
Abstract
Chronic hepatitis B affects over 300 million people globally, and although there is an effective preventative vaccine, there is no cure. The recent discovery of the NTCP entry receptor means that infection models now permit investigation of drugs that target all stages of HBV life cycle. This is important as existing treatment strategies reduce the viral burden but rarely prevent expression of viral antigens associated with disease progression. It is likely this will only be achieved through the destruction or silencing of cccDNA in the hepatocyte nucleus. In this chapter we describe limitations and advantages of current and emerging treatment and cure strategies for chronic hepatitis B. We also discuss future drug targets with the hepatitis B life cycle that we believe are worthy of investigation. Together, these approaches should speed development of new therapeutic regimens that may lead to HBV cure in the foreseeable future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
There is currently no cure for chronic hepatitis B (CHB ), with current treatments limited to a small number of direct acting antivirals (Chap. 17), or interferon (Chap. 16), the latter being poorly efficacious against most HBV genotypes . There is an urgent need for new treatments that augment existing therapies, as well as development of cure strategies for the 300 million people living with CHB worldwide. In this Chapter we discuss the basis and limitations of current antiviral therapy, as well as recent advances leading to new approaches which may form the basis of future treatment and cure regimens.
The HBV Life Cycle
Genomic Replication Pathway
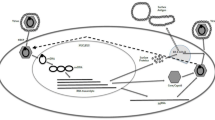
Hepatitis B virus (HBV) is a pararetrovirus with a partially double-stranded relaxed circular DNA genome which replicates via reverse transcription of an RNA intermediate (pregenomic RNA, pgRNA) (Fig. 14.1). Viral entry is mediated first via attachment of the hepatitis B surface antigen (HBsAg) to cell surface heparan sulfate (HS) proteoglycans (HSPGs) [1, 2], followed by viral entry through engagement of the preS1 domain of the large envelope protein to the sodium-taurocholate co-transporting polypeptide (NTCP) receptor [3]. Following this, the viral envelope is removed and viral nucleocapsids comprising the relaxed circular DNA (RC DNA) genome, the endogenous HBV DNA polymerase, and a host protein kinase, all enclosed by the hepatitis B core protein, are released into the cytoplasm. The capsids are transported to the hepatocyte nucleus [4], where the capsid disassembles and releases the viral genome [5]. The partially double-stranded RC DNA is “repaired” by the endogenous HBV DNA polymerase and host cell DNA polymerase II to be fully double-stranded forming a covalently closed circular (ccc) DNA structure [6]. This highly stable nuclear-localized cccDNA is the major transcriptional template for pgRNA and viral mRNA production, which complexes with host cell histones, the viral core protein (HBcAg) and other nuclear proteins and transcriptional factors to form viral minichromosomes [7].

Hepatitis B life cycle, showing nuclear and cytoplasmic stages of HBV infection and sites of action for current and future HBV treatments
Two forms of RNA molecule are transcribed from cccDNA, namely the greater than genome length 3.5 kb pg RNA which also serves as mRNA for the HBV core and polymerase proteins, as well as viral mRNAs encoding the HBV precore (pcRNA, 3.5 kb), envelope (2.4 and 2.1 kb), and HBx (0.7 kb) proteins.
Following the export of pgRNA from the nucleus and translation of the viral core and polymerase proteins, the pgRNA and the polymerase protein are packaged within newly forming viral nucleocapsids. The reverse transcriptase domain within the viral polymerase promotes synthesis of minus sense DNA, which is then copied to plus sense DNA, producing the relaxed circular DNA genome. Two fates await the nucleocapsid-localized RC DNA—it may either be recycled back to the nucleus to continue the cycle of cccDNA generation and formation, mRNA synthesis and RC DNA formation, or it may bud into the Golgi (microvesicular body) where it is enveloped by viral surface proteins, and secreted from the cell. The mature virion is then secreted into the blood, enabling the infection cycle to continue.
Generation of HBV Proteins
mRNAs transcribed from cccDNA encode the core protein (nucleocapsid) and polymerase (translated from the 3.5 kb pgRNA), the precore protein which is processed to form the secreted hepatitis B e antigen (HBeAg, translated from the pcRNA), envelope proteins and the HBx protein. The pcRNA is initially translated as an intracellular 25 kDa protein (p25), which is subsequently processed to a 22 kDa molecule (p22) and finally the 17 kDa HBeAg which is secreted from the cell [8]. The HBeAg in serum exists as dimeric and multimeric forms and delineation of both the HBeAg [9] and HBcAg [10] crystal structures, sheds light on structural differences between these molecules that may facilitate identification of future therapeutic targets for both molecules. Cysteine molecules at positions −7 and 61 are critical for HBeAg stability, whilst core dimers are formed via the core dimer interface of two monomeric core proteins via the cysteines at position 61 [10]. In addition to replication of unspliced RNA molecules, up to 15 different splice derived transcripts have been identified, some of which encode novel proteins, including the hepatitis B splice protein, and have been associated with disease progression [11–15], although they do not appear to be critical for HBV replication.
HBV Genotypes
HBV exists as ten different genotypes (A–J) and multiple sub-genotype with geographically distinct distributions which are becoming increasingly blurred through human migration [16]. CHB should not be thought of as a single disease, but rather as a “disease state” or spectrum with marked diversity in CHB natural history including differences in age of acquisition (neonate vs. adult), modes of transmission, disease progression, replication phenotype, response to therapy, and disease resolution across HBV genotypes [16]. For example, persons infected with genotype A respond better to treatment with IFN-α than do all other genotypes a nd patients infected with genotype B respond much better than genotype C. However interferon is virtually ineffective against patients infected with genotype D. Genotype C is generally associated with more severe liver disease and a higher propensity for liver cancer than most other genotypes, although there are exceptions with subgenotype A1 associated with rapid progression to cancer without prior cirrhosis in young African males [17, 18] via mechanisms that have not been fully identified. HBV sequences differ by at least 8 % across the complete HBV genome between genotypes, and by between 4 and 8 % between sub-genotypes, suggesting that new treatments targeting specific areas of the HBV genome (such as the HBeAg) may need to be tailored to individual genotypes or subtypes.
Molecular Targets: Current and Future HBV Treatments
Current Direct Acting Antiviral Therapies
Current direct acting antiviral therapies (DAAs ) inhibit the reverse transcription of pgRNA to DNA, i.e., the replication stage of HBV that takes place within the viral nucleocapsid (Fig. 14.1). They include the class of nucleos(t)ide analogs (NA) and act as competitive inhibitors of the HBV pol, inhibiting synthesis of viral DNA from the pgRNA template through chain termination. The clinical activity of these molecules is explained in detail in Chap. 16.
DAAs have no direct impact on the initial formation of cccDNA , although in reducing the amount of RC DNA they should indirectly impact cccDNA that is formed following the recycling of RC DNA to the nucleus. However, since this recycling happens early in the HBV infection cycle and DAA treatment routinely commences following the establishment of persistent infection long after initiation of HBV infection, the impact of DAAs on cccDNA levels is essentially minimal. This is important, as the continual presence of cccDNA in the nucleus, even in the absence of productive viral replication, is one of the main impediments to curing HBV infection. In turn, continual production of hepatitis B surface antigen (HBsAg) originating from cccDNA or integrated DNA is associated with disease progression and liver cancer, meaning that eradication of HBV-associated disease may only be achieved when strategies are developed that eliminate or suppress cccDNA.
To date five drugs belonging to the class of NA have been approved for treatment of CHB. These are lamivudine (LMV), adefovir dipivoxil (ADV), entecavir (ETV), telbivudine (LdT), and tenofovir (TDF). These drugs are divided into three groups, the l-nucleosides (LMV and LdT), acyclic phosphonates (ADV and TDF), and d-cyclopentane (ETV) groups. The effectiveness of many of these drugs is limited by the selection of resistant strains of HBV during treatment [19]. This is because the “error prone” nature of the HBV reverse-transcriptase results in a high mutation rate (1 in every 105 nucleotide substitutions for each cycle of replication [20]) leading to a large population of variant HBV sequences, some of which encode preexisting mutations in the HBV polymerase gene which confer resistance to one or more DAAs. These mutant strains of HBV are typically selected on therapy, giving rise to a dominant population of drug resistant viruses. The different DAAs have different propensities for selection of resistance, with the most widely used drug LMV driving selection of resistant HBV in up to 23 % of patients following 12 months of therapy, rising to 80 % by 5 years of treatment [21]. In contrast it is encouraging that there is no evidence to date for selection of HBV strains that encode resistance to TDF, even following 5 years of treatment [22, 23] (Table 14.1). Although rates of resistance are low for adefovir and entecavir monotherapy, in the setting of prior resistance to LMV, high rates of resistance to these drugs is observed because of cross-resistance (Table 14.1, adapted from Zoulim and Locarnini, 2009 [19]).
Resistance Profiles of Current DAAs
The molecular mechanisms of resistance to d rugs for treatment of CHB have been eloquently described previously [19, 30] but are briefly summarized herein.
To date, eight codons within the HBV polymerase are associated with primary drug resistance to NAs. These are codons 169, 180, 181, 184, 202, 204, 236, and 250 [19] (Fig. 14.2).

Primary resistance mutations to NA identified within the HBV polymerase. (Reproduced from Zoulim and Locarnini 2012 [31])
The reason for the strong barrier to selection of resistance to the acyclic phosphonate TDF is unclear. It remains to be determined whether continued use of this drug as monotherapy leads to selection of TDF resistant HBV. Nonetheless, the high degree of potency exhibited by TDF (and related compounds such as TAF—see below), and the high genetic barrier to selection of HBV resistance shows there is a role for these compounds in first line therapy against CHB. These compounds also have an important role when first line therapy with alternative NA treatments (such as LMV) has led t o the select ion of HBV encoding resistance mutations, or in the setting of failed immunomodulatory therapy.
New DAAs in the Pipeline: Tenofovir Alafenamide (TAF)
Although TDF is a very effective antiviral compound with no evidence to date of selecting antiviral resistance, there are reports of small numbers of HBV infected patients who do not respond to TDF therapy [130]. The reasons for this are unclear—it is likely that poor compliance is a contributor in these studies, but where compliance was not an issue it suggests reduced TDF efficacy might be related to quasispecies diversity or coinfection with multiple genotypes, such as genotypes A and G. Tenofovir alafenamide (TAF) is a TDF prodrug that provides efficient delivery of active drug to hepatocytes at reduced dosage, with improved plasma stability [32]. A recent study of 51 CHB subjects with HBeAg negative HBV infection showed that doses as low as 8 mg per day for 4 weeks resulted in similar levels of viral decline as the standard 300 mg daily dose of TDF [33]. Further clinical studies in larger cohorts are currently underway at a dosage of 25 mg per day.
New Compounds Undergoing Clinical Trial
DAAs are limited in their effectiveness in that they only inhibit active viral replication. They have no effect on viral entry, nor does it on the preexisting pool of viral cccDNA. These mRNAs express a continual source of HBV proteins such as the HBsAg and HBx, both associated with persistence and disease progression. There is an unmet need for new HBV treatments which complement existing antiviral therapies. In the following section new strategies currently under phase II clinical development targeting HBV entry, or expression of HBV mRNAs and small molecules targeted to the core protein will be discussed.
Myrcludex B
Although the discovery of the NTCP recept or for HBV entry has been relatively recent [3, 34, 35], it has been known for some time that synthetic peptides derived from the large envelope protein block HBV entry, as well as entry of the related hepatitis delta virus (HDV), which utilizes the HBV envelope protein for viral entry [36–39]. This discovery has led to the production of an HBV “entry inhibitor” Myrcludex B, which is a myristlyated PreS1 peptide currently under clinical trial. It has recently been shown that Myrcludex B not only prevents HBV spreading from infected human hepatocytes in vivo, but also hinders amplification of the cccDNA pool in initially infected hepatocytes [40]. This important finding suggests Myrcludex B could be a useful tool in the treatment of CHB, as well as more obvious applications such as the prevention of reinfection following liver transplantation.
RNA Interference
RNA interference (RNAi) is gaining increasing credence as a treatment strategy for chronic HBV. RNAi is a process by which small interfering RNA molecules of 21–25 nucleotides (short interfering or siRNAs) induce gene silencing at the posttranscriptional level, to effectively knock down gene expression [41]. The overlapping nature of the HBV genome means that multiple HBV RNAs can be targeted by a single siRNA molecule [42]. Cell culture and murine studies have shown that RNAi, delivered as a HBV plasmid, inhibits HBV replication in these models [43]. In transgenic mice, RNAi expression reduced HBsAg secretion in serum, as well as HBV mRNAs and genomic DNA in the liver, and also reduced the number of hepatocytes staining positive for core antigen (HBcAg) to undetectable levels [44].
Although effective at reducing HBV replication in cell culture and murine models, progress towards RNAi as an effective therapy has been limited by difficulties in delivering siRNA molecules to the liver. A recent major advance in the field has overcome the problem of liver-specificity, using cholesterol-conjugated siRNAs [45] which localize the siRNAs to the hepatocyte. Using this approach, researchers at Arrowhead have demonstrated liver-specific knockdown of HBV replication and protein expression in murine models [45]. These studies have now been extended to a chronically infected chimpanzee [46] and Arrowhead has successfully treated patients in a phase 2 multicenter randomized, double-blind placebo controlled dose-escalation study in patients with HBeAg-negative CHB whose viremia was controlled by entecavir [47]. The RNAi ARC-520 was shown to be safe and well-tolerated, with a 50 % drop in HBsAg observed in treated subjects compared to placebo controls. In addition to the Arrowhead molecules, RNAi approaches are being developed by other biotech companies including Alnylam [ALN-HBV: ESC-GalNAc-SiRNA multicomponent lipid nanoparticles] and Tekmira [TKM-HBV: Lipid Nanoparticle (LNP)]. These too show promise, with preclinical evaluation of the Alnylam RNAi demonstrating a 2.3 log reduction in HBsAg in chronically HBV-infected chimpanzees. Since prolonged expression of the HBsAg is associated with increased risk of progression to HCC [48], it remains to be determined whether siRNA-mediated reductio ns in HBsAg levels would positively impact on the long-term risk of progression to liver cancer.
Emerging Viral Targets
HBV Core Protein
The HBV core protein (nucleocapsid) is critical for viral RNA packaging, reverse transcription and intracellular trafficking. It is also an important for cccDNA generation and stability, binding directly to the cccDNA [49, 50]. These properties suggest the core protein is a suitable therapeutic target, enabling regulation of multiple facets of the HBV life cycle. Elucidation of the core crystal structure in 1999 [10] has provided new insights in core protein assembly, including defining the core dimer–dimer interface [10] leading to development of a range of compounds with therapeutic potential.
Packaging and Capsid Assembly Inhibitors
Several non-nucleoside analog (NNA) inhibitors of pgRNA packaging and HBV capsid assembly have been identified that dysregulate or selectively inhibit either pgRNA encapsidation, nucleocapsid assembly, or both. These include the phenylpropenamide derivatives AT-61 and AT-130 developed by Avid Therapeutics (later managed by Gilead Sciences) [51]. These compounds selectively inhibit viral pgRNA packaging [52] and are active against both wild-type and lamivudine-resistant HBVs [53, 54], inducing structural changes in HBV capsids. AT-130 has been shown to bind to a promiscuous pocket at the core dimer–dimer interface that favors a unique binding site in the capsid [55, 56]. This binding decreases viral production by initiating virion assembly prematurely in the replication cycle, resulting in morphologic ally normal but empty capsids that are noninfectious [52].
Hetero-aryl-dihydropyrimidines (HAPS) are a class of antiviral agent which inhibit HBV replication by preferentially stabilizing non-capsid polymers of the core protein [57–62]. In addition to reducing HBV replication and pgRNA levels, they markedly reduce cccDNA [59] and prevent interaction of the core protein with the minichromosome, thereby inhibiting cccDNA transcription and stability [57]. Like the phenylpropenamides, the HAPs are also active against nucleos(t)ide analogs (NA)-resistant strains of HBV [54].
Other compounds targeting the viral nucleocapsid include core inhibitors being developed by Novira Therapeutics (NVR-1221) [63] and 2-amino-N-(2,6-dichloropyridin-3-yl) acetamide which binds in the groove structure within the HBV capsid [64].
HBV cccDNA
The continual presence of cccDNA in the hepatocyte nucleus is a major impediment to HBV treatment and cure, as it is not directly targeted by current treatments and is a continual source of the pgRNA transcriptional template and additional viral mRNAs. Although a proportion of cccDNA molecules are removed during resolution of acute (transient) HBV infection, due to immune-mediated clearance of HBV infected cells during hepatocyte turnover [65–71], reactivation of HBV infection in immunosuppressed patients that may have previously resolved their acute infection [72–76] shows that cccDNA is not completely cleared during disease resolution in all patients. Hence the removal of cccDNA, or the suppression of its transcriptional activity, is a desired aim for treatment and cure regimens.
Epigenetic Regulation of cccDNA
Studies of the related hepadnavirus duck hepatitis B virus (DHBV) show that cccDNA extracted from infected duck livers exists as a heterogeneous pool of viral minichromosome of 20–21 topoisomers, present as either a half-chromatized (transcriptionally active) or fully chromatized (transcriptionally silent) molecules [7]. Studies of cccDNA extracted from HepG2.2.15 cells which are stably transformed with an overlength copy of the HBV genome showed that HBV cccDNA is also present as a minichromosome [77].
HBV replication and gene expression is controlled by epigenetic regulation of cccDNA, by acetylation and methylation of histone proteins which surround the cccDNA minichromosome [49, 61]. Histone deacetylases (HDACs) are a class of enzymes that remove acetyl groups from histones, allowing them to wrap the DNA more tightly, thereby regulating transcription. A second class of enzymes, the histone acetyl transferases (HATs) facilitate binding of transcription factors to DNA through the acetylation of histones bound to cccDNA. Together these molecules play a critical role in transcription of viral mRNAs from the HBV minichromosome, including pgRNA, the major replicative intermediate.
Recent advances in the HIV arena have shown that molecules which inhibit HDAC activity (HDAC inhibitors or HDACi) reactivate latent HIV proviral DNA incorporated within resting CD4 cells. This has led to the development of the “Shock and Kill” approach for HIV cure [78], whereby latent provirus in resting CD4 cells is reactivated by HDACi’s, with subsequent replicating virus then destroyed using DAAs [79, 80]. It is over 20 years since Newbold and colleagues [7] first suggested that HBV chromatin could represent a unique target for novel antiviral therapies. However it is not clear that the aforementioned HIV approach is directly applicable to HBV treatment, since HBV is never truly “latent.” Indeed, acknowledged reactivation of HBV cccDNA would be an outcome, leading to increased viral replication, with elimination or deactivation of cccDNA likely to be the best means of achieving HBV cure. However, such reactivation could be managed clinically with potent NAs such as TDF or ETV. Transcriptionally active cccDNA is associated with histone acetylases (PCAF, p300, CBP), and HBx regulatory protein [58, 81] and it has recently been shown that interferon alpha (IFN-α) represses cccDNA transcription by recruting a range of transcriptional co-repressors to the cccDNA, providing a molecular mechanism for IFN-α mediated repression of HBV transcription.
Stimulation of HDAC activity decreases both pgRNA transcription and HBV replication [82] suggesting that treatment with HDAC inhibitors would increase transcription and replication. However for reasons that are unclear, treating HBV infected cells with the HDAC SIRT1 inhibitor Sirtinol reduced HBV replication and pgRNA transcription [83]. It has also been shown that inhibition of cccDNA bound HAT activity results in detachment of PCAF and p300, decrease HBV replication and pgRNA transcription from cccDNA [82]. Together these findings show that epigenetic regulation of cccDNA is possible, although a complete understanding of factors regulating cccDNA biogenesis and expression will be required before these molecules are further progressed with confidence.
DNA methyltransferases . HBV gene expression is regulated in part by DNA methylation, with transfection of methylated HBV DNA in HepG2 cells leading to reduced HBV mRNA levels, decreased surface and core protein expression and decreased secretion of HBV viral proteins [84]. Methylated cccDNA was also identified in tumor and nonneoplastic human liver tissues [84]. Proof of principal utilizing HBV DNA methylation as a method for reducing gene expression has been provided by Xirong et al. [85], who showed that DNA methyltransferase 3a (Dnmt3a) targeting the HBV X promoter (XP) suppressed HBV replication and HBsAg expression in HepG2 cells and transgenic mice. These studies are yet to be performed using infection models which would demonstrate specificity for HBV cccDNA, but they show promise as an additional mechanism for regulating HBV replication and gene expression.
Selective Removal of cccDNA
The ability to eliminate cccDNA from the infected hepatocyte would be a major advance towards HBV cure. The development of designer targeted endonucleases that specifically recognize and cleave selected DNA sequences [86], resulting in gene disruption due to imprecise DNA repair, suggests this approach is indeed feasible. These methodologies include transcription activator-like effector nucleases (TALENs) [87] as well as the CRISPR (clustered regularly interspaced short palindromic repeats) CAS9 system [88, 89].
Talens. DNA targeting transcription activator-like effect ors (TALEs) derived from the plant pathogen Xanthomonas [90] been coupled with nuclease domains to form transcription activator-like effector nucleases (TALENs), capable of directed cleavage of specific DNA sequences. Recently, Bloom and colleagues reported that TALENS targeting HBV core and envelope gene sequences led to targeted disruption of up to 31 % of cccDNA molecules in HepG2.2.15 cells, with concomitant reductions in other HBV markers such as the secreted HBsAg [87]. This is a promising finding; however, it must be noted that this cell line, similar to other stably transduced cell lines commonly used for analysis of cccDNA (i.e., AD38 cells) contain integrated forms of the HBV genome, and true cccDNA is not the transcriptional driver in these systems. It remains to be determined whether the observed changes in DNA levels were mediated against newly synthesized cccDNA molecules, or had resulted from disruption of HBV DNA integrated into the HepG2.2.15 genomes. The effectiveness of Talens against cccDNA will become clearer when they are tested in the NTCP expressing cells that mimic more closely natural infection. Importantly though, TALENs demonstrate high specificity for HBV sequences [87], with “off-target effects” and toxicity not evident in cell culture or murine studies.
CRISPR. The CRISPR (Clustered Regularly Interspersed Palindromic Repeats) Cas9 (CRISPR associated protein 9) gene editing technology has recently been utilized to target HBV DNA in cell culture [88, 89] and murine models [89]. This approach uses RNA “guide” sequences which target the CAS9 endonuclease to the desired sequence for specific cleavage, which is then disrupted following DNA repair via a nonhomologous end joining (NHEJ) process. Importantly, Seeger and Sohn [88] have recently demonstrated the utility of this approach to specifically target HBV cccDNA, using the NTCP-HepG2 infection model. They showed that CRISPR/CAS9 targeting of the HBV ENII/CP region and the PC regions resulted in deletions in HBV cccDNA up to 2 kb in size, providing proof of principal that this approach can be used for the targeted disruption of cccDNA.
Limitations to Epigenetic Regulation of cccDNA as Therapy
Epigenetic regulation of HBV cccDNA as anti-HBV therapy needs to be carefully considered due to possible off-target effects on acetylation of host DNA and their ability to reactivate HBV infection. Indeed, reactivation of HBV has been observed following treatment of patients with the anticancer HDAC inhibitory drug Romidepsin [91] and in patients receiving immunosuppressive therapy [72–76, 92]. This suggests that not all cccDNA is eliminated in some patients who have previously cleared an acute infection, raising questions about the true definition of HBV cure. With the development of increasingly sensitive diagnostic tools it may become evident that some level of cccDNA persists in all patients, even with the clearance of the majority of infected hepatocytes, meaning that natural eradication of HBV is unachievable. Through specifically targeting cccDNA, it may be possible to induce cure, although this approach is currently not being considered based on current knowledge and clinical experience. A further problem with this approach for controlling HBV infection are “off-target” effects, since they may also affect the epigenome of host chromosomes. Approaches need to be developed that are specific to HBV cccDNA epigenome and not host DNA. Additional important questions include whether such alteration of cccDNA promotes integration of the modified DNA and whether this in turn may lead to expression of novel protein(s) and or a greater propensity for progression to HCC.
Future Targets for HBV Therapies?
HBeAg
The HBeAg is required for the establishment of persistent infection, with CHB rarely establishing in babies borne to HBeAg-negative mothers, in contrast to 90 % of newborns from HBeAg-positive mothers developing CHB [93]. In turn, low levels of HBeAg in patient serum are associated with HBeAg seroconversion in the setting of IFN-α treatment [94], this being an accepted marker of treatment response that usually precedes HBsAg clearance. Taken together these findings suggest that the HBeAg itself may be a suitable therapeutic target, with reductions in the levels of HBeAg perhaps driving the immune response to clear infection, or reducing mother to baby transmission in newborns. Unlike the related HBcAg, the HBeAg crosses the placenta [95], and is thought to establish tolerance to the HBcAg and HBeAg in the developing fetus [96, 97]. The HBeAg protein sequence differs from the HBV core protein by the addition of ten highly conserved [98] amino acids at the N-terminus and removal of the C-terminal arginine rich core domain [99]. Recent delineation of the HBeAg crystal structure [9] shows that the cysteine molecule at position −7 in the N-terminal 10-mer sequence (SKLCLGWLWG) bonds with cysteine 61 to form a dimer whose structure differs markedly from the HBcAg dimer [10]. It has been suggested that these differences may explain some of the biophysical, biochemical and functional properties of the HBeAg and HBcAg molecules [9, 10]. For example, the HBeAgs inability (usually) to form capsids, instead forming high molecular weight soluble forms in the blood [100], its propensity to initiate tolerogenic rather than immunogenic T-cell responses [101–104], inability to bind and activate B cells without T cell support [102–104], and suppression of innate immune signaling pathways [105–107]. It is hoped that the characterization of the HBeAg crystal may lead to development of molecules such as humanized mAbs which specifically target the HBeAg that may in turn drive HBeAg and HBsAg seroconversion. This approach should also be considered for other HBV proteins as it is becoming increasingly important for treatment of chronic conditions such as rheumatoid arthritis and Crohn’s disease [108]. Our group has recently identified a novel single variable domain (VNAR) of the shark immunoglobulin new antigen receptor (IgNAR) antibody which displayed biologically useful affinity for recombinant and native HBeAg, and recognized a unique conformational epitope [109]. This molecule was subsequently engineered for ER-targeted in vitro delivery to function as an intracellular antibody (intrabody), which effectively regulated precore/HBeAg expression, showing potential as a HBeAg therapeutic that is worthy of further investigation, exploiting the possible importance of FcRn expression on hepatocytes and their role in intracellular antibody “neutralization” [110].
HBx
The hepatitis B virus X protein is a non-structural prote in that is essential to initiate and maintain virus replication after infection [111]. Although HBx does not bind directly to cccDNA, its recruitment is mediated through its interaction with a wide range of cellular transcription factors and cofactors [112]. Modulation of cccDNA transcription and HBV replication by “HBx-knockout” strains of HBV which transcribe less pgRNA that wild-type HBV strongly implicates the HBx protein in regulation of cccDNA transcription [58, 111]. These findings suggest that the HBx protein may be a suitable therapeutic target. Indeed numerous studies have shown that siRNA mediated knockdown of the HBx protein reduces HBV replication [113, 114]. The overlapping nature of the HBV genome means that siRNAs targeting the HBX gene also target the major pgRNA transcriptional template, as well as the 3′ end of the HBV polymerase. Consequently it is unclear if HBV replication is reduced in these models due to reduced HBx-mediated transcription of cccDNA, or decreased levels of the pgRNA itself, or both. S tudies using the NTCP infection model as well as in non-transformed hepatocytes are required. It should also be noted that the overlapping nature of the HBV genome means that mutations at positions A176T2/G1764A which frequently occur in the HBV basal core promoter also alter the HBx coding sequence at positions 130 and 131. These changes are associated with increase virulence and disease progression, with Tseng and colleagues showing that the A1762T mutation in particular was associated with increased risk of liver cirrhosis [115]. The role of HBx in this association is unclear. Identifying the crystal structure of the HBx protein may enable identification of the exact location of binding sites with host transcription factors, facilitating the HBx protein as a novel therapeutic target.
HBsAg
HBsAg clearance and seroconversion to anti-HBs is the closest outcome to natural HBV “cure.” However, this rarely occurs during therapy, thought mainly due to the persistence of the cccDNA and associated minichromosome transcriptiona l template which is not affected by the most potent antiviral currently available, TDF. A recent clinical study of HBeAg-positive patients treated with TDF showed that HBsAg loss was observed in 10 % of patients following 5 years of therapy [22, 27]. Independent predictors of HBsAg loss in these patients included high levels of HBsAg at baseline and the log 10 IU/ml decline in HBsAg levels by week 24. This contrasts with studies on patients treated with IFN, where treatment response (HBsAg loss) was associated with low levels of HBsAg at baseline [116]. Since the continued expression of HBsAg in the absence of viral replication is associated with adverse disease outcomes, strategies which target the HBsAg itself may be a useful addition to current NA therapies and reduce the likelihood of progression to liver cancer in CHB patients. One such approach may be therapeutic mAbs. This approach was initially tested by van Nunen and colleagues [117], who evaluated the efficacy of the anti-HBs mAb Tuvirumab in chronic HBV patients, either as monotherapy or in combination with IFN. Although long-term efficacy (neutralization of HBsAg in serum) was not observed, temporary reductions of HBsAg levels by at least 50 % were observed in all patients. In three patients receiving combination therapy, serum HBsAg was reduced to background levels. In addition, loss of serum HBV-DNA was observed in three patients in the combination group, with subsequent HBeAg seroconversion in two patients. Subsequently Lever and colleagues showed that although treatmen t of two patients with an anti-HBs mouse monoclonal (RFHBs1) had no impact on serum HBsAg levels, serum HBeAg levels were reduced to below the level of detection in both patients, and HBV DNA was reduced to below the level of detection in one patient [118]. Taken together, these findings suggest that therapeutic mAbs targeting the HBsAg are worth revisiting.
Host Targets
Toll-Like Receptors
New therapeutic approaches for treatment of CHB are not limited to targeting HBV itself, with promising studies showing upregulation of cell defense mechanisms by TLR agonists activating antiviral signaling pathways that reduce HBV replication [119]. For example, TLR7 and TLR8 agonists limit HBV replication by upregulating IFN-α expression, with these compounds currently in phase II clinical trials [120–122].
APOBEC
APOBEC proteins are host cytidine deaminases that hypermutate single stranded DNA. Lucifora and colleagues recently demonstrated that IFN-α mediates its antiviral effect at least in part by upregulating APOBEC3A enzymes that hypermutate the HBV genome [123]. The antiviral effect was also mediated following stimulation of the lymphotoxin beta receptor, which activated the related APOBEC3B [124]. Interestingly, the HIV Vif protein binds to and downregulates Apobec3, and is thought to be a potential therapeutic target for HIV, although a HBV protein which downregulates Apobec analogous to the HIV Vif protein is yet to be identified. Although the HBV core protein binds Apobec3, it does not downregulate Apobec expression, instead facilitating the interaction of Apobec with viral cccDNA, thought to be critical in mediating the antiviral effect of IFN-α [123]. Since HBV encodes proteins such as the HBeAg that downregulate antiviral signaling pathways [105–107, 125, 126] it would not surprise if it also encodes a suppressor of Apobec3A. Studies determining whether HBV encodes a me diator of Apobec3 expression similar to the HIV Vif protein are worthy of consideration.
Cyclophilin
Cyclophilins are host cofactors essential for replication of a number of viruses including hepatitis C virus (HCV) and HIV [127, 128], although their role in the HBV life cycle is yet to be elucidated. Cyclophilin inhibitors have antiviral activity against HCV, and it has recently been shown in cell culture studies that the cyclophilin inhibitors alisporivir and NIM811 have antiviral activity against HBV, reducing HBV DNA and HBsAg production [129]. The effect was enhanced following co-treatment with the NA telbivudine. The potential of cylophilin inhibitors as an HBV therapy has been recognized by biotechnology companies such as OnCore, with the cyclophilin inhibitor OCB-030 currently under development for this purpose (http://www.oncorebiopharma.com).
Conclusion
There is an urgent need for improved HBV therapies and curative regimens for chronic HBV. Although there is a highly effective preventative HBV vaccine, it has no impact on existing infections affecting over 300 million people globally. The development of new treatments, including more potent DAAs, entry inhibitors, epigenetic or enzymatic manipulation of HBV cccDNA, viral protein targets and host targets, suggests that we are on the dawn of a new era for chronic hepatitis B treatment. The recent identification of the HBV NTCP entry receptor and the establishment of appropriate cell culture and animal models enabling studies of the complete HBV life cycle including cccDNA should speed development of new therapeutic approaches that may lead to HBV cure in the foreseeable future.
References
Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46(6):1759–68. Epub 2007/11/30.
Sureau C, Salisse J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology. 2013;57(3):985–94. Epub 2012/11/20.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1, e00049. Epub 2012/11/15.
Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, et al. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010;6(10), e1001162. Epub 2010/11/10.
Rabe B, Vlachou A, Pante N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A. 2003;100(17):9849–54. Epub 2003/08/12.
Kock J, Schlicht HJ. Analysis of the earliest steps of hepadnavirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J Virol. 1993;67(8):4867–74. Epub 1993/08/01.
Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, et al. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69(6):3350–7. Epub 1995/06/01.
Scaglioni PP, Melegari M, Wands JR. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J Virol. 1997;71(1):345–53.
DiMattia MA, Watts NR, Stahl SJ, Grimes JM, Steven AC, Stuart DI, et al. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure. 2013;21(1):133–42. Epub 2012/12/12.
Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3(6):771–80. Epub 1999/07/08.
Bayliss J, Lim L, Thompson AJ, Desmond P, Angus P, Locarnini S, et al. Hepatitis B virus splicing is enhanced prior to development of hepatocellular carcinoma. J Hepatol. 2013;59(5):1022–8. Epub 2013/07/03.
Soussan P, Garreau F, Arnufl B, Pol S, Kremsdorf D, editors. HBSP protein and related defective particles is associated to liver fibrosis The Molecular Biology of Hepatitis B Viruses. Woodshole, MA: Springer; 2004.
Soussan P, Garreau F, Zylberberg H, Ferray C, Brechot C, Kremsdorf D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J Clin Invest. 2000;105(1):55–60. Epub 2000/01/05.
Soussan P, Pol J, Garreau F, Schneider V, Le Pendeven C, Nalpas B, et al. Expression of defective hepatitis B virus particles derived from singly spliced RNA is related to liver disease. J Infect Dis. 2008;198(2):218–25. Epub 2008/06/06.
Soussan P, Tuveri R, Nalpas B, Garreau F, Zavala F, Masson A, et al. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J Hepatol. 2003;38(3):343–8.
Kim BK, Revill PA, Ahn SH. HBV genotypes: relevance to natural history, pathogenesis and treatment of chronic hepatitis B. Antivir Ther. 2011;16(8):1169–86. Epub 2011/12/14.
Kew MC, Kramvis A, Yu MC, Arakawa K, Hodkinson J. Increased hepatocarcinogenic potential of hepatitis B virus genotype A in Bantu-speaking sub-Saharan Africans. J Med Virol. 2005;75(4):513–21.
Kramvis A, Kew MC. Molecular characterization of subgenotype A1 (subgroup Aa) of hepatitis B virus. Hepatol Res. 2007;37(s1):S27–32. Epub 2007/07/14.
Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137(5):1593–608 e1–2.
Girones R, Miller RH. Mutation rate of the hepadnavirus genome. Virology. 1989;170(2):595–7. Epub 1989/06/01.
Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C, et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis. 2003;36(6):687–96.
Marcellin P, Buti M, Krastev Z, de Man RA, Zeuzem S, Lou L, et al. Kinetics of hepatitis B surface antigen loss in patients with HBeAg-positive chronic hepatitis B treated with tenofovir disoproxil fumarate. J Hepatol. 2014. Epub 2014/07/22.
Marcellin P, Heathcote EJ, Buti M, Gane E, de Man RA, Krastev Z, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359(23):2442–55. Epub 2008/12/05.
Lai CL, Gane E, Liaw YF, Hsu CW, Thongsawat S, Wang Y, et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357(25):2576–88. Epub 2007/12/21.
Liaw YF, Gane E, Leung N, Zeuzem S, Wang Y, Lai CL, et al. 2-Year GLOBE trial results: telbivudine Is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology. 2009;136(2):486–95. Epub 2008/11/26.
Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131(6):1743–51. Epub 2006/11/08.
Marcellin P, Chang TT, Lim SG, Sievert W, Tong M, Arterburn S, et al. Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2008;48(3):750–8. Epub 2008/08/30.
Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, Fang J, et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology. 2009;49(5):1503–14. Epub 2009/03/13.
Lee YS, Suh DJ, Lim YS, Jung SW, Kim KM, Lee HC, et al. Increased risk of adefovir resistance in patients with lamivudine-resistant chronic hepatitis B after 48 weeks of adefovir dipivoxil monotherapy. Hepatology. 2006;43(6):1385–91. Epub 2006/05/27.
Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132(4):1574–85. Epub 2007/04/06.
Zoulim F, Locarnini S. Management of treatment failure in chronic hepatitis B. J Hepatol. 2012;56 Suppl 1:S112–22. Epub 2012/02/04.
Agarwal K, Fung SK, Nguyen TT, Cheng W, Sicard E, Ryder SD, et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol. 2014. Epub 2014/12/03.
Agarwal K, Fung SK, Nguyen TT, Cheng W, Sicard E, Ryder SD, et al. Twenty eight day safety and efficacy of tenofovir alafe-namide (TAF) fumarate in chronic hepatitis B (CHB) patients. Hepatology. 2013;58(51):675.
Tong S, Li J. Identification of NTCP as an HBV receptor: the beginning of the end or the end of the beginning? Gastroenterology. 2014;146(4):902–5. Epub 2014/03/01.
Watashi K, Urban S, Li W, Wakita T. NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci. 2014;15(2):2892–905. Epub 2014/02/22.
Engelke M, Mills K, Seitz S, Simon P, Gripon P, Schnolzer M, et al. Characterization of a hepatitis B and hepatitis delta virus receptor binding site. Hepatology. 2006;43(4):750–60. Epub 2006/03/25.
Glebe D, Urban S, Knoop EV, Cag N, Krass P, Grun S, et al. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology. 2005;129(1):234–45. Epub 2005/07/14.
Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A. 2002;99(24):15655–60. Epub 2002/11/15.
Urban S, Gripon P. Inhibition of duck hepatitis B virus infection by a myristoylated pre-S peptide of the large viral surface protein. J Virol. 2002;76(4):1986–90. Epub 2002/01/19.
Volz T, Allweiss L, Ben MM, Warlich M, Lohse AW, Pollok JM, et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58(5):861–7. Epub 2012/12/19.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–11. Epub 1998/03/05.
Chen Y, Cheng G, Mahato RI. RNAi for treating hepatitis B viral infection. Pharm Res. 2008;25(1):72–86. Epub 2007/12/13.
Klein C, Bock CT, Wedemeyer H, Wustefeld T, Locarnini S, Dienes HP, et al. Inhibition of hepatitis B virus replication in vivo by nucleoside analogues and siRNA. Gastroenterology. 2003;125(1):9–18. Epub 2003/07/10.
McCaffrey AP, Nakai H, Pandey K, Huang Z, Salazar FH, Xu H, et al. Inhibition of hepatitis B virus in mice by RNA interference. Nat Biotechnol. 2003;21(6):639–44. Epub 2003/05/13.
Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21(5):973–85. Epub 2013/02/27.
Lanford R, Wooddell CI, Chavez D, Oropeza CE, Chu Q, Hamilton HL, et al. ARC-520 RNAi therapeutic reduces hepatitis B virus DNA, S antigen and e antigen in a chimpanzee with a very high viral titer. Hepatology. 2013;58(S1):705A–30A.
Yuen M-F, Chan HL-Y, Given B, Hamilton J, Schluep T, Lewis DL, et al. Phase II, dose ranging study of ARC-520, a siRNA-based therapeutic, in patients with chronic hepatitis B virus infection. Hepatology. 2014;60:1267A–90A (LB-21).
Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373(9663):582–92.
Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130(3):823–37. Epub 2006/03/15.
Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. 2001;307(1):183–96. Epub 2001/03/13.
King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, et al. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-l-2′,3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother. 1998;42(12):3179–86. Epub 1998/12/03.
Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 2007;76(2):168–77. Epub 2007/08/22.
Delaney WE, Edwards R, Colledge D, Shaw T, Furman P, Painter G, et al. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob Agents Chemother. 2002;46(9):3057–60. Epub 2002/08/17.
Billioud G, Pichoud C, Puerstinger G, Neyts J, Zoulim F. The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res. 2011;92(2):271–6. Epub 2011/08/30.
Katen SP, Chirapu SR, Finn MG, Zlotnick A. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol. 2010;5(12):1125–36. Epub 2010/09/18.
Katen SP, Tan Z, Chirapu SR, Finn MG, Zlotnick A. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure. 2013;21(8):1406–16. Epub 2013/07/23.
Belloni L, Li L, Palumbo GA, Chirapu SR, Calvo L, Finn MG, et al. HAPS hepatitis B virus (HBV) capsid inhibitors prevent HBC interaction with the viral minichromosome and selected host cells to inhibit transcription and affect cccDNA stability. Dig Liver Dis. 2014;46, e9.
Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A. 2009;106(47):19975–9. Epub 2009/11/13.
Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, et al. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J Virol. 2008;82(20):10262–70. Epub 2008/08/08.
Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299(5608):893–6. Epub 2003/02/08.
Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51(3):581–92. Epub 2009/07/21.
Yang X, Xu X, Guan H, Wang L, Wu Q, Zhao G, et al. A new series of HAPs as anti-HBV agents targetting at capsid assembly. Biorog Med Chem Lett. 2014;24(17):4247–9.
Gane E, Schwabe C, Walker K, Flores L, Hartman G, Klumpp K, et al. Phase 1a safety and pharmacokinetics of NVR 3-778, a potential first-in-class HBV core inhibitor. Hepatology. 2014;60:1267A–90A (LB-19).
Cho MH, Jeong H, Kim YS, Kim JW, Jung G. 2-Amino-N-(2,6-dichloropyridin-3-yl)acetamide derivatives as a novel class of HBV capsid assembly inhibitor. J Viral Hepat. 2014;21(12):843–52. Epub 2014/01/01.
Alison MR, Lin WR. Hepatocyte turnover and regeneration: virtually a virtuoso performance. Hepatology. 2011;53(4):1393–6. Epub 2011/04/12.
Mason WS, Xu C, Low HC, Saputelli J, Aldrich CE, Scougall C, et al. The amount of hepatocyte turnover that occurred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J Virol. 2009;83(4):1778–89. Epub 2008/12/17.
Mason WS, Litwin S, Xu C, Jilbert AR. Hepatocyte turnover in transient and chronic hepadnavirus infections. J Viral Hepat. 2007;14 Suppl 1:22–8. Epub 2007/11/21.
Esposito C, Parrilla B, De Mauri A, Cornacchia F, Fasoli G, Foschi A, et al. Hepatocyte growth factor (HGF) modulates matrix turnover in human glomeruli. Kidney Int. 2005;67(6):2143–50. Epub 2005/05/11.
Summers J, Jilbert AR, Yang W, Aldrich CE, Saputelli J, Litwin S, et al. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc Natl Acad Sci U S A. 2003;100(20):11652–9.
Dandri M, Burda MR, Will H, Petersen J. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology. 2000;32(1):139–46. Epub 2000/06/28.
Fourel I, Cullen JM, Saputelli J, Aldrich CE, Schaffer P, Averett DR, et al. Evidence that hepatocyte turnover is required for rapid clearance of duck hepatitis B virus during antiviral therapy of chronically infected ducks. J Virol. 1994;68(12):8321–30. Epub 1994/12/01.
Blanpain C, Knoop C, Delforge ML, Antoine M, Peny MO, Liesnard C, et al. Reactivation of hepatitis B after transplantation in patients with pre-existing anti-hepatitis B surface antigen antibodies: report on three cases and review of the literature. Transplantation. 1998;66(7):883–6.
Hoofnagle JH. Reactivation of hepatitis B. Hepatology. 2009;49(5 Suppl S1):56–65. Epub 2009/04/29.
Hung HH, Su CW, Wu JC, Lee SD. Reactivation of hepatitis B virus after transarterial chemo-embolization for hepatocellular carcinoma in one patient with negative hepatitis B surface antigen. J Hepatol. 2010;52(3):463–5. Epub 2010/02/05.
Kempinska A, Kwak EJ, Angel JB. Reactivation of hepatitis B infection following allogeneic bone marrow transplantation in a hepatitis B-immune patient: case report and review of the literature. Clin Infect Dis. 2005;41(9):1277–82.
Palmore TN, Shah NL, Loomba R, Borg BB, Lopatin U, Feld JJ, et al. Reactivation of hepatitis B with reappearance of hepatitis B surface antigen after chemotherapy and immunosuppression. Clin Gastroenterol Hepatol. 2009;7(10):1130–7. Epub 2009/07/07.
Bock CT, Schranz P, Schroder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes. 1994;8(3):215–29. Epub 1994/07/01.
Deeks SG. HIV: shock and kill. Nature. 2012;487(7408):439–40. Epub 2012/07/28.
Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158(5):989–99. Epub 2014/08/19.
Lo S. Sharon Lewin: guiding us towards a cure for HIV. Lancet. 2014;384(9939):223. Epub 2014/07/22.
Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, et al. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122(2):529–37. Epub 2012/01/19.
Pallumbo GA, Belloni L, Valente S, Rotili D, Pediconi N, Mai A, et al. Suppression of hepatitis B virus (HBV) transcription and replication by small molecules that target the epigenetic control of nuclear cccDNA minichromosome. J Hepatol. 2013;58:S56.
Ren JH, Tao Y, Zhang ZZ, Chen WX, Cai XF, Chen K, et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J Virol. 2014;88(5):2442–51. Epub 2013/12/18.
Vivekanandan P, Thomas D, Torbenson M. Methylation regulates hepatitis B viral protein expression. J Infect Dis. 2009;199(9):1286–91. Epub 2009/03/24.
Cabuang LM, Shaw T, Littlejohn M, Colledge D, Sozzi V, Soppe S, et al. In vitro replication phenotype of a novel (-1G) hepatitis B virus variant associated with HIV co-infection. J Med Virol. 2012;84(8):1166–76. Epub 2012/06/20.
Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR. Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol. 2012;86(17):8920–36. Epub 2012/06/22.
Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21(10):1889–97. Epub 2013/07/26.
Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic acids. 2014;3, e216. Epub 2014/12/17.
Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic acids. 2014;3, e186. Epub 2014/08/20.
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14(1):49–55. Epub 2012/11/22.
Anderson JL, Fromentin R, Corbelli GM, Ostergaard L, Ross AL. Progress towards an HIV cure: update from the 2014 International AIDS Society symposium. AIDS Res Hum Retroviruses. 2015;31(1):36–44.
Nunes J, Marinho RT, Fonseca JE, Pereira da Silva JA, Velosa J. Prophylaxis of hepatitis B reactivation with immunosuppressive therapy in rheumatic diseases. Orientations for clinical practice. Acta Reumatol Port. 2011;36(2):110–8. Epub 2011/08/16.
Terazawa S, Kojima M, Yamanaka T, Yotsumoto S, Okamoto H, Tsuda F, et al. Hepatitis B virus mutants with precore-region defects in two babies with fulminant hepatitis and their mothers positive for antibody to hepatitis B e antigen. Pediatr Res. 1991;29(1):5–9. Epub 1991/01/01.
Fried MW, Piratvisuth T, Lau GK, Marcellin P, Chow WC, Cooksley G, et al. HBeAg and hepatitis B virus DNA as outcome predictors during therapy with peginterferon alfa-2a for HBeAg-positive chronic hepatitis B. Hepatology. 2008;47(2):428–34.
Schodel F, Peterson D, Zheng J, Jones JE, Hughes JL, Milich DR. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem. 1993;268(2):1332–7. Epub 1993/01/15.
Chen MT, Billaud JN, Sallberg M, Guidotti LG, Chisari FV, Jones J, et al. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A. 2004;101(41):14913–8. Epub 2004/10/08.
Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci U S A. 1990;87(17):6599–603.
Revill P, Yuen L, Walsh R, Perrault M, Locarnini S, Kramvis A. Bioinformatic analysis of the hepadnavirus e-antigen and its precursor identifies remarkable sequence conservation in all orthohepadnaviruses. J Med Virol. 2010;82(1):104–15. Epub 2009/12/02.
Messageot F, Salhi S, Eon P, Rossignol JM. Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J Biol Chem. 2003;278(2):891–5.
Baba K, Ise I, Aihara S, Kishimoto S, Tsuda F, Tachibana K, et al. Small and large forms of hepatitis B e antigen in the serum: determination by two-site sandwich radioimmunoassay with monoclonal antibodies. Clin Exp Immunol. 1986;64(2):295–301. Epub 1986/05/01.
Chen M, Sallberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, et al. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol. 2005;79(5):3016–27.
Milich DR, Chen M, Schodel F, Peterson DL, Jones JE, Hughes JL. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci U S A. 1997;94(26):14648–53. Epub 1998/02/07.
Milich DR, McLachlan A. The nucleocapsid of hepatitis B virus is both a T-cell-independent and a T-cell-dependent antigen. Science. 1986;234(4782):1398–401. Epub 1986/12/12.
Milich DR, Schodel F, Hughes JL, Jones JE, Peterson DL. The hepatitis B virus core and e antigens elicit different Th cell subsets: antigen structure can affect Th cell phenotype. J Virol. 1997;71(3):2192–201. Epub 1997/03/01.
Jegaskanda S, Ahn SH, Skinner N, Thompson AJ, Ngyuen T, Holmes J, et al. Down-regulation of IL-18 mediated cell signalling and IFN-gamma expression by the hepatitis B virus e antigen. J Virol. 2014. Epub 2014/05/30.
Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol. 2011;55(4):762–9. Epub 2011/02/22.
Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45(1):102–10.
Lynch CM, Hart BW, Grewal IS. Practical considerations for nonclinical safety evaluation of therapeutic monoclonal antibodies. MAbs. 2009;1(1):2–11. Epub 2010/01/05.
Walsh R, Nuttall S, Revill P, Colledge D, Cabuang L, Soppe S, et al. Targeting the hepatitis B virus precore antigen with a novel IgNAR single variable domain intrabody. Virology. 2011;411(1):132–41. Epub 2011/01/18.
Schilling R, Ijaz S, Davidoff M, Lee JY, Locarnini S, Williams R, et al. Endocytosis of hepatitis B immune globulin into hepatocytes inhibits the secretion of hepatitis B virus surface antigen and virions. J Virol. 2003;77(16):8882–92. Epub 2003/07/30.
Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55(5):996–1003. Epub 2011/03/08.
Cougot D, Wu Y, Cairo S, Caramel J, Renard CA, Levy L, et al. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J Biol Chem. 2007;282(7):4277–87. Epub 2006/12/13.
Ely A, Naidoo T, Arbuthnot P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009;37(13), e91. Epub 2009/05/29.
Tong WP, Zhou Y, Wang X, Yang F, Wu KL, Wu J, et al. An accurate quantitative method for screening effective siRNA probes targeting a hepatitis B virus transcript in single living cells. Biochem Biophys Res Commun. 2008;367(4):866–73. Epub 2008/01/19.
Tseng TC, Liu CJ, Yang HC, Chen CL, Yang WT, Tsai CS, et al. Higher proportion of viral basal core promoter mutant increases the risk of liver cirrhosis in hepatitis B carriers. Gut. 2014. Epub 2014/04/26.
Marcellin P, Bonino F, Yurdaydin C, Hadziyannis S, Moucari R, Kapprell HP, et al. Hepatitis B surface antigen levels: association with 5-year response to peginterferon alfa-2a in hepatitis B e-antigen-negative patients. Hepatol Int. 2013;7(1):88–97. Epub 2013/03/23.
van Nunen AB, Baumann M, Manns MP, Reichen J, Spengler U, Marschner JP, et al. Efficacy and safety of an intravenous monoclonal anti-HBs in chronic hepatitis B patients. Liver. 2001;21(3):207–12. Epub 2001/06/26.
Lever AML, Waters AJ, Brook MG, Karayiannis P. Treatment of Chronic Hepatitis B Virus Infection With Monoclonal Antibody to the Hepatitis B Virus Surface Antigen in Two Patients With Hypogammaglobuinaemia. In: Zuckerman AJ, editor. Viral hepatitis and liver disease. New York, NY: Alan R Liss; 1988.
Thompson AJ, Colledge D, Rodgers S, Wilson R, Revill P, Desmond P, et al. Stimulation of the interleukin-1 receptor and Toll-like receptor 2 inhibits hepatitis B virus replication in hepatoma cell lines in vitro. Antivir Ther. 2009;14(6):797–808. Epub 2009/10/09.
Fosdick A, Zheng J, Pflanz S, Frey CR, Hesselgesser J, Halcomb RL, et al. Pharmacokinetic and pharmacodynamic properties of GS-9620, a novel Toll-like receptor 7 agonist, demonstrate interferon-stimulated gene induction without detectable serum interferon at low oral doses. J Pharmacol Exp Ther. 2014;348(1):96–105. Epub 2013/10/18.
Menne S, Tumas DB, Liu KH, Thampi L, AlDeghaither D, Baldwin BH, et al. Sustained efficacy and seroconversion with the Toll-like receptor 7 agonist GS-9620 in the Woodchuck model of chronic hepatitis B. J Hepatol. 2015. Epub 2015/01/07.
Wang Y, Chen K, Wu Z, Liu Y, Liu S, Zou Z, et al. Immunizations with hepatitis B viral antigens and a TLR7/8 agonist adjuvant induce antigen-specific immune responses in HBV-transgenic mice. Int J Infect Dis. 2014;29:31–6. Epub 2014/12/03.
Sommer G, Heise T. Posttranscriptional control of HBV gene expression. Front Biosci. 2008;13:5533–47. Epub 2008/05/30.
Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014. Epub 2014/02/22.
Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, et al. The hepatitis B e antigen suppresses IL-1beta-mediated NF-kappaB activation in hepatocytes. J Viral Hepat. 2011;18(10):e499–507. Epub 2011/09/15.
Wu S, Kanda T, Imazeki F, Arai M, Yonemitsu Y, Nakamoto S, et al. Hepatitis B virus e antigen downregulates cytokine production in human hepatoma cell lines. Viral Immunol. 2010;23(5):467–76. Epub 2010/10/05.
Gaither LA, Borawski J, Anderson LJ, Balabanis KA, Devay P, Joberty G, et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology. 2010;397(1):43–55. Epub 2009/11/26.
Zhou D, Mei Q, Li J, He H. Cyclophilin A and viral infections. Biochem Biophys Res Commun. 2012;424(4):647–50. Epub 2012/07/21.
Phillips S, Chokshi S, Chatterji U, Riva A, Bobardt M, Williams R, et al. Alisporivir inhibition of hepatocyte cyclophilins reduces HBV replication and hepatitis B surface antigen production. Gastroenterology. 2015;148(2):403–14 e7. Epub 2014/10/12.
Matthews et al., Clin Infect. Dis. 56 (9). e87-94.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Revill, P., Locarnini, S. (2016). The Basis for Antiviral Therapy: Drug Targets, Cross-Resistance, and Novel Small Molecule Inhibitors. In: Liaw, YF., Zoulim, F. (eds) Hepatitis B Virus in Human Diseases. Molecular and Translational Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-22330-8_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-22330-8_14
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-22329-2
Online ISBN: 978-3-319-22330-8
eBook Packages: MedicineMedicine (R0)