
Abstract
On-surface synthesis of covalently bond nanostructures under ultrahigh vacuum conditions has received increased attention in the recent years. This approach allows to study solvent-free chemical reactions and moreover to use well-defined substrates, which act as a catalyst and/or exerting steric effects leading to kinetic and regioselective control of the chemical process at hand. Recently, successful 1,3-dipolar azide–alkyne cycloaddition reactions were performed on metal substrates with complete regioselectivity of a specific product. This chapter presents a summary of these experimental efforts on different metal substrates, while also focusing on a comprehensive understanding of the catalyst prerequisite for on-surface coupling reactions and a quantitative description of steric effects dominating the coupling mechanism and the regioselectivity of the reaction products. Future perspectives for the bottom-up development of functional nanostructures involving on-surface azide–alkyne cycloadditions are discussed.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Scanning Tunneling Microscopy
- Density Functional Theory Calculation
- Cycloaddition Reaction
- Scanning Tunneling Microscopy Image
- Transition State Energy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Decades of organic synthesis research have accomplished an extensive pool of suitable chemical processes in solution, which are nowadays increasingly investigated in the two-dimensional confinement of surfaces [1–3]. On-surface synthesis under ultrahigh-vacuum (UHV) conditions for the bottom-up growth of covalently bound organic nanostructures with well-defined functionalities has become an attractive field of study in modern nanoscience and nanotechnology. Among the many possible cases of study, cycloaddition reactions [4], commonly referred to as “click” reactions, are highly attractive candidates for on-surface synthesis efforts. Such by-product-free reactions, performed as solution-phase processes, are widely used for the modification of surfaces and nanoparticles [5, 6] and also for the preparation of biologically active compounds in pharmaceutical research [7, 8]. In this field, one of the most widely used reactions is the azide–alkyne 1,3-dipolar cycloaddition [9] leading to 1,4 and 1,5 triazoles (Fig. 1). The so-called uncatalyzed pathway (Huisgen azide–alkyne [3 + 2] cycloaddition) is a thermal process which delivers a mixture of both regioisomers. However, the use of a Cu(I) catalyst provides 1,4-triazoles with high efficiency and regioselectivity under ambient conditions (CuAAC) [9].

Solution-phase 1,3-dipolar cycloaddition reaction between azides and alkynes. a General reaction scheme, including the Cu-catalyzed (top) and the Huisgen uncatalyzed (bottom) reaction pathways. Adapted with permission from [10]. Copyright (2013) American Chemical Society. b Schematic depiction of a “click” reaction investigated for the immobilization of azidomethylferrocene on the alkyne-terminated silicon electrode. Adapted with permission from [6]. Copyright (2008) WILEY-VCH Verlag. c Application of the Cu(I)-catalyzed triazole formation in drug discovery, in particular for multivalent neoglycoconjugates. Adapted with permission from [8]. Copyright (2013) American Chemical Society
Resembling the CuAAC mechanism in solution, a successful cycloaddition between 9-ethynylphenanthrene (alkyne) and 4-azidobiphenyl (azide) was recently accomplished on a Cu(111) surface at room temperature under UHV conditions, with complete regioselectivity toward the formation of the corresponding 1,4-triazoles [10]. In that study, two different precursor molecules were charged with a single alkyne or azide moiety (Fig. 2a), allowing dimerization after successful coupling. As for the solution-phase process, the observed complete regioselectivity and the low activation temperature for the reaction were discussed considering the involvement of a copper acetylide (C–H activation) and bonding of the alkyne group to the Cu(111) surface. However, the Cu surface was shown to be very reactive toward the organic reactants, in particular causing the azide moieties to degrade. Such degradation was found to be the limiting factor and furthermore explained the low yield (~1.1 %) of the coupling reaction on this surface.

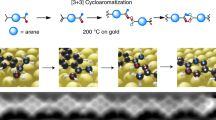
In a different study, the reactivity of N-(4-azidophenyl)-4-ethynylbenzamide (AEB) monomers on a Au(111) surface (AuAAC) under UHV conditions (Fig. 2b) was investigated with a combination of cryogenic scanning tunneling microscopy (STM) and density functional theory (DFT) [11]. In this case, the inert Au(111) surface was chosen since it can be expected to have a weaker interaction with the azide moiety, and thus improve the coupling rate of the on-surface azide–alkyne cycloaddition reaction. Furthermore, the AEB monomers were designed with two phenyl rings connected through an amide linker as backbone. These features allow the reactants to be thermally deposited on the surface, where they can lay flat and diffuse, thus increasing the probability of the alkyne and azide groups to meet and react. In addition, the amide linker could form intermolecular hydrogen bonds perpendicular to the targeted reaction direction. This makes possible to aim for supramolecular ordering after the deposition and therefore enhance the reaction probability accordingly. Moreover, a single AEB reactant is charged with an azide and an alkyne moiety which allows this compound to undergo oligomerization.
In this chapter, a comprehensive comparison between the completely regioselective on-surface CuAAC [10] and AuAAC [11] processes will be presented. Section 2 presents a summary of the experimental efforts for each process. Given that Au complexes have not been shown to catalyze chemical reactions of this kind, detailed DFT calculations for the AuAAC process are presented in Sect. 3. These not only explain the low temperatures required to trigger this chemical process on a metal substrate, but also the role of the substrate in the reaction, and how its careful selection can improve the regioselectivity of the reaction output. Finally, a brief discussion about the future perspectives for this on-surface synthesis approach to develop functional nanostructures on surfaces is presented in Sect. 4.
2 On-Surface Azide–Alkyne Cycloaddition Reactions
In the following section, up-to-date experimental efforts for the on-surface azide–alkyne cycloaddition reactions will be presented. Emphasis is given to critical points involving the relatively low-temperature requirement for C–H activation of the alkyne moiety on both substrates, the reaction yield and the reactivity limitations.
2.1 Deposition and On-Surface Reaction at Room Temperature
As mentioned before, the on-surface azide–alkyne cycloaddition reaction on the Cu(111) surface at room temperature mimics the CuAAC solution-phase process and leads to the exclusive formation of 1,4-triazoles. On the other hand, the Au(111) surface might not act as a catalyst for this specific reaction, and therefore, the evolution of this system at the same experimental conditions was investigated [11]. Molecular deposition of the monomers always resulted in a partially covered surfaces, showing a first layer mixture of reactants along with reacted compounds (Fig. 3a). Around these features, it was always found a disordered molecular phase, which was ascribed to the azide end group degradation occurring on both Cu(111) and Au(111) surfaces.

STM images after the reactants deposition onto the metal surfaces at room temperature. a Deposition onto Cu(111) results in the formation of the corresponding 1,4-triazole (enclosed in green) surrounded by a disordered phase ascribed to the azide degradation. Such a degradation was also observed on Au(111). b Arrangement of AEB monomers and dimers on Au(111) after the reaction. c AEB dimers and trimers formed as a result of the successful azide–alkyne “click” reaction. The inset in (c) shows the molecular structure of the 1,4 regioisomer. Adapted with permission from [10, 11]. Copyright (2013) American Chemical Society
The potential degradation of the azide moiety does not represent a major limiting factor for the coupling reaction on Au(111). High-resolution STM images (Fig. 3b) show a mixture of reacted and unreacted molecules on the substrate. A number of monomers were oriented in a way that the alkyne group of one monomer and the azide group of the other were in close proximity and hence properly positioned to undergo the targeted cycloaddition reaction. Besides the dimerization, the formation of longer linear structures confirmed that the corresponding trimers were also formed, i.e., the targeted oligomerization of AEB monomers can be achieved. Furthermore, the successful formation of trimers (Fig. 3c) strongly supports the occurrence of the proposed azide–alkyne “click” process since the formation of such trimeric structures should only be feasible if the reaction of an AEB monomer and a dimer occurs between the azide and alkyne end groups of these two reaction partners (Fig. 2). It is important to note that Au complexes have not been shown to catalyze azide–alkyne cycloadditions; therefore, the success of the reaction without further thermal annealing is surprising on this surface.
The inset in Fig. 3c represents the molecular structure of a 1,4-triazole regioisomer on Au(111). This structure matches very well the STM observations, since the alternative 1,5-regioisomer would present an L-shaped configuration instead of the observed linear structure (Fig. 2). It is emphasized that for all the experiments on Au(111), only the formation of the 1,4-regioisomer was also observed. However, the thermal azide–alkyne cycloaddition in the absence of a Cu catalyst generally requires high temperatures resulting in a mixture of 1,4 and 1,5 triazoles [9]. If the cycloaddition products were formed in the crucible during the heating process prior to the deposition and subsequently deposited on the surface, a mixture of both regioisomers is to be expected in the STM images. Therefore, also the observed complete regioselectivity strongly pointed toward an on-surface reaction. It is likely that the Au substrate lowers the activation energy and also steers the regioselectivity of the [3 + 2] cycloaddition reaction (mechanistic studies will be discussed in Sect. 3).
DFT calculations further confirmed the correct assignment of the on-surface dimerization products to the 1,4-regioisomer structure. On Au(111) (Fig. 4), the reactants and the proposed products were verified by comparing experimentally determined distances with the theoretical values calculated for the gas phase of the AEB monomer, dimer, and trimer. The center-to-center distance between the two aryl rings of the AEB monomer was found to be 0.65 ± 0.03 nm (calculated, 0.65 nm); for the dimer, the measured distance between the adjacent minima of the 1,4-triazole group is 0.57 ± 0.02 nm (calculated, 0.50 nm); the same distance, measured for the triazoles of the AEB trimer, is 0.55 ± 0.01 and 0.56 ± 0.01 nm (calculated, 0.50 nm), respectively. Evidently, the triazole groups corresponding to the dimers and trimers have the same size. Therefore, not only a good qualitative agreement with the STM images was found, but also a good quantitative agreement between the theoretically and experimentally determined distances. The small differences can be ascribed to the absence of the Au(111) surface in the gas-phase calculations, which restrains out-of-plane movements of the molecular components. As a result, bonding angles can differ leading to deviations of the calculated center-to-center distances.

A good quantitative agreement was found between the experimental and the center-to-center theoretical distances for the three AEB species observed on the Au(111) surface. Adapted with permission from [11]. Copyright (2013) American Chemical Society
2.2 Yield of the On-Surface Azide–Alkyne Cycloaddition Reactions
Another important aspect that should be addressed is to estimate the real amount of products obtained after the reactions (Fig. 5). Therefore, a detailed comparison between the reaction outputs on the Cu(111) and Au(111) substrates is hereinafter presented, with special attention on the monomers’ reactivity and the formation of higher order structures. On one hand, a total of 35 observations of the 1,4-triazoles products were made in 8 independent experiments on the Cu(111) surface (sampling ~3200 deposited molecules). On the other hand, in the study performed on Au(111), a representative sampling from four different deposition experiments of AEB on Au(111) was chosen. From a total of 1083 intact molecules observed in this case, 689 (63.6 %) were AEB monomers. For the AEB 1,4-dimer and the trimers, a total of 310 (28.6 %) and 84 (7.8 %) molecules was identified, respectively.

A substantial amount of monomers remained unreacted on the Cu(111) surface, as compared with the amount on Au(111). This could be ascribed to limited diffusion on the surface. It is known that copper substrates are more reactive toward organic compounds, hence limiting their free diffusion on the surface at room temperature. On the other hand, the dimerization yield for the azide–alkyne cycloaddition on Au(111) is considerably larger than the one observed on Cu(111). Although the large amount of unreacted monomers on the latter is certainly influencing the low yield obtained, the viability of using gold surfaces instead of copper for more efficient on-surface reactions involving azides cannot be neglected. Furthermore, it confirms that the degradation the azide moieties undergo on the Au(111) is not as severe when compared to their degradation on Cu(111), hence not being a major limiting factor for the reaction. Unfortunately, a direct comparison of the trimerization yields is not possible at the moment, given that the design of the reactants in the experiments performed on Cu(111) did not target oligomerization, but more importantly, it was focused on probing the viability of the on-surface coupling reaction (Fig. 2a).
2.3 Controlled STM Tip Manipulations
In Sect. 2.1, the correct assignment of the on-surface reaction products was assessed by direct comparison between experimentally determined and DFT calculated center-to-center distances. To ensure that the observed species are indeed covalently connected monomers, controlled STM tip manipulations were performed. Due to the high mechanical stability of the triazole groups, mechanical perturbations induced by the STM tip must not lead to the dissociation of the products.
Therefore, extensive controlled manipulations with the STM tip were carried out on the dimers and the trimers formed on Au(111). Figure 6 shows a sequence of STM images with consecutive manipulations, where the starting point of the experiment was an arrangement of a dimer and two trimers. After a successful manipulation, the dimer was partially detached from the two linear trimers (Fig. 6b). Subsequent manipulations led to a complete separation of the dimer from the two trimers (Fig. 6c–f). Importantly, the structure of the dimer remained intact after the STM tip manipulations, which confirms the covalent nature of the linkage of the two monomers for the build-up of the 1,4-dimeric structure. The amide functionality between the phenyl rings combined with the triazole group in the dimer provides the product large torsional freedom. The trimers could also be successfully manipulated although a complete detachment from the adjacent molecules was a challenging task. This can be attributed to the stronger van der Waals forces, and probably also to the larger number of hydrogen bonds (between amide bonds), between the linear structures upon switching from the dimer to trimer. These STM manipulations demonstrated that AEB dimers and the corresponding trimers display high mechanical stability, as it is expected for covalently bonded chemical structures, particularly triazoles.

Mechanical manipulation of a 1,4-dimer structure with the STM tip. Manipulations were conducted at T ≤ 5 K with 100 pA ≤ I ≤ 3 nA and V = 5 mV. The white arrows represent the manipulation vectors. Reprinted with permission from [11]. Copyright (2013) American Chemical Society
2.4 Analysis of the Reactivity
Another interesting point when comparing the experiments on Cu(111) and Au(111) is the amount of reactants that did not form the corresponding dimer and trimer products. It is important to note that for a successful on-surface reaction, two reactants must diffuse toward each other so that the reactive alkyne and azide functionalities are close enough for the successful coupling. Therefore, the isolated monomers and/or the self-assembled structures obtained after initial deposition must somehow relate to their reactivity (as discussed in Sect. 2.2). For example, in STM images acquired after the deposition onto Au(111) showed that the AEB monomers that did not react are present in three different configurations (Fig. 7): (i) nucleated along the step edges. This behavior has been also observed on Cu(111) for the alkyne reactant [10], which is in general known to have high affinity for reactive sites on surfaces, such as step edges; (ii) agglomerated adjacent to molecular islands, possibly as a result of diffusion of the molecules on the Au(111) surface and self-assembly steered by van der Waals forces; and (iii) self-assembled in a three-monomers arrangement, forming a stable triangular shaped island. This configuration was observed quite frequently (~27.4 % of the AEB monomers which remain intact after the deposition), being the only configuration where exclusively monomers self-assembled independently (see also Fig. 3b).

Analysis of the AEB monomers reactivity. Unreacted monomers appear in three distinct configurations which probably hinder the reaction. a Nucleated along step edges, in this case the alkyne reactant on Cu(111). b AEB monomers agglomerated adjacent to molecular islands on Au(111). c AEB monomers self-assembled in a triangular arrangement on Au(111). Here, the inset represents one of the possibilities of how similar end groups facing each other would prevent a successful reaction. Adapted with permission from [10, 11]. Copyright (2013) American Chemical Society
It is quite interesting that for molecules which are able to undergo dimerization at room temperature, such stable triangular configuration can be found under similar experimental conditions. However, as for most of the on-surface synthesis reactions, the pre-arrangement of the reactants is crucial [3, 12, 13]. Therefore, one possibility why the reaction does not occur in the triangular configuration is that this particular configuration is too stable and therefore fixed. Moreover, the functional end groups are sterically blocked within this configuration (model structure in Fig. 7c), which leads to reduced reactivity.
3 Reaction Mechanism
The last open question remaining is related to the reaction mechanism dominating the observed azide–alkyne cycloaddition reactions. On the Cu(111) surface, di–\(\sigma\) bonding of the alkyne to the substrate was observed, in analogy to the mechanism involving formation of a Cu(I) \(\pi\) complex with the alkyne triple bond. Only the 1,4-regioisomer was obtained in these experiments under UHV conditions, but when liquid-phase synthesis with the protocol of the uncatalyzed path was performed, only the 1,4-product was obtained as well. Therefore, steric hindrance effects driving the observed regioselectivity were qualitatively assigned as responsible for the reaction to proceed with complete regioselectivity in a surface-confined situation. On the Au(111) experiments, however, di–\(\sigma\) bonding of the alkyne to the bare substrate was not observed; thus, a purely AuAAC-like mechanism should not be feasible at the experimental conditions. Therefore, DFT calculations were carried out for this case, to elucidate the on-surface coupling mechanism and more importantly to clarify the role of the Au substrate on the regioselectivity.
3.1 Involvement of the Surface in the Reaction
To simplify the calculations, a para-alkynylazidobenzene (p-AAB) monomer was chosen as a suitable model compound for the representation of the observed on-surface azide–alkyne cycloaddition reaction on Au(111) [11]. First, the transition-state energies of the dimerizations of AEB and the model compound p-AAB in vacuum were determined. In a second step, the transition state energy of the dimerization of p-AAB on a flat unreconstructed Au(111) surface (see Fig. 8a–c) was calculated and subsequently compared with the calculated vacuum barrier. Interestingly, all three calculated barrier heights for the cycloadditions were comparable: The same value (0.72 eV) was determined for the reaction of the model compound in vacuum and on the Au substrate, whereas a value of 0.69 eV was obtained for the AEB dimerization in vacuum. These calculated reaction barriers are low when compared to other on-surface reactions that require higher temperatures to be triggered [12, 13]. Moreover, the reaction was found to be strongly exothermic by 2.78 eV for the p-AAB in vacuum and on the flat Au(111) substrate. These results explain why the reaction readily proceeded on Au(111) at room temperature without di–\(\sigma\) bonding of the alkyne to the substrate.

On-surface dimerization of the model compound p-AAB on the Au(111) surface. a–c Reaction on a flat Au(111) surface. d–f Reaction with the reacting terminal carbon atom bound to an additional Au atom on the surface. Initial state energies are defined as 0 eV for both reactions. Adapted with permission from [11]. Copyright (2013) American Chemical Society
On the other hand, the flat unreconstructed surface initially used in the DFT calculations does not resemble the actual herringbone-reconstructed Au(111) substrate. To assess the potential catalytic role played by the gold atoms of the surface, the “click” reaction was simulated on Au(111) with one uncoordinated additional Au atom (Fig. 8d–f). To take into account the complete surface reconstruction would require a very large unit cell and therefore resulting in different locations where the molecules could adsorb. The extreme case where a single uncoordinated atom stands out as the most reactive site is more feasible for the calculations and provides valuable information regarding surface catalysis when compared with the counter extreme case, i.e., the flat unreconstructed surface. It was found that the strong binding of the alkyne group to the additional gold atom increases the binding energy of the initial state of this configuration (Fig. 8d) by 0.51 eV when compared to the flat surface (Fig. 8a). In this case, the exothermicity decreases down to 2.24 eV, and more importantly, the transition state energy is 0.71 eV, barely different from the 0.72 eV calculated for the flat surface. The small difference between these transition state energies indicates a negligible catalytic effect of uncoordinated Au atoms for the on-surface cycloaddition of p-AAB at room temperature. It is reasonable to expect that other surface sites of a herringbone reconstruction will show, at best, a catalytic activity as good as the one observed for the extreme case of an additional uncoordinated Au atom on the surface.
3.2 Role of Steric Effects on the Complete Regioselectivity
As described in Sect. 3, the metal surface seems to play only a negligible role in the on-surface azide–alkyne cycloaddition coupling reactions. Therefore, the observed complete regioselectivity for each case could not be explained in terms of surface catalysis. Instead, steric hindrance effects could be responsible for the experimentally observed complete regioselectivity. To quantify this effect on Au(111), the energy difference between the two possible regioisomers after coupling was calculated. The 1,5-regioisomer was found to be less stable than the 1,4 version by 0.72 eV on the Au(111) surface. This significant energy difference induced by steric hindrance between the aryl rings (Fig. 9) affects the reaction considerably and therefore increases the transition state energy of the 1,5-regioisomer formation process accordingly. This clearly shows that the selectivity of this reaction can be strongly enhanced by moving from a solution phase which proceeds in a three-dimensional space, to the analogous on-surface process that occurs restricted to two dimensions. This was the first report where the beneficial effect of two-dimensional confinement on the complete regioselectivity of an on-surface reaction was quantitatively explained.

Representation of the steric hindrance effect affecting the 1,5-regioisomer. Reprinted with permission from [11]. Copyright (2013) American Chemical Society
4 Outlook and Perspectives
The success of azide–alkyne cycloadditions on metal substrates under mild reaction conditions has been demonstrated to be a promising alternative to develop functional nanostructures on surfaces. Furthermore, the complete regioselectivity being tuned by steric hindrance should be considered a more general effect, especially in cases where the surface does not play a catalytic role in the reaction, rendering it as a key factor to control on-surface regioselectivity. Taking all this into consideration, future work could follow two different lines: (i) to systematically probe the viability of the same reactions (including the same reactants) on different substrates, e.g., insulators, semiconductors or oxides; and (ii) to exploit the low activation temperature of this reaction and combine it with reactions with a higher activation barrier.
For the first case, the choice of a more inert surface renders itself promising. By lowering the reactivity of the substrate toward the azide moiety and the monomers in general, i.e., choosing a Au(111) substrate instead of a Cu(111), the efficiency of the on-surface cycloaddition was enhanced by almost 30 %. Such behavior could also be expected if the reaction is performed on an insulating or oxide substrate or thin layer. There has been promising advances in on-surface synthesis via Ulmann coupling on insulators, for example [14]. On the other hand, the second case could be a promising route to obtain advanced supramolecular nanostructures on surfaces. The products of on-surface cycloadditions between terminal azides and alkynes have been proven to be extremely robust, while maintaining considerable flexibility (see Sect. 2.3). Given the low activation temperature required to induce the covalent coupling, orthogonal cross-coupling reactions can be envisioned. To combine the azide–alkyne cycloaddition reaction with a different on-surface reaction may be an alternative to control functionalities in two-dimensional networks at surfaces. This would allow to mix the optimal mechanical properties from the triazoles with, for example, optoelectronic properties. In summary, there is still plenty of room for new research regarding the on-surface azide–alkyne cycloadditions, making it an interesting candidate for future on-surface synthesis studies.
References
Gourdon, A.: On-surface covalent coupling in ultrahigh vacuum. Angew. Chem. Int. Ed. 47(37), 6950 (2008)
Perepichka, D., Rosei, F.: Chemistry: extending polymer conjugation into the second dimension. Science 323(5911), 216 (2009)
Champness, N.: Surface chemistry: making the right connections. Nat. Chem. 4(3), 149 (2012)
Kolb, H., Finn, M., Sharpless, K.: Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40(11), 2004 (2001)
Li, Y., Cai, C.: Chem. Click chemistry-based functionalization on non-oxidized silicon substrates. Asian J. 6(10), 2592 (2011)
Ciampi, S., Le Saux, G., Harper, J.B., Gooding, J.J.: Optimization of click chemistry of ferrocene derivatives on acetylene-functionalized silicon(100) surfaces. Electroanalysis 20(14), 1513 (2008)
Kolb, H., Sharpless, K.: The growing impact of click chemistry on drug discovery. Drug Discovery Today 8(24), 1128 (2003)
Pérez-Balderas, F., Ortega-Muñoz, M., Morales-Sanfrutos, J., Hernández-Mateo, F., Calvo-Flores, F.G., Calvo-Asín, J.A., Isac-García, J., Santoyo-González, F.: Multivalent neoglycoconjugates by regiospecific cycloaddition of alkynes and azides using organic-soluble copper catalysts. Org. Lett. 5(11), 1951 (2003)
Rostovtsev, V., Green, L., Fokin, V., Sharpless, K.: Angew. A stepwise huisgen cycloaddition process: copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Chem. Int. Ed. 41(14), 2596 (2002)
Bebensee, F., Bombis, C., Vadapoo, S., Cramer, J., Besenbacher, F., Gothelf, K., Linderoth, T.: On-surface azide-alkyne cycloaddition on cu(111): does it “click” in ultrahigh vacuum? J. Am. Chem. Soc. 135(6), 2136 (2013)
Díaz Arado, O., Mönig, H., Wagner, H., Franke, J.H., Langewisch, G., Held, P.A., Studer, A., Fuchs H.: On-surface azide-alkyne cycloaddition on au(111). ACS Nano 7(10), 8509 (2013)
Zhong, D., Franke, J.H., Podiyanachari, S., Blömker, T., Zhang, H., Kehr, G., Erker, G., Fuchs, H., Chi, L.: Linear alkane polymerization on a gold surface. Science 334(6053), 213 (2011)
Gao, H.Y., Wagner, H., Zhong, D., Franke, J.H., Studer, A., Fuchs, H.: Glaser coupling at metal surfaces. Angew. Chem. Int. Ed. 52(14), 4024 (2013)
Kittelmann, M., Rahe, P., Nimmrich, M., Hauke, C., Gourdon, A., Kühnle, A.: On-surface covalent linking of organic building blocks on a bulk insulator. ACS Nano 5(10), 8420 (2011)
Acknowledgements
The authors gratefully acknowledge the support of the Deutsche Forschungsgemeinschaft (DFG) through the collaborative research center SFB 858 (project B2) and the transregional collaborative research center TRR 061 (projects B3 and B7).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Díaz Arado, O., Mönig, H., Fuchs, H. (2016). On-Surface Synthesis by Azide–Alkyne Cycloaddition Reactions on Metal Surfaces. In: Gourdon, A. (eds) On-Surface Synthesis. Advances in Atom and Single Molecule Machines. Springer, Cham. https://doi.org/10.1007/978-3-319-26600-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-26600-8_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26598-8
Online ISBN: 978-3-319-26600-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)