
Abstract
Cancer research is emerging as a complex orchestration of genomics, data-sciences, and network-sciences. For improving cancer diagnosis and treatment strategies, data across multiple scales, from molecules like DNA, RNA, metabolites, to the population, need to be integrated. This requires handling of large volumes of high complexity “Omics” data, requiring powerful computational algorithms and mathematical tools. Here we present an integrative analytics approach for cancer genomics. This approach takes the multi-scale biological interactions as key considerations for model development. We demonstrate the use of this approach on a publicly available lung cancer dataset collected for 109 individuals from an 18 years long clinical study. From this data, we discovered novel disease markers and drug targets that were validated using peer-reviewed literature. These results demonstrate the power of big data analytics for deriving disease actionable insight.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Integrative analysis
- Network analysis
- Multi-scale
- Multi-omics
- Patient stratification
- Drug target
- Precision medicine
- Big data
- Lung cancer
1 Introduction
For centuries, diseases have been studied and treated based on their external manifestations. Following the Human genome project, with an improved understanding of the genes and their interactions, the focus of cancer research has shifted to the genetic mechanisms which lead to disease development. The human genome consists of the DNA present within the cell nucleus, and is made up of over six billion nucleotides. These nucleotides code for molecules which make the different cells function properly. Any change in the six billion nucleotides is therefore capable of altering the functioning of cells. Such changes sometimes result in production of proteins with altered functions, or altered levels of proteins in the cells. This can result in loss of the homeostatic balance and uncontrolled growth of the cells. Such cells damage the surrounding tissue, resulting in tumor formation, leading to cancer. Research is now focused on understanding the root cause of the disease, which lies in the alterations in the genome (DNA) or gene expression. In the traditional empirical or reductionist approach, the problems are reduced to a single scale and studied in isolation. But, the connectivity and interdependence between the multiple levels of organization in a living system bestow upon it the unique properties which make it function as a whole [26]. Therefore the reductionist approach ignores many of the key features and complexity of the living system [27]. A more integrated, holistic approach is required for studying cancers.
Cancers are more responsive to treatment in early stages, compared to more advanced stages. This makes it essential to identify appropriate markers for diagnosis of cancer, as early as possible. When a patient is stratified and diagnosed correctly, appropriate treatment can start. However, cancers display a high level of variability between patients. Therefore, the same treatment/drugs may not be suitable for two patients. Precision medicine is based on this concept of individual patient variability [12]. This means that drugs should be highly focused for specific patient profiles right from development to treatment. This will reduce not only the treatment burden on patients, but also improve the efficacy of drugs, and trial success. This is of even more relevance since cancer drugs are expensive and can have severe side-effects. This type of patient variability based drug development and treatment approach, along with timely, high confidence diagnosis, requires an in-depth understanding of cancer. Unraveling these complex features requires an integrated system level approach instead of a reductionist approach [17]. This requires the integration of muti-scale “Omics” information, and is made tractable through big data analytics.
The development of high throughput technologies have made a lot of multi-scale “Omics” data available. These data capture DNA, RNA, protein, and metabolite level information in the form of genomics, transcriptomics, proteomics, phenomics, and metabolomics data, among others [24]. There has been large reduction in the cost and time involved in the generation of these data. It is estimated that over the next 10 years, Omics data will be at par, if not surpass data generated from sources such as astronomy, YouTube, and Twitter in terms of acquisition, storage, distribution and analysis [37]. The way the different forms of biological data are collected and represented makes high variety and variability inherent characteristics of these data. This makes integration across multiple datasets a challenge [36]. In addition, biological problems are usually NP-hard, and are therefore computationally intensive to solve [25]. These problems are further complicated by the high dimensionality of biological data, where the number of features (variables) for which observations are recorded is more than the number of samples by a few orders of magnitude. Therefore, the extraction of actionable biological and clinical insight from these data is riddled with some of the main challenges associated with the analysis of big data.
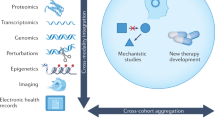
In this paper we show how big data analytics can help in understanding cancer genomics. We describe an integrative analysis framework which uses data-sciences and network-sciences techniques for model creation from multi-scale, multi-omics data. This framework, shown in Fig. 1, is useful for discovering actionable insights in cancer. This framework consists of 4 stages. In the first stage, exploratory data analysis techniques are used for hypothesis creation from the experimental data, which includes DNA and RNA data. Traditional techniques are used to extract information from the individual datasets, to obtain disease biomarkers for diagnosis and prediction of patient survival/response. Next, the results from the exploratory data analysis are combined and filtered, within an appropriate biological context, in the multi-scale integrative analysis. This is a step towards developing a mechanistic model for the disease. Finally, the results from the exploratory data analysis and multi-scale integration steps are combined with information from existing knowledge-bases to obtain a functional understanding of the disease along with high quality biomarkers, and potential drug targets.

Framework for integrative data analysis
To demonstrate this integrative cancer genomics model, we have used a publicly available lung cancer (lung squamous cell carcinoma or SCC) clinical dataset, collected over 18 years [9] as an example. The data for this case study were downloaded from the Array Express database under accession id E-MTAB-1727 (www.ebi.ac.uk/arrayexpress). All analysis were run using the iOMICS platform that has been built by us and deployed in the Google cloud. This is accessible at http://iomics-clinical.interpretomics.co. This paper is organized into 5 main sections. A description of the input data, including experimental data and external knowledge-bases is provided in Sect. 2. Section 3 gives the key aims of the analysis. Section 4 describes the exploratory data analyses step of the analysis framework on the experimental data. Section 5 describes the multi-omics integrative step using results from exploratory data analysis. Finally, the integration of external knowledge-bases and application of network theory to derive actionable insights and biomarkers is described in Sect. 6.
2 Available Data
We have used multi-omics, multi-scale data for a group of lung cancer patients and healthy individuals to illustrate our analytics framework. These data were collected by the original authors [9] and made publicly available. They include DNA, RNA, and clinical data for 93 cancer patients and 16 healthy individuals. Together these make up the experimental datasets. Apart from the experimental datasets, various reference knowledge-bases are available, which have been used in the analytic framework. A description of all these available data is given, followed by a description of the key questions which can be answered using these multiple datasets.
2.1 DNA Level Data
The available DNA level data consist of information regarding DNA sequence alterations for 67 lung cancer patients [9]. This data was not available for the remaining 26 cancer patients and the 16 healthy individuals. For each of the over 300,000 DNA sequence sites captured, the genotype data provide the state of the DNA sequence (alleles) for both copies of DNA (one from each parent). These data were captured from genotyping experiments.
2.2 RNA Level Data
DNA sequence alterations can cause disease by altering the production of proteins in the cells. The first step in the translation of DNA sequence to proteins is the production of mRNA. mRNA levels in the cells are therefore a measure of the expression of DNA to proteins. mRNA levels were captured in the lung cancer study [9] and made available in the form of intensity measures from microarray experiments, for all 109 individuals (93 lung cancer patients and 16 healthy individuals). These intensity values need to first be normalized across samples and converted to expression measures before they can be used for analysis.
2.3 Clinical Data
Clinical data was recorded for all 93 lung cancer patients and 16 healthy individuals by the original authors [9]. These data contained patient information such as age at diagnosis, sex, disease stage, treatments received and other features related to disease risk and condition. A summary of the main sample characteristics for the 93 lung cancer patients from this dataset is given in Table 1. This information provides disease characteristics and can therefore be used to scale the molecular level (DNA, RNA) information, described earlier, with the disease state.
Survival Information. Survival information was also recorded for the 93 lung cancer patients. This information includes information regarding how long the patients survived during the study period. This includes both overall patient survival and survival without disease recurrence. While overall survival was recorded for all 93 patients, recurrence free survival was recorded for 87 cancer patients. One characteristic of survival information of this kind is that it is censored. This means that data is not available for those patients that survived beyond the duration of the clinical study, as well as for those that withdrew from the study. Therefore appropriate modeling algorithms, capable of handling censored data, are required in order to combine the survival data with other types of data from the patients (clinical, DNA, and RNA).
2.4 Background Databases
Vast quantities of biological knowledge, has been collected through biological experiments and is available in the public domain. This knowledge, in the form of reference databases can be used to extend the results of the experimental data, and provide them a functional context. This step is essential to obtain a mechanistic understanding of disease development, and for identifying drug targets. Three types of biological databases, namely functional characterization databases, metabolic databases, and protein interaction databases have been used in the analysis framework to complement the experimental data.
Functional Characterization Databases. Functional characterization databases contain information curated from research studies regarding the various biological properties of the protein products of genes. These include properties such as biological function, cellular localization and the high level pathways and processes. These functional properties provide biological relevance to lists of gene names, leading to the development of an explanation for why and how they are involved in the disease. We have used two such functional characterization databases for our analysis. These are the Gene Ontology database (GO) [5] and KEGG [20].
Metabolic Databases. Metabolic databases contain information regarding the multitude of biochemical reactions taking place in the living system. This information ranges from the small molecules (metabolites) being formed or destroyed, along with the involvement of genes in these processes. These biochemical (metabolic) reactions are responsible for the interaction of a living system with its environment, as well as the various processes taking place within the system. The collection of all these biochemical reactions in humans is called human metabolism. While information regarding human metabolism is growing, models of human metabolism exist which contain the current knowledge of metabolism. Recon X [39] is one such model, which contains information regarding metabolic reactions, their reactants, products, stoichiometry and associated genes, and is available in standard SBML format [18]. We have used Recon X with 7439 reactions and 2626 metabolites in our framework.
Protein Interaction Databases. At a level higher than metabolism, the functional characteristics of a living system arise from the interactions between proteins. Proteins transfer signals within and between cells, and lead to mediation of metabolic reactions based on these signals. The interactions between different proteins are captured in protein interaction databases. This information is represented as interaction networks with proteins forming the nodes, and edges representing the interactions between them. The protein-protein interactions can be directional or undirected, depending on the type of interaction. Our analytics framework uses the protein interaction database available from IntAct [31].
3 Key Aims
Based on the available experimental data, we have explored three main lines of analysis. The basaloid subtype of lung cancer is particularly aggressive and shows poor prognosis for the patients [9]. So, in the first, we aimed to identify the molecular differences between two cancer subtypes based on histology, the basaloid and SCC subtypes (Table 1), along with an understanding of how these molecular differences functionally result in differences in the two cancer subtypes. For the second line of analysis, we aimed to identify the molecular states associated with poor patient survival. In the third line of analysis, we compared the healthy individuals with the cancer patients using their molecular information, to identify therapeutic targets which can be used in drug development. We show how the different datasets and steps in the analysis framework come together to answer the questions posed by these three lines of analysis.
4 Exploratory Data Analysis
The first step of the analysis framework is exploratory data analysis. This involves the analysis of individual experimental data sets, using traditional approaches, for hypothesis creation. The various analyses which can be performed depend on the type of experimental data available, and questions to answer. All the DNA, RNA, and clinical data were used to lay the foundation for the remaining analysis steps.

Karyotype plot showing location of identified point mutations (red lines) along the chromosomes (Color figure online).
4.1 Mutation Association with Cancer State
For the first line of our analysis, we used the DNA level data to identify DNA sequence states which can differentiate the two cancer subtypes, namely the basaloid and the SCC. For each of the DNA sequence sites in the data, we first calculated the frequency of observing the least common sequence state for each cancer subtype. Based on these, odds were calculated for each disease subtype for observing the least common state (p/(1-p)). Then a ratio of these odds, called the odds ratio was taken for each site. Significant deviation of the odds ratio from one for a site signified that that particular site was associated with one or the other disease subtype. This association testing analysis was run using PLINK [32]. In order to identify meaningful results, we used high stringency cut-offs for the odds ratio and significance p-value (p-value \(\leqslant \) 0.001, odds ratio \(\geqslant \) 3). From this analysis, we were able to identify 735 disease subtype associated sites. Figure 2 shows the locations of these 735 sites along the chromosomes. This plot was generated using the quantsmooth R/Bioconductor package [30].
4.2 Differential Gene Expression
Cancer subtype differences can also manifest at the gene expression level, captured by the RNA data. We analyzed the RNA level data to study the differences between the two disease subtypes (basaloid and SCC) by identifying differentially expressed genes. We used the R package LIMMA [34] for analyzing the expression level differences between the two disease subtypes. This algorithm is able to make statistical inferences even with a small number of samples [34]. It uses a linear model to model the expression values across samples, as a function of the disease subtypes. A separate model is fitted for each gene. This is followed by an empirical Bayes step across genes to identify the p-value and FDR (False Discovery Rate) adjusted p-value [34]. The log fold change between the disease subtypes is also estimated as the base 2 logarithm ratio of expression in the two states. Finally, we identified the differentially expressed genes which showed absolute log fold change \(>\) 0.6 with differential expression p-value \(<\) 0.0001. These cut-offs are variable and affect the stringency of the results. From this analysis, we identified 106 differentially expressed genes between the basaloid and SCC subtypes. Figure 3 shows the mRNA expression levels and hierarchical clustering of the 93 lung cancer patients for the identified differentially expressed genes. A clear separation in the expression values can be seen for the two subtypes.

Heirarchical clustering of expression for identified differentially expressed genes
For our third line of analysis, we needed to compare healthy individuals with cancer patients to identify potential drug targets. Only RNA data was available for the healthy patients, and therefore was used for this comparison. We identified the differentially expressed genes between cancer and healthy individuals. The same steps and parameters were used for this analysis, as for the differential expression analysis between the cancer subtypes.
4.3 Patient Stratification
The second line of analysis aims at identifying markers of patient survival. Patients’ molecular profiles influence their response to treatment and disease progression. In the case of cancer, patient survival time is a reasonable measure of patient response. In a treatment context, markers associated with treatment response can stratify patients into groups of responsive and non-responsive patients. These markers will then be able to identify which group a new patient belongs to and facilitate precision medicine through most effective treatment. Since gene expression is an intermediary between DNA and protein, it can be used to connect patient response with the molecular profile.
In this analysis, we integrate gene expression RNA level data with recurrence free survival information for the cancer patients, to answer the questions posed by the second line of analysis. We used the Cox regression to model survival time as a function of gene expression. The Cox regression model was used because of its ability to handle censored data [28]. Apart from the censored nature of survival data, another problem for the analysis is the high data dimensionality. Expression information is available for over 20,000 genes for the 87 samples with survival information. Therefore in order to identify high confidence genes as markers of patient survival, an appropriate dimensionality reduction technique needs to be applied. For this, we used the semi-supervised principle components based dimensionality reduction technique implemented in the R package SuperPC [6, 7] to calculate the adjusted Cox regression coefficients. We used a training set constructed from a random set of 2/3rd of the samples to build the adjusted Cox regression model, and built a reduced model with the genes with the highest coefficients. We then tested the resulting model on the remaining 1/3rd samples (test set). This procedure was repeated 10 times, to obtain the best fitting model.

Survival probability curves for identified molecular subgroups
We used the genes from the final model to cluster all 93 lung cancer patients into 2 groups, with 78 and 9 patients each. While one of these groups showed a good survival probability (84 patients), the survival probabilities for the other group were very poor (Poor prognosis group: 9 patients). We then reapplied this analysis on the 78 patients who were part of the good survival probability group. This resulted in further subgrouping of the patients into two groups, both of which had better survival probabilities compared to the poor prognosis group. The survival probabilities for the resulting 3 groups of patients is shown in Fig. 4. These curves were generated using the R package ggplot2 [40]. Interestingly, all 9 patients in the poor prognosis group belong to the pure basaloid subtype. This indicates that these patients represent a particularly aggressive molecular profile seen in the pure basaloid patients.

Mapping of differentially expressed genes and SNPs. Genes are in blue/green and mutations are in pink. The sizes of gene nodes represent fold change, while the blue and green shades represent direction of change (Color figure online)
5 Multi-scale Integrative Analysis
The next stage of the analysis framework involves integration of the results from exploratory data analysis within a biologically meaningful context to improve our understanding of cancer and how the disease state develops. The type of multi-scale integration, and resulting inferences, depends on the available experimental datasets. With varied multi-scale datasets, the resulting disease model becomes richer, providing improved insights. For the lung cancer dataset, only DNA and RNA level molecular data are available, therefore we demonstrate the applicability of this step through DNA-RNA integration. From this analysis, we can identify the disease subtype associated DNA sequence mutations which lead to changes in RNA expression, thereby providing a mechanism for how these sequence changes are pathogenic.
We first annotated the 735 identified disease subtype associated DNA sequence alterations with genes based on their chromosomal location. The human genome build GrCH38 was used for the gene locations. This provided a list of 558 unique genes with disease associated mutations. We compared these to the differential gene expression analysis results between the basaloid and SCC subtypes, and identified genes which were also differentially expressed. This gene-mutation mapping, as visualized in Cytoscape (a tool for dynamic network visualization [1]), is shown in Fig. 5. Many of the genes discovered in this part of the analysis such as CLCA2, TIAM2 and BCL2 have been associated with progression and metastasis in various tumors [11, 14, 23].
6 Data Integration and Network Analysis
The final step of the analysis framework takes the results of the exploratory data analysis and multi-scale integration steps and combines them with existing biological knowledge-bases to finally answer the questions posed in the three analysis lines. We used network analysis to obtain insights from this stage of the analysis. This is because the complex interactions in the biological system are better understood when modeled as networks [22]. The networks provide information regarding the biological interactions and flow of information. When analyzing functional networks, key nodes and interactions are identified using centrality measures such as degree, betweenness, connectedness and eigenvector centrality. Node clustering is used to identify functional clusters. Node neighborhoods are analyzed for identifying interactions network interactions. We have used the R package igraph [13] for studying network properties.
6.1 Functional Characterization Databases
We used the information in the functional characterization databases for all the gene lists from the 3 types of exploratory data analysis, as well as the multi-scale integration analysis, for the first two lines of analysis. We used the R package GO.db [10] and 168 cancer and metabolic KEGG pathways [35] to annotate genes with their functional properties. Since these are non-random lists of genes, the functions they perform will be linked with the development of the disease state. In other words, these disease state associated functions will be over-represented for the gene list, than expected by chance. To identify these overrepresented gene functions, we used the Fisher’s exact test, as implemented in XomPathways [38]. We modeled the results as a bipartite gene-function (pathway/GO) network, and the resulting gene-gene and function-function networks. The key functional properties and genes involved in the disease state were identified from the most central nodes in these networks. For the gene-biological processes functional annotation, the identified key processes were related to epithelial morphology, consistent with histology based subgrouping of the disease subtypes (Table 2).
6.2 Metabolic Network Reconstruction and Protein Interactions
The functional properties of genes provides a high level view of the contribution of genes to the disease state. At the core of these properties lies the metabolism. Metabolism is centered around the processes of energy and biomass production, as these are the two core requirements for the cell. Some of the core metabolic reactions involved in energy production are depicted in Fig. 6. Diseases such as cancer develop due to metabolic changes brought about by protein changes. In order to identify potential drug targets for disease treatment, a mechanistic model is essential which considers the metabolite and protein interactions. Therefore, for our third line of analysis, we combined RNA based gene expression data, which is indicative of protein expression, with metabolic and protein interaction data, from reference databases.

Subset of human metabolic network. This image has been taken from [21]
Using the approach of the GIMME algorithm [8], we used expression data to identify reactions which were occurring in the disease and healthy states. For this we used an expression threshold such that about 25 % of the genes were assumed to be switched off. This information was used to initialize the metabolic model from RECON X. The metabolic models for both state were constructed using flux balance analysis, a type of constraint based modeling. Since the cancer cells show extensive growth and proliferation, the disease state network was optimized for maximum biomass production. The healthy lung cells are differentiated and primarily use energy for carrying out their functions. Therefore the healthy state network was optimized for maximizing energy production. Thermodynamically unfeasible cycles were removed from the models and the fluxes through all metabolic reactions were calculate. The R package sybil, sybilEFBA, sybilSBML and sybilcyclefreeflux were used for this analysis, along with glpkAPI [3, 4, 15, 16].
We compared the fluxes through the reactions in both disease and healthy state metabolic models to identify the reactions with the most change in flux. From the information contained in Recon X, we identified the genes associated with these reactions. From a therapeutics point of view, these genes are potential targets for lung cancer. However, not all of them may be druggable. Additionally, targeting these genes may lead to high toxicity, or alteration in tumor properties rendering the drug ineffective. To solve these problems, we extended the identified gene list to include the other genes which directly interacted with these genes in the protein interaction network. These extra genes can indirectly modulate the metabolic reactions. The resulting gene list had 214 genes.

Results from the analysis of cancer and healthy metabolic networks. The first step identifies altered reactions, the second step lists out the genes involved directly in the reactions. The final step identifies putative drug targets based on the protein interaction network.
Out of these, the genes which interact with many other genes in the human protein interaction network (PIN) are likely to be inappropriate drug targets due to high toxicity. Therefore we calculated a degree score for each gene based on its degree centrality in the PIN. We also calculated a reaction score for each gene by summing its interactions with target reactions. Indirect interactions were weighted 0.5. Finally, we looked at whether the identified genes were differentially expressed between the healthy and tumor state, since this provided a mechanism by which the change in metabolic reactions was affected. Many well known cancer genes such as MYC, ERBB2, STAT3 and GSR, along with novel genes, were identified as therapeutic targets. Figure 7 gives the results from this analysis.
7 Conclusions
Here we described and illustrated the use of a big data multi-scale multi-omics framework for the identification of gene level biomarkers associated with lung cancer. Using this approach, we were able to identify diagnostic and prognostic biomarkers for the cancer subtypes, therapeutic targets for lung cancer, and even identified a hidden molecular subtype having dismal prognosis. We showed how different stages of the analysis framework come together to answer complex disease associated questions such as mechanism of disease development, processes influencing patient survival, and putative therapeutic targets. The results were all validated using bibliomic data (peer reviewed publications). While a basic meta-analysis framework for integrative analysis was described here, more comprehensive mathematical techniques can also be applied to get better results [33]. The identified target genes can be further validated as potential drug targets using an in-silico knockouts approach. This type of analysis uses iterative constraint based modeling on the metabolic network model, to study the effect of altering specific reactions on both the healthy and disease state [19]. We used the iOMICS platform in a semi-automated fashion for running the analysis. The input parameters and experimental data types were provided as input. User intervention was required for providing a biologically meaningful direction to the analyses, bringing them in line with the type of available data and hypotheses. This is an essential feature for the analysis of biological data.
While we have demonstrated the use of this analysis framework for cancer, it can be extended to other complex diseases such as neurological and heart diseases. It provides a general framework which can be used to combine multi-omics data to derive cross-scale inferences. For this purpose, the type of input experimental data decides the following analytics. Depending on the type of experiments conducted, the aspect of the disease state interactions unveiled by the analysis may vary. AlQuraishi et al. [2] looked at a complex disease such as cancer at the genomic scale, where they integrated biophysical data with genomic data to study tumor vs. normal state. On the other hand, an organ and system level view of drug interactions can provide useful insights regarding the efficacy and toxicity of drugs [29]. However, it does not depend on the specific type of experiment used to capture the same information. Although we have used data collected from genotyping and mRNA microarray experiments, the analytics approach can also be applied to cases with much larger quantities of data, collected from sequencing experiments, and can be integrated with data from knowledge-bases other than those used here.
References
Cytoscape. http://www.cytoscape.org/
AlQuraishi, M., Koytiger, G., Jenney, A., MacBeath, G., Sorger, P.K.: A multiscale statistical mechanical framework integrates biophysical and genomic data to assemble cancer networks. Nat. Genet. 46, 1363–1371 (2014)
Amer Desouki, A.: sybilcycleFreeFlux: cycle-Free Flux balance analysis: Efficient removal of thermodynamically infeasible cycles from metabolic flux distributions (2014). R package version 1.0.1
Amer Desouki, A.: sybilEFBA: Using Gene Expression Data to Improve Flux Balance Analysis Predictions (2015). R package version 1.0.2
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al.: Gene ontology: tool for the unification of biology. Nat. Genet. 25(1), 25–29 (2000)
Bair, E., Hastie, T., Paul, D., Tibshirani, R.: Prediction by supervised principal components. J. Am. Stat. Assoc. 101(473), 119–137 (2006)
Bair, E., Tibshirani, R.: Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2(4), E108 (2004)
Becker, S.A., Palsson, B.O.: Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4(5), e1000082 (2008)
Brambilla, C., Laffaire, J., Lantuejoul, S., Moro-Sibilot, D., Mignotte, H., Arbib, F., Toffart, A.C., Petel, F., Hainaut, P., Rousseaux, S., et al.: Lung squamous cell carcinomas with basaloid histology represent a specific molecular entity. Clin. Cancer Res. 20(22), 5777–5786 (2014)
Carlson, M.: GO.db: A set of annotation maps describing the entire Gene Ontology, R package version 3.1.2
Chen, J.S., Su, I.J., Leu, Y.W., Young, K.C., Sun, H.S.: Expression of t-cell lymphoma invasion and metastasis 2 (tiam2) promotes proliferation and invasion of liver cancer. Int. J. Cancer 130(6), 1302–1313 (2012)
Collins, F.S., Varmus, H.: A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015)
Csardi, G., Nepusz, T.: The igraph software package for complex network research. Int. J. Complex Syst. 1695(5), 1–9 (2006)
Del Bufalo, D., Biroccio, A., Leonetti, C., Zupi, G.: Bcl-2 overexpression enhances the metastatic potential of a human breast cancer line. The FASEB J. 11(12), 947–953 (1997)
Gelius-Dietrich, G.: glpkAPI: R Interface to C API of GLPK (2015). R package version 1.3.0
Gelius-Dietrich, G., Desouki, A.A., Fritzemeier, C.J., Lercher, M.J.: sybil-efficient constraint-based modelling in R. BMC Syst. Biol. 7(1), 125 (2013)
Hansen, J., Iyengar, R.: Computation as the mechanistic bridge between precision medicine and systems therapeutics. Clin. Pharmacol. Ther. 93(1), 117–128 (2013)
Hucka, M., Finney, A., Sauro, H.M., Bolouri, H., Doyle, J.C., Kitano, H., Arkin, A.P., Bornstein, B.J., Bray, D., Cornish-Bowden, A., et al.: The systems biology markup language (sbml): a medium for representation and exchange of biochemical network models. Bioinformatics 19(4), 524–531 (2003)
Jerby, L., Ruppin, E.: Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 18(20), 5572–5584 (2012)
Kanehisa, M., Goto, S.: Kegg: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000)
Khazaei, T., McGuigan, A., Mahadevan, R.: Ensemble modeling of cancer metabolism. Front. Physiol. 3, 135 (2012)
Kitano, H.: Systems biology: a brief overview. Science 295(5560), 1662–1664 (2002)
Li, X., Cowell, J.K., Sossey-Alaoui, K.: CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene 23(7), 1474–1480 (2004)
Li, Y., Chen, L.: Big biological data: challenges and opportunities. Genomics, Proteomics Bioinform. 12(5), 187–189 (2014)
Martın H, J.A., Bourdon, J.: Solving hard computational problems efficiently: asymptotic parametric complexity 3-coloring algorithm. PloS One 8(1), e53437 (2013)
Mazocchi, F.: Complexity in biology. Exceeding the limits of reductionism and determinism using complexity theory. EMBO Rep. 9, 10–14 (2008)
Mazocchi, F.: Complexity and the reductionism-holism debate in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 413–427 (2012)
Miller, R., Halpern, J.: Regression with censored data. Biometrika 69(3), 521–531 (1982)
Moreno, J.D., Zhu, Z.I., Yang, P.C., Bankston, J.R., Jeng, M.T., Kang, C., Wang, L., Bayer, J.D., Christini, D.J., Trayanova, N.A., et al.: A computational model to predict the effects of class i anti-arrhythmic drugs on ventricular rhythms. Sci. Transl. Med. 3(98), 98ra83 (2011)
Oosting, J., Eilers, P., Menezes, R.: quantsmooth: Quantile smoothing and genomic visualization of array data. R package version 1.35.0 (2014)
Orchard, S., Ammari, M., Aranda, B., Breuza, L., Briganti, L., Broackes-Carter, F., Campbell, N.H., Chavali, G., Chen, C., Del-Toro, N., et al.: The mintact projectintact as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, 358–363 (2013)
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., De Bakker, P.I., Daly, M.J., et al.: Plink: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81(3), 559–575 (2007)
Ritchie, M.D., Holzinger, E.R., Li, R., Pendergrass, S.A., Kim, D.: Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 16(2), 85–97 (2015)
Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W., Smyth, G.K.: limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015)
Segrè, A.V., Groop, L., Mootha, V.K., Daly, M.J., Altshuler, D., Consortium, D., Investigators, M., et al.: Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 6(8), e1001058 (2010)
Sîrbu, A., Ruskin, H.J., Crane, M.: Cross-platform microarray data normalisation for regulatory network inference. PLoS One 5(11), e13822 (2010)
Stephens, Z.D., Lee, S.Y., Faghri, F., Campbell, R.H., Zhai, C., Efron, M.J., Iyer, R., Schatz, M.C., Sinha, S., Robinson, G.E.: Big data: astronomical or genomical? PLoS Biol. 13(7), e1002195 (2015)
Talukder, A.K., Ravishankar, S., Sasmal, K., Gandham, S., Prabhukumar, J., Achutharao, P.H., Barh, D., Blasi, F.: Xomannotate: analysis of heterogeneous and complex exome-a step towards translational medicine. PLoS ONE 10, e0123569 (2015)
Thiele, I., Swainston, N., Fleming, R.M., Hoppe, A., Sahoo, S., Aurich, M.K., Haraldsdottir, H., Mo, M.L., Rolfsson, O., Stobbe, M.D., et al.: A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 31(5), 419–425 (2013)
Wickham, H.: ggplot2: Elegant Graphics for Data Analysis. Springer Science & Business Media, New York (2009)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Agarwal, M., Adhil, M., Talukder, A.K. (2015). Multi-omics Multi-scale Big Data Analytics for Cancer Genomics. In: Kumar, N., Bhatnagar, V. (eds) Big Data Analytics. BDA 2015. Lecture Notes in Computer Science(), vol 9498. Springer, Cham. https://doi.org/10.1007/978-3-319-27057-9_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-27057-9_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27056-2
Online ISBN: 978-3-319-27057-9
eBook Packages: Computer ScienceComputer Science (R0)