
Abstract
Aging is a biological process characterized by the progressive deterioration of physiological functions that occurs through the accumulation of macromolecular and cellular damage. This phenomenon impairs tissue function and is a risk factor for many disorders including cardiovascular disease, neurodegenerative disorders, and cancer. A recent study has enumerated nine cellular and molecular hallmarks that represent common denominators of aging and together determine the aging phenotype, highlighting the concept of aging plasticity. Among the multiple molecular mechanisms which may contribute to aging modulation, microRNAs (miRNAs) are raising enormous interest due to their ability to affect all the “Hallmarks of Aging.” In this chapter, we will focus on the description of the diverse functional roles of geromiRs, the large and growing subgroup of miRNAs implicated in aging. We will also address the molecular mechanisms underlying miRNA function in aging and discuss potential strategies for managing aging and extending longevity based on geromiR modulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Inevitably, all of us will experience a progressive deterioration of our body’s fitness due to the biological process known as aging. This dramatic phenomenon has long interested scientists, and with the extraordinary life-span increase of human populations over the last century, there is a growing interest for understanding this condition at the molecular level [1]. Aging is characterized by a progressive loss of physiological integrity that occurs through the accumulation of macromolecular and cellular damage, leading to impaired fitness and increased vulnerability to death. This deterioration is the primary risk factor for major human pathologies, including cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases. One of the main advances in aging research was the discovery of aging plasticity, which implies that this process can be modulated by genetic, nutritional, and pharmacological factors. The first evidence of aging plasticity came from the discovery that caloric restriction—underfeeding without malnutrition—extended life-span in many model organisms [2]. Likewise, other external perturbations, such as temperature or oxygen levels, were found to influence life-span in several organisms. A recent study [3] has enumerated nine cellular and molecular hallmarks that represent common denominators of aging and together determine the aging phenotype. These “hallmarks of aging” have been divided into three categories: primary hallmarks, antagonistic hallmarks, and integrative hallmarks. Primary hallmarks include genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis. These are the main culprits of the molecular damage associated with aging and, therefore, they are all unequivocally negative hallmarks. The second category involves the compensatory or antagonistic responses to this damage, and includes three hallmarks: deregulated nutrient-sensing, mitochondrial dysfunction, and cellular senescence. Antagonistic hallmarks can be positive or negative depending on their intensity. At low levels they mediate beneficial effects, but at high levels are deleterious. Finally, stem cell exhaustion and altered intercellular communication fall into the category of integrative hallmarks that are the end result of the previous two groups and are ultimately responsible for the functional decline associated with aging [3].
The discovery of miRNAs in 1993 [4] has considerably changed the classical view of gene expression regulation, revealing a new group of molecules that can contribute to the complex process of aging. As addressed in other chapters of this book, miRNAs participate in virtually all biological processes within the cell as well as in numerous pathological conditions [5, 6]. Accordingly, it is tempting to speculate that aging-related miRNAs that we have previously termed geromiRs [7] are able to widely repress target genes driving the abovementioned hallmarks. In this chapter, we will summarize the significant changes in miRNA expression during aging in invertebrate and mammalian model organisms. In addition, we will present the regulatory roles of miRNAs in relation to the cellular and molecular hallmarks of aging and will discuss their experimental manipulation in order to improve healthspan and life-span (Fig. 11.1).

Micro-managing the hallmarks of aging. Schematic representation of miRNAs that regulate the nine hallmarks: genomic instability, telomere attrition, epigenetic regulation, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication
MicroRNA Expression Profiles in Aging
Several miRNAs appear to play a key role in aging. First, numerous works have reported significant age-related changes in miRNA expression during animal life-span. Additionally, a growing number of miRNAs have been demonstrated to influence most of the well-known longevity and senescence pathways. In this section, we will address the effect of aging on miRNA profiles in humans and animal models.
The first landmark study that identified a geromiR was carried out by Boehm and Slack, who found that lin-4 miRNA loss-of-function Caenorhabditis elegans mutants had a shortened longevity compared to wild-type animals, while overexpression of this miRNA extended organism life-span [8]. To date, we know that a large number of C. elegans miRNAs display changes in their expression during adulthood. Thus, the adult-specific disruption of argonaute-like gene-1 (alg-1) , which is necessary for miRNA maturation and function, results in abnormal longevity, suggesting that miRNAs are essential for normal aging [9]. Further studies characterized the abundance of miRNAs expressed in individual worms at different ages and noticed that expression variability increased with age. One of these miRNAs, miR-71, is required for normal longevity. Accordingly, transgenic worms over-expressing mir-71 were found to be long-lived [10].
Similar to works on C. elegans, studies on Drosophila melanogaster have also provided evidence for the contribution of miRNAs to the aging process. For example, flies harboring a hypomorphic mutation in loquacious (loqs) , a key component of the miRNA machinery, develop normally but show late-onset brain degeneration and a reduced life-span, thus implicating miRNA functions in age-associated pathologies [11]. A detailed analysis of brain miRNAs in this organism has also revealed that the loqs mutation causes an accelerated aging phenotype that is characterized by progressive neurodegeneration, increased stress sensitivity, and locomotion alterations. A global analysis of miRNA expression levels singled out miR-34 as the most likely mediator of this phenotype [11]. Notably, this work went on to identify translational repression of E74A as the key event responsible for miR-34 effects on aging. E74A is a component of steroid hormone signaling pathways, a molecular network that had been previously associated with aging modulation [12]. In an elegant example of antagonistic pleiotropy [11], the authors have proposed that although E74A is required during juvenile development, silencing of E74A by miR-34 in adulthood is critical to avoid the harmful effects of this protein, which exhibits sharply opposing functions on animal fitness at different life stages. In addition, a different study identified a novel miRNA, named miR-282, and provided evidence of its involvement on viability, longevity, and egg production. A preliminary expression analysis of computationally predicted targets of miR-282 suggested that its effects on aging may be mediated by the modulation of a nervous system-specific adenylate cyclase (rutabaga) during metamorphosis [13].
In contrast to invertebrates, current knowledge about the role of miRNAs in mammalian aging is still very limited due to the increased complexity and longer life-span of these organisms. To date, no study has been able to demonstrate the ability of a single miRNA to modulate the rate of aging in rodents by loss- or gain-of-function modifications. However, cumulative observations during the last decade of miRNA research strongly indicate that these molecules contribute to mammalian aging. Profiling studies evaluated the miRNA expression levels of young, adult, and old rat brains, and reported 547 known and 171 candidate novel miRNAs that were differentially expressed among these groups [14]. Aged brains exhibit a predominant upregulation of approximately 70 miRNAs and a downregulation of their respective target genes [15]. Apart from the brain, muscle also degenerates with aging resulting in loss of muscle mass (sarcopenia) over time. miRNA expression profiling from mouse muscle at two different ages shows 34 miRNAs differentially expressed with age, including miR-206 and miR-434 [16]. Perhaps the strongest evidence supporting the putative role of miRNAs in mammalian aging is the activity of specific miRNAs in both short- and long-life animal models. Thus, a study in Ames dwarf mice, which live 70% longer than wild-type mice due to deficiencies in pituitary hormones, has suggested a critical role for miR-27a in their characteristic life-span extension [17]. This miRNA is significantly increased in the liver of Ames mice, whereas its target genes, ornithine decarboxylase and spermidine synthase, are downregulated. Interestingly, both are important enzymes of the aging-associated polyamine biosynthetic pathway. Conversely, the upregulation of miR-1 and the miR-29 family have also been linked to the progeroid phenotype of a mouse model of Hutchinson-Gilford progeria syndrome by controlling multiple overlapping aging pathways [18, 19].
Several transcriptional studies in humans and other primates have also supported this notion of miRNA modulation of the aging process. Thus, the analysis of mRNA, miRNA, and protein expression in human and macaque brain evidenced regulatory relationships between miRNAs and mRNAs during aging of both species [20]. Further studies of miRNA expression in human serum from young and old individuals have found that the expression of miR-151a-5p, miR-181a-5p, and miR-1248 is significantly decreased in older individuals. Consistently, these miRNAs also show decayed levels in the serum of elderly rhesus monkeys [21]. Recently, one work has reported the first comparison of miRNAs expression profiles of cells from centenarians, octogenarians, and young individuals. Surprisingly, centenarians showed a narrow upregulation of miRNA levels compared to young individuals, but wide upregulated miRNA levels compared to octogenarians [22]. In addition, 15 platelet miRNAs were differentially regulated by age in a study performed in 154 healthy subjects, while their respective target mRNAs were inversely expressed [23].
Micro-managing the Hallmarks of Aging
Over the last years, advances in genetics and molecular biology have led to the identification of a subset of genes whose deregulation affects the aging process. In fact, the study of these genes has been instrumental in defining the hallmarks of aging [3]. In this section, we will discuss the functional relevance of miRNA-mediated regulation of these evolutionary conserved molecular and cellular processes.
Genomic Instability
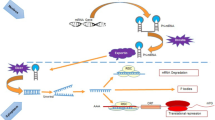
Even under the most controlled environmental conditions a mortal organism ages, illustrating how exogenous and endogenous sources of damage strongly influence the aging process through the generation of DNA lesions. Cells display a broad repertoire of macromolecule turnover and repair systems that are matched with the variety and frequency of DNA damage. These mechanisms, responsible for genome integrity, include molecular circuits that detect damage and activate pathways aimed at repairing the damage and/or preventing abnormal cellular behaviors, in a process termed DNA damage response (DDR) [24]. Recent studies have unveiled different miRNAs that contribute to the modulation of DDR to try to maintain genome integrity (Fig. 11.2).

Functional relevance of miRNAs in genomic instability. miRNAs regulate the DNA damage response that is triggered by genomic instability in aging
The miR-34 family members were the first group of miRNAs described to be transcriptionally regulated by p53 in response to DNA damage [25]. Thus, miR-34c expression is induced by p53 and inhibits c-Myc following DNA damage, preventing aberrant DNA synthesis and activating the S-phase checkpoint [26]. Additionally, miR-34 inhibits HDM4, a negative regulator of p53, and establishes a positive feedback loop [27]. In contrast to this protective role, miR-34a can act as a promoter of DNA damage and mitotic catastrophe, because it inhibits 53BP1 and counteracts its recruitment to DNA double-strand breaks, impairing DNA reparation [28]. In Zmpste24-deficient mice, a model of Hutchinson-Gilford progeria syndrome [29], altered chromatin architecture mediates the transcription of components of the miR-29 family in a p53-dependent manner [19]. Similar to the p53-dependent miR-34 feedback loop, miR-29 targets and represses PPM1D, a phosphatase that fine-tunes the DDR through inhibition of the activity and stability of p53. Similarly, Werner syndrome (WS), a premature aging disorder caused by mutations in a RecQ-like DNA helicase, can be phenocopied in both mice and worms by loss of function of the corresponding orthologous genes: WRN and wrn-1, respectively. This study also revealed that worms with mutations in wrn-1 show a reduced expression of miR-124, and that, surprisingly, the loss of miR-124 phenocopies the wrn-1 mutation [30].
Accumulating evidence also suggests control of miRNA biogenesis as an important regulatory element of the DDR pathway. For example, knockdown of DICER, a nuclease complex that contributes to miRNA maturation, reduced the ATM-dependent DDR through downregulation of miRNAs at damaged sites [31]. By contrast, DICER knockdown increases the resistance to camptothecin-induced DNA damage by reduction of let-7, which increased p21/p27 levels, again impairing DNA reparation [32]. Also in response to DNA damage, p53 interacts with the Drosha processing complex and coordinates the maturation of several miRNAs with growth-suppressive function, including miR-16-1, miR-143, and miR-145 [33]. Finally, DICER levels are also fine-tuned in response to DNA damage. TAp63, a member of the p53 family, directly binds to the Dicer promoter and activates its transcription in mice [34].
Telomere Attrition
Telomeres are repetitive DNA sequences located at the end of chromosomes that protect them from degradation. Telomeres recruit a multiprotein complex named shelterin [35] to form a chromatin structure that prevents its recognition as double strand breaks and the access of DNA repair proteins [36]. Only telomerase, a special DNA polymerase, can replicate telomeres. Adult cells do not express telomerase, which results in a progressive shortening of these protective fragments throughout aging until cells lose their replicative capacity.
miRNAs regulate diverse components of the shelterin or telomerase protein complexes. For example, miR-155 targets a conserved sequence motif in the 3′ UTR of the shelterin component TRF1, thereby mediating its translational repression and triggering telomere fragility and chromosome alterations [37]. As mentioned above, other miRNAs are able to regulate telomerase, for example, miR-498 regulates the catalytic subunit of telomerase by targeting the 3′ UTR of the encoding mRNA [38]. Conversely, shortening of telomeres may change the miRNA expression profile of different cells. Thus, miRNA expression profiles in cells with intact or with shortened telomeres revealed that 47 miRNAs were differentially expressed between these cell types. Some of these miRNAs are implicated in growth arrest or act as oncogene repressors [39]. In addition, chromosome instability driven by telomere shortening dramatically alters the pattern of miRNA expression in cells [40]. In summary, genomic instability and telomere attrition represent two molecular hallmarks that influence cancer and aging biology. This genetic damage is counteracted by the DDR systems, which in turn are heavily regulated by miRNAs. Therefore, modulating the activity of these miRNAs could either accelerate or decelerate tissue aging and age-related carcinogenesis [41].
Epigenetic Alterations
Sirtuins are important NAD-dependent protein deacetylases and ADP- ribosyltransferases implicated in histone modification, an essential epigenetic mechanism. The sirtuin pathway, whose upregulation extends longevity in yeast, worms, and flies [2], is also influenced by miRNAs. Among the different sirtuins, SIRT1 is widely recognized as a crucial regulator of metabolism, stress responses, replicative senescence, and inflammation [42]. In fact, SIRT1 is an important mediator of the beneficial metabolic effects of caloric restriction (CR) and the target effector of the antiaging molecule resveratrol. Among the miRNAs that might regulate SIRT1 during aging, the aforementioned age-related miR-34 is one of the best examples, as assessed by its ability to directly target SIRT1 mRNA in several in vitro and in vivo experiments [43]. Likewise, miR-217 upregulation in human endothelial cells during aging reduces SIRT1 activity and promotes senescence [44]. There is also evidence that several miRNAs, including miR-181a/b, miR-9, miR-204, miR-135a, and miR-199b, downregulate SIRT1 during differentiation of mouse embryonic stem cells into different tissues [45]. Alternatively, other miRNAs modulate SIRT1 activity indirectly. For instance, miR-519 contributes to human fibroblasts senescence by decreasing the protein levels of SIRT1 through targeting the RNA binding protein HuR [46].
Apart from histone deacetylation, miRNAs are able to control other epigenetic mechanisms, like methylation of DNA. Thus, it has been shown that Dicer1 deficiency in mice leads to decreased DNA methylation, along with increased telomere recombination and telomere elongation [47]. This methylation defect is a consequence of the reduced expression of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Further analyses have identified miR-290 as an agent in this decrease of methyltransferase levels, through repression of the retinoblastoma-like 2 protein (Rbl2). Moreover, miRNAs can also be methylated, as illustrated by the finding in Drosophila of an increase in the 2′-O-methylation at the 3′ end of miRNAs with age. These epigenetic modifications guide the preferential loading of miRNAs into Ago2, and not in Ago1. Loss of methylation leads to accelerated neurodegeneration and shorter life-span, suggesting the role of methylation of miRNAs in age-associated events [48].
Loss of Proteostasis
Protein folding and degradation of misfolded proteins are key processes that are closely related to aging. In addition, their experimental manipulation can precipitate or ameliorate this unavoidable process. Therefore, protein homeostasis or proteostasis is a bona fide aging hallmark [3, 49]. The main mechanisms implicated in proteostasis (autophagy, proteasomal degradation, and chaperone-mediating protein folding) are subjected to miRNA regulation.
In mammals, the regulation of the proteasome is especially important in the nervous system, as illustrated by the severe neurodegenerative diseases related to the accumulation of protein aggregates during aging [50]. In patients suffering from the spinocerebellar ataxia type 1 neurodegenerative disorder (SCA1), a group of upregulated miRNAs defines a pathological expression pattern. The targets of these miRNAs are enriched in members of the ubiquitin-proteasome system, suggesting that this system is deregulated in SCA1 [51]. More recently, another work has identified DNA damage as a mechanism that modifies the repertoire of proteasome-associated miRNAs, suggesting that in stress conditions these regulatory components modify proteasome function [52].
Autophagy is an evolutionarily conserved mechanism that allows the cell to digest its own components [53]. The activity of different factors belonging to this pathway can be fine-tuned by miRNAs after transcription. Thus, the tumor suppressor miR-101 inhibits autophagy by targeting three different genes: STMN1, RAB5A, and ATG4D [54]. miR-376b modulates human autophagy by regulating intracellular levels of ATG4C and BECN1 [55], while miR-34a targets ATG4B [56]. Alvarez-Erviti et al. have demonstrated that at least eight different miRNAs can regulate chaperone-mediated autophagy, which has a key role in the pathogenesis of a wide range of diseases, especially in the nervous system [57]. These miRNAs decrease chaperone-mediated autophagy through downregulation of hsc70 and LAMP-2A protein levels in the brain [57]. Finally, miR-216a impairs the autophagic function in aging endothelial cells through inhibition of two autophagy-related genes: Beclin1 and ATG5 [58]. Likewise, chaperones, a group of proteins implicated in the correct protein folding, are also under miRNA regulation. Thus, miR-17-5p targets multiple endoplasmic reticulum stress-related chaperones during chronic oxidative stress [59].
In summary, these first four hallmarks of aging, genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis, are included in the category of primary hallmarks because they represent the main causes of cellular damage that underlie the aging process. As described above, miRNAs regulate all these four processes and the molecular mechanisms that repair or counteract this damage, including those mediated by telomerase, chaperones, and epigenetic enzymes [3].
Deregulated Nutrient-Sensing
There is a close relationship between nutrient-sensing and aging. Among nutrient-sensing pathways, IGF-1 signaling stands out as a highly conserved regulatory module that influences longevity and also coordinates growth, development, and metabolism [2]. Like other aging hallmarks, nutrient-sensing pathways are also susceptible to miRNA regulation [60]. For example, the anomalous upregulation of miR-1, which targets the Igf-1 mRNA, is associated with the systemic deregulation of the somatotroph axis in premature aging mice [18]. Conversely, miR-470, miR-669b, and miR-681 are significantly upregulated in brain of long-lived Ames dwarf mice and their expression inversely correlates with the expression of several genes of the IGF-1/insulin pathway. Functional studies have demonstrated that these miRNAs target the IGF-1 receptor and contribute to reduce the levels of phosphorylated AKT and FOXO3a, two downstream targets of this signaling pathway, in the brain of mutant mice [61]. In addition, human miR-145 also represses the expression of IGF-1 receptor and its ligand, IRS-1 [62], while miR-206 and miR-320 target this somatotroph axis in rats [63, 64]. Similarly, miR-182 and miR-223 downregulation enhances IGF-1 signaling and mediates the estrogen impact on skeletal muscle. It has been proposed that IGF-1R and FOXO3a are the main targets whose overexpression triggers this positive effect [65]. In addition to regulating IGF-1, miR-17, miR-19b, miR-20a, and miR-106a target PTEN and inhibit the AKT-mTOR pathway [66], emphasizing the diversity of miRNAs which can contribute to modulate nutrient-sensing mechanisms implicated in the control of aging and longevity.
Mitochondrial Dysfunction
Mitochondrial dysfunction is another hallmark of aging [3]. The decline of autophagic clearance during aging affects the equilibrium between mitochondrial fusion and fission, leading to a build-up of dysfunctional mitochondria, oxidative stress, inflammation, and apoptosis. The miRNAs that exert their roles in the mitochondria are called mitomiRs and could act as vectors that sense and respond dynamically to the changing microenvironment in this organelle. These regulatory elements modulate nuclear and mitochondrial encoded targets [67]. In a recent study, let-7b, miR146a, miR-133b, miR-106a, miR-19b, miR-20a, miR-34a, miR-181a, and miR-221 have been identified as mitomiRs. Further analysis has revealed that targets of these miRNAs include genes that play important roles in processes like energy metabolism, mitochondrial transport, and apoptosis, whose deregulation is linked to aging [68]. Mitochondrial dynamics also plays a key role in some age-related pathologies, such as Parkinson’s disease [69]. In addition, miR-34b and miR-34c, whose expression is reduced in brain from Parkinson’s disease patients, alter the mitochondrial function in neuronal cells through the inhibition of DJ1 and parkin, two proteins associated with familial forms of the pathology [69]. An overview of miRs and mitochondrial function is available in Chap. 3.
Cellular Senescence
A growing number of miRNAs are arising as important modulators of senescence. This irreversible state of cellular growth arrest is an important effector of the cellular response against DNA damage that prevents the malignant proliferation of cells harboring oncogenic DNA mutations. However, as in the case of DDR, some aspects of cellular senescence have led to the consideration that it has a dual role through life-span, protecting from cancer development but promoting inflammation and tissue exhaustion, which prompts age-related alterations [70]. Numerous works have revealed functional roles for miRNAs in senescence through a variety of mechanisms. For example, miR-21 was reported as the first oncomiR that is able to produce a hyper oncogenic signal sufficient to limit cell proliferation. Accordingly, miR-21 is upregulated in senescent cells and its overexpression leads to cell-cycle arrest [71]. Another well-known oncogene, H RAS (V-12), induces senescence in primary fibroblasts through the production of reactive oxygen species (ROS). Yang et al. first identified protein tyrosine phosphatase 1B (PTP1B) as a major target of RAS-induced inhibition by ROS [72]. In turn, phospho-Tyr 393 of argonaute 2 is a direct substrate of PTP1B and its phosphorylation impairs the loading of miRNAs and the formation of the RNA-induced silencing complex (RISC) [72]. Furthermore, a recent work has identified 22 senescence-associated miRNAs in human mammary epithelial cells. In this subset, miR-26b, miR-181a, miR-210, and miR-424 repress Polycomb group proteins CBX7, embryonic ectoderm development (EED), enhancer of zeste homologue 2 (EZH2) and suppressor of zeste 12 homologue (Suz12), and activate p16, a key regulator of cellular senescence [73]. Moreover, macrophage polarization plays a key role in developing age-associated diseases such as macular degeneration. miR-33 impairs the ability of macrophages to efflux cholesterol, and this intracellular lipid polarizes older macrophages to an abnormal, alternatively activated phenotype that promotes pathologic vascular proliferation. miR-33 exerts this pathological role by downregulating the ATP binding cassette transporter ABCA1 and, consequently, mice deficient for Abca1 exhibit accelerated aging [74]. Another geromiR, let-7, contributes to prevent senescence through the inhibition of the retinoblastoma/E2F repressor complex, allowing the expression of proliferation-promoting genes [75]. The biogenesis pathway of miRNAs plays a crucial role in the regulation of cellular senescence. Thus, in a recent work, the synthesis of canonical miRNAs was disrupted by knockdown of the microprocessor complex subunit DGCR8 [76]. In this experiment, DGCR8 inactivation results in a dramatic antiproliferative response, with the acquisition of a senescent phenotype [76].
In summary, antagonistic hallmarks (deregulated nutrient-sensing, mitochondrial dysfunction, and cellular senescence) establish the biological response to primary hallmarks and, at low doses, mediate positive effects. However, when they become very strong they can be deleterious. miRNAs contribute to fine-tune the intensity of these responses and keep them under physiological limits [3].
Stem Cell Exhaustion
In addition to senescence, the decline of adult stem cell self-renewal and pluripotency is also considered a key determinant in the age-associated deterioration of tissue homeostasis and maintenance. Notably, several reports have described senescence or age-related changes in miRNAs of human or rhesus macaque mesenchymal stem cells [77, 78]. Furthermore, numerous studies have reported essential roles for miRNAs in processes such as renewal, pluripotency, quiescent state maintenance, proliferation, and differentiation of adult stem cells in several tissues and organisms. For example, the loss of self-renewal potential in old neural stem cells has been linked to age-dependent upregulation of let-7b, which in turn inhibits HMGA2 expression, a repressor of the INK4a/ARF locus [79]. Another illustrative example involving let-7 is the testis stem-cell niche. In Drosophila testis, aging results in a marked decrease in the self-renewal factor Unpaired (Upd), leading to a loss of germline stem cells. The RNA binding protein Imp protects Upd from degradation by let-7. In the absence of Imp, Upd mRNA becomes unprotected and susceptible to degradation [80]. Hematopoietic stem cell self-renewal in mice is also regulated by miR-33 repression of TP53 [81], and the maintenance of quiescent state in human muscle adult stem cells has been shown to be highly dependent on miRNA activity, being miR-489 one of the most prominent effectors [82]. Stem cell proliferation and neuronal differentiation in mice are also finely regulated by miRNAs of the miR-106b-25 cluster, which are in turn controlled by the aging-associated FoxO transcription factors [83]. Additionally, the expression of miR-486-5p and miR-598 in human adipose tissue-derived mesenchymal stem cells (hADSCs) progressively increases with aging and regulates the expression of SIRT1, inducing a premature senescence-like phenotype. In the case of miR-486-5p, this mechanism blocks adipogenic and osteogenic differentiation [84, 85]. Studies of human embryonic stem cells (hESCs) have described a nonfunctional p53-p21 axis at the G1/S checkpoint, which has recently been reported to be regulated by the miR-302 family [86]. A progressive increase of miR-335 in ex vivo cultures of hMSCs, as well as forced expression of miR-335, resulted in early senescence-like alterations including senescence-associated secretory phenotype (SASP). Also in hMSCs, miR-141-3p direct binding to the 3′ UTR of ZMPSTE24, which is involved in maturation of lamin A, leads to accumulation of prelamin A in the nuclear envelope [87].
Altered Intercellular Communication
Aging also involves changes at the level of intercellular communication, be it endocrine, neuroendocrine, or neuronal. miRNAs influence intercellular communication by being included in exosomes or directly circulating through body fluids [88, 89]. In C. elegans, life-span is controlled by signaling between the germline and the soma. Different approaches have confirmed that miRNAs influence this process by targeting well-known aging signaling pathways. For example, germ cell removal extends life-span by triggering the activation of the DAF-16/FOXO transcription factor in the intestine, and it was reported that miR-71 and let-7 function to mediate this increase in longevity [90, 91]. In recent years, several lines of research have converged on the concept of inflammaging: an age-related systemic chronic inflammation that represents a prominent alteration in intercellular communication. miRNAs can fine-tune this activation of immune cells by regulating the SASP and the Toll-like receptors, two putative mechanisms underlying inflammaging [92, 93]. For example, miR-21 and miR-29a are able to bind to TLR8, activating TLR-mediated NF-kB signaling that leads to increased secretion of the proinflammatory cytokines interleukin-6 and TNF-a [94].
There are several possibilities for restoring defective intercellular communication underlying aging processes. These strategies include nutritional interventions such as caloric restriction, a condition that can promote longevity and protect against age-associated disease across species. An elegant example in this regard has been reported in C. elegans through the identification of miRNA-80 as a major regulator during CR. Thus, miR-80 deletion confers system-wide healthy aging in a mechanism that involves the factors cbp-1, daf-16/FOXO, and hsf-1 [95].
In summary, stem cells exhaustion and altered intercellular communication fall into the category of integrative hallmarks, as they are the main mediators of the aging phenotype and together influence the aging rate [3]. miRNAs regulate self-renewal, inflammation, and other important conditions concerning this group of integrative hallmarks of aging.
Conclusions and Perspectives
The discovery of miRNAs has opened a new chapter in aging research that could help to achieve a deeper knowledge of the molecular network underlying this complex process. Although we are far from understanding the precise involvement of these molecules in age-related alterations, solid evidence from the literature, discussed in this chapter, supports an important role for the growing group of geromiRs in aging modulation. miRNA-mediated regulation impacts all nine molecular and cellular hallmarks of aging, modifying the aging rate and age-related diseases both in invertebrates and mammals [96]. Developing mammalian in vivo models for ablation and gain of specific miRNAs will undoubtedly help us to answer many important questions pertaining to how individual geromiRs regulate tissue aging and organismal life-span. Nevertheless, diverse hallmarks are interconnected, which will necessitate broader, integrative strategies. For example, cells that enter into senescence acquire a SASP that leads to production of proinflammatory cytokines and the triggering of inflammaging, a status with altered intercellular communication associated with aging [92]. In addition, a single miRNA may regulate several processes affecting more than one hallmark. On top of this, the regulation of miRNA expression, epigenetically or by transcription factors during aging, will also be a topic of interest for future investigations. The recent advances in strategies to effectively block or delivery specific miRNA in vivo may also facilitate new therapeutic opportunities to delay or ameliorate age-related alterations as well as premature aging syndromes [97]. Alternatively, a promising new area for miRNAs is in diagnostics, where miRNAs have great potential as molecular biomarkers of longevity with ability to predict individual longevity better than chronological age [98].
Abbreviations
- DDR:
-
DNA damage response
- IGF-1:
-
Insulin-like growth factor 1
References
Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–71.
Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–12.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54.
Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–88.
Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–39.
Ugalde AP, Espanol Y, Lopez-Otin C. Micromanaging aging with miRNAs: new messages from the nuclear envelope. Nucleus. 2011;2:549–55.
Boehm M, Slack F. A developmental timing microRNA and its target regulate life span in C. elegans. Science. 2005;310:1954–7.
Kato M, Chen X, Inukai S, Zhao H, Slack FJ. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA. 2011;17:1804–20.
Lucanic M, Graham J, Scott G, Bhaumik D, Benz CC, Hubbard A, Lithgow GJ, Melov S. Age-related micro-RNA abundance in individual C. elegans. Aging. 2013;5:394–411.
Liu N, Landreh M, Cao K, Abe M, Hendriks GJ, Kennerdell JR, Zhu Y, Wang LS, Bonini NM. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature. 2012;482:519–23.
Simon AF, Shih C, Mack A, Benzer S. Steroid control of longevity in Drosophila melanogaster. Science. 2003;299:1407–10.
Vilmos P, Bujna A, Szuperak M, Havelda Z, Varallyay E, Szabad J, Kucerova L, Somogyi K, Kristo I, Lukacsovich T, Jankovics F, Henn L, Erdelyi M. Viability, longevity, and egg production of Drosophila melanogaster are regulated by the miR-282 microRNA. Genetics. 2013;195:469–80.
Yin L, Sun Y, Wu J, Yan S, Deng Z, Wang J, Liao S, Yin D, Li G. Discovering novel microRNAs and age-related nonlinear changes in rat brains using deep sequencing. Neurobiol Aging. 2015;36:1037–44.
Li N, Bates DJ, An J, Terry DA, Wang E. Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging. 2011;32:944–55.
Kim JY, Park YK, Lee KP, Lee SM, Kang TW, Kim HJ, Dho SH, Kim SY, Kwon KS. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging. 2014;6:524–44.
Bates DJ, Li N, Liang R, Sarojini H, An J, Masternak MM, Bartke A, Wang E. MicroRNA regulation in Ames dwarf mouse liver may contribute to delayed aging. Aging Cell. 2010;9:1–18.
Marino G, Ugalde AP, Fernandez AF, Osorio FG, Fueyo A, Freije JM, Lopez-Otin C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc Natl Acad Sci U S A. 2010;107:16268–73.
Ugalde AP, Ramsay AJ, de la Rosa J, Varela I, Marino G, Cadinanos J, Lu J, Freije JM, Lopez-Otin C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011;30:2219–32.
Somel M, Guo S, Fu N, Yan Z, Hu HY, Xu Y, Yuan Y, Ning Z, Hu Y, Menzel C, Hu H, Lachmann M, Zeng R, Chen W, Khaitovich P. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010;20:1207–18.
Noren Hooten N, Fitzpatrick M, Wood 3rd WH, De S, Ejiogu N, Zhang Y, Mattison JA, Becker KG, Zonderman AB, Evans MK. Age-related changes in microRNA levels in serum. Aging. 2013;5:725–40.
Serna E, Gambini J, Borras C, Abdelaziz KM, Belenguer A, Sanchis P, Avellana JA, Rodriguez-Manas L, Vina J. Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci Rep. 2012;2:961.
Simon LM, Edelstein LC, Nagalla S, Woodley AB, Chen ES, Kong X, Ma L, Fortina P, Kunapuli S, Holinstat M, McKenzie SE, Dong JF, Shaw CA, Bray PF. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood. 2014;123:e37–45.
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94.
Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014;6:214–30.
Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, Elia A, Kress TR, Dickens M, Clemens MJ, Heery DM, Gaestel M, Eilers M, Willis AE, Bushell M. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci U S A. 2010;107:5375–80.
Okada N, Lin CP, Ribeiro MC, Biton A, Lai G, He X, Bu P, Vogel H, Jablons DM, Keller AC, Wilkinson JE, He B, Speed TP, He L. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014;28:438–50.
Kofman AV, Kim J, Park SY, Dupart E, Letson C, Bao Y, Ding K, Chen Q, Schiff D, Larner J, Abounader R. microRNA-34a promotes DNA damage and mitotic catastrophe. Cell Cycle. 2013;12:3500–11.
Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–8.
Dallaire A, Garand C, Paquel ER, Mitchell SJ, de Cabo R, Simard MJ, Lebel M. Down regulation of miR-124 in both Werner syndrome DNA helicase mutant mice and mutant Caenorhabditis elegans wrn-1 reveals the importance of this microRNA in accelerated aging. Aging. 2012;4:636–47.
Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–5.
Liu B, Liu M, Wang J, Zhang X, Wang X, Wang P, Wang H, Li W, Wang Y. DICER-dependent biogenesis of let-7 miRNAs affects human cell response to DNA damage via targeting p21/p27. Nucleic Acids Res. 2015;43:1626–36.
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33.
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, Wistuba I, Flores ER. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–90.
Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34.
Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, Herbig U, Longhese MP, d’Adda di Fagagna F. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012;14:355–65.
Dinami R, Ercolani C, Petti E, Piazza S, Ciani Y, Sestito R, Sacconi A, Biagioni F, le Sage C, Agami R, Benetti R, Mottolese M, Schneider C, Blandino G, Schoeftner S. miR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res. 2014;74:4145–56.
Kasiappan R, Shen Z, Tse AK, Jinwal U, Tang J, Lungchukiet P, Sun Y, Kruk P, Nicosia SV, Zhang X, Bai W. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J Biol Chem. 2012;287:41297–309.
Uziel O, Yosef N, Sharan R, Ruppin E, Kupiec M, Kushnir M, Beery E, Cohen-Diker T, Nordenberg J, Lahav M. The effects of telomere shortening on cancer cells: a network model of proteomic and microRNA analysis. Genomics. 2015;105:5–16.
Castro-Vega LJ, Jouravleva K, Liu WY, Martinez C, Gestraud P, Hupe P, Servant N, Albaud B, Gentien D, Gad S, Richard S, Bacchetti S, Londono-Vallejo A. Telomere crisis in kidney epithelial cells promotes the acquisition of a microRNA signature retrieved in aggressive renal cell carcinomas. Carcinogenesis. 2013;34:1173–80.
Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579–90.
Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95.
Lee J, Kemper JK. Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging. 2010;2:527–34.
Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa-Nicotera M, Ippoliti A, Novelli G, Melino G, Lauro R, Federici M. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. 2009;120:1524–32.
Saunders LR, Sharma AD, Tawney J, Nakagawa M, Okita K, Yamanaka S, Willenbring H, Verdin E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging. 2010;2:415–31.
Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging. 2010;2:333–43.
Benetti R, Gonzalo S, Jaco I, Munoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, Li E, Serrano M, Millar S, Hannon G, Blasco MA. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–79.
Abe M, Naqvi A, Hendriks GJ, Feltzin V, Zhu Y, Grigoriev A, Bonini NM. Impact of age-associated increase in 2′-O-methylation of miRNAs on aging and neurodegeneration in Drosophila. Genes Dev. 2014;28:44–57.
Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1:S33–8.
Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–48.
Persengiev S, Kondova I, Bontrop RE. Functional annotation of small noncoding RNAs target genes provides evidence for a deregulated ubiquitin-proteasome pathway in spinocerebellar ataxia type 1. J Nucleic Acids. 2012;2012:672536.
Tsimokha AS, Kulichkova VA, Karpova EV, Zaykova JJ, Aksenov ND, Vasilishina AA, Kropotov AV, Antonov A, Barlev NA. DNA damage modulates interactions between microRNAs and the 26S proteasome. Oncotarget. 2014;5:3555–67.
Fernandez AF, Lopez-Otin C. The functional and pathologic relevance of autophagy proteases. J Clin Invest. 2015;125:33–41.
Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, Jaattela M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41.
Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–76.
Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM, Jiang X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622–34.
Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013;4, e545.
Menghini R, Casagrande V, Marino A, Marchetti V, Cardellini M, Stoehr R, Rizza S, Martelli E, Greco S, Mauriello A, Ippoliti A, Martelli F, Lauro R, Federici M. MiR-216a: a link between endothelial dysfunction and autophagy. Cell Death Dis. 2014;5, e1029.
Wang Q, Hu W, Lei M, Wang Y, Yan B, Liu J, Zhang R, Jin Y. MiR-17-5p impairs trafficking of H-ERG K+ channel protein by targeting multiple ER stress-related chaperones during chronic oxidative stress. PLoS One. 2013;8, e84984.
Jung HJ, Suh Y. Regulation of IGF-1 signaling by microRNAs. Front Genet. 2014;5:472.
Liang R, Khanna A, Muthusamy S, Li N, Sarojini H, Kopchick JJ, Masternak MM, Bartke A, Wang E. Post-transcriptional regulation of IGF1R by key microRNAs in long-lived mutant mice. Aging Cell. 2011;10:1080–8.
La Rocca G, Badin M, Shi B, Xu SQ, Deangelis T, Sepp-Lorenzinoi L, Baserga R. Mechanism of growth inhibition by MicroRNA 145: the role of the IGF-I receptor signaling pathway. J Cell Physiol. 2009;220:485–91.
Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, Liu XY, Zhang YY, Li Y, Lin SG, Yu XY. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem Biophys Res Commun. 2009;381:597–601.
Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2009;36:181–8.
Olivieri F, Ahtiainen M, Lazzarini R, Pollanen E, Capri M, Lorenzi M, Fulgenzi G, Albertini MC, Salvioli S, Alen MJ, Kujala UM, Borghetti G, Babini L, Kaprio J, Sipila S, Franceschi C, Kovanen V, Procopio AD. Hormone replacement therapy enhances IGF-1 signaling in skeletal muscle by diminishing miR-182 and miR-223 expressions: a study on postmenopausal monozygotic twin pairs. Aging Cell. 2014;13:850–61.
Patel M, Gomez NC, McFadden AW, Moats-Staats BM, Wu S, Rojas A, Sapp T, Simon JM, Smith SV, Kaiser-Rogers K, Davis IJ. PTEN deficiency mediates a reciprocal response to IGFI and mTOR inhibition. Mol Cancer Res. 2014;12:1610–20.
Bandiera S, Mategot R, Girard M, Demongeot J, Henrion-Caude A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic Biol Med. 2013;64:12–9.
Rippo MR, Olivieri F, Monsurro V, Prattichizzo F, Albertini MC, Procopio AD. MitomiRs in human inflamm-aging: a hypothesis involving miR-181a, miR-34a and miR-146a. Exp Gerontol. 2014;56:154–63.
Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I, Estivill X, Marti E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet. 2011;20:3067–78.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56.
Dellago H, Preschitz-Kammerhofer B, Terlecki-Zaniewicz L, Schreiner C, Fortschegger K, Chang MW, Hackl M, Monteforte R, Kuhnel H, Schosserer M, Gruber F, Tschachler E, Scheideler M, Grillari-Voglauer R, Grillari J, Wieser M. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell. 2013;12:446–58.
Yang M, Haase AD, Huang FK, Coulis G, Rivera KD, Dickinson BC, Chang CJ, Pappin DJ, Neubert TA, Hannon GJ, Boivin B, Tonks NK. Dephosphorylation of tyrosine 393 in argonaute 2 by protein tyrosine phosphatase 1B regulates gene silencing in oncogenic RAS-induced senescence. Mol Cell. 2014;55:782–90.
Overhoff MG, Garbe JC, Koh J, Stampfer MR, Beach DH, Bishop CL. Cellular senescence mediated by p16INK4A-coupled miRNA pathways. Nucleic Acids Res. 2014;42:1606–18.
Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, Chowers I, Edwards PA, Baldan A, Parks JS, Ory DS, Apte RS. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–61.
Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol. 2012;14:266–75.
Gomez-Cabello D, Adrados I, Gamarra D, Kobayashi H, Takatsu Y, Takatsu K, Gil J, Palmero I. DGCR8-mediated disruption of miRNA biogenesis induces cellular senescence in primary fibroblasts. Aging Cell. 2013;12:923–31.
Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, Saffrich R, Benes V, Blake J, Pfister S, Eckstein V, Ho AD. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS One. 2008;3, e2213.
Yu JM, Wu X, Gimble JM, Guan X, Freitas MA, Bunnell BA. Age-related changes in mesenchymal stem cells derived from rhesus macaque bone marrow. Aging Cell. 2011;10:66–79.
Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell. 2008;135:227–39.
Toledano H, D’Alterio C, Czech B, Levine E, Jones DL. The let-7-Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature. 2012;485:605–10.
Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9:3277–85.
Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482:524–8.
Brett JO, Renault VM, Rafalski VA, Webb AE, Brunet A. The microRNA cluster miR-106b~25 regulates adult neural stem/progenitor cell proliferation and neuronal differentiation. Aging. 2011;3:108–24.
Kim YJ, Hwang SH, Lee SY, Shin KK, Cho HH, Bae YC, Jung JS. miR-486-5p induces replicative senescence of human adipose tissue-derived mesenchymal stem cells and its expression is controlled by high glucose. Stem Cells Dev. 2012;21:1749–60.
Shin KK, Kim YJ, Hong CP, Yang JW, Bae YC, Jung JS. Retracted article: miR-598 induces replicative senescence in human adipose tissue-derived mesenchymal stem cells via silent information regulator 1. Mol Cell Biochem. 2013;372(1–2):285.
Dolezalova D, Mraz M, Barta T, Plevova K, Vinarsky V, Holubcova Z, Jaros J, Dvorak P, Pospisilova S, Hampl A. MicroRNAs regulate p21(Waf1/Cip1) protein expression and the DNA damage response in human embryonic stem cells. Stem Cells. 2012;30:1362–72.
Yu KR, Lee S, Jung JW, Hong IS, Kim HS, Seo Y, Shin TH, Kang KS. MicroRNA-141-3p plays a role in human mesenchymal stem cell aging by directly targeting ZMPSTE24. J Cell Sci. 2013;126:5422–31.
Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403.
Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208.
Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab. 2012;15:439–50.
Shen Y, Wollam J, Magner D, Karalay O, Antebi A. A steroid receptor-microRNA switch regulates life span in response to signals from the gonad. Science. 2012;338:1472–6.
Olivieri F, Rippo MR, Monsurro V, Salvioli S, Capri M, Procopio AD, Franceschi C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev. 2013;12:1056–68.
Olivieri F, Rippo MR, Prattichizzo F, Babini L, Graciotti L, Recchioni R, Procopio AD. Toll like receptor signaling in “inflammaging”: microRNA as new players. Immun Ageing. 2013;10:11.
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, Zanesi N, Crawford M, Ozer GH, Wernicke D, Alder H, Caligiuri MA, Nana-Sinkam P, Perrotti D, Croce CM. MicroRNAs bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109:E2110–6.
Vora M, Shah M, Ostafi S, Onken B, Xue J, Ni JZ, Gu S, Driscoll M. Deletion of microRNA-80 activates dietary restriction to extend C. elegans healthspan and lifespan. PLoS Genet. 2013;9, e1003737.
Dimmeler S, Nicotera P. MicroRNAs in age-related diseases. EMBO Mol Med. 2013;5:180–90.
Gordon LB, Rothman FG, Lopez-Otin C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156:400–7.
Pincus Z, Smith-Vikos T, Slack FJ. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet. 2011;7:e1002306.
Acknowledgments
We thank Dr. Víctor Quesada for critical reading of the manuscript. Our work is supported by grants from Ministerio de Economía y Competitividad and Instituto de Salud Carlos III (RTICC), Spain. The Instituto Universitario de Oncología is supported by Obra Social Cajastur-Asturias. C.L.-O. is an Investigator of the Botin Foundation supported by Banco Santander through its Santander Universities Global Division.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Caravia, X.M., López-Otín, C. (2015). Regulatory Roles of miRNAs in Aging. In: Santulli, G. (eds) microRNA: Basic Science. Advances in Experimental Medicine and Biology, vol 887. Springer, Cham. https://doi.org/10.1007/978-3-319-22380-3_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-22380-3_11
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22379-7
Online ISBN: 978-3-319-22380-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)