
Abstract
Neuropsychiatric symptoms affect up to half of the patients with SLE. The effect on disease severity, quality of life, and prognosis is extremely heterogeneous. Symptoms of neuropsychiatric SLE (NPSLE) range from mild diffuse conditions to acute life-threatening events. Although the underlying mechanisms are still largely unraveled, several pathogenic pathways have been identified, such as antibody-mediated neurotoxicity, vasculopathy due aPL, and cytokine-induced neurotoxicity.
The chapter focuses on the central nervous system manifestations of NPSLE, with special emphasis on classification, diagnostic, and treatment aspects.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
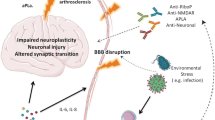


Keywords
1 Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder, which can present itself with a wide spectrum of clinical and immunological manifestations. Treatment advances in SLE have resulted in an increased survival of patients with SLE, and clinicians have become more aware of neuropsychiatric SLE (NPSLE) as an important manifestation of SLE [1]. NPSLE is defined as the neurological syndromes of the central, peripheral, and autonomic nervous system and the psychiatric syndromes observed in patients with SLE in whom other causes have been excluded [2]. It is the current understanding that the underlying pathology of NPSLE is a result of multifactorial sources including the presence of autoantibodies, changes in the microvasculature, and the intracranial production of inflammatory mediators, either alone or in combination [3].
NPSLE involvement is associated with different degrees of morbidity, varies in presentation and severity, may overlap, and can be of particular challenge for the clinician as symptoms may be difficult to distinguish from other neuropsychiatric conditions with different etiologies.
The major challenge with regard to diagnosis, treatment, and research within the field of NPSLE has been the lack of consensus in classifying the disease, due to the fact that the vast majority of the available literature on NPSLE long has been based on individual clinical interpretations. With the view of improving the definition and classification of NPSLE, the American College of Rheumatology agreed in 1999 on case definitions for 19 different central, peripheral, and autonomic nervous system syndromes in patients with SLE for which other causes have been excluded [4].
The central nervous system (CNS) symptoms, as defined by the ACR include focal neurological manifestations (cerebrovascular disease, seizures, myelopathy, aseptic meningitis, movement disorder, and demyelinating syndrome) or diffuse psychiatric/neuropsychological syndromes such as cognitive dysfunction, mood and anxiety disorders, psychosis, acute confusional state, and headaches. Peripheral neurologic conditions include cranial neuropathy, polyneuropathy, mononeuropathy, acute inflammatory demyelinating polyradiculoneuropathy (Guillain–Barré syndrome), myasthenia gravis, plexopathy, and autonomic disorders as summarized in Table 9.1.
This chapter provides an overview of NPSLE including aspects of the ACR classification and the current understanding of the various manifestations of NPSLE with a special emphasis on practical aspects of the diagnosis and management of neurological manifestations based on the EULAR management guidelines for the treatment guidelines for NPSLE [5]. The chapter focuses on the central nervous system manifestations of NPSLE, and peripheral nervous system manifestations will not be discussed in detail.
2 Epidemiology of NPSLE
SLE is an autoimmune disorder, which potentially can affect any organ and present itself with a wide spectrum of clinical and immunological manifestations. Specific classification criteria for SLE were updated by the American College of Rheumatology (ACR) in 1997 and include the serial or simultaneous presence of 4 of 11 defined criteria as shown in Table 9.2 [6]. The Systemic Lupus International Collaborating Clinics (SLICC) group has recently revised and validated a new set of classification criteria. The SLICC criteria require at least four of the proposed criteria, including at least one clinical and one immunological criterion for an SLE classification (Table 9.2) resulting in fewer misclassifications compared to the ACR criteria (Table 9.3) [7].
The annual incidence (per 100,000) of SLE in Europe ranges between 3.3 per year in Iceland and 4.7 in Sweden, compared to 21.9 in the Afro-Caribbeans. The prevalence varies according to the studied population, but studies suggest ranges between 26 per 100,000 in the UK (including all races) and 42 per 100,000 in Sweden, whereas the overall prevalence in the USA is reported lying between 14.6 and 50 cases per 100,000 persons [8].
NPSLE consists of a heterogeneous variety of neurological and psychiatric syndromes, none of which are exclusive or specific for SLE. The reported prevalence of NPSLE varies from 12 to 95 % between studies in which NPSLE criteria were applied to SLE patients. The wide range has been suggested to indirectly reflect the variation in study design, definition of neurological involvement, and ethnicity or geography, but may also be attributable to the availability of neurological expertise and investigations [9–11].
At least half of NPSLE manifestations occur at disease onset or within the first year after SLE onset, mainly in the presence of generalized disease activity [12].
Manifestations such as headache, mood disorders, anxiety, and mild cognitive dysfunction are common, but do not usually reflect overt CNS lupus activity [12]. Cognitive dysfunction and cerebrovascular events correlate with advancing age based on data from the general population, but this question has yet to be answered for patients with underlying SLE [13].
3 Pathogenic Mechanisms of NPSLE
The current understanding of the underlying pathological mechanisms which result in the multifaceted clinical presentations of NPSLE is suggested to be caused by a variety of mechanisms, including vascular injury mediated by mainly antiphospholipid antibodies (aPL) and immune complexes (mainly leading to transient ischemic attacks or strokes and seizures) and diffuse neuropsychiatric manifestations (such as cognitive impairment) in combination or alone [14].
With regard to thrombotic ischemic cerebral events, aPL have in the past been associated with focal neurological syndromes due to their ability to cause thrombotic events within vessels of different calibers leading to tissue ischemia [15]. In particular, the role of anticardiolipin (aCL) and lupus anticoagulant (LAC) has been investigated in NPSLE. A strong correlation between aPL and the overall frequency of neuropsychiatric manifestations was reported in a range of studies [16–19], but was questioned in other studies [20–23].
Menon et al. found the persistent presence of aPL to be associated with cognitive impairment [24]. aCL has been found to be associated with an overall NPSLE involvement more often than LAC [17, 19, 25]; however, on investigating cerebrovascular disease, predominantly stroke, LAC has been proved to be the most strongly associated aPL [26–30]. Despite the fact that most data have demonstrated an association between aCL and/or LAC and NPSLE, the role of anti-β2 glycoprotein I (anti-b2GPI) is less clear.
In vitro studies have also suggested a direct modulatory effect of aPL on neuronal cell function [31] and a pathogenic effect on neuronal cells [32].
Anti-ribosomal P protein antibodies (aRP) have a high specificity for SLE and have been found to be present in 6–46 % of subjects with SLE [33]. Elevated titers of aRP have been found in patients during SLE flares and may be associated with particular clinical manifestations including NPSLE [34]. Data from the Systemic Lupus International Collaborating Clinics (SLICC) inception cohort on 1,710 patients confirmed the association between elevated titers of aRP and psychosis [29]. However, a meta-analysis evaluating the diagnostic accuracy of anti-RP for NPSLE for psychosis, mood disorders, or both and for other diffuse manifestations did not confirm an association between aRP and any manifestation of NPSLE. Karassa et al. reported a sensitivity and specificity for the diagnosis of NPSLE of 26 % and 80 %, respectively. For psychosis, mood disorder, or both, the sensitivity and specificity were 27 % and 80 %, respectively. For other diffuse manifestations, the sensitivity was 24 % and the specificity 80 % [35]. Consequently, aRP testing does not discriminate between patients with NPLSE manifestations compared to those without NPSLE, and a role in clinical practice is yet to be fully defined. However, high titers of aRP are suggested to play a role in patients with suspected SLE psychosis [36].
Other autoantibodies of interest in the setting of NPSLE are antineuronal antibodies [37]. This subset of antibodies has been identified in the cerebrospinal fluid (CSF) and postmortem neuronal tissues of patients with NPSLE [38, 39]. Circulating anti-NR2 antibodies have been associated with NPSLE; however, studies on circulating anti-NR2 antibodies are inconsistent [40], whereas their presence in the CSF seem to be more consistent [14, 41, 42]. Subsets of the commonly occurring anti-dsDNA antibodies in SLE patients have also been suggested to be able to cross react with NMDA receptors in the CNS causing diffuse neuropsychiatric manifestations [37].
4 Classification of NPSLE
The first description of neuropsychiatric involvement in a patient with SLE (NPSLE) goes back to the nineteenth century [43]. In details, NPSLE was first described in the nineteenth century by Kaposi and Osler in a patient with pleurisy, pneumonia, disturbed neurologic function, and rapid progression to death [43]. As the neurological symptoms of NPSLE can present focally, diffusely, centrally, peripherally, and psychiatric, in isolation or simultaneously, they remain a challenge for the treating clinician. Due to this complexity, it is therefore not surprising that the definition of a uniform terminology and classification long has kept clinicians’ and scientists’ minds busy. Prior to 1999, several classifications had been proposed to describe the diverse clinical presentations of CNS involvement in SLE, and discrepancies existed among recommended methods of evaluation [36, 44, 45].
In 1985, How et al. established a range of neurological and psychiatric manifestations to support a diagnosis of NPSLE. Accordingly, a classification of NPSLE required the presence of one major criterion alone or one minor criterion (such as an abnormal finding on electroencephalography, nuclear brain scanning, CSF examination, or cerebral angiography). It was, however, recommended to rule out other causes such as infection, drugs, metabolic causes (such as uremia), or hypertension [45].
Two years later, Singer et al. published a consensus document with the aim to ascertain the level of agreement on neuropsychiatric SLE manifestations among a group of international experts in autoimmune diseases [46]. The majority of the participating experts felt that the ACR criteria for SLE were “insufficient for clinical usage” in the setting of NPSLE. Starting from a list of more than 50 possible clinical, laboratory findings and imaging manifestations of neuropsychiatric SLE, only four items were selected. These included atypical psychosis, several categories of seizures, transverse myelitis, and global cognitive dysfunction. This approach would have represented the basis for further studies and could have possibly expanded the ACR classification criteria for SLE; however, subsequent validation studies were never performed [14].
In 1997, the ACR Research Committee convened an ad hoc multidisciplinary committee consisting of 35 members across specialties, such as rheumatology, neurology, psychiatry, neuropsychiatry, and hematology, with the aim of developing a standard nomenclature for neuropsychiatric SLE. The ACR committee developed neuropsychiatric SLE case definitions with diagnostic criteria, exclusions, associations, and ascertainment. The implementation of standards and recommendations for essential laboratory evaluations and imaging techniques was a main advancement in the attempts to classify NPSLE [36]. The ACR NPSLE case definitions include 12 central nervous system syndromes and 7 syndromes of the peripheral nervous system compatible with the disease (Table 9.1). According to the ACR definition, the central nervous system symptoms are relatively common in patients with NPSLE, accounting for around 93 % of cases [9].
The defined clinical ACR manifestations can be divided in neurological manifestations affecting the CNS (focal or diffuse) and the peripheral syndromes. The first group includes aseptic meningitis, cerebrovascular disease, myelopathy, seizures, acute confusional state, cognitive dysfunction, movement disorders (chorea), psychosis, mood and anxiety disorders, and headaches. The peripheral syndromes encompass the remaining 7 % of NPSLE cases and include acute inflammatory demyelinizing polyradiculoneuropathy (Guillain–Barré syndrome), cranial neuropathy, mono- or polyneuropathy (single/multiplex), myasthenia gravis, autonomic disorders, and plexopathy (Table 9.1). NPSLE classification criteria provide an operational framework for the study of NP manifestations in SLE; however, when applied to the general population, their specificity is low due to the occurrence of such manifestations in the general population [47].
The European League against Rheumatism (EULAR) has recently published a guideline for the diagnosis and management of NPSLE manifestations [12]. One of the core statements of the EULAR consensus document is the recommendation, that the initial management of these patients should not differ to those without SLE. The treatment recommendations will be mentioned in each subsection of this chapter [12].
5 Clinical Manifestations, Diagnosis, and Management of NPSLE
5.1 Aseptic Meningitis
Aseptic meningitis refers to patients who have clinical and laboratory evidence for meningeal inflammation with negative routine bacterial cultures. The most common cause is enterovirus. Additional etiologies include other infections, medications, and malignancy. Aseptic meningitis is a rare finding in SLE patients. If meningitis is suspected, any underlying infectious cause must be ruled out. In patients receiving immunosuppressive therapy, opportunistic pathogens, such as Listeria monocytogenes, reactive tuberculosis, and Cryptococcus neoformans, should be considered. Kim et al. reported from a retrospective cohort of 1,420 SLE patients that 20 (1.4 %) were identified with meningitis. In over half of these patients, microorganisms were identified, and the most common organism was Cryptococcus neoformans, and a diagnosis of aseptic meningitis was made in nine patients [48]. Thus, a lumbar puncture and analysis of the cerebrospinal fluid (CSF) are generally indicated if meningitis is suspected. CSF results with a white cell count of less than 500 cells/mm3, over 50 % CSF lymphocytes, total protein less than 80–100 mg/dL, and normal glucose, and a negative Gram stain may suggest aseptic meningitis.
A variety of drugs can induce aseptic meningitis which therefore may be considered if a patient presents with a clinical picture suggesting aseptic meningitis. In patients with connective tissue disease, the use of nonsteroidal anti-inflammatory drugs (especially ibuprofen) has in the past been linked to aseptic meningitis [49, 50]. The treatment of aseptic meningitis is based on supportive management [12].
5.2 Cerebrovascular Disease
Cerebrovascular disease in SLE is in over 80 % of the cases attributable to transient ischemic attacks (TIA) or ischemic strokes. The main risk factors are high and persistent SLE activity, cumulative corticosteroid dosage, the persistent presence of moderate-to-high titers of aPL, heart valve disease, systemic hypertension, and age in itself [12]. The less commonly seen are hemorrhagic strokes (7–12 %), subarachnoid hemorrhage (3–5 %), and sinus thrombosis (2 %) [12, 51].
In comparison to the acute onset of focal neurological disease, the mechanisms underlying diffuse, multifocal CNS manifestations are less defined and less clear in presentation and may develop slowly over time not necessarily in association with SLE disease activity [52]. Magnetic resonance imaging and CT angiography can be useful to rule out cerebral hemorrhage, may inform on the localization and the extent of the ischemic brain injury, and help to characterize the brain lesion [12].
The acute management of a stroke in patients with SLE is not different to the management in the general population. Stroke teams are available in most centers and should be made aware of any patient with a suspected stroke according as, for example, described in the UK National Institute of Clinical Excellence (NICE) guideline on stroke management [53]. Thrombolysis may subsequently be considered in eligible patients, and therapy is otherwise based on anti-aggregation. A full work-up for secondary stroke prevention includes the modification of cardiovascular risk factors (such as hypertension, hypercholesterolemia, diabetes mellitus, etc). An electrocardiogram (ECG) and ultrasound of the carotid arteries with or without carotid endarterectomy should be performed as part of the work-up. In case of underlying SLE activity, this may be managed with steroids and/or immunosuppressive therapy [12].
In patients with persistent aPL and stroke fulfilling the classification criteria for APS [54], long-term anticoagulation should be initiated [55]. The dosage of anticoagulation is an ongoing subject of debate. Two randomized controlled trials have compared the standard anticoagulant treatment (target INR 2–3) with high-intensity treatment (target INR 3.5), and both studies did not show an advantage of high-intensity vitamin K antagonist (VKA) for the prevention of recurrent thrombotic events. However, in both studies, patients randomized to the high-intensity group frequently did not achieve adequate anticoagulation targets. In one of these studies, patients were only in target 43 % of the time and included a relatively low number of patients with arterial events [56]. However, in a systematic review of sixteen studies, Ruiz-Irastorza et al. recommended high-intensity warfarin therapy for patients with recurrent events while on VKA (target 2–3) [57]. The role of new oral anticoagulants such as direct factor Xa and thrombin inhibitors still remains to be determined in this setting awaiting results from randomized controlled trials.
5.3 Myelopathy
Myelopathy in NPSLE is defined as a disorder of the spinal cord characterized by rapidly evolving paraparesis and/or sensory loss, with a demonstrable motor and/or sensory cord level (to include transverse) and/or sphincter involvement. Usually, the myelopathy has rapid onset (hours or days) of one or more of the following diagnostic manifestations: (1) bilateral weakness of legs with or without arms (paraplegia/quadriplegia), which may be asymmetric, and (2) sensory impairment with cord level similar to that of motor weakness, with or without bowel and bladder dysfunction [58]. The underlying pathomechanism may be caused by ischemia/thrombosis and/or inflammation, and patients may present with signs of grey matter dysfunction (which includes flaccidity and hyporeflexia) or white matter dysfunction (such as spasticity and hyperreflexia) [52, 59].
Neuromyelitis optica (NMO), also known as Devic syndrome, is a severe demyelinating disorder of the central nervous system causing longitudinal transverse myelitis of at least three vertebral segments, recurrent optic neuritis, and normal brain MRI. NMO has been reported in patients with SLE and is associated with the presence of anti-aquaporin antibodies (IgG subtype) [14].
Myelitis is estimated to affect 1–2 % of patients with SLE which is more than 1,000 times greater than the prevalence of idiopathic myelitis in the general population [60, 61].
Transverse myelitis has also been associated with aPL and, in atypical presentations, an important differential diagnosis remains multiple sclerosis [62]. Anecdotal reports of transverse myelitis associated with aPL have been described since 1985 when Harris et al. reported it in a 45-year-old woman with a lupus-like illness and high-titer aCL of the IgM isotype [63].
Current views suggest that underlying prothrombotic mechanisms related to aPL play a key role in the development of acute transverse myelitis in patients with SLE [64–66]. D’Cruz et al. described a series of 15 patients with transverse myelitis as the presenting manifestation of SLE. Seventy percent of the patients were aPL positive, supporting the view of a strong association of transverse myelitis with aPL [67].
Contrast-enhanced MRI is the imaging method of choice [12]. A CSF analysis is useful to rule out underlying infection. Immunosuppressive therapy with intravenous methylprednisolone and intravenous cyclophosphamide can be effective in SLE myelitis, particularly when instituted within the first few hours of presentation. More than half of the patients have relapses; steroids should therefore be tapered cautiously and maintenance immunosuppression may be indicated. Plasma exchange therapy has been used in severe refractory cases. In case of persistent aPL, anticoagulation may be indicated [68, 69].
5.4 Seizures
Generalized primary seizures and partial seizures are a common neurological manifestation found in up to 20 % of patients with SLE (compared with 0.5–1.0 % in the general population). Seizures have been described among the early CNS manifestations and may precede a diagnosis of SLE [51, 70, 71]. Cerebrospinal fluid (CSF) pleocytosis is common, suggesting that a low-grade lupus-related encephalopathy may be a possible underlying cause. Cerebral atrophy has also been described in patients with SLE and may predispose to seizures [52, 72].
Results from the Systemic Lupus International Collaborating Clinics (SLICC group) prospective inception cohort of 1,631 SLE patients showed that 4.6 % of patients had at least one seizure, most of which occurred around the time of their SLE diagnosis. This finding is in conjunction with our own findings [71].
Interestingly, there was some indication that the regular use of antimalarial drugs reduced the risk of seizures. A higher risk of seizure was seen within three groups, patients with lower education status forslag, patients with more organ damage since the diagnosis of SLE, and lupus patients of African ethnicity forslag. There was an association with disease activity but not with autoantibodies [70]. The group also found that seizures due to SLE frequently got better without long-term seizure medication and without decreasing quality of life.
The treatment is based on anticonvulsive therapy, which may be considered in patients with high recurrence risk, brain MRI structural abnormalities causally linked to seizures, focal neurological signs, partial seizure, and epileptiform EEG [12]. Seizures secondary to SLE disease activity may be treated with glucocorticoids and immunosuppression, and in case of refractory seizures, cyclophosphamide has been used in anecdotal cases [73].
5.5 Acute Confusional State
Acute confusional state (delirium) has been described in up to 7 % of patients with SLE in whom other underlying pathology has been excluded. Characterized by an acute-onset variation (or fluctuation) of the level of consciousness, acute confusional states may at worse progress to a coma [12]. Milder forms of acute confusional state include the reduced ability to focus attention, mood disturbances, and impaired cognition.
The initial acute management requires exploration of underlying causes. CSF examination is required to exclude any underlying infection. The imaging of choice is SPECT; however, possible limitations in expertise and availability may restrict clinical practice to CT or MRI in order to rule out ischemic events, underlying hemorrhage or malignancy [13]. Benzodiazepines or antipsychotics may be required in the acute setting. Glucocorticoid and immunosuppression play some role and may in selected cases have to be escalated to plasma exchange and cyclophosphamide. Rituximab has been used in refractory cases [68, 69].
5.6 Cognitive Dysfunction
Cognitive dysfunction ranges from mild to moderate or severe impairment and manifests itself by reduced cognitive function (such as memory problems or the reduced ability of abstract thinking) and is a common finding among patients with SLE. In up to 80 % of SLE patients, mild to moderate cognitive dysfunction has been reported [51, 74], whereas severe cognitive dysfunction is a rare complication found in up to 5 % of patients with SLE [12]. It has been reported to occur in the absence of SLE disease activity and fluctuates over the course of the disease, often independently of depression or anxiety [14]. In patients with persistently aPL, anecdotal evidence suggest cognitive impairment to improve on anticoagulation [55].
A major challenge in diagnosing cognitive impairment remains the fact that other common manifestations of SLE, such as fatigue, widespread pain, and depression are associated with cognitive impairment [14]. A study of SLE outpatients showed that SLE patients complaining of cognitive dysfunction generally performed normally on neuropsychological tests but had traits of depression whereas actual poor neuropsychological performance not always was noticed by the patient [75].
The management of cognitive dysfunction is supportive, and exacerbating factors such as anxiety and depression should be managed accordingly. Bertsias et al. have recommended psychoeducational group interventions as being useful. Equally may steroids and/or immunosuppressive therapy be considered to control concurrent SLE or other NPSLE activity [76].
5.7 Movement Disorders (Chorea)
Movement disorders, such as chorea, ataxia, choreoathetosis, dystonia, and hemiballismus, occur in roughly 1 % patients with SLE [29]. The existing literature mainly consists of anecdotal reports, case reports, and small case series and has been described as juvenile SLE onset, associated with the use of contraceptives and in patients with aPL [77–79]. Chorea has in the past been associated with the persistent presence of aPL [80]. The underlying pathological mechanisms have been suggested as multifactorial; there does not seem to be an exclusive ischemic underlying pathology [77].
In addition to symptomatic therapy for persistent symptoms (dopamine antagonists), antiplatelet agents may be considered in SLE patients with aPL according to the recent EULAR guidelines. Glucocorticoids and immunosuppressive and/or anticoagulation therapy may be considered in severe cases when generalized disease activity and/or thrombotic manifestations are present [12].
5.8 Psychosis
In the context of severe psychiatric manifestations, the WHO has defined acute psychosis as an “acute psychotic disorder in which ‘hallucinations, delusions, and perceptual disturbances are obvious but markedly variable, changing from day to day or even from hour to hour. Emotional turmoil, with intense transient feelings of happiness and ecstasy or anxieties and irritability, is also frequently present” [81]. According to the EULAR task force report, any patient presenting with possible NPSLE should receive the standard of care to rule out underlying causes organic systemic disease, metabolic abnormalities, etc. in case of a presentation of any NPSLE manifestation, such as acute psychosis. Corticosteroid-induced psychiatric disease occurs in 10 % of patients treated with prednisone 1 mg/kg (or more) and manifests itself primarily as mood disorder rather than psychosis but remains an important differential diagnosis [82]. NPSLE has been reported to present with paranoia with visual and auditory hallucinations [83]. Recovery is usually complete, but relapses are not rare, and the treatment may include antidepressive agents, steroids, and/or immunosuppressive agents if SLE activity is suspected. In a subgroup analysis from a large single-center study on 751 patients, cyclophosphamide followed by azathioprine maintenance therapy has shown a significant effect [84].
5.9 Mood and Anxiety Disorders
Despite epidemiological controversies, anxiety and depression consistently are one of the most commonly reported NPSLE manifestations. The most common psychological symptom in patients with SLE is depression [85–88]. Depressive symptoms may begin acutely accordingly with disease onset [89], possibly reflecting the patient’s reaction to chronic illness and the associated lifestyle limitations, including fatigue, limited sun exposure, and chronic medication use [87, 90].
Some studies have postulated an organic cause. An association has been reported between severe depression and aRP antibodies, and antibodies to NMDA receptors, but not with other antibodies [89, 91–93]. Elevated levels of aRP antibodies have been found up to 88 % of these patients [91, 92].
As reported for depression, following the initial diagnosis of SLE, or after an acute exacerbation, some patients display symptoms of anxiety, either instead of or in addition to depression. The patient may become anxious about a variety of possible consequences of their illness, including disfigurement, disability, dependency, loss of a job, social isolation, or death. Ishikura et al. showed that prevalence and intensity of anxiety in the course of SLE positively correlated with insufficient knowledge about disease and its therapy, perceived by the patient at the beginning of disease, and did not correlate with SLE activity [94]. Furthermore, Hawro et al. reported a shorter SLE duration in patients with anxiety disorder [95]. Thus, one may speculate that patients anxiety may be caused by inadequate knowledge about their chronic illness and its treatment options.
5.10 Headaches
The term “lupus headache” is used for a particular type of headache directly attributable to SLE and is a stand-alone variable with comparable definitions in at least two composite indices of global SLE disease activity: firstly, the British Isles Lupus Assessment Group (BILAG) 2004 index [96], which defines lupus headache as a disabling headache that is unresponsive to narcotic analgesia and lasts >3 days, and secondly, the SLEDAI-2K [97], which defines lupus headache as a severe, persistent headache (which may be migrainous, but must be nonresponsive to narcotic analgesia).
However, the specificity of this term is under debate as headaches are common in SLE patients but probably not more frequent than in the general population of similar age and gender.
In a review of 50 studies and 115,000 participants in 17 European countries [32], Stovner and Andree reported that the 1-year prevalence of headache in the general population was 55 % (62 % in women and 45 % in men) and the lifetime prevalence of headache was 77 %. In addition, Stovner et al. reported a 1-year prevalence of migraine of 15 % (19 % in women and 8 % in men), and the lifetime prevalence rates were 16 % overall (20 % in women and 11 % in men) [98]. The 1-year prevalence of tension headache was 80 %. Data from the SLICC cohort showed that the frequency of headache at the enrollment visit was comparable to the 1-year prevalence rates in the general population [99]. In the SLICC cohort of a total of 308 patients, 17.8 % had some type of headache. The specific headache types were migraine in 187 patients (60.7 %), tension in 119 (38.6 %), intractable nonspecific in 22 (7.1 %), cluster in 8 (2.6 %), and intracranial hypertension in 3 (1.0 %) [99]. The occurrence of headache is not related to overall SLE disease activity and is not associated with changes in lupus medications. The majority of headaches in SLE patients are unlikely caused by a direct effect of SLE. Regardless of the cause, SLE patients with headaches report a lower quality of life. Most headaches in SLE patients get better and resolve over time.
6 Conclusions
Neuropsychiatric symptoms affect up to half of the patients with SLE. The effect on disease severity, quality of life, and prognosis is extremely heterogeneous. Symptoms of NPSLE range from mild diffuse conditions to acute life-threatening events. Although the underlying mechanisms are still largely unraveled, several pathogenic pathways have been identified, such as antibody-mediated neurotoxicity, vasculopathy due aPL, and cytokine-induced neurotoxicity.
A diagnosis of NPSLE requires the exclusion of other conditions, and clinical assessment directs the selection of appropriate investigations, including neuroimaging to evaluate brain structure and function, analysis of CSF, electrophysiological studies, and neuropsychological assessment. Treatment includes the use of symptomatic therapies and specific interventions with either anticoagulation or immunosuppressive agents, according to the underlying pathogenetic mechanism. The management of comorbidities contributing to the neuropsychiatric event is also crucial.
References
Ginzler EM, Dvorkina O (2005) Newer therapeutic approaches for systemic lupus erythematosus. Rheum Dis Clin North Am 31(2):315–328
Nived O, Sturfelt G, Liang MH, De Pablo P (2003) The ACR nomenclature for CNS lupus revisited. Lupus 12(12):872–876
Hanly JG (2005) Neuropsychiatric lupus. Rheum Dis Clin North Am 31(2):273–298, vi
The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes (1999) Arthritis Rheum. 42(4):599–608
Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C et al (2008) EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis 67(2):195–205
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25(11):1271–1277
Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. (2012) Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 64(8):2677–2686.
D’Cruz DP, Khamashta MA, Hughes GR (2007) Systemic lupus erythematosus. Lancet 369(9561):587–596
Hanly JG, Su L, Farewell V, McCurdy G, Fougere L, Thompson K (2009) Prospective study of neuropsychiatric events in systemic lupus erythematosus. J Rheumatol 36(7):1449–1459
Mok CC, To CH, Mak A (2006) Neuropsychiatric damage in Southern Chinese patients with systemic lupus erythematosus. Medicine (Baltimore) 85(4):221–228
Sanna G, Bertolaccini ML, Cuadrado MJ, Laing H, Khamashta MA, Mathieu A et al (2003) Neuropsychiatric manifestations in systemic lupus erythematosus: prevalence and association with antiphospholipid antibodies. J Rheumatol 30(5):985–992
Bertsias GK, Ioannidis JP, Aringer M, Bollen E, Bombardieri S, Bruce IN et al (2010) EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis 69(12):2074–2082
Kelly-Hayes M (2010) Influence of age and health behaviors on stroke risk: lessons from longitudinal studies. J Am Geriatr Soc 58(Suppl 2):S325–S328
Hanly JG (2014) Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol 10(6):338–347
Meroni PL, Borghi MO, Raschi E, Tedesco F (2011) Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol 7(6):330–339
Mok CC, Lau CS, Wong RW (2001) Neuropsychiatric manifestations and their clinical associations in southern Chinese patients with systemic lupus erythematosus. J Rheumatol 28(4):766–771
Afeltra A, Garzia P, Mitterhofer AP, Vadacca M, Galluzzo S, Del Porto F et al (2003) Neuropsychiatric lupus syndromes: relationship with antiphospholipid antibodies. Neurology 61(1):108–110
Yu HH, Lee JH, Wang LC, Yang YH, Chiang BL (2006) Neuropsychiatric manifestations in pediatric systemic lupus erythematosus: a 20-year study. Lupus 15(10):651–657
Borowoy AM, Pope JE, Silverman E, Fortin PR, Pineau C, Smith CD et al (2012) Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 faces of lupus cohort. Semin Arthritis Rheum 42(2):179–185
Houman MH, Smiti-Khanfir M, Ben Ghorbell I, Miled M (2004) Systemic lupus erythematosus in Tunisia: demographic and clinical analysis of 100 patients. Lupus 13(3):204–211
Kamen DL, Barron M, Parker TM, Shaftman SR, Bruner GR, Aberle T et al (2008) Autoantibody prevalence and lupus characteristics in a unique African American population. Arthritis Rheum 58(5):1237–1247
Singh S, Gupta MK, Ahluwalia J, Singh P, Malhi P (2009) Neuropsychiatric manifestations and antiphospholipid antibodies in pediatric onset lupus: 14 years of experience from a tertiary center of North India. Rheumatol Int 29(12):1455–1461
Kozora E, Filley CM, Zhang L, Brown MS, Miller DE, Arciniegas DB et al (2012) Immune function and brain abnormalities in patients with systemic lupus erythematosus without overt neuropsychiatric manifestations. Lupus 21(4):402–411
Menon S, Jameson-Shortall E, Newman SP, Hall-Craggs MR, Chinn R, Isenberg DA (1999) A longitudinal study of anticardiolipin antibody levels and cognitive functioning in systemic lupus erythematosus. Arthritis Rheum 42(4):735–741
Mikdashi J, Handwerger B (2004) Predictors of neuropsychiatric damage in systemic lupus erythematosus: data from the Maryland lupus cohort. Rheumatology 43(12):1555–1560
Brey RL, Holliday SL, Saklad AR, Navarrete MG, Hermosillo-Romo D, Stallworth CL et al (2002) Neuropsychiatric syndromes in lupus: prevalence using standardized definitions. Neurology 58(8):1214–1220
Hanly JG, Urowitz MB, Siannis F, Farewell V, Gordon C, Bae SC et al (2008) Autoantibodies and neuropsychiatric events at the time of systemic lupus erythematosus diagnosis: results from an international inception cohort study. Arthritis Rheum 58(3):843–853
Hanly JG, Urowitz MB, Jackson D, Bae SC, Gordon C, Wallace DJ et al (2011) SF-36 summary and subscale scores are reliable outcomes of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis 70(6):961–967
Hanly JG, Urowitz MB, Su L, Bae SC, Gordon C, Clarke A et al (2011) Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis 70(10):1726–1732
Sciascia S, Bertolaccini ML, Roccatello D, Khamashta MA, Sanna G (2014) Autoantibodies involved in neuropsychiatric manifestations associated with systemic lupus erythematosus: a systematic review. J Neurol 261(9):1706–1714
Chapman J, Cohen-Armon M, Shoenfeld Y, Korczyn AD (1999) Antiphospholipid antibodies permeabilize and depolarize brain synaptoneurosomes. Lupus 8(2):127–133
Martinez-Cordero E, Rivera Garcia BE, Aguilar Leon DE (1997) Anticardiolipin antibodies in serum and cerebrospinal fluid from patients with systemic lupus erythematosus. J Investig Allergol Clin Immunol 7(6):596–601
Eber T, Chapman J, Shoenfeld Y (2005) Anti-ribosomal P-protein and its role in psychiatric manifestations of systemic lupus erythematosus: myth or reality? Lupus 14(8):571–575
Zandman-Goddard G, Chapman J, Shoenfeld Y (2007) Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin Arthritis Rheum 36(5):297–315
Karassa FB, Afeltra A, Ambrozic A, Chang DM, De Keyser F, Doria A et al (2006) Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus: an international meta-analysis. Arthritis Rheum 54(1):312–324
Sciascia S, Bertolaccini ML, Baldovino S, Roccatello D, Khamashta MA, Sanna G (2013) Central nervous system involvement in systemic lupus erythematosus: overview on classification criteria. Autoimmun Rev 12(3):426–429
Weiner SM, Klein R, Berg PA (2000) A longitudinal study of autoantibodies against central nervous system tissue and gangliosides in connective tissue diseases. Rheumatol Int 19(3):83–88
Bluestein HG, Williams GW, Steinberg AD (1981) Cerebrospinal fluid antibodies to neuronal cells: association with neuropsychiatric manifestations of systemic lupus erythematosus. Am J Med 70(2):240–246
Zvaifler NJ, Bluestein HG (1982) The pathogenesis of central nervous system manifestations of systemic lupus erythematosus. Arthritis Rheum 25(7):862–866
Lauvsnes MB, Omdal R (2012) Systemic lupus erythematosus, the brain, and anti-NR2 antibodies. J Neurol 259(4):622–629
Yoshio T, Onda K, Nara H, Minota S (2006) Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum 54(2):675–678
Arinuma Y, Yanagida T, Hirohata S (2008) Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum 58(4):1130–1135
Osler W (1895) On the visceral complications of the erythema exudativum multiforme. Am J Med Sci 110:629–646
Kassan SS, Lockshin MD (1979) Central nervous system lupus erythematosus. The need for classification. Arthritis Rheum 22(12):1382–1385
How A, Dent PB, Liao SK, Denburg JA (1985) Antineuronal antibodies in neuropsychiatric systemic lupus erythematosus. Arthritis Rheum 28(7):789–795
Singer J, Denburg JA (1990) Diagnostic criteria for neuropsychiatric systemic lupus erythematosus: the results of a consensus meeting. The Ad Hoc Neuropsychiatric Lupus Workshop Group. J Rheumatol 17(10):1397–1402
Ainiala H, Hietaharju A, Loukkola J, Peltola J, Korpela M, Metsanoja R et al (2001) Validity of the new American College of Rheumatology criteria for neuropsychiatric lupus syndromes: a population-based evaluation. Arthritis Rheum 45(5):419–423
Kim JM, Kim KJ, Yoon HS, Kwok SK, Ju JH, Park KS et al (2011) Meningitis in Korean patients with systemic lupus erythematosus: analysis of demographics, clinical features and outcomes; experience from affiliated hospitals of the Catholic University of Korea. Lupus 20(5):531–536
Bernstein RF (1980) Ibuprofen-related meningitis in mixed connective tissue disease. Ann Intern Med 92(2 Pt 1):206–207
Karmacharya P, Mainali NR, Aryal MR, Lloyd B (2013) Recurrent case of ibuprofen-induced aseptic meningitis in mixed connective tissue disease. BMJ Case Rep pii: bcr2013009571
Hanly JG, Harrison MJ (2005) Management of neuropsychiatric lupus. Best Pract Res Clin Rheumatol 19(5):799–821
Jeltsch-David H, Muller S (2014) Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol 10(10):579–596
Swain S, Turner C, Tyrrell P, Rudd A, Guideline DG (2008) Diagnosis and initial management of acute stroke and transient ischaemic attack: summary of NICE guidance. BMJ 337:a786
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R et al (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4(2):295–306
Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR (1995) The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 332(15):993–997
Crowther M, Crowther MA (2010) Intensity of warfarin coagulation in the antiphospholipid syndrome. Curr Rheumatol Rep 12(1):64–69
Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA (2010) Antiphospholipid syndrome. Lancet 376(9751):1498–1509
Cikes N, Bosnic D, Sentic M (2008) Non-MS autoimmune demyelination. Clin Neurol Neurosurg 110(9):905–912
Birnbaum J, Petri M, Thompson R, Izbudak I, Kerr D (2009) Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis Rheum 60(11):3378–3387
Theodoridou A, Settas L (2006) Demyelination in rheumatic diseases. J Neurol Neurosurg Psychiatry 77(3):290–295
Kaplin AI, Krishnan C, Deshpande DM, Pardo CA, Kerr DA (2005) Diagnosis and management of acute myelopathies. Neurologist 11(1):2–18
Karussis D, Leker RR, Ashkenazi A, Abramsky O (1998) A subgroup of multiple sclerosis patients with anticardiolipin antibodies and unusual clinical manifestations: do they represent a new nosological entity? Ann Neurol 44(4):629–634
Harris EN, Gharavi AE, Mackworth-Young CG, Patel BM, Derue G, Hughes GR (1985) Lupoid sclerosis: a possible pathogenetic role for antiphospholipid antibodies. Ann Rheum Dis 44(4):281–283
Alarcon-Segovia D, Deleze M, Oria CV, Sanchez-Guerrero J, Gomez-Pacheco L, Cabiedes J et al (1989) Antiphospholipid antibodies and the antiphospholipid syndrome in systemic lupus erythematosus. A prospective analysis of 500 consecutive patients. Medicine (Baltimore) 68(6):353–365
Lavalle C, Pizarro S, Drenkard C, Sanchez-Guerrero J, Alarcon-Segovia D (1990) Transverse myelitis: a manifestation of systemic lupus erythematosus strongly associated with antiphospholipid antibodies. J Rheumatol 17(1):34–37
Ruiz-Arguelles GJ, Guzman-Ramos J, Flores-Flores J, Garay-Martinez J (1998) Refractory hiccough heralding transverse myelitis in the primary antiphospholipid syndrome. Lupus 7(1):49–50
D’Cruz DP, Mellor-Pita S, Joven B, Sanna G, Allanson J, Taylor J et al (2004) Transverse myelitis as the first manifestation of systemic lupus erythematosus or lupus-like disease: good functional outcome and relevance of antiphospholipid antibodies. J Rheumatol 31(2):280–285
Neuwelt CM (2003) The role of plasmapheresis in the treatment of severe central nervous system neuropsychiatric systemic lupus erythematosus. Ther Apher Dial Off Peer-Rev J Int Soc Apher Jpn Soc Apher Jpn Soc Dial Ther 7(2):173–182
Bartolucci P, Brechignac S, Cohen P, Le Guern V, Guillevin L (2007) Adjunctive plasma exchanges to treat neuropsychiatric lupus: a retrospective study on 10 patients. Lupus 16(10):817–822
Hanly JG, Urowitz MB, Su L, Gordon C, Bae SC, Sanchez-Guerrero J et al (2012) Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Ann Rheum Dis 71(9):1502–1509
Jacobsen S, Petersen J, Ullman S, Junker P, Voss A, Rasmussen JM et al (1998) A multicentre study of 513 Danish patients with systemic lupus erythematosus. I. Disease manifestations and analyses of clinical subsets. Clin Rheumatol 17(6):468–477
Joseph FG, Scolding NJ (2010) Neurolupus. Pract Neurol 10(1):4–15
Barile-Fabris LA (2005) Treatment of neuropsychiatric manifestations of systemic lupus erythematosus. Reumatol Clin 1(Suppl 2):S42–S45
Ainiala H, Loukkola J, Peltola J, Korpela M, Hietaharju A (2001) The prevalence of neuropsychiatric syndromes in systemic lupus erythematosus. Neurology 57(3):496–500
Vogel A, Bhattacharya S, Larsen JL, Jacobsen S (2011) Do subjective cognitive complaints correlate with cognitive impairment in systemic lupus erythematosus? A Danish outpatient study. Lupus 20(1):35–43
Hanly JG, Cassell K, Fisk JD (1997) Cognitive function in systemic lupus erythematosus: results of a 5-year prospective study. Arthritis Rheum 40(8):1542–1543
Baizabal-Carvallo JF, Alonso-Juarez M, Koslowski M (2011) Chorea in systemic lupus erythematosus. J Clin Rheumatol 17(2):69–72
Caramelli P, Toledo SM, Marchiori PE, Barbosa ER, Scaff M (1993) Chorea as a sign of systemic lupus erythematosus activity. Case report. Arq Neuropsiquiatr 51(2):267–269
Mathur AK, Gatter RA (1988) Chorea as the initial presentation of oral contraceptive induced systemic lupus erythematosus. J Rheumatol 15(6):1042–1043
Orzechowski NM, Wolanskyj AP, Ahlskog JE, Kumar N, Moder KG (2008) Antiphospholipid antibody-associated chorea. J Rheumatol 35(11):2165–2170
World Health Organization (1993) The ICD-10 classification of mental and behavioural disorders: diagnostic criteria for research. Geneva: World Health Organization: 263 p
Chau SY, Mok CC (2003) Factors predictive of corticosteroid psychosis in patients with systemic lupus erythematosus. Neurology 61(1):104–107
Segui J, Ramos-Casals M, Garcia-Carrasco M, de Flores T, Cervera R, Valdes M et al (2000) Psychiatric and psychosocial disorders in patients with systemic lupus erythematosus: a longitudinal study of active and inactive stages of the disease. Lupus 9(8):584–588
Rahman P, Humphrey-Murto S, Gladman DD, Urowitz MB (1997) Cytotoxic therapy in systemic lupus erythematosus. Experience from a single center. Medicine 76(6):432–437
Kozora E, Ellison MC, West S (2006) Depression, fatigue, and pain in systemic lupus erythematosus (SLE): relationship to the American College of Rheumatology SLE neuropsychological battery. Arthritis Rheum 55(4):628–635
Goodwin JM, Goodwin JS, Kellner R (1979) Psychiatric symptoms in disliked medical patients. Jama 241(11):1117–1120
Kozora E, Ellison MC, Waxmonsky JA, Wamboldt FS, Patterson TL (2005) Major life stress, coping styles, and social support in relation to psychological distress in patients with systemic lupus erythematosus. Lupus 14(5):363–372
Stoll T, Kauer Y, Buchi S, Klaghofer R, Sensky T, Villiger PM (2001) Prediction of depression in systemic lupus erythematosus patients using SF-36 Mental Health scores. Rheumatology 40(6):695–698
Shortall E, Isenberg D, Newman SP (1995) Factors associated with mood and mood disorders in SLE. Lupus 4(4):272–279
Jump RL, Robinson ME, Armstrong AE, Barnes EV, Kilbourn KM, Richards HB (2005) Fatigue in systemic lupus erythematosus: contributions of disease activity, pain, depression, and perceived social support. J Rheumatol 32(9):1699–1705
West SG, Emlen W, Wener MH, Kotzin BL (1995) Neuropsychiatric lupus erythematosus: a 10-year prospective study on the value of diagnostic tests. Am J Med 99(2):153–163
Schneebaum AB, Singleton JD, West SG, Blodgett JK, Allen LG, Cheronis JC et al (1991) Association of psychiatric manifestations with antibodies to ribosomal P proteins in systemic lupus erythematosus. Am J Med 90(1):54–62
Lapteva L, Nowak M, Yarboro CH, Takada K, Roebuck-Spencer T, Weickert T et al (2006) Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum 54(8):2505–2514
Ishikura R, Morimoto N, Tanaka K, Kinukawa N, Yoshizawa S, Horiuchi T et al (2001) Factors associated with anxiety, depression and suicide ideation in female outpatients with SLE in Japan. Clin Rheumatol 20(6):394–400
Hawro T, Krupinska-Kun M, Rabe-Jablonska J, Sysa-Jedrzejowska A, Robak E, Bogaczewicz J et al (2011) Psychiatric disorders in patients with systemic lupus erythematosus: association of anxiety disorder with shorter disease duration. Rheumatol Int 31(10):1387–1391
Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN et al (2005) BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group’s disease activity index for patients with systemic lupus erythematosus. Rheumatology 44(7):902–906
Romero-Diaz J, Isenberg D, Ramsey-Goldman R (2011) Measures of adult systemic lupus erythematosus: updated version of British Isles Lupus Assessment Group (BILAG 2004), European Consensus Lupus Activity Measurements (ECLAM), Systemic Lupus Activity Measure, Revised (SLAM-R), Systemic Lupus Activity Questionnaire for Population Studies (SLAQ), Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2 K), and Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index (SDI). Arthritis Care Res 63(Suppl 11):S37–S46
Stovner LJ, Andree C (2010) Prevalence of headache in Europe: a review for the Eurolight project. J Headache Pain 11(4):289–299
Hanly JG, Urowitz MB, O’Keeffe AG, Gordon C, Bae SC, Sanchez-Guerrero J et al (2013) Headache in systemic lupus erythematosus: results from a prospective, international inception cohort study. Arthritis Rheum 65(11):2887–2897
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Schreiber, K., Jacobsen, S. (2016). Neuropsychiatric Systemic Lupus Erythematosus. In: Roccatello, D., Emmi, L. (eds) Connective Tissue Disease. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-319-24535-5_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-24535-5_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24533-1
Online ISBN: 978-3-319-24535-5
eBook Packages: MedicineMedicine (R0)