
Abstract
The physicochemical and biological properties of biopharmaceuticals are, in many aspects, different from small molecule drugs. These differences must also be taken into account when evaluating the risk of carcinogenicity in humans. For example, because of their expected biological activity, growth factors or immunomodulators present an inherent risk for potentially enhancing tumor incidence in humans.
The present chapter reviews the background for this position of biotechnologically-derived pharmaceuticals. Growth factors can be seen as oncogenes, as these proteins will stimulate cell surface receptors related to cell proliferation. In this respect, ICH S6(R1) deviates from the common approach for carcinogenicity testing, as generally 2-year bioassay studies are not expected for these products. Also, for immunomodulators, the regulatory guidance acknowledges an inherent risk for cancer when immunosuppressive activity can be expected based on the pharmacology of the compound (e.g., impaired immune surveillance).
In this chapter, a few case studies are presented, illustrating different approaches in evaluating the carcinogenic potential of biopharmaceuticals. Furthermore, approaches to the translation of these findings to the human situation are discussed. Insulin-like growth factor and insulin are different in mitogenic and metabolic activity by stimulation of IGF1- and Insulin receptor A or B, respectively. Insulin analogues such as Insulin AspB10 and insulin glargine have been analyzed in this respect by novel in vitro and in vivo strategies, and this approach reveals its usefulness from a regulatory point of view.
GLP1-agonists induce thyroid C-cell tumors by a direct action at the C-cell, and we have described a pharmacodynamic/pharmacokinetic approach to model the relationship between exposure and the induction of thyroid hyperplasia or adenoma (dependent on the compound).
By virtue of their pharmacology, some monoclonal antibodies are also known to be associated with occurrence of tumors in humans, and an overview of these reported cases is also included in this chapter. The concerns of an increased cancer risk associated with medicines may arise at any time during a drug’s life cycle: in early phases during development, or after many years of use in clinical practice. Pharmacovigilance represents the science and activities related to the detection, assessment, understanding and prevention of adverse effects or other drug-related problems. In this section, we review a series of medicines for which cancer has been a suspected or actual risk detected, as well as the problems that are encountered in studying and communicating such cancer risks or the uncertainties about these risks.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Carcinogenicity evaluation
- Biotechnology-derived pharmaceuticals
- Non-clinical evaluation
- Insulin
- GLP-1 agonists
- Immunomodulators
8.1 Introduction
While new pharmaceuticals are constantly being developed to alleviate disease in humans, it is recognized that these pharmaceuticals can also be associated with adverse effects due to their chemical structure and/or inherent pharmacologic activity. Therefore, non-clinical and clinical evaluation of human pharmaceuticals is required to demonstrate both efficacy and safety in the intended clinical therapeutic situation. Carcinogenicity is one of the major safety concerns that is largely de-risked in the non-clinical studies. In this chapter, we focus on the carcinogenic risk of proteins produced by recombinant biotechnology, generally called biopharmaceuticals, as specific aspects make them different from conventional small molecules.
8.1.1 Carcinogenicity Risk Assessment in General: Initiators and Promoters
Experimental models in the past have helped to differentiate classes of compounds as being either tumor initiators (often detected as causing cancer by a single or at least short-term administration) or tumor promoters (usually given continuously and chronically over a long period). Initiators became known as compounds usually interacting directly with DNA, damaging its sequence, i.e. toxic for the gene. Promoters appear to need a proliferation step in the cell cycling, either by direct stimulation of proliferation, or by indirect cell multiplication e.g. as a repair of damaged tissue after a toxic phenomenon (irritation or other types of cell damage).
In the risk assessment for potential carcinogenicity of human pharmaceuticals genotoxic and non-genotoxic compounds can be differentiated. Regulators assume that genotoxic compounds have an inherent carcinogenic potential and accept these compounds as human pharmaceuticals only if their benefit outweighs their risk [1]. This is the case for some cytostatic anticancer drugs and a few anti-HIV antiviral drugs [2]. Most genotoxic compounds will therefore not be further developed as human pharmaceuticals. The field of molecular oncology is rapidly advancing and it is expected that new insights based upon this increasing knowledge will contribute to improved carcinogenic assessments of biopharmaceuticals as well as small molecules [3].
It is now well accepted that the process of cancer development is usually not a one-hit process and that the development of tumors likely requires multiple steps. Exceptions might be the hereditary retinoblastoma, also called von Hippel-Lindau disease, which has been described as a two-hit model [4], and glioneuroblastoma based on a single change in Wilms tumor1 gene [5]. Other research revealed the differentiation between “gatekeeper” genes and “caretaker” genes. Gatekeeper genes are characterized by their control of net cellular proliferation, whereas caretaker genes are involved in maintaining the integrity of the genome, and consist of repair-genes [6]. Predisposition to cancer might be related to inherited mutation of these repair genes, e.g. in Xeroderma Pigmentosa patients. Human disease characteristics are seen also in animal models, e.g. genetically modified mouse models [7, 8].
Other types of genes are called oncogenes where genetic alterations would lead to an increase in protein function and activity, and suppressor genes, where the loss of functionality is the crux, with an important point that both alleles need to be affected (loss of heterozygosity).
Examples of oncogenes are H-ras and K-ras genes. One of the existing transgenic models for standard carcinogenicity testing mentioned in the ICH guidelines is the TgRasH2 mouse, tested at the 1990s ILSI-HESI initiative Alternatives to Carcinogenicity Testing [ILSI-HESI ACT] [9].
For suppressor genes, the presence of just one intact gene might be important for resistance against cancer. An example for a suppressor gene is the p53-gene. A p53-heterozygous mouse model was established in the 1990s [10], and has been tested extensively, but was found mainly sensitive to genotoxic agents in the ILSI-HESI ACT and the EPA program.
There is now a growing insight in the types of damage that might lead to human cancer as is clear from the paper on Hallmarks on Cancer [11]. These authors have given an updated overview of all (at least a high number) of processes that are involved in the induction and progress in cancer.
Important elements among these components are the processes indicating growth and proliferation, self-sufficiency in growth signals and limitless replicative potential. In addition, insensitivity to anti-growth signals and evading apoptosis can be seen as growth-stimulating factors. These components explain on the one hand that proliferation is an important factor as a non-genotoxic phenomenon, but is on its own insufficient to lead to cancer. It should be emphasized that human epithelial cancers do not always follow a predictable histopathological sequential pattern from hyperplasia to adenoma and then to carcinoma. While this sequence is not uncommon, differences might depend on the number of spontaneous mutations.
This also clear from our large database work, that compounds with a similar pharmacological action e.g. β2-agonists in some cases will induce benign adenomas, whereas in other cases only hyperplasia was observed (Van der Laan et al., manuscript in preparation). A Vitamin D analogue induced cell proliferation in adrenals after 6 months of administration, while pheochromocytomas were observed after 57 weeks [12].
Non-genotoxic compounds commonly enhance proliferation, either by direct receptor stimulation or by enhancing the release of proliferating factors. Even indirect stimulation of cell growth as a compensatory mechanism for cell damage can be seen as a non-genotoxic mechanism leading to cancer, e.g. by damaging bladder mucosa [13].
The methodology of modern Next Generation Sequencing allows us to assess mutations in tumor tissue, and compare the pattern of mutations among tumors from the same organ in different animals which helps explain the specificity of the mutations. Bronchud [3, 14] showed that increased number of mutations correlates with the increasing premalignant changes.
Non-genotoxic compounds might act by proliferation. Vogelstein et al. [15] recently described that spontaneous mutations occur in a variety of places, and some mutations occur in “driver-genes”, whereas other mutations (the vast majority) in “passenger genes”, with no direct result on the tumor-character of a cell. The number of mutations is also dependent on the age of the individual as well as the organ.
What we learned from a recent study with insulin AspB10 and IGF1, is that non-genotoxic compounds may accelerate tumor formation (in a transgenic breast cancer model in mice), and may stimulate a specific pattern of what has been called before “spontaneous mutations”. See Sect. 8.3 Case studies, Insulins and IGF-1.
8.1.2 Growth Factors and Other Biopharmaceuticals
Although proliferation of cell growth cannot be seen as the single cause for cancer, its impact is high in the list of causes as explained by Hanahan and Weinberg [11]. This is recognized in the risk assessment of growth stimulating factors as medicines.
Growth factors can be seen as a category of oncogenes, as these proteins stimulate cell surface receptors [for a review see Pan and Godwin] [16] leading to signal transduction relating to cell proliferation. Classically the definition of an oncogene is a gene that will transform the cell with some attributes of malignancy. With insight that is more recent we now know that the effect of proliferation by a growth factor depends on the cellular context. Growth factors and extracellular mitogenic signals are identified as Platelet-Derived Growth Factors (e.g. PDGFβ), Fibroblast Growth Factors (e.g. FGF-3/INT-2, FGF4/HST), WNT (e.g. WNT-1, WNT-2), Epidermal growth factor (e.g. EGF, TGF-a) or cytokines (e.g. Interleukin-2, Granulocyte-Macrophage-Colony Stimulating Factor). Another class of oncogenes can be identified as cell surface receptor, such as the EGF receptor family (EGFR, ERBB2 [HER-2/neu]), PDGF receptor family, VEGF receptor family, but also the insulin-receptor family. These receptor families are receptor tyrosine kinases. The receptor domain is located extracellularly and binds the growth factors, whereas the kinase domain is located intracellularly. This kinase domain is now a target for numerous modern anticancer agents, known as more- or less specific tyrosine kinase inhibitors.
Growth factors are polypeptides stimulating cell surface receptors very specifically with high affinity. Unlike endocrine hormones the specific growth factors usually have a local target, autocrine, paracrine or juxtacrine in character. Keratinocyte growth factor (KGF) is such a paracrine growth factor. Kepivance® is on the market as recombinant KGF, and has been specifically evaluated with epithelial cell lines and human carcinoma xenografts. While the results of these nonclinical studies confirmed a potential tumorigenic risk, they also provided important insight that there was a low likelihood that this would occur in humans [see below for further details] [17].
It is important to keep in mind that with systemic administration there might be barriers for growth factors to reach their targets, e.g. the extracellular matrix. Sometimes matrix components are actively involved in the interaction between growth factor and receptor. For example, heparin is involved in the interaction between fibroblast growth factor (FGF) and FGF-receptors [18].
These aspects of growth factors and their receptors are what create inherent risks for a carcinogenic potential.
8.2 Regulatory Guidance
The ICH S6(R1) guideline [19] is the primary source of advice for biopharmaceuticals and provides recommendations for the types of non-clinical studies with which to evaluate potential for toxicity. Because of their unique biological and physiochemical characteristics, ICH S6 recommends a scientifically based case-by-case approach. As discussed in detail above, although biopharmaceuticals are not genotoxic and therefore not expected to be ‘complete carcinogens’, chronic administration could potentially result in an increased risk of tumor promotion and/or growth based on their expected pharmacologic activity [20]. Evaluation of the carcinogenic potential for any new chemical entity depends on both its intended clinical duration of use, type of disease and specific concerns based on its pharmacological properties including genotoxicity as recommended in ICH S1A [1]. Although ICH S1A primarily addresses small molecular weight compounds, several scenarios are presented when a rodent 2-year bioassay should be considered for biopharmaceuticals. These include (1) different biological effects observed between the recombinant protein and the endogenous product; (2) structural differences between the recombinant product and natural product; and (3) recombinant products administered at pharmacologic doses greater than expected endogenous levels. These scenarios focus on recombinant proteins (e.g. growth factors, hormones and interferons) intended for replacement or augmentation therapy and do not pertain to other biopharmaceuticals such as mAbs and fusion proteins.
In the original ICH S6 guideline published in 1997, it was recognized that depending on the duration of clinical dosing, patient population and/or exaggerated pharmacology, an assessment of carcinogenic potential may need to be considered. The guidance suggests that a 2 year rodent bioassay assuming that relevant pharmacological activity can be sustained could provide useful data if the accumulated safety database is not sufficient to determine the potential for carcinogenicity. Since the pharmacology of certain classes of drugs, such as growth factors (see above) and immunosuppressive agents [19, 21] could represent a potential carcinogenic risk following chronic administration, the purpose of the S6 guideline was to offer alternatives rather than to default to the rodent bioassay to provide an appropriate carcinogenic risk assessment.
Differences in interpretation and implementation of the original ICH S6 guideline as confirmed by the increasing number of examples of opposing recommendations from the global regulatory regions led to an addendum of the guideline [19, 22]. The purpose of the addendum was to clarify several topics, including the carcinogenicity section while still maintaining the flexibility, and the case-by-case approach mandated in the original guideline. In the S6 addendum, with respect to the carcinogenicity, the section was expanded to provide more detail and to offer suggestions for different scenarios. Similar to the original guideline, a product-specific assessment of carcinogenic potential should be considered based on the duration of dosing and/or mechanism of action of the biopharmaceutical and when there is a potential concern, a number of approaches should be considered. This product-specific assessment should be based on accumulated nonclinical data and knowledge of the intended mechanism of action with the product. Literature data (from knock out animals, human genetic diseases), information from similar targets or class effects, and clinical data can provide useful information with which to base a risk assessment [19].
8.2.1 General Practical Advice
An advantage with respect to nonclinical safety strategy for biopharmaceuticals distinct from small molecular weight compounds is their type of toxicity, i.e. toxicity associated with biopharmaceuticals is primarily limited to exaggerated pharmacology and therefore potential toxicity should theoretically be easier to predict [23, 24]. Accordingly, a specific carcinogenicity risk-assessment strategy should be defined early in a development program and be updated periodically as non-clinical information accumulates. This risk assessment can be added to briefing documents submitted to Health Authorities and in addition to communicating risk, suggestions for a risk management plan which may include clinical or post-marketing monitoring and labeling proposals. A good example of a specific product carcinogenic strategy is a critical assessment from both a scientific and practical point of view to appropriately assess the potential carcinogenicity of Interleukin-10 [25]. Their assessment of the known biological activity across different species concluded that chronic administration of IL-10 would not be expected to be associated with a carcinogenic risk. In addition, a critique of the various other models such as transgenic mice and xenograft models were unlikely to provide relevant data.
Products will generally fall into one of three categories, those in which there are sufficient data for assessing potential carcinogenicity, those where there are insufficient data and those in which the mechanism of action infers a potential for carcinogenic risk.
Those products that enable an appropriate risk assessment without the need for additional nonclinical studies are those in which no data from either the repeat dose toxicity studies or alerts from a review of the literature (including knock-out animals, target biology) indicate that the candidate pharmaceutical is involved in either growth potential or cell proliferation. Recombinant proteins that are identical to the native protein sequences such as coagulation factors used for replacement therapy could be examples where additional studies may not be required. IL-10 discussed above is another example. Agents such as antagonists to growth factors, e.g. anti-VEGF mAbs meant to inhibit angiogenesis are also examples and in fact are used as an anti-cancer therapy.
Chronic administration of growth factors and immunosuppressive agents, on the other hand represent a potential concern for carcinogenic potential. For some of these types of products, i.e. growth factors, evaluation of transformed cells or xenograft models may be useful alternatives to the longer term in vivo repeat dose toxicity studies. Recombinant human keratinocyte growth factor (rHuKGF) for example was evaluated using human tumor cell lines (using both KGF+ and KGF- cell lines), a mouse xenograft model and a modified transgenic rasH2 (Tg.rasH2) model. In vitro results were not completely consistent; some of the tumor cell lines were positive and some were negative. Some of those positive cell lines were further evaluated in the xenograft model and one; possibly two of the six/seven that were positive showed a modest dose-dependent increase in growth [17, 20, 26, 27]. The rasH2 transgenic assay, however, was negative. In the repeat dose toxicity studies in rats, gastric hyperplasia and hypertrophy provided evidence of the expected pharmacologic activity of rHuKGF. Although the rasH2 transgenic model was negative, since increased proliferation was observed in at least one xenograft model, the USPI label (US packet insert) states under Warnings and Precautions “the effects of Kepivance® on stimulation of KGF receptor-expressing, non-hematopoietic tumors in patients are not known. Kepivance® has been shown to enhance the growth in human epithelial tumor cell lines in vitro and to increase the rate of tumor cell line growth in a human carcinoma xenograft model” [27]. A similar strategy was followed for recombinant human erythropoietin (rHuEPO) in which rHuEPO was incubated with various cell lines and evaluated in numerous xenograft models and in one mouse surrogate carcinogenicity study [28]. Erythropoietin produced no effect in any of these models [20]. Despite all of the negative data, the USPI carries a black box warning based on shortened overall survival in patients with certain types of cancers [29]. The Summary of Product Characteristics (SmPC) added that “erythropoietin receptors may be expressed on a variety of tumor cells” [20]. Similarly for immunosuppressive agents an increased risk of malignancy is generally accepted, which is why ICH S6(R1) recommends that with these types of compounds, the potential hazard is best addressed through appropriate product labeling and clinical risk management practices.
There will be targets, particularly novel ones, in which there are insufficient data available with which to conclude that no additional non-clinical data are needed. In this case, a more extensive evaluation may be necessary and may include the option of additional non-clinical studies which might include a 2-year rodent bioassay.
In summary, it is good practice to begin to consider the long term consequences of a particular target early in the discovery process. At this time, a review of literature, in conjunction with in vitro and in vivo efficacy data can provide knowledge of what level of potential concern exists for carcinogenic risk. For a target that has an obvious risk (i.e. growth factor agonist), additions to repeat dose toxicity studies can be included such as proliferation indicators. In addition, transformed cell lines, xenograft models, transgenic mouse models and of course the 2-year rodent bioassay can all be considered. However, these models need to be accurately characterized and scrutinized as to their relevance to the patient population. For example, questions such as whether target deficient mice which have reduced levels of, but are not depleted of target cells are a relevant model. Are there basic differences in physiology between human and rodents? The lack of cross reactivity with rodents could push for the need to use surrogate models and how representative are those models to human. Finally the lack of background data for many of the transgenic mouse models could lead to misinterpretation of findings. Therefore decisions about the type of studies need to consider the relevance of the animal data to human and that the conduct of the study should be designed to mitigate the concern or the label should reflect the concern.
8.3 Case Studies
8.3.1 Insulins and IGF-1
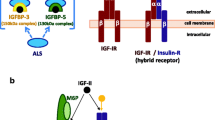
Insulin is a naturally occurring compound and also a known growth factor essential for normal functioning of metabolic processes. For a long time it was thought that insulin binds the insulin receptor and induces only metabolic effects, whereas IGF1R activation by IGF1 and IGF2 would induce mitogenic activities. With the discovery of the different isoforms of the insulin receptor it was found that activation of the insulin receptor A (IRA) could also induce mitogenic effects [30].
Like IGF2, IGF1 is able to activate the IGF1R but it has a low affinity for IRA and IRB. Insulin can only bind to IRA and insulin receptor (IRB) (Fig. 8.1).

Schematic overview of crosstalk of the insulin and IGF receptors. The red box in the α-subunit of the IRB represents exon 11
While the intended pharmacological action of insulin is mediated through IRB, the mitogenic potential of insulin and insulin analogues are related to their affinity for and downstream effect via the IRA and IGF1R.
There are two distinct and well studied signaling cascades, the PI3K/Akt and Erk/MAPK. PI3K/Akt is thought to have a major role in metabolism, whereas Erk/MAPK leads to the more mitogenic effects.
The phosphatidylinositol 3-kinase (PI3K) protein complex gets phosphorylated at the regulatory subunit p85α, the catalytic subunit of this complex produces PI(3,4,5)P3 (PIP3), a molecule that recruits phosphatidylinositol dependent protein kinase 1 (PDK1) to plasma membrane. PDK1 phosphorylates and activates the kinase Akt. Akt activates and inactivates a whole range of different proteins by phosphorylation including p27kip (which inhibits cell cycle inhibition), PDE3B (which induces lipolysis through PKA), FOXO1 (induces gluconeogenesis), Foxa2 (inhibits gluconeogenesis), AS160 (induces translocation of Glut-4, which induces glucose uptake), GSK3 (reduces glycogen synthesis and lipogenesis), AMPK (induces lipogenesis), TSC1, TSC2 (induces mTORC1 activity which include mitogenic effects), BAD (apoptosis), transcription factor FKHR (which induces mitogenic effects and c-Jun and JNK.
The Erk/MAPK signaling cascade is initiated by phosphorylation of IRS1/2 and Shc which recruit the SOS/Grb2 complex. This complex trigger activation of the membrane bound GTPase Ras, which in turn activates raf, which will phosphorylate Mek, which will phosphorylate Erk1 and Erk2. Finally the activated Erk will phosphorylate numerous substrates (Elk1, c-myc, SRC1, Pax6, STAT3 and c-FOS), these substrates are involved in the onset of the transcription machinery that will lead to the mitogenic effects (angiogenesis, cell proliferation, cell survival, protein synthesis and cell growth).
Insulin analogues are widely used to control the blood glucose levels in a more steady and precise manner than it would be possible with regular human insulin injections. Small variations have been incorporated in the insulin molecular structure to change its ADME (administration, distribution, metabolism and excretion) characteristics. One of the first insulin analogues developed was insulin AspB10, a fast acting insulin analogue with its histidine-B10 residue replaced by an aspartic acid residue [38]. This molecule was known to have an increased binding activity for IR, but harbored also a 7–10 times higher binding affinity for IGF1R [39]. Several studies reported an enhanced proliferative behavior of cancer cells after stimulation with AspB10 compared to regular insulin [40–44]. It is thought that this mitogenic activity of AspB10 was caused by up regulation of the p70S6K signaling pathway [45].
Also in vivo studies have been performed to study the mitogenic actions of insulin AspB10 and directly after the first in vivo study reported an increased incidence of mammary tumors in female rats; the development of AspB10 was discontinued [46]. Also some recent studies could confirm the increased carcinogenic potential of insulin AspB10 using different mouse and rat models [42, 47, 48].
This finding of mammary tumors for AspB10-insulin also drew the attention of the regulatory authorities for human medicines, and they prepared a position paper to deal with this issue [49]. In fact, the Position paper emphasized that sponsors should scientifically support their approach to evaluate the carcinogenic potential of new insulin analogues, without choosing by default to conduct a 2-year rat study. A stepwise approach starting with in vitro receptor pharmacology is proposed, and the approach chosen by Ter Braak et al. [43, 48] reflects this.
Since AspB10, many other insulin analogues have been developed and all of them have been thoroughly tested for possible carcinogenic side effects.
One insulin analogue (insulin Glargine) has obtained extra attention since, like insulin AspB10, this compound has an increased binding affinity towards the IGF1R [50, 51]. Some in vitro studies showed increased mitogenic effects for insulin glargine [43, 52–54], where others found no increased mitogenic potential of insulin glargine compared to regular human insulin [55, 56]. A likely explanation for these contradictory findings is that in the presence of serum, glargine is rapidly metabolized by endopeptidases into M1 and M2 (M1 after removal of the two arginines, M2 with additional deamination of threonine at position B30) which have a low mitogenic potency [57]. Therefore, depending on the experimental set-up either the mitogenic potency of glargine or M1 and M2 has been measured.
The results from in vivo studies regarding the carcinogenic potential of glargine have generally shown negative results [47, 58, 59] although the most recent study using a conditional breast cancer mouse model did reveal up regulation of mitogenic MAPK-signalling pathway similar to AspB10 and IGF1 [48].
The examples described above of insulin Asp B10 and insulin glargine, underline the importance of appropriate testing for carcinogenic potential of new insulin analogues. Currently, the focus lies on receptor binding and functional effects for the IR isoforms and IGF1R, but characterization of downstream signaling pathway activation as shown in Fig. 8.2, also seems to have important predicting potential. The transcriptomic changes downstream of these pathways can be used as a tool to predict the biological direction of cells and tissues. A microarray provides a high-throughput platform for the development of such genetic classifiers as described [60], and for drug screening purposes quantitative PCR is a cheap alternative to quickly evaluate the mitogenic potential of these growth factors. Additionally chronic in vivo experiments could be useful to evaluate the in vivo carcinogenic effects of insulin analogues. While the use of humanized cancer models might improve the accuracy of carcinogenicity assessments and reduce animal numbers, further evaluation is needed to demonstrate clinical relevance of these models. Since cancer is a heterogeneous disease it is essential that the tumors in these models are properly characterized preferably by combining different omics approaches (as was done in ter Braak et al. 2015, [NGS] (manuscript in preparation).

8.3.2 GLP1-Agonists
In the last few years, GLP-1 receptor agonists have been on the market for treatment of type II diabetes. These drugs are designed to improve the balance between insulin and glucagon secretion, to lower gastric emptying and to reduce appetite. Several products are currently on the market AstraZeneca has marketed Byetta® (exenatide fast release [FR]), and Bydureon® (exenatide slow release [SR]), Novo-Nordisk has marketed Victoza® (liraglutide) and Sanofi has marketed Lyxumia® (lixisenatide) [61–64]. Eli Lilly received a marketing authorisation for Trulicity® (dulaglutide) in the summer of 2014 [65]. In addition, Eperzan® (albiglutide) was approved in 2014 [66].
Exenatide and lixisenatide are synthetic peptides based on solid phase peptide synthesis and are not biopharmaceuticals in the sense of a recombinant biotechnologically-derived product [61, 64]. Liraglutide is a recombinant protein produced in Saccharomyces cerevisiae [63]. Dulaglutide is a recombinant protein, generated in Chinese Hamster Ovary (CHO) cells, and consists of two chains in one molecule, with IgG4 Fc-parts as the bases [65].
Carcinogenicity studies in rats and mice showed a special effect of these GLP-1 agonists with respect to induction of tumors, i.e. an increased risk for thyroid C-cell carcinogenicity. This was seen for the first in class, exenatide, with a small risk with the fast-release form Byetta, but with an enhanced risk in the slow release form, Bydureon. It was also observed with liraglutide (Victoza), which has slower elimination than exenatide. The latter two products thus have higher chronic exposure then Byetta.
A mode of action (MOA) was identified relating GLP-1r agonist exposure to C-cell carcinogenicity (Fig. 8.3). This MOA was confirmed to be GLP-1 receptor specific. Basically, the MOA tells us that binding of the agonist to GLP-1 receptors on the surface of the C-cell leads to increased cAMP concentrations in the cell. Apparently this stimulation of the cAMP concentration leads to an increase of the mRNA for calcitonin, and the production of calcitonin itself by the C-cells. This increase in mRNA is associated with hyperplasia of the C-cells, and eventually some cells are transformed to adenoma and carcinoma (see section 8.1.1).

Schematic representation of the mechanism of action relating GLP-1 receptor agonist exposure to C-cell carcinogenicity
The question arose as to which underlying factors determine C-cell carcinogenicity and how these factors may explain the differences among the various GLP-1 analogues. An additional question is whether the effects seen in rodent are relevant to humans. Indeed, generally a linear concentration-effect relationship is assumed when scaling the animal observations to the human situation.
However, receptor-mediated effects usually have non-linear concentration-effect sigmoid relationships. In addition, animals may have a different sensitivity that is not quantified in the standard approach of carcinogenicity assessment. A technique to answer such questions is mechanism based pharmacokinetic-pharmacodynamic (PK-PD) modeling [67–70]. We developed such a framework for assessment of GLP-1 receptor induced C-cell carcinogenicity by producing a PK model for the various GLP-1 products in animal studies, initially for exenatide FR [Byetta®] and liraglutide [Victoza®] [71]. In a following step, the liraglutide PK model was successfully extended with a PD model, describing the concentration-effect relationship with plasma calcitonin as a biomarker.
Based on this framework a PKPD model was developed for both exenatide FR and exenatide SR (Bydureon®). Given the lack of information on calcitonin levels in this case, a logistic regression model was developed linking the chronic exposure of exenatide directly to adenoma incidence.
The modeling approach provides a promising method to investigate the underlying mechanisms of the exposure response relationship in toxicological problems regarding a single drug or a drug class. The analysis conducted thus far also illustrates the importance of applying this approach from the very beginning of the development of a pharmaceutical candidate. During the application of the modeling approach some data gaps or weaknesses in the design of a study program (e.g. lack of calcitonin data) became clear. Further steps have still to be explored to apply this approach for the translation to the human situation.
GLP-1 receptors have been proven to be co-localized with thyroid C-cells in humans, but the density is much lower, although not absent. Data are available from long-term treatment with liraglutide in nonhuman primates. After 52 weeks no effects of liraglutide on C-cell proliferation was observed [63], suggesting that primate C-cells are less sensitive to proliferation induced by stimulation of GLP-1 receptors. The relevance of the rodent tumors for humans is likely to be low but cannot be completely excluded (See Sect. 8.4. Pharmacovigilance, risk management and regulatory actions taken).
8.3.3 Immunosuppressive and Immunomodulatory Agents
Immunomodulatory therapeutic monoclonal antibodies currently comprise a large portion of biopharmaceuticals available for clinical use and are widely prescribed. The theoretical risk for long term use of these agents is the risk of malignancy, in particular lymphoma which could result from a disruption of the immune system’s host defense [20, 72]. Increased tumor risk, primarily lymphomas has been associated with genetic immunodeficiencies such as severe combined immunodeficiency (SCID) and Wiskott-Aldrich syndrome [72, 73]. In addition, an increased incidence of tumor types has been observed as a result of viral infections. For example, infection with HIV is associated with an increase in tumors such as Kaposi’s sarcoma and Burkitt’s lymphoma and in the case of HPV infection; an increased risk of cervical cancer has been observed [74, 75].
Cyclosporine, a commonly prescribed medicine for diseases such as psoriasis, has been shown to induce lymphomas in monkeys [76]. In addition, clinical use with azathioprine and cyclosporine in renal transplant patients have been associated with an increase in a number of tumor types, the majority being lymphoma and skin tumors with lower incidences in other organs such as lung, cervix, brain, etc. In one data set from the Human Kidney Transplant Registry (1971–1976), incidences of lymphomas and skin tumors could be as high as 30–40 and 4.2 times the general population, respectively [74, 77].
With respect to biopharmaceuticals, OKT3, the first therapeutic monoclonal antibody available in 1986 was used as an anti-rejection drug for organ transplantation [78, 79]. Its primary in vivo action is to opsonize the circulating lymphocytes by binding to the CD3 receptor. These cells are subsequently removed by the reticuloendothelial cells in the liver and spleen and are non-functional when they reappear [80]. Because OKT3 cross reacts only with human, chimpanzee and gorilla CD3, no animal chronic toxicity or carcinogenicity data are available, clinical experience with OKT3 provides the bulk of the safety database [78]. Shortly after the introduction of OKT3, a sharp increase in post-transplantation lymphoproliferative disorder (PTLD), a well recognized complication of immunosuppression became apparent in cardiac transplant patients [81]. The incidence of PTLD was higher in OKT3-treated patients than in patients who did not receive OKT3, 11.4 and 1.3 %, respectively. According to Swinnen’s multivariate analysis [81], the only factor that was significantly associated with PTLD was the use of OKT3. A dose response relationship was also evident in that 35.7 % of patients that received a cumulative dose of more than 75 mg OKT3 had PTLD versus 6.2 % patients that received a cumulative dose less than 75 mg OKT3. In addition, the interval between OKT3 treatment and PTLD emergence was shorter, often 1–2 months in patients with the higher cumulative dose although definitive conclusions cannot be confirmed given the low number of patients [81]. Primary infection or reactivation of Epstein-Barr virus (EBV) is also thought to play a role in the pathogenesis based on its presence in tumor tissue obtained from lymphomas and the development of PTLD is thought to result from an inadequate T-cell control over EBV-driven B cell proliferation [72, 81]. Although various lymphomas are associated with EBV there are differences in disease burden. For example EBV can be detected in >90 % of cases from patients that develop lymphoma within 1 year following transplantation whereas EBV was detected in approximately 50 % of patients who developed lymphomas after 1 year following transplantation [72, 82, 83]. In addition, there was no difference in the frequency of reactivation of EBV between those patients dosed with OKT3 that exhibited PTLD and those that did not [81].
The potential association with reactivation of viruses has also been observed in animal studies. Administration of alefacept, a fusion protein which it binds to CD2 inhibits the CD2/LFA-3 interaction thus resulting in T cell depletion. In the 12 month monkey toxicity study, a lymphoma was noted in one female. In addition, B cell hyperplasia was observed in some of the other monkeys. Reactivation of lymphocryptovirus (LCV) was thought to be related to the lymphoproliferative changes as it is known that LCV infection can lead to B-cell lymphomas in immune suppressed monkeys [20, 84].
Abatacept, a fusion protein which inhibits T-cell activation by blocking the interaction the antigen presenting cell with CD28, produced an increase in the incidence of malignant lymphomas at all dose levels and mammary gland tumors in female mice at the mid and high dose levels in a 2 year bioassay. Further analysis of the mice showed that they were infected with murine leukemia and mouse mammary tumor viruses. Similar to the example discussed above, it is known that reactivation of these viruses can occur in immunosuppressed mice. However, administration of abatacept to monkeys for 12 months did not result in lymphoproliferative disease even though there was evidence of immunosuppression (depletion of germinal centers in lymph nodes and spleen) and the monkeys were known to be infected with LCV. Therefore, although there appears to be an association between immunosuppression and reactivation of latent viruses leading to an increased risk in malignancy, assessment of the relevant clinical risk are difficult due to the variability in results [20].
Host defense mouse models have also been used to evaluate immunomodulatory agents. In one published example, keliximab (anti-CD4 mAb) was tested in a B16 melanoma experimental metastasis model. Although administration of the positive control (a pan T-cell antibody) increased the number of lung metastases, keliximab, a selective CD4+ mAb had no effect [85].
In general, given the variability in results in animal studies and the lack of confidence that a negative finding in a non-clinical model can mitigate or eliminate the theoretical risk, the conduct of these animal studies is not recommended and hence few published examples exist. Instead the theoretical risk is typically outlined in the product label under Warnings and Precautions including a defined Risk Management plan. Therefore for many of the immunomodulatory agents the current clinical practice is to monitor patients post-marketing as is the case with the anti-TNF therapies.
8.3.3.1 Tumor Necrosis Factors (TNF) Inhibitors
TNF was initially isolated as a key cytokine involved with the necrosis of tumors, hence its name [86, 87]. Current understanding of the biology of TNF is that it is believed to play a regulatory role in inflammation and host defense [72, 88]. However, TNF has also been shown to exhibit diverse effects on tumor biology which are not completely understood. In contrast to inhibition of tumor growth, locally produced TNF (i.e. within the tumor microenvironment) has been shown to promote DNA aberrations resulting in maintenance of cancer growth and spread [88, 89]. In clinical trials, high levels of TNF in patients with chronic lymphocytic leukemia were associated with increased tumor spread [89, 90]. Therefore, depending on the situation, TNF can be viewed as either an anti-cancer agent or as a tumor promoter. In fact, data from several Phase I studies have shown a stabilization of disease in some of the patients with progressing advanced cancer [88, 91]. Although its exact function still needs to be clarified, anti-TNF mAbs have demonstrated effective control of auto-immune diseases such as rheumatoid arthritis [RA] and inflammatory bowel disease [IBD] [92, 93]. Because of its involvement in host defense, a number of studies have been conducted in RA patients to evaluate whether an increased risk in malignancy is associated with anti-TNF therapies. Conflicting results however have been observed (Table 8.1). The primary reason for this discrepancy could be due to the method of data analysis. Some of the investigations analyzed data using randomized clinical trials (RCTs) whereas national clinical, health, and demographic country registers were used to collect data in other studies [94–104]. Analyzed data from early RCTs in RA patients showed a dose-dependent increased risk for malignancy following the use of infliximab and adalimumab and a trend for an increase for etanercept [95, 96]. A subsequent study that evaluated a greater number of RCTs (63 versus 9), additional anti-TNF therapies to infliximab, etanercept and adalimumab (golimumab, certolizumab), and other mAbs (rituximab [anti-CD20] and tocilizumab [anti-IL6]) did not observe an increased risk of malignancy in patients treated for at least 6 months [104].
In addition, increased malignancy was not elevated following anti-TNF treatment in cohort studies using country registries [89, 97–100, 105]. In one of these studies, 6366 RA patients who had recently begun anti-TNF therapy were followed in some patients for up to 6 years for a total of 25,693 person-years and except for the first year of follow up, no differences were noted among the 3 anti-TNF drugs (adalimumab, etanercept and infliximab). During that first year, as compared with the cohort of unselected, biologics-naïve patients, patients receiving adalimumab exhibited an increased risk in malignancy (relative risk of 1.91) whereas etanercept usage resulted in a decreased risk (relative risk of 0.43) and infliximab was associated with a relative risk of 1.23. Beyond the first year follow up, the relative risks for all three were similar (0.80–0.83) [100]. Although no association was observed between solid tumors and lymphomas, an apparent increase risk in skin cancers, both nonmelanoma and melanoma skin cancer has been observed in two studies [94, 98].
While it is important to understand the potential risk of new malignancies, other questions such as whether anti-TNF treatment is associated with an increased risk of recurrent malignancies or associated with a worse prognosis of cancer that occurs either during or after treatment. For example, should physicians treat patients with anti-TNF who have a history of malignancy or if so how long should they wait following recovery from malignancy to start treatment? Data for two separate observational studies (German and British registries) did not demonstrate an increased risk of malignancy. Although in one study there was a slightly higher recurrence rate in those patients with prior malignancy in the anti-TNF treatment group as compared with patients treated with DMARDs, it was not statistically significant. In addition, patients without prior malignancy did not show an increased risk as compared with the unexposed patients [105]. While these data are encouraging, it should be noted that they are based on relatively small sample sizes most likely due to the reluctance of physicians to prescribe anti-TNF treatment to a patient with prior malignancy [86, 105, 106].
The conflicting results observed among these studies are most likely due to differences in study type (RCT vs. cohort), duration of exposure to anti-TNF treatment, composition of control or cohort group and sample sizes. Other limitations include the rarity of lymphomas and other cancers which complicate statistical analysis in that relatively small changes in the numerator can result in a major change in estimated risk [96]. Another complication is that lymphoma is increased in the general RA population [107]. In some of these studies it appeared that the increased risk observed after the first year was not maintained which could be due to differences in cancer detection not cancer causation [100]. Another bias is called “channeling” in which the patients with more severe disease are the ones that receive the anti-TNF therapy earlier and who may already be at a higher risk of developing lymphoma [98].
8.4 Pharmacovigilance, Risk Management and Regulatory Actions Taken
8.4.1 Detecting and Assessing Cancer Risks
As discussed previously, the intended pharmacology of some biopharmaceuticals can be expected to lead to an increased carcinogenic risk such as immunmodulators and growth factors. As animal models have a low predictive value for these biopharmaceuticals, it has been the recommendation by ICH S6(R1) that post marketing follow-up for biotechnologically-derived proteins may be more informative. In such situations the problem may be dealt with by including it in the Risk Management Plan (RMP) as a potential risk, sometimes with obligatory additional studies. Cancer risks have been associated with a considerable number of medicines. Amongst these are medicines used for the actual treatment of cancer itself, but also a large number of medicines for chronic diseases. TNF-inhibitors intended for diseases such as rheumatoid arthritis, erythropoietin used in cancer patients, insulin glargine or GLP-1-analogs in diabetes mellitus.
The concerns of a cancer risk can arise at any time before or after approval. In other cases concerns rise after adverse events have been spontaneously reported or published in the scientific literature, or when the results of interventional or observational clinical studies have been published.
Whatever the source, a suspicion of higher occurrence of malignant or premalignant disease needs further assessment. An evaluation of the strength of the evidence includes the plausibility, biological mechanisms, dose-response relation, strength of the association, time-to-onset, consistency, specificity [108]. When causality is considered possible and relevant, additional action may be needed to further study, minimise, and communicate the risk. The impact on the benefit-risk balance and public health in general must be assessed.
Assessing cancer risks in patients is often difficult. While time may be pressing, due to the severity of the adverse event, data are frequently not available or of insufficient quality. Often it takes several years before the first data becomes available. Pharmacoepidemiological studies, using observational data may suffer from possible confounding by indication or disease severity (e.g. in the study of insulin or erythropoietin and cancer risks). The choice of suitable reference groups can be challenging, or non-users of comparable disease severity may simply not exist in case of debilitating/life-threatening or orphan indications (e.g. in insulin, somatropin or TNF-inhibitors). Induction and latency times need to be distinguished, but are often impossible to separate [109].
8.4.2 Examples from Post-marketing Experience
We list here a few examples of biopharmaceuticals for which malignancies were reported post marketing in the recent past. The overview is based on the European Medicines Agency (EMA) or FDA safety labelling updates and European public assessment reports (EPAR) and illustrates the challenges associated with justifying regulatory action (e.g. updating the product information, including communication of the risk) based on often a limited number (but serious) case reports, or difficult to interpret and sometimes contradictory results from large and lengthy studies.
Calcitonine (long term use) Miacalcin (calcitonin-salmon) Injection and Nasal Spray).
On 19 July 2012, the EMA completed a review of the benefits and risks of calcitonin-containing medicines, concluding that there was evidence of a small increased risk of cancer with long-term use of these medicines. The Agency’s Committee for Medicinal Products for Human Use (CHMP) recommended that they should only be authorised for short-term use in Paget’s disease, acute bone loss due to sudden immobilisation and hypocalcaemia caused by cancer. The CHMP also concluded that the benefits of calcitonin-containing medicines did not outweigh their risks in the treatment of osteoporosis and that they should no longer be used for this condition [110, 111].
In March 2014 the FDA issued a label update on Malignancy: In a meta-analysis of 21 randomized, controlled clinical trials with calcitonin-salmon (nasal spray or investigational oral formulations), the overall incidence of malignancies reported was higher among calcitonin salmon-treated patients (4.1 %) compared with placebo-treated patients (2.9 %). Among the tumor types, basal cell carcinoma was the most common type of tumor. Other types included breast cancer and non-melanoma skin cancers. The malignancy risk in individual studies was generally not statistically significant; however in CT 320 [112], a large vertebral fracture prevention trial in postmenopausal women, a statistically significant increase in risk of malignancy was observed (Odds-ratio = 1.62, 95 % CI: 1.00, 2.61). There was no excess of malignancies with Miacalcin for treatment up to 6 months, while at longer treatment durations more malignancies were reported with Miacalcin treatment than with placebo. The FDA advised that the benefits for the individual patient should be carefully considered against possible risks.
Tumor necrosis factor (TNF) inhibitors include infliximab (Remicade), adalimumab (Humira/Trudexa) or certolizumab pegol (Cimzia), or with a circulating receptor fusion protein such as etanercept (Enbrel) [113–116].
In Jan 2005 the CHMP revised the product information for infliximab (Remicade; EU approval in 1999) and adalimumab (Humira, EU approval in 2003) to include details of the post-marketing experience on malignancies and lymphoproliferative disorders, including incidence. At that time possible risk for the development of lymphomas or other malignancies in patients treated with a TNF-antagonist could not be excluded.
In 2006, post marketing reports of hepatosplenic T-cell lymphoma (HSTCL) were identified for the anti-TNF agent Remicade (infliximab). Since launch in 1998 to about early 2006, six cases of hepatosplenic T-cell lymphoma had been reported in patients with Crohn’s disease treated with infliximab. Five of them were in the age range of 12–19 years. All patients were on concomitant treatment with azathioprine or 6-mercaptopurine. Based on the data presented, a causal relationship of hepatosplenic T-cell lymphoma and infliximab therapy cannot be excluded. The relevant sections of the SmPC were updated to include the information on this finding, and a Direct Healthcare Professional Communication (DHPC) was sent out.
In May 2008 the product information of adalimumab (Humira/Trudexa) was revised related to the reports of rare cases of hepatosplenic T-cells lymphoma in patients treated with adalimumab. It was not considered that the reports of these rare cases of HSTCL alter the positive benefit/risk balance for adalimumab in the approved indications, when the PI is updated. Nevertheless, a DHPC was recommended. The target groups of prescribers for the DHPC should be the same as for the DHPC sent out about 2 years before for Remicade, due to the identification of HSTCL.
In June 2008 after three spontaneous reports of hepatosplenic T-cell lymphoma that prompted the update of section 4.4 and 4.8 of the SmPC of Humira®, a DHPC to alert prescribers about these very rare events was distributed. The CHMP Pharmacovigilance Working Party (PhVWP) suggested that a RMP of anti-TNF drugs should contain educational material aimed at the diagnosis of this extremely rare event.
Additionally, it was noted that this issue had been the subject of a recent communication from the FDA.
In November 2009 the FDA issued a label update (Boxed warning) on Malignancy: Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, of which Humira is a member.
In March 2010 based on a post marketing cumulative review of all malignancies in paediatric and young adult patients with infliximab a causal relationship between infliximab and the development of paediatric malignancies cannot be established. It is possible that concomitant exposure to other immunosuppressants and/or presence of underlying autoimmune diseases were contributory factors. Nevertheless, given its mechanism of action as TNF-blocking agent it cannot be excluded that infliximab may be also a contributing factor in the development of the observed malignancies.
In March 2010 cumulative reviews of the cases of leukaemia in adult and malignancies in paediatric patients reported with use of adalimumab did not allow establishing a causal relationship between the development of these malignancies and adalimumab. It is possible that concomitant exposure to other immunosuppressants and/or presence of underlying autoimmune diseases were contributory factors. Nevertheless, given its mechanism of action as TNF-blocking agent it cannot be excluded that adalimumab may be also a contributing factor in the development of the observed malignancies.
In November 2012 the cumulative review of registries, clinical trials and post marketing cases of Merkel cell carcinoma (MCC, or neuroendocrine carcinoma of the skin) coincident with infliximab or golimumab use identified 19 reports for infliximab and none for golimumab. All 19 reports were post marketing cases. No MCC cases were observed in registries and clinical trials. Of the 19 reports there were 2 fatalities reported in patients either taking multiple immunosuppressants concomitantly with infliximab or with limited information regarding medical history. Of the 19 reports, most of them had confounding factors (i.e. one or more risk factors for MCC such as prior immunosuppressant history, concomitant immunosuppressant therapies, and/or a history of malignancy) limiting the causality assessment with infliximab. Based on this review, MCC is considered causally associated with the use of infliximab, and a drug class effect to TNF inhibitors. Key factors supporting this conclusion include the biological plausibility based on immunosuppression by TNF-α inhibitors, the apparent sensitivity of MCC to immunosuppression, and the elevated reporting rate compared with the background rate of this type of cancer, all which suggest an association of MCC with this drug class. MCC is therefore added to section 4.8, with a frequency category of “Not known” for both infliximab and golimumab, as the frequency of the event cannot be estimated from the available data. The severity and seriousness of the event of MCC also justify its addition to section 4.4 warning prescribers that cases of MCC have been reported in patients treated with TNF blocker therapy and recommending periodic skin examination, particularly for patients with risk factors for skin cancer.
Based on the cumulative review of melanoma cases, coincident with infliximab or golimumab use it remains unclear whether a causal relationship exists between infliximab or golimumab use and the development of melanoma, however the possible contribution of infliximab or golimumab use to the risk cannot be excluded.
In Nov 2012 the cumulative search of the company clinical and post marketing databases for reports of possible Merkel Cell Carcinoma (MCC) or neuroendocrine carcinoma of the skin coincident with adalimumab therapy identified 15 reports of MCC. One report was from clinical trials and there were 14 postmarketing reports. Of the 14 post marketing reports, most of them had confounding factors and/or limited information to fully assess causality with adalimumab and 1 report had no confounding factors or alternative etiology reported. The 1 report of MCC from a clinical trial also had confounding factors. There were no fatalities due to MCC among the total of 15 reports. Although it is not clear whether the appearance of MCC in patients receiving adalimumab might be due to a number of factors such as other TNF inhibitor therapy, the underlying autoimmune diseases, sun exposure, the patient’s age, or exposure to other non-biologic immunosuppressant therapy, the possible contribution of adalimumab use to the risk cannot be excluded.
In Jan 2013 for adalimumab (Humira) an index case of glioblastoma in a 28 year old female for the indication of psoriasis was reported. Unknown and very rapid onset of malignant grade 4 glioblastoma developed with fatal outcome 6 weeks after diagnosis. Up to Jan 2013 the MHRA had received the following UK cases; 5 cases of glioblastoma, 5 cases of brain neoplasm and 2 cases of brain neoplasm malignant. There is limited information on the onset from first dose in 5 of the other UK cases. 5 have an onset of >6 months. In two of the cases with onset >6 months it is possible that the neoplasm is a recurrence of previous brain cancer or has metastasised from a different primary malignancy. The MHRA have received the following Non-UK reports; 11 cases of glioblastoma, seven (7) cases of glioblastoma multiforme, fifteen (15) cases of brain neoplasm, and two cases of brain neoplasm malignant. The SmPC currently labels solid organ neoplasm including breast cancer, lung neoplasm and thyroid neoplasm.
“Other malignancies” is an important potential risk in the RMP of adalimumab. An increased risk of cancer is a known risk with all TNF inhibitors, although there is variation in which types are specifically mentioned in the product information. In the Pharmacovigilance Risk Assessment Committee (PRAC) it was discussed whether European registries are available to examine for further evidence, however none were known to be currently running except for the Rheumatoid Arthritis registry, which would be of limited use as it is open to all drug substances. It has therefore been decided to raise this issue.
In May 2013 the FDA issued a label update on Malignancies: The potential risk with the combination of azathioprine or 6-mercaptopurine and HUMIRA should be carefully considered.
A recently published Swedish cohort study Raaschou et al. [117] in 120 TNFi-treated and 120 biologics-naive individuals concluded that among patients with RA and a history of breast cancer, those who started TNFi-treatment did not experience more breast cancer recurrences than patients with RA treated otherwise.
Taken together the current pre and post-marketing results from studies, registries and spontaneous reporting is sometimes conflicting and many uncertainties remain regarding the contribution of risk factors and actual baseline risk with the patient under TNF inhibitor treatment (also see section E Table 1). Our post-marketing overview illustrates that past regulatory actions have been considered justified based the seriousness (although rare) of the cancer risk. Nevertheless, the remaining uncertainties present challenges for timing and content of risk minimisation and communication.
Known risks:
Erythropoietin (Epogen/Procrit (epoetin alfa) and Aranesp (darbepoetin alfa)
In October 2006 section 5.1 of the SmPC of Aranesp was updated to modify the language relating to influence of darbepoetin alfa on tumour progression and survival for lymphoproliferative disease patients treated with darbepoetin alfa.
In July 2011 the FDA issued Safety Labeling Changes on risk on tumor progression or recurrence [118].
Somatropin (growth hormone) (high dose) risk on tumour progression or recurrence. In general, somatropin is contraindicated in the presence of active malignancy. Any pre-existing malignancy should be inactive and its treatment complete prior to instituting therapy with somatropin. Somatropin should be discontinued if there is evidence of recurrent activity. Since growth hormone deficiency may be an early sign of the presence of a pituitary tumour (or, rarely, other brain tumours), the presence of such tumours should be ruled out prior to initiation of treatment. Somatropin should not be used in patients with any evidence of progression or recurrence of an underlying intracranial tumour [119].
On 10 December 2010, the European Commission initiated a procedure under Article 20/Article 107 referral for somatropin-containing medicinal products and requested the CHMP to assess all the available data and its impact on the risk benefit balance for somatropin-containing medicinal products. The scope of the review was to assess the long-term safety of growth hormone treatments in light of the emerging safety data from the French ‘Santé Adulte GH Enfant’ (SAGHE) study. In particular the assessment regarded the potential increased risk of mortality due to diseases of the circulatory system, bone tumours and subarachnoid or intracerebral haemorrhage in children and when high doses are used. It looked at data on 10,000 adults who started treatment between 1985 and 1996, using a mandatory national registry. An analysis in approximately 7000 of those patients who were treated for growth hormone deficiency and for gestational or idiopathic short stature showed a possible increased risk of mortality with somatropin compared with the general population. In particular, an increased risk of mortality due to bone tumours and cardiovascular events (such as bleeding in the brain) was seen. The risk appeared to be highest when doses higher than the ones approved were used.
Based on the evaluation of the currently available data and the scientific discussion within the Committee, the CHMP concluded that the benefit-risk balance of somatropin-containing medicines remains positive when used in the approved indications at the approved doses. However, to ensure that somatropin-containing medicines are used appropriately, the CHMP recommended that specific wording be included in the product information of all somatropin-containing medicines. In particular, the harmonised wording will emphasise that somatropin must not be used if there is any evidence of tumour activity, and that the maximum recommended daily dose should not be exceeded.
Unknown risks:
Insuline glargine (also see section C. Animal case studies a. Insulins and IGF-1)
Conclusions of large population based studies were not consistent with each other. Problems with these patient based studies arise because investigators are fully dependent on the information that is provided in the database. Often patient information lacks for example, BMI, smoking habits, familial cancer incidence etc. while these factors might have a significant impact on the development of cancer (Table 8.2).
In addition, prescription information including the dose or duration of the treatment is not always known or taken into account. The follow-up duration of these studies is often short, less than 5 years, whereas it is doubtful whether this timeframe is long enough for a tumor to develop de novo. Further, there is the problem of causality; doctors might prescribe specific insulin analogues to patients with specific health related problems, so we might observe an increased carcinogenic risk of a certain treatment whereas the treatment itself does not cause such an effect. Lastly, some of these studies are severely criticized because of a lack of statistical analysis [130–132].
In May 2010, based upon the data that have become available since the granting of the initial Marketing Authorisation, the CHMP considered that the benefit-risk balance of Lantus remains positive, but determined that its safety profile should be closely monitored for the following reasons: Following the publication of four epidemiological studies on the risk of (breast) cancer with the use of insulin glargine in the journal Diabetologia, concerns were raised about the safety of insulin glargine in this respect. At the time of the first renewal (May 2010), three post-marketing pharmacoepidemiology studies were initiated by the MAH to further investigate the possible increased risk of cancer associated with the use of insulin glargine.
In June 2012 the results from the ORIGIN trial (Outcomes Reduction with an Initial Glargine INtervention) were published. This was a multinational 7-year randomized clinical study that investigated the effect of Lantus on cardiovascular (CV) morbidity and mortality in patients with pre diabetes (impaired fasting glucose [IFG], impaired glucose tolerance [IGT]) or early Type 2 diabetes mellitus (T2DM) who had evidence of CV disease.
Based on review of the data the CHMP concluded in December 2013 that a cancer relationship to insulin glargine was not demonstrated in any cancer subtype (e.g. breast, colon, prostate, lung), or for new or recurrent cancers, or deaths from cancer, over 6.2 years of median follow-up. Kaplan-Meier curves for the first cancer diagnosed during the trial, the first new cancer diagnosed, and death due to cancer were practically super imposable between the insulin glargine and standard care groups. Although this is important information, it has not been included in the SmPC because, (i) the design of the ORIGIN study was not anticipated in order to assess the risks of cancer (ii) it is questionable whether these results could be extrapolated to long-standing diabetes with high doses of insulin (iii) available epidemiological results regarding the risk of breast cancer for longer exposures to glargine are not fully consistent across different studies.
GLP1-agonists (exenatide (Byetta/Bydureon), approved in 2005/2012; liraglutide (Victoza), approved 2010]; lixisenatide (Lyxumia), approved in EU 2013; albiglutide (Eperzen), approved in 2014 by GSK). (Also see section C. Animal case studies b. GLP1-agonists) [61–64, 66].
In September 2011 (first renewal) based upon the data that have become available since the granting of the initial Marketing Authorisation, the CHMP considers that the benefit-risk balance of Byetta (exenatide) remains positive, but stated that its safety profile should be closely monitored for the following reasons: A number of safety issues have been identified for Byetta, in particular the potential association between exenatide and pancreatic cancer and thyroid neoplasms. The latter will be further investigated in a new epidemiological study. Also the possible drug interaction between exenatide and tacrolimus and exenatide and lamotrigine needs further evaluation.
References
International Conference on Harmonisation. ICH Guideline S1A: Need for Carcinogenicity Studies of Pharmaceuticals. November 1995. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S1A/Step4/S1A_Guideline.pdf. Accessed 23 Dec 2014
Van Oosterhout JP, Van der Laan JW, De Waal EJ, Olejniczak K, Hilgenfeld M, Schmidt V, Bass R (1997) The utility of two rodent species in carcinogenic risk assessment of pharmaceuticals in Europe. Regul Toxicol Pharmacol 25(1):6–17
Bronchud MH (2007) Molecular oncology. In: Meyers RA (ed) Cancer. From mechanisms to therapeutic approaches. Wiley-VCH Verlag, Weinheim, pp 3–54
Maher ER, Neumann HPH, Richard S (2011) von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 19(6):617–623. doi:10.1038/ejhg.2010.175, Epub 2011 Mar 9
Somasundaram A, Ardanowski N, Opalak CF, Fillmore HL, Chidambaram A, Broaddus WC (2014) Wilms tumor 1 gene, CD97, and the emerging biogenetic profile of glioblastoma. Neurosurg Focus 37(6):E14. doi:10.3171/2014.9.FOCUS14506
Kinzler KW, Vogelstein B (1997) Gatekeepers and caretakers. Nature 386:761–763. doi:10.1038/386761a0
DeVries A, Van Oostrom CTM, Hofhuis FMA, Dortant PM, Berg RJW, DeGruijl FR, Wester PW, VanKreijl CF, Capel PJA, VanSteeg H, Verbeek SJ (1995) Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature 77:169–173. doi:10.1038/377169a0
Melis JPM, Wijnhoven SW, Beems RB, Roodbergen M, vanden Berg J, Moon H, Friedberg E, van der Horst GTJ, Hoeijmakers JHJ, Vijg J, van Steeg H (2008) Mouse models for Xeroderma pigmentosum group A and group C show divergent cancer phenotypes. Cancer Res 68(5):1347–1353. doi:10.1158/0008-5472.CAN-07-6067
Robinson DE, MacDonald JS (2001) Background and framework for ILSI’s collaborative evaluation program on alternative models for carcinogenicity assessment. International Life Sciences Institute. Toxicol Pathol 29(Suppl):13–19
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CAJ, Butel JS, Bradley A (1992) Mice deficient for p53 are developmental normal but susceptible to spontaneous tumors. Nature 355(6366):215–221
Hanahan D, Weinberg RA (2011) 2011 Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Tischler AS, Powers JF, Pignatello M, Tsokas P, Downing JC, McClain RM (1999) Vitamin D3-induced proliferative lesions in the rat adrenal medulla. Toxicol Sci 51(1):9–18
Silva Lima B, Van der Laan JW (2000) Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard. Regul Toxicol Pharmacol 32(2):135–143. doi:10.1006/rtph.2000.1427
Bronchud MH (2002) Is cancer really a ‘local’ cellular clonal disease? Med Hypotheses 59(5):560–565
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339(6127):1546–1558. doi:10.1126/science.1235122
Pan Z-Z, Godwin AK (2007) Oncogenes. In: Meyers RA (ed) Cancer. From mechanisms to therapeutic approaches. Wiley-VCH Verlag, Weinheim, pp 55–114
EMA EPAR Kepivance 2005. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000609/WC500040536.pdf
Sterner E, Meli L, Kwon SJ, Dordick JS, Linhardt RJ (2013) FGF-FGFR signaling mediated through glycosaminoglycans in microtiter plate and cell-based microarray platforms. Biochemistry 52(50):9009–9019. doi:10.1021/bi401284r
International Conference on Harmonisation. ICH Guideline S6(R1): Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. June 2011. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf. Accessed 23 Dec 2014
Vahle JL, Finch GL, Heidel SM, Hovland DN Jr, Ivens I, Parker S, Ponce RA, Sachs C, Steigerwalt R, Short B, Todd MD (2010) Carcinogenicity assessments of biotechnology-derived pharmaceuticals: a review of approved molecules and best practice recommendations. Toxicol Pathol 38:522–553. doi:10.1177/0192623310368984
Bugelski PJ, Volk A, Walker MR, Krayer JH, Martin P, Descotes J (2010) Critical review of preclinical approaches to evaluate the potential of immunosuppressive drugs to influence human neoplasia. Int J Toxicol 29(5):435–466. doi:10.1177/1091581810374654
International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. http://www.ich.org
Terrell TG, Green JD (1994) Issues with biotechnology products in toxicologic pathology. Toxicol Pathol 22(2):187–193
Inoue T (1998) Biotechnologically-derived pharmaceuticals in Japan: present and future prospects. In: Griffiths SA, Lumley CE (eds) Safety evaluation of biotechnologically-derived pharmaceuticals: facilitating a scientific approach. Kluwer Academic Publishers, Dordrecht, pp 51–63
Rosenblum IY, Dayan AD (2002) Carcinogenicity testing of IL-10: principles and practicalities. Hum Exp Toxicol 21(7):347–358
Ning S, Shui C, Khan WB, Benson W, Lacey DL, Knox SJ (1998) Effects of keratinocyte growth factor on the proliferation and radiation survival of human squamous cell carcinoma cell lines in vitro and in vivo. Int J Radiat Oncol Biol Phys 40(1):177–187
FDA Kepivance, Prescribing information 2004, 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125103s146lbl.pdf
EMA EPAR Neorecormon (2004) http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000116/WC500024975.pdf
FDA Epoietin Alfa, Prescribing information 1989. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/103234s5323lbl.pdf
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R (1999) Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 19(5):3278–3288
Taguchi A, White MF (2008) Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol 70:191–212
Pollak M (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 8(12):915–928. doi:10.1038/nrc2536
Pollak M (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12(3):159–169. doi:10.1038/nrc3215
Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A (2012) Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology 153(5):2152–2163. doi:10.1210/en.2011-1843, Epub 2012 Feb 21
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Cheng Z, Tseng Y, White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 21(10):589–598. doi:10.1016/j.tem.2010.06.005, Epub 2010 Jul 16
Schwartz GP, Burke GT, Katsoyannis PG (1989) A highly potent insulin: des-(B26-B30)-[AspB10, TyrB25-NH2]insulin(human). Proc Natl Acad Sci U S A 86(2):458–461
Milazzo G, Sciacca L, Papa V, Goldfine ID, Vigneri R (1997) ASPB10 insulin induction of increased mitogenic responses and phenotypic changes in human breast epithelial cells: evidence for enhanced interactions with the insulin-like growth factor-I receptor. Mol Carcinog 18(1):19–25
Glendorf T, Knudsen L, Stidsen CE, Hansen BF, Hegelund AC, Sorensen AR, Nishimura E, Kjeldsen T (2012) Systematic evaluation of the metabolic to mitogenic potency ratio for B10-substituted insulin analogues. PLoS One 7:e29198
Hansen BF, Kurtzhals P, Jensen AB, Dejgaard A, Russell-Jones D (2011) Insulin X10 revisited: a super-mitogenic insulin analogue. Diabetologia 54(9):2226–2231. doi:10.1007/s00125-011-2203-8, Epub 2011 Jun 3
Gallagher EJ, Alikhani N, Tobin-Hess A, Blank J, Buffin NJ, Zelenko Z, Tennagels N, Werner U, LeRoith D (2013) Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes 62(10):3553–3560. doi:10.2337/db13-0249, Epub 2013 Jul 8
Ter Braak B, Siezen CL, Kannegieter N, Koedoot E, van de Water B, van der Laan JW (2014) Classifying the adverse mitogenic mode of action of insulin analogues using a novel mechanism-based genetically engineered human breast cancer cell panel. Arch Toxicol 88(4):953–966. doi:10.1007/s00204-014-1201-2, Epub 2014 Jan 25
Sciacca L, Cassarino MF, Genua M, Vigneri P, Giovanna Pennisi M, Malandrino P, Squatrito S, Pezzino V, Vigneri R (2014) Biological effects of insulin and its analogs on cancer cells with different insulin family receptor expression. J Cell Physiol 229(11):1817–1821. doi:10.1002/jcp.24635
Oleksiewicz MB, Bonnesen C, Hegelund AC, Lundby A, Holm GM, Jensen MB, Krabbe JS (2011) Comparison of intracellular signalling by insulin and the hypermitogenic AspB10 analogue in MCF-7 breast adenocarcinoma cells. J Appl Toxicol 31(4):329–341. doi:10.1002/jat.1590, Epub 2010 Oct 8
Dideriksen LH, Jørgensen LN, Drejer K (1992) Carcinogenic effect on female rats after 12 months administration of the insulin analogue B10 Asp. Diabetes 41:143A
Tennagels N, Welte S, Hofmann M, Brenk P, Schmidt R, Werner U (2013) Differences in metabolic and mitogenic signalling of insulin glargine and insulin aspart B10 in rats [corrected]. Diabetologia 56(8):1826–1834. doi:10.1007/s00125-013-2923-z, Epub 2013 May 8
Ter Braak B, Siezen CLE, Speksnijder EN, Koedoot E, van Steeg H, Salvatori DCF, van de Water B, van der Laan JW (2015) Mammary gland tumor promotion by chronic administration of IGF1 and the insulin analogue AspB10 in the p53R270H/+WAPCre mouse model. Breast Cancer Res 17:14. doi:10.1186/s13058-015-0518-y
EMA (2001) Points to consider on the non-clinical assessment of the carcinogenic potential of human insulin analogues. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003252.pdf
Sciacca L, Cassarino MF, Genua M, Pandini G, Le Moli R, Squatrito S, Vigneri R (2010) Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia 53(8):1743–1753. doi:10.1007/s00125-010-1760-6, Epub 2010 Apr 28
Kurtzhals P, Schaffer L, Sorensen A, Kristensen C, Jonassen I, Schmid C, Trub T (2000) Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 49(6):999–1005
Yehezkel E, Weinstein D, Simon M, Sarfstein R, Laron Z, Werner H (2010) Long-acting insulin analogues elicit atypical signalling events mediated by the insulin receptor and insulin-like growth factor-I receptor. Diabetologia 53(12):2667–2675. doi:10.1007/s00125-010-1899-1, Epub 2010 Sep 12
Sommerfeld MR, Muller G, Tschank G, Seipke G, Habermann P, Kurrle R, Tennagels N (2010) In vitro metabolic and mitogenic signaling of insulin glargine and its metabolites. PLoS One 5(3):e9540. doi:10.1371/journal.pone.0009540
Teng JA, Hou RL, Li DL, Yang RP, Qin J (2011) Glargine promotes proliferation of breast adenocarcinoma cell line MCF-7 via AKT activation. Horm Metab Res 43:519–523. doi:10.1055/s-0031-1280780, Epub 2011 Jul 19
Kellerer M, Haring HU (2001) Insulin analogues: impact of cell model characteristics on results and conclusions regarding mitogenic properties. Exp Clin Endocrinol Diabetes 109(1):63–64
Staiger K, Hennige AM, Staiger H, Haring HU, Kellerer M (2007) Comparison of the mitogenic potency of regular human insulin and its analogue glargine in normal and transformed human breast epithelial cells. Horm Metab Res 39(1):65–67
Pierre-Eugene C, Pagesy P, Nguyen TT, Neuille M, Tschank G, Tennagels N, Hampe C, Issad T (2012) Effect of insulin analogues on insulin/IGF1 hybrid receptors: increased activation by glargine but not by its metabolites M1 and M2. PLoS One 7(7):e41992. doi:10.1371/journal.pone.0041992, Epub 2012 Jul 26
Stammberger I, Bube A, Durchfeld-Meyer B, Donaubauer H, Troschau G (2002) Evaluation of the carcinogenic potential of insulin glargine (LANTUS) in rats and mice. Int J Toxicol 21(3):171–179
Stammberger I, Essermeant L (2012) Insulin glargine: a reevaluation of rodent carcinogenicity findings. Int J Toxicol 31(2):137–142. doi:10.1177/1091581811431111, Epub 2012 Jan 3
Ter Braak SJ, Wink S, Koedoot E, Pont C, Siezen CLE, Van der Laan JW, van de Water B (2015) Alternative signaling network activation through different insulin receptor family members caused by promitogenic antidiabetic insulin analogues in human mammary epithelial cells. Breast Cancer Res 17:97. doi:10.1186/s13058-015-0600-5
EMA EPAR Byetta 2006. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000698/WC500051842.pdf
EMA EPAR Bydureon 2011. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002020/WC500108239.pdf
EMA EPAR Victoza 2009. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001026/WC500050016.pdf
EMA EPAR Lyxumia 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002445/WC500140449.pdf
EMA EPAR Trulicity 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002825/WC500179473.pdf
EMA EPAR Eperzan 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002735/WC500165119.pdf
Sheiner LB, Steimer JL (2000) Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol 40:67–95
Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G (2005) Mechanism-based pharmacokinetic-pharmacodynamic modeling-a new classification of biomarkers. Pharm Res 22(9):1432–1437, Epub 2005 Aug 24
Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA (2008) Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci 29(4):186–191. doi:10.1016/j.tips.2008.01.007, Epub 2008 Mar 18
Derendorf H, Meibohm B (1999) Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm Res 16(2):176–185
Knudsen BL, Madsen LW, Andersen S, Almholt K, de Boer AS, Drucker DJ, Gotfredsen C, Egerod FL, Hegelund AC, Jacobsen SD, Moses AC, Mølck AM, Nielsen HS, Nowak J, Solberg H, Thi TD, Zdravkovic M, Moerch U (2010) Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing clacitonin release and C-cell proliferation. Endocrinology 151(4):1473–1486. doi:10.1210/en.2009-1272, Epub 2010 Mar 4
Ponce RA, Gelzleichter T, Haggerty HG, Heidel S, Holdren MS, Lebrec H, Mellon RD, Pallardy M (2014) Immunmodulation and lymphoma in humans. J Immunotoxicol 11(1):1–12. doi:10.3109/1547691X.2013.798388
Salavoura K, Kolialexi A, Tsangaris G, Mavrou A (2008) Development of cancer in patients with primary immunodeficiencies. Anticancer Res 28:1263–1269
Weaver JL (2012) Establishing the carcinogenic risk of immunomodulatory drugs. Toxicol Pathol 40:267–271. doi:10.1177/0192623311427711
Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F (2005) Carcinogenicity of human papillomaviruses. Lancet Oncol 6(4):204
Gaschen L, Schuurman HJ (2001) Ultrasound detection of non-Hodgkin’s lymphoma in three cynomolgus monkeys after renal transplantation and cyclosporine immunosuppression. J Med Primatol 30(2):88–93
Kauffman HM, Cherikh WS, McBride MA, Cheng Y, Hanto DW (2006) Post-transplant de novo malignancies in renal transplant recipients: the past and present. Trans Internatl 19:607–620
Dempster AM (2000) Nonclinical safety evaluation of biotechnologically derived pharmaceuticals. In: El-Gewely MR (ed) Biotechnology annual review. Elsevier Science, Amsterdam, pp 221–258
Wordell CJ (1991) Biotechnology update. Hosp Pharm 26:897–900
Goldstein G (1987) Overview of the development of orthoclone OKT3: monoclonal antibody for therapeutic use in transplantation. Transplant Proc 19(2 Suppl 1):1–6
Swinnen LJ, Costanzo-Nordin MR, Fisher SG, O’Sullivan EJ, Johnson MR, Heroux AL, Dizikes GJ, Pifarre R, Fisher RI (1990) Increased incidence of lymphoproliferative disorder after immunosuppression with the monoclonal antibody OKT3 in cardiac-transplant recipients. N Engl J Med 323(25):1723–1728
Macsween KF, Crawford DH (2003) Epstein-Barr virus-recent advances. Lancet Infect Dis 3:131–140
Young LS, Rickinson AB (2004) Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi:10.1038/nrc1452
Clarke J, Hurst C, Martin P, Vahle J, Ponce R, Mounho B, Heidel S, Andrews L, Reynolds T, Cavagnaro J (2008) Duration of chronic toxicity studies for biotechnology-derived pharmaceuticals: is 6 months still appropriate? Reg Toxicol Pharmacol 50:2–22. doi:10.1016/j.yrtph.2007.08.001
Bugelski PJ, Herzyk DJ, Rehm S, Harmsen AG, Gore EV, Williams DM, Maleeff BE, Badger AM, Truneh A, O’Brien SR, Macial RA, Wier PJ, Morgan DG, Hart TK (2000) Preclinical development of keliximab, a primatized™ anti-CD4 monoclonal antibody, in human CD4 transgenic mice: characterization of the model and safety studies. Hum Exp Toxicol 19:230–243. doi:10.1191/096032700678815783
Strangfeld A, Zink A (2010) Are we playing it safe? Tumor necrosis factor alpha inhibition and the risk of solid malignancies. Rheumatologist 1–7. http://www.the-rheumatologist.org/details/article/867933/Are_We_Playing_It_Safe.html
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 72(9):3666–3670
Balkwill F (2009) Tumor necrosis factor and cancer. Nat Rev Cancer 9:361–371. doi:10.1038/nrc2628
Raaschou P, Simard JF, Neovius M, Askling J (2011) Does cancer that occurs during or after anti-tumor necrosis factor therapy have a worse prognosis? A national assessment of overall and site-specific cancer survival in rheumatoid arthritis patients treated with biologic agents. Arthritis Rheum 63(7):1812–1822. doi:10.1002/art.30247
Ferrajoli A, Keating MJ, Manshouri T, Giles FJ, Dey A, Estrov Z, Koller CA, Kurzock R, Thomas DA, Faderi S, Lerner S, O’Brien S, Albitar M (2002) The clinical significance of tumor necrosis factor-α plasma level in patients having chronic lymphocytic leukemia. Blood 100(4):1215–1219
Brown ER, Charles KA, Hoare SA, Rye RL, Jodrell DI, Aird RE, Vora R, Prabhakar U, Nakada M, Corringham RE, DeWitte M, Sturgeon C, Propper D, Balkwill FR, Smyth JF (2008) A clinical study assessing the tolerability and biological effects of infliximab, a TNF-α inhibitor, in patients with advanced cancer. Ann Oncol 19:1340–1346. doi:10.1093/annonc/mdn054
Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ (1999) Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology 116(1):22–28
FDA Etanercept, Summary basis of approval (1998) http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm088697.pdf
Raaschou P, Simard JF, Holmqvist M, Askling J (2013) Rheumatoid arthritis, anti-tumor necrosis factor therapy, and risk of malignant melanoma: nationwide population based prospective cohort study from Sweden. BMJ 346:1–12. doi:10.1136/bmj.f1939, Published 8 April 2013
Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V (2006) Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies. Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295(19):2275–2285. doi:10.1001/jama.295.19.2275
Bongartz T, Warren FC, Mines D, Matteson EL, Abrams KR, Sutton AJ (2009) Etanercept therapy in rheumatoid arthritis and the risk of malignancies: a systematic review and individual patient data meta-analysis of randomised controlled trials. Ann Rheum Dis 68(7):1177–1183. doi:10.1136/ard.2008.094904
Wolfe F, Michaud K (2004) Lymphoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 50(6):1740–1751. doi:10.1002/art.20311
Wolfe F, Michaud K (2007) Biologic treatment of rheumatoid arthritis and the risk of malignancy. Analyses from a large US observational study. Arthritis Rheum 56(9):2886–2895. doi:10.1002/art.22864
Askling J, Fored CM, Brandt L, Baecklund E, Bertilsson L, Feltelius N, Cöster L, Geborek P, Jacobsson LT, Lindblad S, Lysholm J, Rantapää-Dahlqvist S, Saxne T, Klareskog L (2005) Risks of solid cancers in patients with rheumatoid arthritis and after treatment with tumor necrosis factor antagonists. Ann Rheum Dis 64:1421–1426. doi:10.1136/ard.2004.033993
Askling J, van Vollenhoven RF, Granath F, Raaschou P, Fored CM, Baecklund E, Dackhammar C, Feltelius N, Cöster L, Geborek P, Jacobsson LT, Lindblad S, Rantapää-Dahlqvist S, Saxne T, Klareskog L (2009) Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor α therapies: does the risk change with the time since start of treatment? Arthritis Rheum 60(11):3180–3189. doi:10.1002/art.24941
Haynes K, Beukelman T, Curtis JR, Newcomb C, Herrinton LJ, Graham DJ, Solomon DH, Griffin MR, Chen L, Liu L, Saag KG, Lewis JD (2013) Tumor necrosis factor alpha inhibitor therapy and cancer risk in chronic immune mediated diseases. Arthritis Rheum 65(1):48–58. doi:10.1002/art.37740
Setoguchi S, Solomon DH, Weinblatt ME, Katz JN, Avorn J, Glynn RJ, Cook EF, Carney G, Schneeweiss S (2006) Tumor necrosis factor α antagonist use and cancer in patients with rheumatoid arthritis. Arthritis Rheum 54(9):2757–2764
Dreyer L, Mellemkjaer L, Andersen AR, Bennett P, Poulsen UE, Ellingsen TJ, Duijnhoven RG, Straus SM, Raine JM, de Boer A, Hoes AW, De Bruin ML (2013) Number of patients studied prior to approval of new medicines: a database analysis. PLoS Med 10(3):e1001407. doi:10.1371/journal.pmed.1001407, Epub 2013 Mar 19
Lopez-Olivo MA, Tayar JH, Martinez-Lopez JA, Pollono EN, Cueto JP, Gonzales-Crespo MR, Fulton S, Suarez-Almazor ME (2012) Risk of malignancies in patients with rheumatoid arthritis treated with biologic therapy: a meta-analysis. JAMA 308(9):898–908. doi:10.1001/2012.jama.10857
Strangfeld A, Hierse F, Rau R, Burmester G-R, Krummel-Lorenz B, Demary W, Listing J, Zink A (2010) Risk of incident or recurrent malignancies among patients with rheumatoid arthritis exposed to biologic therapy in the German biologics register RABBIT. Arthritis Res Ther 12(1):R5. doi:10.1186/ar2904, Epub 2010 Jan 8
Dixon WG, Watson KD, Lunt M, Mercer LK, British Society for Rheumatology Biologics Register Control Centre Consortium, Hyrich KL, Symmons DPM (2010) Influence of anti-tumor necrosis factor therapy on cancer incidence in patients with rheumatoid arthritis who have had a prior malignancy: results from the British Society for Rheumatology Biologics Register. Arthritis Care Res 62(6):755–763. doi:10.1002/acr.20129
Baecklund E, Iliadou A, Askling J, Ekbom A, Backlin C, Grannath F, Catrina AI, Rosenquist R, Feltelius N, Sundström C, Klareskog L (2006) Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 54(3):692–701. doi:10.1002/art.21675
Hill AB (1965) The environment and disease: association or causation? Proc R Soc Med 58:295–300
Stricker BH, Stijnen T (2010) Analysis of individual drug use as a time-varying determinant of exposure in prospective population-based cohort studies. Eur J Epidemiol 25(4):245–251. doi:10.1007/s10654-010-9451-7, Epub 2010 Apr 1
WHO. http://www.who.int/medicines/areas/quality_safety/safety_efficacy/pharmvigi/en/
Miacalcin team Novartis Pharmaceuticals Corporation Available for Public Disclosure without Redaction Miacalcin® (calcitonin-salmon) FDA Joint Reproductive Health Drugs and Drug Safety and Risk Management Advisory Committee Meeting on the Benefit/Risk of Salmon Calcitonin for the Treatment of Postmenopausal Osteoporosis – Briefing Book, 29 Jan 2013
Chesnut CH III, Silverman SL, Andriano K, Genant H, Gimona A, Harris S, Kiel D, LeBoff M, Maricic M, Miller P, Moniz C, Peacock M, Richardson P, Watts N, Baylink D (2000) A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the PROOF Study. Am J Med 109(4):267–276
EMA EPAR Cimzia 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/001037/WC500069736.pdf
EMA EPAR Remicade 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000240/WC500050890.pdf
EMA EPAR Enbrel 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000262/WC500027366.pdf
Raaschou P, Frisell T, Askling J (2014) TNF inhibitor therapy and risk of breast cancer recurrence in patients with rheumatoid arthritis: a nationwide cohort study. Ann Rheum Dis 0:1–7. doi:10.1136/annrheumdis-2014-205745
EMA EPAR Aranesp 2006. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000332/WC500026145.pdf
EMA EPAR Somatropin referral 2011. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/Somatropin/human_referral_000287.jsp&mid=WC0b01ac05805c516f
Andersson C, Vaag A, Selmer C, Schmiegelow M, Sørensen R, Lindhardsen J, Gislason GH, Køber L, Torp-Pedersen C (2012) Risk of cancer in patients using glucose-lowering agents: a nationwide cohort study of 3.6 million people. BMJ Open 2(3). pii: e000433. doi:10.1136/bmjopen-2011-000433. Print 2012
Colhoun HM (2009) Use of insulin glargine and cancer incidence in Scotland: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 52(9):1755–1765. doi:10.1007/s00125-009-1453-1, Epub 2009 Jul 15
Currie CJ, Poole CD, Gale EA (2009) The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 52(9):1766–1777. doi:10.1007/s00125-009-1440-6, Epub 2009 Jul 2
Hemkens LG, Grouven U, Bender R, Gunster C, Gutschmidt S, Selke GW, Sawicki PT (2009) Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia 52(9):1732–1744. doi:10.1007/s00125-009-1418-4, Epub 2009 Jun 30
Jonasson JM, Ljung R, Talback M, Haglund B, Gudbjornsdottir S, Steineck G (2009) Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia 52(9):1745–1754. doi:10.1007/s00125-009-1444-2, Epub 2009 Jul 9
Kostev K (2012) Risk of breast cancer in patients on long-acting insulin analogues in comparison with those on human insulin. Diabetologia 55(5):1554–1555. doi:10.1007/s00125-012-2497-1, Epub 2012 Feb 19
Mannucci E, Monami M, Balzi D, Cresci B, Pala L, Melani C, Lamanna C, Bracali I, Bigiarini M, Barchielli A, Marchionni N, Rotella CM (2010) Doses of insulin and its analogues and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care 33(9):1997–2003. doi:10.2337/dc10-0476, Epub 2010 Jun 14
Ruiter R, Visser LE, van Herk-Sukel MP, Coebergh JW, Haak HR, Geelhoed-Duijvestijn PH, Straus SM, Herings RM, Stricker BH (2012) Risk of cancer in patients on insulin glargine and other insulin analogues in comparison with those on human insulin: results from a large population-based follow-up study. Diabetologia 55(1):51–62. doi:10.1007/s00125-011-2312-4, Epub 2011 Sep 29
Home PD, Lagarenne P (2009) Combined randomised controlled trial experience of malignancies in studies using insulin glargine. Diabetologia 52(12):2499–2506. doi:10.1007/s00125-009-1530-5, Epub 2009 Sep15
Rosenstock J, Fonseca V, McGill JB, Riddle M, Halle JP, Hramiak I, Johnston P, Davis M (2009) Similar risk of malignancy with insulin glargine and neutral protamine Hagedorn (NPH) insulin in patients with type 2 diabetes: findings from a 5 year randomised, open-label study. Diabetologia 52(9):1971–1973. doi:10.1007/s00125-009-1452-2, Epub 2009 Jul 16
Nagel JM, Mansmann U, Wegscheider K, Rohmel J (2010) Insulin resistance and increased risk for malignant neoplasms: confounding of the data on insulin glargine. Diabetologia 53(1):206–208. doi:10.1007/s00125-009-1535-0, Epub 2009 Sep 24
Pocock SJ, Smeeth L (2009) Insulin glargine and malignancy: an unwarranted alarm. Lancet 374(9689):511–513. doi:10.1016/S0140-6736(09)61307-6, Epub 2009 Jul 17
Bronsveld H, ter Braak B, Karlstad Ø, Vestergaard P, Starup-Linde J, Bazelier MT, De Bruin ML, de Boer A, Siezen CLE, van de Water B, van der Laan JW, Schmidt MK (2015) Treatment with insulin (analogues) and breast cancer risk in diabetics; a systematic review and meta-analysis of in vitro, animal and human evidence. Breast Cancer Res 17(1):100. doi:10.1186/s13058-015-0611-2
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dempster, M. et al. (2015). Carcinogenicity of Biopharmaceuticals. In: Graziano, M., Jacobson-Kram, D. (eds) Genotoxicity and Carcinogenicity Testing of Pharmaceuticals. Springer, Cham. https://doi.org/10.1007/978-3-319-22084-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-22084-0_8
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22083-3
Online ISBN: 978-3-319-22084-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)