytes compared to neurons relative to the cortex), although there was a great deal of variability across subjects. Statistical correlations (Spearman’s rho) were calculated between histological findings and molecular measures. Significant positive correlations were present between the percentages of both neurons and astrocytes with inclusions in several brain regions and the number of CGG repeats. However, correlations between percentage inclusions and peripheral blood leukocyte FMR1 mRNA or FMRP levels were not statistically significant. This last observation is not surprising in view of the large differences between expression levels in brain and blood and the region-specific differences in FMR1 mRNA levels in brain (Tassone et al. 2004a). Most striking was the clinical–molecular correlation that showed a significant decrease in age of death with increasing CGG repeat length (i.e., the greater the CGG repeat number, the earlier the age of death; (Greco et al. 2006)).
White Matter Pathology
White matter changes seen on MRI studies include nonspecific, subcortical, patchy regions of increased T2 signal intensity in the cerebrum. In a high percentage of FXTAS cases, increased T2 signal intensity is present in the middle cerebellar peduncles (see Chap. 4) (Brunberg et al. 2002) and can also be seen in the deep cerebellar white matter and brainstem.
When these regions are examined microscopically using histologic and immunochemical stains, abnormal areas of white matter show spongiosis, axonal degeneration, and myelin loss. The same histological features are identified in damaged white matter of the cerebellum (Greco et al. 2002, 2006). In cases with the most severe cerebral white matter changes, scattered fibrillary astrocytes are greatly enlarged by irregular expansion of cytoplasm that contains lysosomal debris. These same cells may also contain intranuclear inclusions. Rare axonal spheroids have been identified in spongiotic middle cerebellar peduncles on H&E and neurofilament stains. The middle cerebellar peduncles may also show myelin pallor on LFB–PAS stain (Greco et al. 2006).
Peripheral Nervous System
While the intranuclear inclusions of FXTAS were first identified in neurons and astrocytes of the brain in 2002 (Greco et al. 2002), systemic locations of the inclusions in the peripheral nervous system and other tissues are rapidly being cataloged and published in the medical literature.
Autonomic System
Inclusions have been observed in paraspinal sympathetic ganglion, ganglion cells of adrenal medulla, ganglion cells of the myenteric plexus of the stomach, and ganglion cells of a subepicardial ganglion. Also, intranuclear inclusions have been identified in dorsal root ganglion neurons in the spinal cord (autonomic neurons), but not in the ventral root (Gokden et al. 2008). Symptoms corresponding to this autonomic pathology may include mega-esophagus, constipation, bladder spasms, orthostasis, hypertension, and sexual dysfunction (see Chaps. 1 and 9).
Peripheral Nerve
Nonspecific features of axonal degeneration have been seen in nerve examined at autopsy. Inclusions have not been observed by light microscopy. Clinically, neuropathic features are seen in male premutation carriers (Berry-Kravis et al. 2007), and peripheral neuropathy of variable severity is noted in individuals with FXTAS with reduced peripheral nerve conduction velocity (Soontarapornchai et al. 2008). The possible causes of this dysfunction are unknown.
Skeletal Muscle
Light microscopy, including histochemical and enzyme staining, has shown no pathological changes. Ultrastructural examination has yielded no distinctive abnormalities.
Neuroendocrine
In a limited number of cases (one male and one female), intranuclear inclusions within the anterior and posterior pituitary have been identified and may be associated with dysregulation of neuroendocrine function (Gokden et al. 2008; Greco et al. 2007; Louis et al. 2006). Similar findings have been made for the CGG KI mouse (see Chap. 8). This observation is of particular interest in view of the elevated cortisol levels found in FXTAS, as well as an increased incidence of anxiety disorders and depression (Bourgeois et al. 2009; Hunter et al. 2008; Rodriguez-Revenga et al. 2008).
Testicular pathology has been documented in two cases of FXTAS stained with H&E, including tubular fibrosis, decreased numbers of Leydig cells, and decreased spermatogenesis. Sertoli cells were abundant in the tubules along with a scant number of germ cells and spermatozoa that were remnants of germ maturation, but these changes were comparable to those seen in normal age-matched controls. There were also eosinophilic intranuclear inclusions in a small percentage of the Leydig cells in both cases as well as in the myoid cells of the tubular walls. Inclusions within the Leydig cells may be related to decreased levels of testosterone in some younger males with FXTAS who suffer premature erectile dysfunction. The presence of intranuclear inclusions in myoid cells in testicular connective tissue compartments in FXTAS is intriguing. The tunica propria of the testicle is a component of the tunica albuginea, and it is the middle of the three layers of the fibrous capsule beneath the scrotal skin that protects and supports the testes. Among other cellular components, the tunica albuginea contains myofibroblasts. In the tunica propria smooth muscle cells are involved in contractile and transport functions. Inclusions in these cells suggest that other cell populations outside of the nervous system may also have inclusions. This finding raises the possibility of identifying easily accessible diagnostic tissue for biopsy, and such tissue samples could be used for diagnostic purposes or for monitoring therapeutic responses to treatment (Greco et al. 2007). No data is available about ovary pathology in FXTAS; however, premutation carrier mice show a faster loss of follicles, many oocytes with aberrant nuclear accumulation of FMRP and elevated levels of ubiquitination. Furthermore, follicles are smaller and have fewer granulosa cells (GCs) than normal (Hoffman et al. 2012). Studies in human should follow up to better understand the pathology of the ovary in FXTAS. For more details, see Chap. 10 and 11.
Other Related Pathology
Other pathology of the FXTAS brain include prominent iron accumulation in the choroid plexus and putamen, and a milder iron accumulation in cerebellum, together with an alteration of the expression levels of proteins related to iron metabolism (Ariza et al. 2015; Rogers et al. 2016). Iron depositions are present in postmortem choroid plexus in FXTAS. In addition, transferrin levels are decreased in the epithelial cells, and transferrin receptor 1 distribution is shifted from the basolateral membrane (control) to a predominantly intracellular location (FXTAS). In addition, ferroportin and ceruloplasmin are markedly decreased within the choroid epithelial cells. These data indicate an alteration in iron transport and metabolism in the brain in FXTAS. In addition, these alterations have implications not only for understanding the pathophysiology of FXTAS, but also for the development of new clinical treatments that may incorporate selective iron chelation (Ariza et al. 2015).
FXTAS brain also presents with a reduction of proteins related to glutamate signaling (Pretto et al. 2014). The expression levels of the metabotropic glutamate (Glu) receptor 5 and the Glu transporter excitatory amino acid transporter 1 are reduced in the postmortem cerebellum of FXTAS, with higher CGG repeat number having greater reductions in both proteins. These data suggest a dysregulation of Glu signaling in premutation carriers, which could likely contribute to the development and severity of FXTAS (Pretto et al. 2014).
Summary
Since the initial discovery in 2002 that male premutation carriers with a clinical syndrome of tremor, ataxia, and cognitive decline showed a unique intranuclear inclusion disorder in pathological studies, there have been further studies elucidating the histologic, molecular, and biochemical features of the inclusions. The peripheral nervous system, specifically the autonomic system, is clearly involved, as is the neuroendocrine system. Cellular dysfunctions that underlie these pathologic features are currently under intense investigation with the goal of prevention and treatment of this devastating disorder.
Abbreviations
- DRPLA:
-
Dentatorubropallidoluysian atrophy
- SBMA:
-
X-linked spinobulbar muscular atrophy (Kennedy’s disease)
- NIID:
-
Neuronal intranuclear inclusion disease
- SCA:
-
Spinocerebellar ataxia
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- 5′ UTR:
-
5′ untranslated region
- MS:
-
Multiple sclerosis
- FXTAS:
-
Fragile X-associated tremor/ataxia syndrome
- FMR1 :
-
Fragile X mental retardation gene
- FMRP:
-
Fragile X mental retardation protein
- H&E:
-
Hematoxylin and eosin stain
- HD:
-
Huntington’s disease
- PAS:
-
Periodic acid-Schiff stain
- LFB:
-
Luxol fast blue stain
- MCP:
-
Middle cerebellar peduncle
- IF:
-
Intermediate filament
- NF:
-
Neurofilament
References
Al-Hinti JT, Nagan N, Harik SI (2007) Fragile X premutation in a woman with cognitive impairment, tremor, and history of premature ovarian failure. Alzheimer Dis Assoc Disord 21(3):262–4
Allen EG, Sherman S, Abramowitz A, Leslie M, Novak G, Rusin M, Scott E, Letz R (2005) Examination of the effect of the polymorphic CGG repeat in the FMR1 gene on cognitive performance. Behav Genet 35(4):435–45
Ariza J, Steward C, Rueckert F, Widdison M, Coffman R, Afjei A, Noctor SC, Hagerman R, Hagerman P, Martinez-Cerdeno V (2015) Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res 1598:88–96
Ariza J, Rogers H, Monterrubio A, Reyes-Miranda A, Hagerman PJ, Martínez-Cerdeño V. (2016) A Majority of FXTAS Cases Present with Intranuclear Inclusions Within Purkinje Cells. Cerebellum. Apr 23.
Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, Grigsby J, Bourgeois JA, Finucane B, Jacquemont S, Brunberg JA, Zhang L, Lin J, Tassone F, Hagerman PJ, Hagerman RJ, Leehey MA (2007) Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 22(14):2018–30
Bourgeois JA, Coffey SM, Rivera SM, Hessl D, Gane LW, Tassone F, Greco C, Finucane B, Nelson L, Berry-Kravis E, Grigsby J, Hagerman PJ, Hagerman RJ (2009) A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry 70(6):852–62
Bowman AB, Yoo S-Y, Dantuma NP, Zoghbi HY (2005) Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin–proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14(5):679–691
Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, Tassone F, Brown WT, Greco CM, Hagerman PJ (2002) Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol 23(10):1757–66
Buijsen RA, Visser JA, Kramer P, Severijnen EA, Gearing M, Charlet-Berguerand N, Sherman SL, Berman RF, Willemsen R, Hukema RK (2015) Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum Reprod 31(1):158–68
De Pablo-Fernandez E, Doherty KM, Holton JL, Revesz T, Djamshidian A, Limousin P, Bhatia KP, Warner TT, Lees AJ, Ling H (2015) Concomitant fragile X-associated tremor ataxia syndrome and Parkinson’s disease: a clinicopathological report of two cases. J Neurol Neurosurg Psychiatry 86(8):934–6
Gokden M, Al-Hinti JT, Harik SI (2008) Peripheral nervous system pathology in fragile X tremor/ataxia syndrome (FXTAS). Neuropathology 29(3):280–284
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125(Pt 8):1760–71
Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ (2006) Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 129(Pt 1):243–55
Greco CM, Soontarapornchai K, Wirojanan J, Gould JE, Hagerman PJ, Hagerman RJ (2007) Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J Urol 177(4):1434–7
Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, Brunberg JA, Hagerman PJ, Hagerman RJ (2008) Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol 65(8):1114–6
Hagerman PJ, Hagerman RJ (2004) The fragile-X premutation: a maturing perspective. Am J Hum Genet 74(5):805–816
Hoffman GE, Le WW, Entezam A, Otsuka N, Tong ZB, Nelson L, Flaws JA, Mcdonald JH, Jafar S, Usdin K (2012) Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J Histochem Cytochem 60(6):439–56
Hunsaker MR, Greco CM, Tassone F, Berman RF, Willemsen R, Hagerman RJ, Hagerman PJ (2011) Rare intranuclear inclusions in the brains of 3 older adult males with fragile x syndrome: implications for the spectrum of fragile x-associated disorders. J Neuropathol Exp Neurol 70(6):462–9
Hunter JE, Allen EG, Abramowitz A, Rusin M, Leslie M, Novak G, Hamilton D, Shubeck L, Charen K, Sherman SL (2008) Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav Genet 38(5):493–502
Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain 129(Pt 1):256–71
Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, Herman K, Grigsby J, Greco CM, Berry-Kravis E, Tassone F, Hagerman PJ (2004) Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291(4):460–469
Kenneson A, Zhang F, Hagedorn CH, Warren ST (2001) Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet 10(14):1449–54
Louis E, Moskowitz C, Friez M, Amaya M, Vonsattel JPG (2006) Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: a clinical–pathological study. Mov Disord 21(3):420–425
Martinez-Cerdeno V, Lechpammer M, Lott A, Schneider A, Hagerman R (2015) Fragile X-associated tremor/ataxia syndrome in a man in his 30s. JAMA Neurol 72(9):1070–3
Pretto DI, Hunsaker MR, Cunningham CL, Greco CM, Hagerman RJ, Noctor SC, Hall DA, Hagerman PJ, Tassone F (2013) Intranuclear inclusions in a fragile X mosaic male. Transl Neurodegener 2(1):10
Pretto DI, Kumar M, Cao Z, Cunningham CL, Durbin-Johnson B, Qi L, Berman R, Noctor SC, Hagerman RJ, Pessah IN, Tassone F (2014) Reduced excitatory amino acid transporter 1 and metabotropic glutamate receptor 5 expression in the cerebellum of fragile X mental retardation gene 1 premutation carriers with fragile X-associated tremor/ataxia syndrome. Neurobiol Aging 35(5):1189–97
Rodriguez-Revenga L, Madrigal I, Alegret M, Santos M, Mila M (2008) Evidence of depressive symptoms in fragile-X syndrome premutated females. Psychiatr Genet 18(4):153–5
Rogers H, Ariza J, Monterrubio A, Hagerman P, Martínez-Cerdeño V. (2016) Cerebellar mild iron accumulation in a subset of FMR1 premutation carriers with FXTAS. The Cerebellum (in press).
Soontarapornchai K, Maselli R, Fenton-Farrell G, Tassone F, Hagerman PJ, Hessl D, Hagerman RJ (2008) Abnormal nerve conduction features in fragile X premutation carriers. Arch Neurol 65(4):495–8
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ (2000) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66(1):6–15
Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ (2004a) Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 41(4):e43
Tassone F, Iwahashi C, Hagerman PJ (2004b) FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol 1(2):103–105
Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, Jacquemont S, Basuta K, Jin LW, Hagerman PJ, Hagerman RJ (2012) Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav 11(5):577–85
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78(3):440–55
Woulfe J (2008) Nuclear bodies in neurodegenerative disease. Biochim Biophys Acta 1783(11):2195–206
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cerdeno, V.M., Greco, C. (2016). The Pathology of FXTAS. In: Tassone, F., Hall, D. (eds) FXTAS, FXPOI, and Other Premutation Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-33898-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-33898-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33896-5
Online ISBN: 978-3-319-33898-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)