
Abstract
This chapter provides a nice overview of primary sclerosing cholangitis (PSC). It brings the reader up to date on current state of knowledge; however it also points out a number of areas in which further work is necessary to answer currently unresolved questions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
This chapter provides a nice overview of primary sclerosing cholangitis (PSC). It brings the reader up to date on current state of knowledge; however it also points out a number of areas in which further work is necessary to answer currently unresolved questions.
In the epidemiology of the disease it remains uncertain why the disease seems to be one of northern climates. There are very few studies from southern Europe, the southern USA, and even less from countries nearer the equator. Furthermore, there are very few studies from the southern hemisphere. The reason for this is unknown, but certainly it may provide some clues as to potential etiologies or may be a reflection of awareness of the disease and rigor of case finding efforts. This question remains unresolved [1].
Similarly the geographic differences in the association of inflammatory bowel disease with PSC remain unexplained. Studies from the more northern parts of the world, including Scandinavia and the upper Midwest of the USA, suggest an association of colitis in about 70 % of the patients with PSC. However, in other studies from the warmer parts of the world the association is found in fewer than 50 % of PSC patients.
One of the other unexplained findings that relates to the association of colitis and primary sclerosing cholangitis is the increased risk of developing colorectal cancers in patients with inflammatory bowel disease who have coexisting PSC. In patients with colitis this risk increases over time as it does in patients with PSC; however in patients with PSC, this risk is usually five times greater than at any point in time than if a patient simply has colitis. The reason for this is unexplained, but it is certainly worthy of further evaluation in the hope that there may be intervention to prevent this high risk of colon cancer.
In the discussion regarding pathogenesis , the authors introduce the concept that the etiology may not be due to simply one cause but may be multifactorial. Perhaps recognition of this will help further our understanding of the causes of the disease and eventual treatment strategies. Currently the approaches to treatment have considered the disease to be of a single cause and have not explored the possibility that agents with different mechanisms of action may have efficacy for diseases of varying causes. One area that the authors allude to and is ripe for much further study has to do with the role of the gut microbiome. This is an important and increasingly recognized area in gastroenterology and hepatology. Clearly the relationship of the gut microbiome and the etiology of PSC is important to explore particularly given the association of inflammatory bowel disease in many PSC patients.
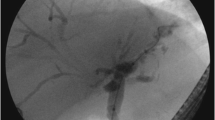
Diagnosis of PSC has become simpler with MR cholangiography which avoids the morbidity associated with endoscopic retrograde cholangiopancreatography. However as the authors point out MR cholangiography is also subject to interobserver variability, which can impair the diagnostic utility of this cost-effective test. [2]
One of the variants of PSC recently described is IgG4-associated disease. Autoimmune pancreatitis which is also an IgG4 associated disease has a number of diagnostic schema that have been proposed; whereas IgG4 associated cholangitis does not have clear diagnostic criteria established, some have used elevations of serum IgG4 levels above normal, others have used serum IgG4 levels above certain multiples of the upper limit of normal, whereas others have required histology. Histologic sampling of the biliary tract is problematic and is rarely diagnostic, and so we are left with a need for standardized criteria to establish a diagnosis of IgG4-associated disease. Once these criteria are established this will help us to better define the disease and then make treatment trials more feasible. At present, the diagnosis is uncertain, hampering development of adequate treatment trials beyond empiric immunosuppressive based therapy [3].
One of the other important questions related to therapy has to do with value of ursodeoxycholic acid , which the authors discuss. Randomized control trials have failed to disclose the benefit of a dose of 13–15 mg/kg/day. There is biochemical improvement, but not clinical improvement, whereas a dose of double that led to clinical worsening and increased risk of colorectal neoplasia. Intermediate doses have not yet been adequately studied. Recently, data suggests that patients on ursodeoxycholic acid with PSC who had the drug withdrawn underwent clinical deterioration. Several other studies have recently shown the patients who achieved biochemical normalization, whether spontaneously or with ursodeoxycholic acid have a better outlook of their disease course. This opens the door for strategies in which ursodeoxycholic acid is administered for a predefined period of time of 6–12 months to see if biochemical normalization can be achieved, and if so the drug would be continued. However, there are no controlled data supporting this approach [4].
Other therapies that are being evaluated include other derivatives of bile acids such as obeticholic acid which is 6-ethyl-chenodeoxycholic acid, an FXR inhibitor as well as well as norursodeoxycholic acid. Results are not yet available from these drugs. Anti-fibrotic drugs such as lysyl oxidase-like 2 cross-linking inhibitor studies are underway with the results expected shortly. Finally, antibiotics such as vancomycin have been explored particularly in children and hold some promise but much more work is needed before this approach can be considered as recommended therapy.
The authors introduce the concept of the management of dominant strictures . Like IgG4 associated disease, we do not yet have agreed-upon criteria to define what a dominant stricture is. This is because these are difficult to define and therefore reports of attempted therapy are difficult to place into context.
Also, an important area that is not often times given adequate attention is cancer surveillance for patients with PSC who are at a substantially increased risk for developing cholangiocarcinoma. Some data suggests that regular cross-sectional imaging with ultrasound or MR along with measure of serum levels of CA19-9 might identify patients early enough to find patients eligible for liver transplantation. However, liver transplantation is not always used for patients with primary sclerosing cholangitis. Programs using neo-adjuvant radiation therapy have achieved excellent results. Hopefully, this approach will begin to spread and become more widely available. In institutions in which this approach is available, earlier detection with surveillance does appear to lead to improved overall patient survival.
Finally, liver transplantation is extremely successful in patients with PSC but it is estimated that 20–40 % of patients with PSC will develop recurrent disease after liver transplantation. Clearly, therapy to prevent the reoccurrence of disease is necessary. Perhaps in the absence of effective therapy for the disease itself, it is not surprising that recurrence cannot be prevented also [5].
Primary sclerosing cholangitis is an important disease with many remaining questions and much need for further research as nicely outlined in this chapter.
References
Molodecky NA, Kareemi H, Parab R, et al. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology. 2011;53(5):1590–9.
Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40(1):39–45.
Rupp C, Rossler A, Halibasic E, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther. 2014;40(11–12):1292–301.
Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: If withdrawal is bad, then administration is good (right?). Hepatology. 2014;60(3):785–8.
Gores GJ, Darwish Murad S, Heimbach JK, Rosen CB. Liver transplantation for perihilar cholangiocarcinoma. Dig Dis. 2013;31(1):126–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Lindor, K.D. (2015). Commentary: Primary Sclerosing Cholangitis. In: Dixon, E., Vollmer Jr., C., May, G. (eds) Management of Benign Biliary Stenosis and Injury. Springer, Cham. https://doi.org/10.1007/978-3-319-22273-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-22273-8_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22272-1
Online ISBN: 978-3-319-22273-8
eBook Packages: MedicineMedicine (R0)