
Abstract
Many biotechnological processes such as biogas production or defined biotransformations are carried out by microorganisms or tightly cooperating microbial communities. Process breakdown is the maximum credible accident for the operator. Any time savings that can be provided by suitable early-warning systems and allow for specific countermeasures are of great value. Process disturbance, frequently due to nutritional shortcomings, malfunction or operational deficits, is evidenced conventionally by process chemistry parameters. However, knowledge on systems microbiology and its function has essentially increased in the last two decades, and molecular biology tools, most of which are directed against nucleic acids, have been developed to analyze and diagnose the process. Some of these systems have been shown to indicate changes of the process status considerably earlier than the conventionally applied process chemistry parameters. This is reasonable because the triggering catalyst is determined, activity changes of the microbes that perform the reaction. These molecular biology tools have thus the potential to add to and improve the established process diagnosis system. This chapter is dealing with the actual state of the art of biogas process analysis in practice, and introduces molecular biology tools that have been shown to be of particular value in complementing the current systems of process monitoring and diagnosis, with emphasis on nucleic acid targeted molecular biology systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Biogas process parameters
- Molecular biology tools
- Quantitative Real-Time PCR
- Next generation sequencing
- Meta-omics
- Fluorescence-in situ hybridization
- Metabolic quotient
1 Introduction
Biogas production by anaerobic digestion of organic matter is a bio-technology with very long tradition for some 2,000–3,000 years. It was applied initially for sanitation purposes and only later additionally for energy production. The issue sanitation with its beneficial effects for the society is presented within this book in Chap. 3. All of the process steps are performed in a food chain by different microorganisms, governed by process engineering in a suitable technical environment. Some of these microbes have to cooperate extremely efficiently in syntrophic dependency in order to be able to thrive and proliferate at the minimum limit of possible energy gain [1, 2].
Methanogenic archaea, and among these particularly the acetoclastic Methanosaetaceae, appear to be most sensitive in biogas processes to stress factors such as short retention times, high ammonia, oxygen and short-chain fatty acid (SCFA) concentration, lack of certain trace elements and increased temperature [3, 4]. Due to their relatively low apparent maximum turnover number (Km) for acetate and long doubling times [5], the acetoclastic methanogens are disfavored at short retention times and increasing acetate concentration in the fermenter [6]. They are increasingly washed out if their proliferation cannot compensate out-dilution. This effect is even pronounced at additional stress conditions, favoring the activity and growth of syntrophic associations with hydrogenotrophic methanogens to the detriment of active Methanosaetaceae and acetoclastic activity [3, 4, 7]. It is incorporated as a central point in the bioindicator concept of process diagnosis [4] (see also Sects. 3 and “Microbial Guilds, Bioindicators and Transcriptional Profiling”).
The second bottleneck is the thermodynamically difficult hydrogen, formate or electron-releasing conversion of short chain fatty acids (SCFAs), alcohols and other intermediates of the biogas process. Most of these reactions are endergonic at standard conditions but can be realized by syntrophic associations involving product-scavenging methanogens [8, 9]. Methanogenic archaea are able to remove the reaction products by converting them finally to biogas, predominantly CH4 and CO2, which segregates from the fermenter sludge to the gas headspace and is further withdrawn by gas utilization. Syntrophic bacteria or anaerobic fungi partners of methanogens are difficult to cultivate and to study without their product-consuming associate. Modern characterization is typically initiated by genome or metagenome analysis, possibly leading to insights about special requirements that allow cultivation of pure isolates and studying their special physiological performances [10–12].
A third recognized bottleneck is the initial rate-limiting hydrolysis of recalcitrant substrates such as lignocellulose-rich biomass (LCB). When compared to aerobic degradation of lignocellulose, considerably less is known on the corresponding anaerobic process and the organisms involved. Besides bacteria, other organisms such as anaerobic fungi may be involved in efficient initial LCB attack and degradation [13]. Chapter 2 in this book is dedicated to anaerobic fungi and recent perceptions of their role in anaerobic LCB digestion. For some of these cellulolytic organisms, the genome has been sequenced [14, 15]. Such genome information is an invaluable data basis for process optimization and further biotechnological exploitation.
Microbial processes in anaerobic digestion are driven by both, biotic and abiotic factors. The physical and chemical environment (e.g. nutritional factors and redox status) are basic to and determine the biotic activity, the substrate conversion by the microbes. Biotic measures to regulate the process (e.g. bioaugmentation) however are scarce as briefly discussed in Sect. 2.7.
The most important issue in process optimization is to avoid the worst case, process disturbance or even breakdown. This requires a process control strategy that includes reliable process diagnosis based on meaningful analytical data. Since the activity of bioindicator microbes, organisms that are typical for certain process conditions, does react before conventionally used process chemical parameters indicate process failure, a promising approach for successful process control is to assess the activity of these bioindicators as integral part of an early-warning system [4]. The relevant actors, i.e. bioindicators performing the crucial biogas process steps, must hence be identified, and suitable analysis tools must be used or developed to track these key organisms and their activity quantitatively.
In the following chapters, microbiology and molecular biology tools for biogas process diagnosis and control are compiled and discussed. Since several important process dynamics such as SCFA and total solid (TS) turnover as well as gas quality/quantity are the result of microbial activity, and respective wet chemistry and physico-chemical analyses are and will be indispensable part of conventional practice but have revealed limitations, recent experience with these conventional applications for agricultural single-stage biogas processes is presented in the following Sect. (2). Molecular biology approaches have only recently emerged and may be introduced into practice after comparison or along with established physico-chemical routines. Some of these molecular tools, however, are promising candidates to be implemented in a holistic suite of analytical tools for process diagnosis and control.
2 Physico-Chemical and Biochemical Process Parameters
The spectrum of physico-chemical parameters actually employed for process diagnosis of agricultural and category 2 biowaste (untreated non-infectious to humans, animals or plants), biogas plants has originally been adopted from anaerobic sewage sludge digestion. Many of these parameters and respective benchmarks are listed and discussed in a review by Weiland [16]. Important aspects for diagnosis and control of single-stage processes are presented in the following.
2.1 Gas Production
In order to evaluate the efficiency of the process it is indispensable to determine the volumetric gas production and the gas quality or at least estimate these parameters from the generated electricity and the actual adjustment of the combined heat and power unit. Different equipment is on the market, ranging from simple manually operated lab instruments to fully automated industry scale online devices. Combined with data on the fed organic dry matter (oDM or volatile solids [VS]), the gas production and quality data inform on the specific methane production or methane yield (m3 CH4/kg VS). Comparison with benchmarks for given substrates and interpretation of the recent methane yield development allows to estimate the actual process efficiency at least roughly and to reveal up- and downward trends. Particular attention should be paid to decreasing CH4 and increasing H2 concentrations in the produced biogas. CH4 concentrations falling below ca. 48 % and H2 concentrations exceeding ca. 100 ppm in single stage processes are alarming and should give rise to counteractive measures.
Isotope ratio mass spectrometry (IRMS) based methods analyzing isotope discrimination by the biogas producing microbial community were recently proposed to detect methanogenic pathway shifts [17, 18]. The switch from acetoclastic to hydrogenotrophic methanogenesis is interpreted as signal of stress conditions which may allow plant operators to adjust their feeding strategy. Since the IRMS equipment is expensive, a laser-assisted online analysis technique was described for this purpose as a more practice-oriented alternative [19]. However, a pathway shift does not necessarily indicate imminent process failure, and interpretation problems with data from variable feedstock composed of C4 and C3 plant material still need to be resolved. Although gas analysis using stable isotope ratios has potential to reveal biogas production pathway changes, and online monitoring is possible, its contribution to process diagnosis is thus confined to basic research in its current state.
2.2 Process Intermediates, SCFA, Total and Volatile Solids, and Specific Determinants
The determination of SCFA (also referred to as volatile fatty acids [VFA], or volatile organic acids [VOA]) is a highly important component of process diagnosis. The SCFA spectrum is typically assessed using liquid or gas chromatography (LC/GC) based routines on suitable extracts in an external specialized lab and should include the iso-forms of butyrate and valerate. Increased levels of these SCFA exceeding ca. 50 mg/L as well as propionic acid concentrations above ca. 1 g/L along with a propionic/acetic acid ratio >1 typically indicate process disturbance in single-stage systems [16], but exceptions have become known as well. Reduced activity of methanogens due to substrate overload or limited availability of essential nutrients such as trace elements is frequently the reason, giving rise to “acid jam”, i.e. accumulation of upstream produced intermediates.
Attention must also be paid to the development of the dry matter (or total solids [TS]) content in the digester. TS can easily be determined on-site in an oven at 105 °C, whereas for analysis of VS, a muffle furnace is required. Information on VS contents in the substrates is important for determining the methane yield, and the VS/TS trend in the digester can anticipate eventually problematic ash accumulation. Increasing TS values over time in the fermenter sludge indicate a problem at the hydrolysis/acidogenesis step leading to compromised process efficiency and incomplete digestion. TS values exceeding 15 % can lead to stirring problems in conventional continuously stirred tank reactors (CSTRs). Reducing the organic loading rate (OLR), i.e. increasing the hydraulic retention time (HRT), might be helpful, otherwise substrate conditioning by physical/mechanical or (bio)-chemical means could be considered. More specific information of TS and VS in the fermenter sludge can be obtained by the fractionated analyses according to Weende and Van Soest [20].
Recently, an online near-infrared spectroscopy (NIRS) application to evaluate the process state with potential for the practice was reported [21]. The authors presented acceptable estimations of VS, ammonium, total inorganic carbon (TIC) and total VFA even in short-term process dynamics of a mesophilic pilot-scale maize silage fed biogas digester. Further extension of the model to include different substrates and analysis parameters, and experience in long-term operation is needed before online NIRS systems can be recommended for process control.
Similarly, techniques involving flow-assisted cell sorting (FACS) [22], matrix-assisted laser desorption/ionization—time of flight mass spectrometry (MALDI-TOF/MS) and Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR/MS) [23, 24] as well as secondary-ion mass spectrometry (SIMS) based systems [25, 26] can be helpful to separate or identify distinct microbes or consortia, or track their specific metabolic activities. Such methods prove to be useful in basic microbiology research and taxonomy and allow identifying microbes with resolution at the subspecies or strain level [23], given a suitable reference database is available. However, “dirty” environmental samples can pose considerable problems. Although there is some potential, application for monitoring the process status of the black-box biogas fermenter actually does not appear to be a realistic option. Physical methodologies basing on or coupled with fluorescence-in situ-hybridization (FISH) are itemized in Sect. 3.2.1.
2.3 Early Warning—The TVA/TIC Ratio
The ratio of total volatile acids to total inorganic carbon (TVA/TIC, also referred to as FOS/TAC or VOA/TIC) is determined by 2-point titration (pH 5.0, pH 4.4) and can easily be performed on-site [27]. It beats out the pH value as early indicator of process acidification due to its much higher sensitivity. Currently, the TVA/TIC ratio is the most used process chemical early warning system of acidification. TVA/TIC ratios of 0.15–0.45 are typical for a stable process without major acid accumulation, whereas rising ratios exceeding 0.45 reflect process disturbance, and values above 0.6–0.7 indicate acidosis. This can be associated with TIC depletion, e.g. in case of trace element deficiency.
However, in cases of atypical process conditions such as at higher NH4 + concentrations and pH-values, considering only TVA/TIC as a process indicator can be risky. The NH4 +/NH3 buffer system can trap protons masking possible acidification events. Obtaining low TVA/TIC values can thus be misleading at higher free ammonia-nitrogen (FAN; see also Sect. 2.4) if distinct SCFA (possibly not measured) may already be at alarming level. Above ca. 1 g NH4 +-N/L, it is therefore suggested to monitor the SCFA spectrum and/or molecular biology parameters (see Sect. 3) as well in order to perceive process perturbation and acid build-up.
2.4 Nutrients, Toxic and Disturbing Agents
Since nutrient composition of the substrates governs and limits microbial process performance, respective analysis should be performed occasionally and particularly if a plant is operated with atypical substrates or such operation is planned. Modern elementary analysis involves Inductively Coupled Plasma (ICP) equipment with detection by Optical Emission Spectroscopy (OES) or even more sensitive Mass Spectrometry (MS). Experience from practice suggests that the C:N ratio should be about 15–45 and the C:N:P:S ratio about 300-600:15:5:3 [4, 16, 28].
Several toxic agents are known that can impede the anaerobic digestion process. One of the most relevant is free ammonia [29] (see also Sect. 2.3) which can be calculated from the NH4 +-N concentration, the process temperature and the pH value [30]. Nitrogen seems to be lacking only in exceptional cases, but reduced N-compounds typically accumulate in anaerobic digestion of protein-rich feedstock and can become toxic [6]. It is thus important to determine these parameters periodically. NH3 diffuses unspecifically inside the cell, can capture protons and hamper proton-dependent ATP generation leading to activity loss and possibly cell death. The typical ammonia toxicity threshold is about 400–500 mg NH3-N/L, but a higher margin is possible in case of adaptation [4, 29]. Particularly microbes relying on H+ pumps are susceptible whereas those with Na+-pumps are favored in the presence of sufficient Na+. Predominantly microbes involved in the hydrogenotrophic metabolism of biogas intermediates are using Na+ pumps for ATP generation [5, 9]. Na+ appears to limit methanogenesis and elicit acidification at values decreasing to about 10 mg/L in the fermenter sludge and must therefore be provided at higher concentration in the substrate mix [31]. This can explain stabilizing, stimulating effects of Na+ addition at limiting, constrained or stress conditions such as in high-performance biogas production from grass silage in the practice [4].
Requirement for trace elements (TEs) in biogas production from biomass, particularly of Co, Ni, Se, Fe, and possibly of Mo, B and W, has been described in many publications, e.g. [16, 32–34] and in Chap. 7 of this book. Since their presence in suitable, available concentrations is a precondition of efficient process performance, TE contents should be determined occasionally and particularly if the feedstock composition is changed. Trace elements, however, can become toxic in higher concentration. Other compounds with toxic or disturbing potential are found among e.g. antibiotics, mycotoxins, detergents and heavy metals [35, 36], and some phenolic compounds appear to have inhibitory properties [37]. Cu and Zn loads are of particular importance for agricultural biogas plants. They can originate in higher concentrations from animal husbandry, and according to several practice reports, can be the cause of process disturbance and efficiency loss [38].
Several devices allow the measurement of O2 and H2S in the biogas. O2 can enter the process by leakages or actively during biological desulfurization. CO2 reduction becomes unfavorable in the presence of better electron scavengers such as O2, SO4 2− and NO3 − [6]. The redox potential is increased in their presence, and the activity of most anaerobic microbes is impeded. O2 should therefore be kept below 1 % (better 0.1 %) in the gas phase, and feeding substrates with high SO4 2− and NO3 − contents minimized, for similar reasons. H2S typically originates from sulfur containing organic matter. Since it is highly toxic for most living beings, maximum working place concentrations have to be respected. Moreover, corrosive acids such as H2SO4, H2SO3 or HNO3 can be formed in the presence of O2 and S- or N-containing compounds. They can damage mechanical devices and constituents of the biogas plant.
2.5 Biological Methane Potential, and Activity, Toxicity and Supplementation Tests
BMP (biological methane potential) or SMA (specific methanogenic activity) tests are typically applied to determine the methane potential of given substrates. By variation of these batch-mode assays in ATS (activity, toxicity and supplementation) tests [39], they can be employed to diagnose and control the actual state of the biogas process. It is attempted to assess e.g. the capacity of inocula to be activated, to evaluate the potential of added or endogenous compounds to exert toxic effects, or to test supplements for process stimulation.
Depending on the type of inoculum and its degree of adaptation, the potential of the biocenosis is tested to produce methane and/or react to changed process conditions. It is emphasized that results of these assays cannot always be used to predict the performance such as the methane yield in flow-through operation. This is mainly due to operational differences, particularly in the effective organic loading rate and the actual microbial retention time. Moreover, these tests are labor- and time consuming, typical test periods vary between several days to weeks.
2.6 Enzyme Tests and Applications
Enzymatic tests are important in research and have some potential for practice application. For example, hydrolytic enzymes are of high interest, since enzymatic saccharification (hydrolysis) is a rate-limiting step in anaerobic digestion (AD) from solid substrates and especially undigested lignocellulosic biomass (LCB) material, e.g. floating layers can pose considerable operational problems [40–42]. Hydrolases must accommodate heterogeneous plant cell wall residing polymers with various degrees of polymerization (DPs), side chain branching patterns and several altering substitutes [43]. Because of this chemical inhomogeneity and substrate specificity corresponding enzymes that act upon them are generally difficult to isolate and characterize. The quantitative determination of enzymatic activities is commonly based on accumulated products after hydrolysis including reducing sugars, total sugars and chromophores. Other assays measure the reduction in substrate quantity or the change in the physical properties of substrates. However, the production of reducing sugars is assayed using alkaline dinitrosalicylic acid (DNS), copper-arsenomolybdate using the 4-hydroxy-benzoylhydrazine (PAHBAH) method, 2,2′-bicinhroninate (BCA) and ferricyanide or directly using anthrone- or phenol-H2SO4. Focusing monomeric products, i.e. glucose as major product, commercial enzymatic glucose kits using coupled hexokinase and glucose-6-phosphat dehydrogenase are available. The main drawback of these methods is a poor stoichiometric relationship between reaction products (e.g. cellodextrins, malto- or xylodextrin) and pure D-glucose standards [44], which may result in an underestimation or overestimation of cellulase and hemicellulase activities [45, 46]. However, substrates used for hydrolysis assays should therefore always be as similar to native polymer structures as possible in matters of DP, solubility and crystallinity.
The esterase activity was suggested as indicator for the overall fermentation process, representing a sum parameter for bacterial heterotrophic activity in general [47]. In this context a positive correlation between esterase activity and substrate conversion rate towards methane was observed, revealing that process disruption is reflected by decreased enzyme activities [48, 49]. Furthermore, a negative correlation of esterase as well as aminopeptidase activities and substrate quality was observed, providing fermentability indications regarding silage as substrates [48]. Therefore, enzyme assays can be a useful tool for monitoring the overall anaerobic digestion process. Modifications towards an all-in-one testing kit like available for other chemical parameters (e.g. Merck Spectroquant® for COD, TOC, nitrate, ammonium etc.) is desirable to provide plant operators with an activity specific easy-to-use monitoring instrument. Compared to photometric tests, lower detection limits (factor 20–500) and shorter reaction times can be reached by the use of fluorimetric determination on the basis of fluorigenic compounds (e.g. fluoresceine diacetate, azocasein) [50]. Thereby, a precise study of catabolic enzyme activities such as esterase, phosphatase, aminopeptidase and glucosidase activities in samples with low biomass density is possible. In sum, non-methanogenic (hydrolytic, acidogenic) and methanogenic activity tests in combination with molecular tools seem to be essential for a better characterization and monitoring of full-scale anaerobic digesters [51].
Beyond measuring the activity or amount of enzymes available in the process, enzymes can be also applied to stimulate the AD process. The utilization of enzymes for environmental and industrial applications have been described to be stable in a large range of even quickly changing conditions, i.e. pH, temperature, presence of inhibitors or interspecies competition [52, 53], although controversially observations have been described by other studies on the use of commercial enzymes and enzyme mixtures considering related costs in AD processes, rather suggesting a specific application with respect to optimum conditions and the source of substrate [54, 55]. It has been shown that a combination of chemical and enzymatic pretreatment of bamboo waste, using commercial cellulase and alkaline, can lead to significantly enhanced chemical oxygen demand (COD) solubilization and substrate saccharification in BMP tests, which not necessarily translates to high methane yields as compared to alkaline pretreatment alone [56], suggesting to re-think the role of enzymes in multiple-pretreatment settings. The application of natural endogenous hydrolases such as amylase and protease from fermentation sludge for pretreatment of wastewater sludge resulted in improved sludge solubilization and acidification regarding the COD and VFA upturn [57], whereas a positive effect on anaerobic biodegradability, hydrolysis, digestion rates as well as maintaining a healthy microbial population were not indicated [58]. However, enzyme treatment can improve the economic production of biogas from agricultural residues, municipal solid and animal wastes by enhancing the fluidity of fibrous feedstock mixtures [59], solubilization and deflocculation of wastewater and sewage sludge biomass towards anaerobic digestibility [60–62].
The instability and time-limited effect of free enzymes can be overcome by immobilization using suitable carriers such as alginate or minerals [52, 63–66]. Moreover, the improvement of high-solid substrate degradation can be also achieved by inoculation of beneficial bacteria, which produce corresponding hydrolases in response to the given feedstock and operating conditions [49, 67, 68]. This has been demonstrated for mixed hemicellulolytic bacteria cultures [69, 70] and isolated bacterial species obtained from natural biogas-producing consortia as well, i.e. hydrogen-producing cultures of Caldicellulosiruptor saccharolyticus and Enterobacter cloacae [71, 72] or Clostridium cellulolyticum, which was successfully adopted to enhance the hydrolysis of wheat straw leading to increased BMP tests improving the utilization of lignocellulosic substrates [73].
An improved understanding of the catalytic potential of the AD ecosystem can be attained by mechanistic models based on kinetic data capturing important details of enzyme-substrate interactions, key substrate surface properties and individual enzyme adsorption and complexation characteristics as demonstrated for cellulose/cellulase interactions [57, 74, 75]. These catalytic information might be implemented into existing complex dynamic models and simulations such as IWA’s Anaerobic Digestion Model (ADM No. 1, 2) or novel Process Simulation Models (PSMs), which are validated against a variety of lab and industrial data on anaerobic digestion to predict the applicability of any substrate for biogas production at any given process condition [76–79].
2.7 Classical Microbiology Approaches
Classical cultivation-based microbiological methods have not gained major importance in the analysis of the biogas process status in practice. This is owed to the fact that most anaerobic microbes have long duplication times. Cultivation of anaerobes is not only tedious and difficult in many instances. It can cause biased results if specific growth and activity requirements of investigated microbes or associations are not known. By applying next generation sequencing (see Sect. 3.1.4), metagenomics, genome analysis and mapping, specific genetic capacities of investigated microbes or associations can be identified which can help to meet unrecognized cultivation requirements and eventually grow hitherto uncultured organisms [80]. Classical light microscopy reveals its limits given the highly turbid sample matrix and the low portion of known and described microorganisms [81].
Bioaugmentation is a classical microbiology measure to counteract process imbalances and a key component of biotechnology routines. Virtually every biogas plant and biotechnological process has been or is started up by inoculation, a special form of bioaugmentation. Numerous experiences show that once a stable process and biocenosis is established, newly introduced strains will encounter enormous difficulties to colonize and propagate in this process [82], although such success was announced in a few reports [71]. Bioaugmentation might be helpful for the case that a disturbed process should be stabilized or re-established by the (re)introduction of certain strains or consortia which had been recognized to be relevant for proper function but were washed out.
3 Molecular Biology Approaches
Biogas process failure can have several reasons. Technical reasons include stirring problems, leakages and temperature changes. At too short microbial retention time and unbalanced or insufficient nutrition (see Sect. 2.4) slowly growing, but possibly important microbes are diluted out. If a process-relevant guild is washed out and no functional substitutes can grow up, this results in process failure or even breakdown. Such bioindicators of the process state are ideally tracked by specific molecular biomarkers. Since these react earlier than the conventional physico-chemical parameters [83], molecular biology bioindicator tracking does not only allow for diagnosing the process, it provides more time to plant operators for specific counteraction.
This chapter is subdivided into several sections where molecular biology methods with more or less potential for process diagnosis are described. Some are used only in basic research and others have started to be applied in practice.
3.1 PCR Based Approaches and Nucleic Acid Sequencing
Since the invention of the Polymerase Chain Reaction (PCR) in 1983, PCR based techniques have conquered the field in molecular biology diagnostics. With the recent progress in affordable next generation sequencing techniques [84] (see Sect. 3.1.4), sequence information in databanks has substantially boosted. On this basis, group-specific primers and probes can be designed with much higher dependability. Diagnostic PCR assays are quickly performed and highly sensitive if suitable (e.g. fluorescence based) detection systems are included. In this chapter, emphasis is therefore on PCR-based methods and among these particularly on quantitative Real-Time PCR (qPCR) assays. PCR applications typically need a specialized laboratory environment but developments for on-site use are emerging. On-line systems, however, are far from being conceivable.
3.1.1 Crucial Prerequisites: Sampling and Nucleic Acid Extraction
A prerequisite for reliable results is that the samples taken are representative of the fermenter sludge. This is not trivial since the fermenter sludge typically is not visually examinable. Bleeders may be partially clogged and act as filters or other phenomena such as floating or sediment layers may cause inhomogeneities. Results should therefore be checked for plausibility and possible sampling bias. Transport and storage of samples is another major source of errors. If samples can be processed within a few hours, they should be kept at process temperature in (almost filled up) closed Polyethylene (PE) or polypropylene (PP) bottles with a cannula for degassing. For longer transport/storage it depends, if DNA as the most stable, rRNA as intermediate or mRNA as the least stable nucleic acid (NA) is the target [85, 86]. In our experience, samples can be stored at ca. 4 °C for 1–2 weeks for analyses on DNA and for a few days for analyses on rRNA level. It is not finally shown for these NA species whether freezing at −20 or −80 °C respectively, and gentle thawing (at ca. 4 °C) does affect the microbial community composition. For mRNA analysis from stored samples it must be considered that this RNA species is in a highly dynamic equilibrium. Both production and degradation must be stopped immediately, e.g. by immersion in liquid N2, acid phenol or other effective preservatives. Respective research is currently being carried out.
It must also be shown that the used NA extraction and purification system is suitable and efficient for the specific type of sample and analysis, and for quantitative analyses (see Sect. 3.1.2), the corresponding NA recovery rate has to be known. From numerous comparative studies dealing with NA extraction and purification systems it is turning out that combined physical cell disruption and chemical lysis is most suitable for environmental samples with a high portion of particulate organic matter such as fermenter sludge samples. Washing the sample prior to extraction is suggested because this substantially reduces inhibitors such as water soluble humic compounds [87]. NA purity in extracts is therefore of major concern, but as pointed out below, current guide values are not always conclusive and helpful for PCR-based assays.
Physical disruption of cells to release NAs is another crucial factor for obtaining suitable extracts. Due to velocity, ease of handling and performance efficiency, bead beating (BB) is used most frequently. Rigid cell walls must be broken, but too harsh BB can shear NAs and lead to detection failure [88]. Physical disruption must therefore be optimized for the targeted type of cells along with the particulate organic matter (OM) content in the sample sheltering the targeted cells. The higher the OM content, the more intense BB must be chosen. If differently recalcitrant cells are present, a fractionated protocol with increasing BB force and pooling of subsampled extracts can be applied [89]. It is essential to further adapt the protocol to the downstream type of analysis. If relatively short fragments such as for qPCR are suitable, relatively strong BB is of advantage. For applications requiring longer NA stretches such as functional transcriptome or genome analysis, strong BB can be counterproductive.
For RNA extracts, efficient DNase treatment and Reverse Transcription (RT) reaction with -RT controls must be performed, otherwise downstream reactions are contaminated and results biased. It must be considered that DNases degrade RNA to a certain extent, as well. This can introduce uncertainty and may only partially be overcome by method standardization leaving the possibility of a systematic error. The produced cDNA is further used just like genomic DNA but its single-stranded nature must be considered for quantitative aspects.
Downstream, extract purification is a trade-off between inhibitor removal and NA loss. PCR inhibitor removal is frequently seen as equal with matching traditional absorbance ratios (A260/230, A260/280). However, these had originally been developed for DNA-DNA hybridization and turned out to be of limited value for PCR applications. Quantitative Real-Time PCR (qPCR) was not inhibited at A260/230 and A260/280 ratios as low as 0.02 and 1.4, respectively [32]. Cell-lysing Guanidinium-Isothiocyanat (GITC) present in some NA extraction kits absorbs at 230 nm but does not seem to compromise PCR. However, (partial) inhibition was obtained if the A320 value (humic compounds absorb at 320 nm) surpassed a level of 0.02–0.03. The A320 value thus appears to be a major indicator of PCR inhibition by samples containing humic compounds.
Between 40 % and over 80 % of extracted DNA was lost by conventional silica column post-purification [90, 91], which considerably compromises the sensitivity of quantitative assays. Optimization of the extraction/purification protocol requires that the number of treatment steps is minimized while inhibitor removal and NA recovery rates are maximized. With optimized kit-based DNA and RNA extraction/purification systems and optimized (RT)qPCR biochemistry such as inclusion of a highly processive polymerase and adjusted Mg2+ concentration [87], about 90 % of spiked DNA and 30–70 % of spiked viral RNA was recovered from cattle manure or biogas fermenter samples with an optimized kit-based total RNA extraction procedure [83]. However, the RNA recovery rate may actually have been lower because no DNase digestion was performed and DNase I can degrade RNA unspecifically. It is thus strongly suggested to report the method detection limit [91] of the given assay along with the DNA and/or RNA recovery rates.
3.1.2 Conventional and (Reverse-Transcription) Quantitative Real-Time PCR: Applications for Process Diagnosis
Conventional PCR is an integral step of several applications such as amplicon sequencing (see 3.1.4) and community fingerprinting (see Sect. 3.1.3). For diagnostic purposes, however, conventional PCR has lost importance in the last years in favor of Real-Time PCR (qPCR) assays. Applications, advantages and limitations of qPCR and RT-qPCR are compiled and discussed in many reviews and book chapters, e.g. [92–94]. (RT)qPCR assays avoid laborious gel-electrophoresis, are performed more quickly, are suitable for high throughput, are less prone to contamination, and provide superior specificity particularly if an additional (e.g. hydrolysis) probe or different chemistry for the same purpose is integrated [95].
The reliability of PCR assays has significantly been improved. DNA-polymerases with a very low error rate (for Taq ca. 3 × 10−5, still much lower e.g. for Pfu, [96, 97]) and suitable reaction environment are available, and primer specificity can significantly be improved due to the enormously grown sequence data in databanks. A major issue, however, is the formation of chimaeras during PCR amplification which can seriously bias community composition analysis. Several programs and online applications can be used to check for chimaeras even in sets with relatively short amplicons [98]. Avoiding the formation of chimaeras, e.g. by analyzing templates with relatively homogenous melting temperatures (Tm) over the region of interest, would be even more straightforward than post-purifying datasets. However, it can be difficult to find template regions that provide sufficient phylogenetic resolution.
Moreover, problems or uncertainty still exist particularly with quantitative analyses of prokaryotic mRNA. This is not only due to unspecific DNase activity (see Sect. 3.1.2), but (partial) inhibition of RT-reactions that typically remain undetected. The RT efficiency at the given reaction conditions typically is not documented and may be subjected to interfering compounds introducing variability. Although these imponderabilia may not be of crucial importance for qualitative approaches such as community analyses, further methodological development is required for reliable quantification of prokaryotic mRNA.
3.1.2.1 Microbial Guilds, Bioindicators and Transcriptional Profiling
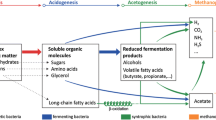
The bioindicator approach (Fig. 1) aims at analyzing and predicting distinct process states, shifts and perturbances, e.g. in biogas reactors. On the molecular level, genes encoding key enzymes of important metabolic pathways in the biogas process such as methyl-coenzyme M reductase (isogenes mcr and mrt encoding coenzyme-B sulfoethylthiotransferase, EC 2.8.4.1, the key enzyme of methanogenesis, which is present in all and exclusively in methanogenic Archaea), formyl-tetrahydrofolate synthetase (or formate-tetrahydrofolate ligase, fhs, EC 6.3.4.3, key enzyme of the Wood-Ljungdahl-pathway) or certain hydrogenases (e.g. ech, hyd) and their transcripts are ideal bioindicators and targets of molecular biomarkers [4], and even more will be identified in the near future [11, 99]. (RT)qPCR markers can be tailored to determine bioindicator organisms of such guilds and their transcription activity (Fig. 1). For the design of specific biomarker systems for defined bioindicator organisms, it is essential that these had been identified previously by community composition analyses at relevant fermentation process conditions. However, the design of specific (RT)qPCR systems for heterogeneous microbial groups or clades in environmental samples can be difficult. This is particularly true if guilds are to be tracked by targeting signatures on functional genes exhibiting wobble bases in the third codon position. Respective primers, so-called “protein primers”, typically are highly degenerated to provide the desired specificity, which complicates functional PCR based assays (e.g. [100]).

The bioindicator/biomarker approach to assess process-relevant microbial guilds and their characteristic (transcriptional) activity. Once bioindicators are identified and biomarkers constructed (1), guilds and their (transcriptional) activity can be quantified in high-throughput assays (2)
Due to the high stability of DNA, as compared to mRNA (see Sect. 3.1.1), DNA based assays will detect live and inactive organisms as well as residual DNA e.g. of dead organisms [101, 102]. Since for realtime process analysis, the active organisms are of particular interest, transcriptional profiling is supposed to identify more meaningful bioindicators than conventional analyses on DNA level, but changes in environmental conditions can not only induce quantitative transcription changes (see Sect. “cDNA/DNA Ratios and the Metabolic Quotient”). Typically, first metabolic activity and subsequently propagation of the populations that are best adapted to the new conditions are encouraged, whereas unadapted populations are losing competitiveness. The activity of inadequately adapted microbes is cut down first, and subsequently they are diluted out in flow-through processes. In microbial successions initiated by organic loading rate (OLR) increase in biogas processes with renewable resources, different bioindicators of the process status have been identified:
Methanosaeta spp. were present only at long microbial retention times, low acetate and ammonia levels [17, 32, 103] or/and at a feeding regime with a substrate mixture containing manure e.g. from husbandry [104, 105]. A potential bioindicator, tentatively named Methanosaeta concilii 2, was identified first from mesophilic maize silage digesters [4]. It is different from the mcrA sister clade (M. concilii 1) encompassing the type strain and most of the Methanosaeta sequences recovered from animal manure environments. Recent sequencing confirmed the presence of M. concilii 2 also in mesophilic grass silage digesters. This guild, probably originating from the cattle manure inoculum, soon lost transcription activity in the grass silage digestion process and was washed out subsequently (B. Munk, unpublished) at increased loading rates. M. concilii 2 and its activity is thus an example of a specific bioindicator of relaxed digestion conditions.
Other methanogens appear to have a similar potential to be used as indicators of the biogas process status. Results of several studies performed in different environments on the DNA and on the transcription level [4, 83, 106–108] suggest that with increasing strain to stress conditions such as shorter microbial retention times with increased SCFA concentrations and critical ammonia contents, Methanosaetaceae and their activity are replaced by Methanosarcinaceae and Methanobacteriaceae, with the latter appearing to be the most resistant. More specifically, certain Methanosarcina genospecies, hitherto undescribed Methanosarcinaceae (tentatively classified as genus II) and strictly hydrogenotrophic methanogens, particularly Methanobacteriaceae, certain Methanobacterium genospecies and hitherto undescribed Methanobacteriaceae (tentatively classified as genus IV) were increasing in maize and grass silage digestion processes on the DNA and on the transcription level [4, 83]: at aggravated strain or stress conditions, very short retention times, high SCFA or ammonia contents, at the onset of process failure, the diversity of methanogens remained almost unchanged at the DNA level, but mcrA/mrtA was transcribed exclusively by certain Methanobacterium genospecies (particularly Methanobacterium III sp. 3a) at mesophilic and Methanothermobacter wolfeii at thermophilic conditions. For these bioindicators, more specific (RT)qPCR based biomarker systems are being developed in order to track their presence and activity and provide a meaningful process diagnosis.
3.1.2.2 cDNA/DNA Ratios and the Metabolic Quotient
In principle, cDNA/DNA ratios can be calculated for any physiological performance of interest by relating the actual net concentration resulting from RNA transcription and transcript degradation to the concentration of the corresponding gene in a given sample. It appears to be most meaningful to determine the cDNA/DNA ratio of selected functional genes of key enzymes as activity parameter to assess the specific activity of certain guilds (see Sect. “Microbial Guilds, Bioindicators and Transcriptional Profiling”, Fig. 1). Respective necessary information for designing specific (RT)qPCR system can be derived from alignments containing relevant sequences deposited in databases and extracted sequences from metagenomes and metatranscriptomes (see Sect. 3.1.4).
For mcrA/mrtA, cDNA/DNA ratios have already been reported, e.g. for peat soil and biogas fermenters [31, 109, 110]. The cDNA/DNA ratios reacted to activating stimuli such as temperature or substrate, whereas the gene concentrations remained almost constant, and they were correlated with the methane production rate within certain limits, indicating the potential of this molecular biology approach to track the activity of the guild of methanogenic Archaea.
Similar approaches may be envisaged to track distinct microbial activities. Concerning biogas processes, cDNA/DNA ratios, e.g. for fhs or ech subunits or other important genes of key enzymes could be very informative on the activity status of the corresponding metabolic pathways. Such information would be very helpful for process diagnosis also for e.g. biorefineries, and monitoring could provide operators with necessary information for process engineering and to decide on possible intervening measures.
However, it has to be considered that prokaryotic mRNA analysis still is delicate and error-prone, particularly if quantitative results are to be obtained (see Sects. 3.1.1 and 3.1.2). At the current state of the art, respective results should therefore be treated with precaution. RTqPCR and upstream sample preparation still need methodological development until interlaboratory comparison will create consistent and reliable results.
A second ecophysiological parameter is the Metabolic Quotient (MQ). The MQ has been developed by Munk et al. [31] and was further explained in more detail [4]. In contrast to the entirely molecular biological parameter cDNA/DNA ratio, the MQ needs concomitant physiological data. For the MQ, the methane productivity (mL CH4 per mL fermenter sludge) is related to the concentration of methanogenic Archaea, as determined by mcrA/mrtA targeted qPCR [100] on the DNA level, regardless if they are dead or alive, in the fermenter sludge at a given time, resulting in the actual specific methanogenic activity (SMAact). SMAact is compared to a reference standard dataset (SMAstd) obtained for efficient process performance at various OLRs without any symptoms of process disturbance. If SMAact/SMAstd is >1, the methanogenic guild of interest is metabolizing at strain or stress conditions, and if SMAact/SMAstd is <1, the methanogens are less active than at the standard reference conditions.
The MQ was measured in different maize silage digestion processes along with conventional indicators of the process state (see Sect. 2) in time series [31, 83]. It turned out that the MQ passed a threshold of about 3 ca. 2 weeks before changes were detected by the conventional chemical process indicators such as noticeable increases of the TVA/TIC ratio or SCFA concentrations. At this process stage, less methanogens than at standard conditions performed the same metabolic task, indicating metabolic strain or even stress of the given methanogenic population. When the TVA/TIC ratio and/or critical SCFAs such as propionic acid had increased to an alarming level of about 0.7 or 1 g/L, respectively, the MQ began to decline or had already decreased, indicating serious process failure and collapse. The methanogenic population was obviously seriously affected and not able anymore to fully accomplish the metabolic task of methane formation, as evidenced by the sudden decrease of methane productivity and the methane yield. When no substrate was fed to the process, the MQ was significantly below 1. According to the observations, a threshold of ca. 0.1 was defined, indicating the lowest level of normal physiologic activity.
The MQ thus allows, over the complete range of tested OLRs, to determine the metabolic state of the resident methanogenic population. A single MQ determination, however, does not necessarily mean very much. Just like with the TVA/TIC ratio, the recent development has explanatory power. An increasing MQ indicates increasing strain or stress. A decreasing MQ can indicate relaxed conditions or process breakdown. If an MQ of 1 was measured, it can be a sign of normal process operation, but it can also be symptom of a collapsed process if it turned down from values exceeding 3.
In most recent experiments with grass silage as substrate and measurements of practice biogas plants operated predominantly with grass silage, the MQ reacted similarly as in maize silage processes and stood within the bandwidth of 0.1–3 at normal process conditions without symptoms of process disturbance (B. Munk, personal communication). Since the TVA/TIC ratio is losing informative value at the high ammonia contents typically found in grass silage digestion, this is of particular importance and demonstrates the potential of the MQ as an early warning tool of process failure in practice (Fig. 1). It is expected that the MQ will become an important ecophysiological molecular microbial parameter and find application in practice monitoring of biogas plants and process diagnosis.
3.1.3 Community Fingerprinting Assays
An ideal method for microbial community analysis would allow the detection of different groups and enumerate all microbial species present in a sample from an ecosystem or habitat. Basically two approaches are used for community analyses: (1) cultivation-dependent analysis (CDA) aiming at the detection of selected groups and species of microorganisms and (2) cultivation-independent analysis (CIA), which are RNA/DNA based and are used to assess the complexity and dynamics of microbial communities. CDA relies on several selective and non-selective culture media that supply different growth conditions for specific or non-specific microbial population targeting. Traditional methods require a vast knowledge of phenotypic features to characterize microorganisms, which is often inaccurate and also leads to an underestimation of the diversity of species. However, the main drawback of conventional cultivation methods to recover less than 1 % of the total microbial species present in environmental samples remains problematic [111]. Thus CDA is nowadays complemented by molecular methods such as polymerase chain reaction (PCR) and fingerprinting techniques to assess shifts in microbial composition by small subunit ribosomal RNA gene analyses [112–114]. CIA is principally based on molecular techniques (Table 1), applying PCR and oligonucleotide probe hybridization in order to identify microbes directly from sample material [115]. Therefore, total genomic DNA or RNA must be extracted from collected microbial cells, avoiding co-extraction of sample matrix-inherent compounds that can totally inhibit the PCR (see Sect. 3.1.1). Cell lysis is accomplished by several methods: mechanically using bead-beating, freeze-boil cycles, chemically by the use of detergents or enzymatically using cell wall degrading enzymes, e.g. lysozyme, lyticase or proteinase [116–118].
Environmental microbiological studies are often based on ribosomal DNA or RNA sequences, because these sequences are functionally and evolutionary conserved and present in all organisms. Here, 16S rDNA and 23S rDNA sequence regions have already been determined for a large number of reasonably described bacterial, archaeal and fungal species. Thus 16S rDNA sequences can be used to investigate phylogenetic relationships and for the identification of unknown microbes via comparisons with database collection entries. The largest reference databases exist for conserved marker gene 16S rRNA [112, 136]. In contrast to rDNA, rRNA targeted techniques rely on high-copy numbers per cell and are specifically used to assess changes in metabolically active microbial populations [137], although extraction and handling procedures are much more complicated due to the rRNAs instability (v. [138]). The intergenic spacer region (ISR) between 16S and 23S rDNA often shows species specific sequence variations by primers binding to conserved nucleotide stretches at the 5′ 23S and 3′ 16S rDNA gene end respectively [139]. In ISR-directed ribosomal intergenic spacer analyses (RISA) it is used to describe phylogenetic microbial diversity (Bacteria and Archaea) by creating RISA profiles. Although it is foremost used in diagnostic PCR-amplifications [140], ISR amplicons as targets for qPCR assays have also been discussed to reflect the metabolic status of key microbes more accurate than 16S based fragment comparisons [141]. Numerous broad-range and group-specific primers are available, targeting many bacterial and archaeal species of interest in AD processes, including fermentative and methanogenic representatives, covering low diversity selective cultivation sample structures up to full-scale agricultural biogas plant complex mixed community fingerprints [24, 69, 142].
The fingerprinting techniques range from simple length heterogeneity PCRs (LH-PCR) depending on different primers, targeting several variable regions in combination [143] up to more sophisticated genetic fingerprinting techniques such as amplified ribosomal DNA restriction analysis (ARDRA) or terminal restriction fragment length polymorphism (T-RFLP), which are all well established and vastly exploited to characterize whole microbial communities, providing pattern profiles of the community diversity [129, 144, 145].
T-RFLP fingerprints can give quantitative insights into communities by using a combination of fluorescence labelled primers and enzymatic digestion of resulting PCR products to generate terminal restriction fragments (T-RFs) from DNA templates. The taxonomic resolution can be improved by combining several fluorochromes and restriction enzymes simultaneously [146, 147]. Problems ascend from incomplete restriction digestion due to e.g. missing restriction sites or fragment length discrepancies caused by different fluorochromes used to estimate in silico yields, which reduces the reproducibility [130]. However, T-RFLP has not only been applied to describe bacterial communities, but also to monitor methanogenic populations and temporal shifts of archaeal communities in bioreactors [148, 149].
Further fingerprinting techniques are denaturing-gradient gel electrophoresis (DGGE), temperature-gradient gel electrophoresis (TGGE) and single-strand conformation polymorphism analysis (SSCP), which detect sequence variations of rRNA gene fragments or other functional genes from total community DNA or cDNA [150]. Complex microbial communities can thus be resolved into single members through band separation by gel electrophoresis. DNA sequence information is obtained from excised bands, which represent operational taxonomic units (OTUs) or even single species, but do often require further preparation and time consuming cloning steps. Co-mitigation or poor separation of bands representing small fragments less than 500 bp and restricted sensitivity to OTUs with a minimum abundance of 10 % (SSCP) lead to limitations of phylogenetic identifications and incomplete microbial profiles [151], which can be partly overcome by e.g. nested PCR, widening the spectrum of detectable phylogenetic groups in direct comparison to dominant members of the bacterial community [152]. In order to reach higher throughput numbers than Sanger Sequencing can provide at this stage, next generation sequencing technologies are used alternatively to analyze thousands of OTUs from different functional guilds. Yet, for complex environmental samples such as soil samples, DGGE, SSCP as well as T-RFLP provide similar compelling results on bacterial community composition [153] and microbial dynamics [152, 154], but a major drawback of ribosomal DNA based fingerprinting methods is that all DNA present in a sample is amplified, regardless the metabolic activity of bacteria, thus being less usable to reflect acute process dynamics such as crises in anaerobic digesters alone (see 3.1.2). Furthermore, molecular fingerprinting methods are not considered quantitative, but can include quantitative matrices as basis for dendrograms or can be related to multivariate analyses including process parameters, hierarchical clustering and specific microbial activities should be combined with genomic/fingerprinting data and incorporated into multivariate ordination methods such as Principal Coordinate or Principal Component Analysis (PCoA/PCA) in order to complete the whole picture drawn from a biogas biocenosis [26, 112, 155].
To evaluate environmental and process derived ecosystems, diversity is a suitable parameter that is measured by the number of different species (also from phylogenetic identification of OTUs from clone libraries assuming that one OTU corresponds to one species) including the inequality in relative abundance (Fig. 2). Therefore, diversity indices include abundance, richness and evenness as well as the Shannon index (H’). Abundance is the relative representation of a species in a community, i.e. number of a specific organism. Richness is defined as the number of different species or OTUs obtained from fingerprinting or cloning methods. Evenness is a measure of the equitability of abundance. The Shannon index (H’) is calculated by the relative abundance and richness of each species (OTU or T-RFLP peak) respectively. The higher the number of phylotypes evenly distributed, the higher the H’ index, which is specifically appropriate for the evaluation of low abundant, but important species (indicator species) due to its high sensitivity by proportional weighting [156]. Recent community studies regarding mesophilic and thermophilic co-digestion [157] and CSTR feeding pattern comparisons [158] indicate that high initial evenness (more dynamic populations) favors the microbial functionality under selective stress conditions, suggesting the microbial community to be more flexible.

Diversity—a complex parameter to describe the microbial composition of a given ecosystem defined by the indices richness (increases with the number of different species), evenness (distribution of present species) and abundance (number of a certain species), each colored hexagon represents one species (1); linking community data with process parameters, different hexagon sizes reflect the number of each species (abundances) ideal-theoretically correlated with typical process parameters and their occurrences (2)
To include activity analysis, microarrays based on the hybridization of oligonucleotides or PCR products can be used to generate gene expression profiles and signatures and have been applied to investigate bacterial communities of composts as well as methanogenic communities by specifically designed microarray-chips, i.e. COMPOCHIP and ANAEROCHIP [159, 160]. This technique has also been applied in combination with real-time PCR to investigate and quantify specific targets of organic waste associated microbial communities [161]. However, the traditional microarray approach cannot detect novel genes since the device construction only involves known nucleotide sequences [162]. Therefore, metatranscriptomics described in the following section are currently preferred to enable gene expression identification without a priori sequence knowledge [162, 163] (see 3.1.4).
3.1.4 Next Generation Sequencing and Meta-Omics
The advent of affordable high-throughput Next Generation Sequencing (NGS) [84, 164] has boosted the number of sequence entries in databanks. This information has not only significantly enlarged our knowledge in systems biology; it represents an invaluable basis for further developments and exploitation. Different NGS technologies using emulsion or bridge PCR are available and can generate millions of parallel reads in small volume reactions with average read lengths between ca. 40 and about 1,100 bp. Illumina platforms are currently the most frequently used. 454 pyrosequencing will no longer be sustained. Originary Pacific Biosciences’ (PacBio) RS sequencers typically generate long reads of >1 kb but the cost per base and the raw error rate (>10 % on average) are relatively high. Cost and error rates are actually lowest for Illumina and in between for Ion Torrent PGM systems [164, 165], but PacBio RS sequencing can be particularly useful e.g. by resolving problematic genomic areas such as AT-rich regions.
As compared to Sanger sequencing, error rates of these NGS systems are high, and problematic (GC-, AT-rich) regions can cause bias. Since this can result in erroneously high diversity, as observed in some ecosystem analyses, high coverage of parallel reads is required to generate reliable NGS results. Including data processing, particularly sequence assembly, all of these issues necessitate massive biocomputing efforts [166]. Respective bioinformatics pipelines and their maintenance are not affordable for any lab. Many limitations, however, will soon be overcome. Cheap annotation via cloud computing is already feasible [167], and developments towards increased read length and accuracy are going on. For example, PacBio recently introduced the RS II sequencers which are based on single molecule, real-time (SMRT) technology. PacBio claims that half of the data are in reads >14,000 base pairs with accuracy equal to Sanger sequencing. In a recent report (Mosher et al. 2014), PacBio RS II sequencing using P4/C2 chemistry surpassed the accuracy of Roche/454 pyrosequencing and generated longer reads.
For application of PCR based NGS approaches, it must additionally be considered that such amplicon sets are typically interspersed with chimaeras (see Sect. 3.1.2). These dissemble higher diversity than actually present and must be eliminated [98]. In addition, possible bias associated with Reverse Transcription or DNase treatment (see Sect. 3.1.1) must be considered [168]. However, if these challenges are adequately met, amplicon sequencing and metagenomics can produce highly comparable results, as this was shown for samples from different biogas processes by parallel analyses of curated V6-V8 (similarly as the V3-V5) 16S rDNA amplicon libraries and extracted 16S rDNA sequences from metagenomes (without interspersed selective PCR step) [169]. Although the NGS sequence numbers were much higher in this comparison, the PCR approach that was directed against the highly variable V6-V8 region provided substantially more profound insight into the bacterial community structures, occasionally even below the genus level.
NGS analysis of (complete) microbial genomes is another approach of inestimable value. It not only deepens our knowledge on microbial capacities, with the rising number of sequenced genomes and improved annotation, a more and more solid reference database is created for metagenomics and metatranscriptomics [170], leading e.g. to more reliable reference matches and improved binning accuracy. Metatranscriptomics currently is the most straightforward approach to investigate (key) metabolic pathways of interest at the transcription level, and RNAseq-based approaches allow quantitative transcriptome profiling, if suitable reference genomes or transcriptomes are available (Mutz et al. 2013). Although transcriptional activity is mostly regarded as equivalent with expression and activity, subcellular compartmentalization or excretion of enzymes and regulation are occurring at the protein level, and posttranslational modification can alter protein location and function. Additionally cross-linked metabolomics and metaproteomics might thus better reflect functional protein expression and activity in future (Vanwonterghem et al. 2014).
Although all of these approaches are providing an increasingly indispensable information background and data mining repository, they will not be applied for production scale monitoring and real-time process assays since equipment costs are too high for routine analysis, and highly skilled personnel is required. However, based on the compiled background, more meaningful and informative bioindicators may be identified and respective specifically targeted, e.g. (RT)qPCR based biomarkers could be developed (see Sect. “Microbial Guilds, Bioindicators and Transcriptional Profiling”). Such assays are much better suited for labs performing routine analyses.
3.2 Microscopy Based Detection of Microorganisms: Specific and Non-specific Imaging
For observations of bacterial and archaeal cells and biofilms, granules or flocs several microscopy techniques are useful, reaching from simple light microscopy (LM) with limited resolution to high-resolution scanning electron microscopy (SEM), transmission electron microscopy (TEM) and confocal laser scanning microscopy (CLSM). Amongst them, fluorescence-coupled microscopy is highly sophisticated due to its ability to detect selected groups or specific species within complex mixed communities. It is therefore widely used in microbial ecology studies allowing the visualization of spatial distribution of cells in a sample. For an in situ hybridization, a labelled probe, i.e. a fluorochromes or radioactive signal joined denatured DNA fragment is annealed to a sequence homologous to a certain target DNA (genomic DNA or PCR-amplicons). Using group- or species-specific staining, the differentiation between distinct populations is permitted leading to deep insights into the organization of biofilms and flocs [171], but strongly depends on the type of microscope used.
3.2.1 Fluorescence in Situ Hybridization-Based Confocal Laser Scanning Microscopy (FISH-CLSM)
Whereas epifluorescent imaging gives optical information from only one layer in two-dimensions [172], confocal scanning laser microscopy (CLSM) is capable of imaging a specimen via successive expositions of thin sections that can be reconstructed by computational assistance for 3D and 4D image visualization and analysis (IMARIS, Bitplane, Oxford Instruments). This allows the determination of multi-dimensional relationships of cells and their surroundings [173, 174]. The specimen is focused with a laser beam and pinhole selected fluorescent signals are detected by a photomultiplier, which results in high sensitive, high detailed and non-destructive image acquisition [175]. Fluorescence in situ hybridization targets genera or species specific ribosomal RNA fragments via probes available for Eubacteria (EUB) and Archaea (ARC). These fragments are specifically labeled with fluorescent dyes (Cy3, Cy5, FITC or FLUOS) that have individual emission wavelength optima to detect and identify multiple populations of target organisms in one sample at the same time [176]. A vast assortment of organism specific probes has already been described [177–179] and the list is constantly expanding in databases such as ‘probeBase’ [180], which provides currently over 1,300 rRNA-targeted oligonucleotide probe entries. The FISH-CLSM derived image also allows the rapid quantification of fluorescence signals, i.e. number of specific cells or percentage of area covered by biofilms [181]. Minimal statistical evaluation requires three independent samples and the observation of three individual specimen spots, when samples are homogeneous and evenly distributed [182]. At this juncture, flow cytometry (FCM) combines the advantages of microscopy and biochemical analysis for the measurement of biochemical and physical characteristics of individual cells moving in a fluid stream passing an optical sensor [177, 183, 184] (Fig. 3). In this regard, cytometric fingerprints have been reported to enable the decoding of microbial community dynamics in managed anaerobic microbial systems [185]. CLSM can also be combined with Raman spectroscopy to e.g. examine extracellular polymeric substances (EPS) producing biofilms and thereof distributed polysaccharides such as cellulose, alginate, sodium alginate, dextran, or nucleic acids during the development of the whole biofilm [186].

Single-cell identification and quantification by either epifluorescence microscopy or quantitative flow cytometry on the basis of fluorescence in situ hybridization according to Amann and Fuchs [177]. The sample preparation involves the fixation of microbial cells to stabilize and permeabilize their membranes to allow labelled oligonucleotide probes to access and hybridize to certain intracellular targets. Adapted from Jul 20, 2015, Nature Publishing Group
Numerous fluorescent dyes for DNA or RNA specific staining such as acridine orange or 4′,6-diamidino-2-phenylindole (DAPI) are used in addition to probe specific labeling, e.g. to assess the total number of bacteria against specific signals from fluorescein-labeled species [187]. Commercially available viability kits for fluorescence microscopy, e.g. Live/Dead BacLightTM (Molecular Probes®, Life Technologies) can be used to discriminate between viable and non-viable cells. Furthermore, a broad range of fluorescein-coupled molecules such as polyanionic dextrans or lectins of various molecular masses, redox-sensitive chemical probes (e.g. resofurin and fluorescein) and other fluorogenic substances (e.g. fluorescein diacetate) can be used in live cell imaging experiments to analyze (i) chemical interactions of defined molecules, (ii) cellular physiological conditions about membrane potential or permeability and (iii) microzonal variations in biofilm chemistry regarding pH, redox potential or ion concentrations [188–190]. FISH is also performed in combination with fingerprinting methods (see Sect. 3.1.3) or cloning experiments as full-cycle rRNA approach to quantitatively determine the relevance and spatial distribution of given operational taxonomic units (OTUs) [191].
Microautoradiography-coupled FISH (MAR-FISH) is another tool for structure and function analyses in microbial ecology [192] that links phylotypic characteristics with metabolic activities to reveal microbial species responsible for key physiological processes [193, 194]. The microbial in situ uptake and incorporation of radioactively labelled substrates can be visualized and enumerated this way [195], but the method is limited to elements with radioactive isotopes (e.g. 13/14C, 3H, 15N, 34S, 33P, 18O), which makes secondary ion mass spectrometry (SIMS) become a constitutive alternative for MAR-FISH. However, in anaerobic digesters it has been used to elucidate metabolic functions of minor phylogenetic groups like Chloroflexi, Syntrophomonas Spirochaeta and Synergistes as well as Methanosaeta spp. in sugar and short fatty acid such as acetate, butyrate, and propionate utilization [196, 197], and led to the determination of degradation rates of glucose, acetate and propionate as well [198, 199]. Although not providing quantitative data, the complementary combination of MAR-FISH with quantitative real-time PCR can be useful to investigate active key functional microbial groups [112].
FISH probing and CLSM have thus been used to show, how microbial communities involved in the anaerobic biodegradation process are organized regarding biofilm formation, immobilization and attachment to solid substrate material [64, 200–202], but also to study bioreactor and full-scale biogas plant performances [203–205]. There are some drawbacks using hybridization based fluorescence microscopy that include fading or photo-bleaching of the fluorochromes, fluorescence quenching, the loss of fluorescence due to sample derived molecules interacting with the fluorochromes, limited archaeal cell wall permeability and inefficient or incorrect hybridization. Many of these problems can be overcome by modifications of the preparation protocol towards sample and organism (i.e. gram-positive/gram-negative Bacteria and methanogenic Archaea) optimized hybridization conditions such as temperature or formamide concentrations [206], enzymatic pretreatment as routinely applied for catalyzed reporter deposition FISH (CARD-FISH) [207], up to double labeling (DOPE-FISH) for improved signal intensity and rRNA accessibility [208, 209], or even individual probe design [177, 210]. FISH-CLSM is clearly a valuable technique for AD processes to analyze microbial dynamics, since both, qualitative and quantitative information can be obtained, but specialized personnel and laboratory equipment is required to perform these analyses.
3.2.2 High-Resolution Microscopy: Scanning Electron Microscopy (SEM)
Apart from molecular biology depending light microscopy techniques, biofilms and single cells can be also investigated by scanning electron microscopy with unequalled magnifications of up to 500,000-fold (Carl Zeiss Ultra 55, Hitachi S-3000 N). New microscope-generations like RISA even integrate correlative, confocal Raman imaging with scanning electron microscopy (Raman-SEM), permitting a direct link between ultra-structural surface properties and molecular compound information (WITec, TESCAN). Therefore, scanning and transmission electron microscopy (SEM/TEM) have not only been used to study overall biofilm organization patterns (see below), but to investigate cell-to-cell interactions of anaerobic digestion process innate syntrophic microbial partners on a nano-scale level such as the interspecies electron transfer [211]. Direct interspecies electron transfer (DIET) depends on hydrogen and carbon source such as ethanol or formate, which was recently discovered for Geobacter metallireducens and Methanosaeta harundinacea or Methanosarcina barkeri interactions to lastly reduce carbon dioxide to methane [212]. It has also been shown, that growth of fermentative and methanogenic microbes on conductive carriers is tangible, suggesting Bacteria (most likely Clostriaceae) and methanogenic Archaea (most likely Methanobacteriaceae) can transfer electrons from a stainless steel support even without the involvement of hydrogen or formate [213].
Considering the low growth rate of methanogenic Archaea, immobilization on support material such as polymers (e.g. polyurethane, acrylonitrile-acrylamide, nylon) is a potential strategy to allow longer residence times in bioreactors for the adjustment to unstable conditions and varying feeding regimes [214] as shown by SEM for lab-scale reactors continuously operated with vinasse waste to keep COD removal rates constant at decreased retention times and various organic loading rates [215]. SEM was also used to study the natural biofilm formation on zeolite particles during in sacco incubation in semi-continuously, completely stirred lab-scale fermenters fed with grass silage [64, 202], or comparing several other carriers for Bacteria and Archaea such as activated carbon, polyvinyl alcohol or glass fibers in anaerobic digesters treating cattle manure [216], demonstrating that specific materials can selectively support methanogens to avoid co-cultivation of unwanted sulfate-reducing bacteria (SRB) during anaerobic wastewater treatment and methane production from molasses [217]. Focusing feedstock for AD, SEM can be part of efficiency evaluations of pretreatment methods for specific substrates such as that steam explosion induces significant morphological changes in treated lignocellulosic materials [218, 219].
In addition to the direct observation of sputtered organic matter, energy dispersive X-ray spectroscopy (SEM-EDS/EDX) allows element analyses of inorganic sample components of carrier materials or to characterize stable and active catalysts for hydrogen production from biogas, using SEM-TEM in combination with other microscopy methods to evaluate the deposition or arrangement of hollow carbon nanotubes and nanofibers [220]. Furthermore, EDS and TEM can be used for the localization of substrates or electron donors and acceptors or characterization of e.g. metal transformation in metal-reducing bacteria. However, the major drawback of electron microscopy is that it is an invasive method, which requires sample fixation and preparation including consecutive dehydration steps for specimen observations in high vacuum. Biological structures can be maintained by critical point drying, lyophilisation or high-pressure freezing. Instead of SEM, environmental SEM using lower vacuum pressures can be used alternatively as well. Pinpoint extraction and ultra-thin layer observations by consecutive cryosectioning are further techniques to investigate certain regions of interest and cellular aspects respectively based on SEM/TEM or focused ion beam (FIB)-SEM that can be also combined with CLSM 3D imaging for real 3D correlations of one and the same biological event in an identical sample [221, 222].
Abbreviations
- BB:
-
Bead-beating
- BMP:
-
Biological/biochemical methane potential
- BLAST:
-
Basic local alignment search tool
- Bp:
-
Base pair(s)
- cDNA:
-
Complementary DNA (transcribed from RNA species)
- CLSM:
-
Confocal laser scanning microscopy
- COD:
-
Chemical oxygen demand
- DGGE:
-
Denaturing-gradient gel electrophoresis
- DNA:
-
Deoxyribonucleic acid
- FISH:
-
Fluorescence in situ hybridization
- LCB:
-
Lignocellulosic biomass
- LM:
-
Light microscopy
- MQ:
-
Metabolic quotient
- mRNA:
-
Messenger RNA
- NA:
-
Nucleic acid(s)
- NGS:
-
Next generation sequencing
- OLR:
-
Organic loading rate
- PC(o)A:
-
Principal coordinate/Principal component analysis
- PCR:
-
Polymerase chain reaction
- PSM:
-
Process simulation model
- qPCR:
-
Quantitative Real-Time PCR
- rDNA:
-
Ribosomal deoxyribonucleic acid
- RNA:
-
Ribonucleic acid
- rRNA:
-
Ribosomal ribonucleic acid
- RT:
-
Reverse transcription
- SCFA:
-
Short-chain fatty acid(s) or also VFA
- SEM:
-
Scanning electron microscopy
- SMA:
-
Specific methanogenic activity
- TEM:
-
Transmission electron microscopy
- TGGE:
-
Temperature-gradient gel electrophoresis
- TVA/TIC:
-
Total volatile acids/total inorganic carbon
- VFA:
-
Volatile fatty acids
- VOA:
-
Volatile organic acids
References
Schink B (2006) Syntrophic associations in methanogenic degradation. In: Molecular basis of symbiosis, vol 41. Springer, Berlin, pp 1–19
McInerney MJ, Struchtemeyer CG, Sieber JR, Mouttaki H, Stams AJM, Schink B, Rohlin L, Gunsalus RP (2008) Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann N Y Acad Sci 1125:58–72
Demirel B, Scherer P (2008) The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: a review. Rev Environ Sci Biotechnol 7(2):173–190
Lebuhn M, Munk B, Effenberger M (2014) Agricultural biogas production in Germany—from practice to microbiology basics. Energy Sustain Soc 4(10):21
Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R (2008) Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6(8):579–591
Stams AJM, Oude Elferink SJWH, Westermann P (2003) Metabolic interactions between methanogenic consortia and anaerobic respiring bacteria. Adv Biochem Eng Biotechnol 81:31–56
Fotidis IA, Karakashev D, Angelidaki I (2014) The dominant acetate degradation pathway/methanogenic composition in full-scale anaerobic digesters operating under different ammonia levels. Int J Environ Sci Technol 11(7):2087–2094
Schink B (1997) Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61(2):262–280
Stams AJM, Plugge CM (2009) Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol 7(8):568–577
McInerney MJ, Sieber JR, Gunsalus RP (2009) Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol 20(6):623–632
Worm P, Koehorst JJ, Visser M, Sedano-Núñez VT, Schaap PJ, Plugge CM, Sousa DZ, Stams AJM (2014) A genomic view on syntrophic versus non-syntrophic lifestyle in anaerobic fatty acid degrading communities. Biochim Biophys Acta—Bioenerg 1837(12):2004–2016
Solli L, Håvelsrud OE, Horn SJ, Rike AG (2014) A metagenomic study of the microbial communities in four parallel biogas reactors. Biotechnol Biofuels 7(1):146
Kazda M, Langer S, Bengelsdorf FR (2014) Fungi open new possibilities for anaerobic fermentation of organic residues. Energy Sustain Soc 4(1):6
Koeck DE, Wibberg D, Maus I, Winkler A, Albersmeier A, Zverlov VV, Liebl W, Pühler A, Schwarz WH, Schlüter A (2014) Complete genome sequence of the cellulolytic thermophile Ruminoclostridium cellulosi wild-type strain {DG}5 isolated from a thermophilic biogas plant. J Biotechnol 188:136–137
Gruninger RJ, Puniya AK, Callaghan TM, Edwards JE, Youssef N, Dagar SS, Fliegerova K, Griffith GW, Forster R, Tsang A, McAllister T, Elshahed MS (2014) Anaerobic fungi (phylum Neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol Ecol 90(1):1–17
Weiland P (2010) Biogas production: current state and perspectives. Appl Microbiol Biotechnol 85(4):849–860
Nikolausz M, Walter RFH, Sträuber H, Liebetrau J, Schmidt T, Kleinsteuber S, Bratfisch F, Günther U, Richnow HH (2013) Evaluation of stable isotope fingerprinting techniques for the assessment of the predominant methanogenic pathways in anaerobic digesters. Appl Microbiol Biotechnol 97(5):2251–2262
Lv Z, Hu M, Harms H, Richnow HH, Liebetrau J, Nikolausz M (2014) Stable isotope composition of biogas allows early warning of complete process failure as a result of ammonia inhibition in anaerobic digesters. Bioresour Technol 167:251–259
Polag D, Krapf LC, Heuwinkel H, Laukenmann S, Lelieveld J, Keppler F (2014) Stable carbon isotopes of methane for real-time process monitoring in anaerobic digesters. Eng Life Sci 14(2):153–160
Van Soest PJ, McQueen RW (1973) The chemistry and estimation of fibre. Proc Nutr Soc 32(3):123–130
Krapf LC, Heuwinkel H, Schmidhalter U, Gronauer A (2013) The potential for online monitoring of short-term process dynamics in anaerobic digestion using near-infrared spectroscopy. Biomass Bioenergy 48:224–230
da Silva TL, Roseiro JC, Reis A (2012) Applications and perspectives of multi-parameter flow cytometry to microbial biofuels production processes. Trends Biotechnol 30(4):225–232
Rosselló-Móra R (2012) Towards a taxonomy of Bacteria and Archaea based on interactive and cumulative data repositories. Environ Microbiol 14:318–334
Stantscheff R, Kuever J, Rabenstein A, Seyfarth K, Dröge S, König H (2014) Isolation and differentiation of methanogenic Archaea from mesophilic corn-fed on-farm biogas plants with special emphasis on the genus Methanobacterium. Appl Microbiol Biotechnol 98(12):5719–5735
Musat N, Foster R, Vagner T, Adam B, Kuypers MMM (2012) Detecting metabolic activities in single cells, with emphasis on nanoSIMS. FEMS Microbiol Rev 36:486–511
Vanwonterghem I, Jensen PD, Ho DP, Batstone DJ, Tyson GW (2014) Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr Opin Biotechnol 27:55–64
Voß E, Weichgrebe D, Rosenwinkel KH (2009) FOS/TAC: Herleitung, Methodik, Anwendung und Aussagekraft. In: Proceedings of the international scientific conference biogas science 2009, vol 3, pp 675–682
Quist-Wessel PMF, Langeveld J (2014) Bioenergy production from waste agricultural biomass. In: Langeveld H, Dixon J, Van Keulen H (eds) Biofuel cropping systems carbon, land and food. Taylor and Francis, Hoboken
Yenigün O, Demirel B (2013) Ammonia inhibition in anaerobic digestion: a review. Process Biochem 48(5–6):901–911
Hansen KH, Angelidaki I, Ahring BK (1998) Anaerobic digestion of swine manure: inhibition by ammonia. Water Res 32(1):5–12
Munk B, Bauer C, Gronauer A, Lebuhn M (2011) A metabolic quotient for methanogenic Archaea. Water Sci Technol 66(11):2311–2317
Munk B, Bauer C, Gronauer A, Lebuhn M (2010) Population dynamics of methanogens during acidification of biogas fermenters fed with maize silage. Eng Life Sci 10(6):496–508
Demirel B, Scherer P (2011) Trace element requirements of agricultural biogas digesters during biological conversion of renewable biomass to methane. Biomass Bioenergy 35(3):992–998
Vintiloiu A, Lemmer A, Oechsner H, Jungbluth T (2012) Mineral substances and macronutrients in the anaerobic conversion of biomass: an impact evaluation. Eng Life Sci 12(3):287–294
Chen Y, Cheng JJ, Creamer KS (2008) Inhibition of anaerobic digestion process: a review. Bioresour Technol 99(10):4044–4064
Mudhoo A, Kumar S (2013) Effects of heavy metals as stress factors on anaerobic digestion processes and biogas production from biomass. Int J Environ Sci Technol 10(6):1383–1398
Hecht C, Griehl C (2009) Investigation of the accumulation of aromatic compounds during biogas production from kitchen waste. Bioresour Technol 100(2):654–658
Sayder B, Vitz H, Mohring S, Merrettig-Bruns U, Kabasci S, Hamscher G, Tuerk J (2009) Gehemmte Biologie. Biogas-J 12(2):44–45
Young JC (2004) Respirometry for environmental science and engineering. SJ Enterprises, Springdale, Arkansas
Noike T, Endo G, Chang JE, Yaguchi J, Matsumoto J (1985) Characteristics of carbohydrate degradation and the rate-limiting step in anaerobic digestion. Biotechnol Bioeng 27(10):1482–1489
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol Biol Rev 66,(3):506–577
Merlin Christy P, Gopinath LR, Divya D (2014) A review on anaerobic decomposition and enhancement of biogas production through enzymes and microorganisms. Renew Sustain Energy Rev 34(C):167–173
Arantes V, Saddler JN (2010) Access to cellulose limits the efficiency of enzymatic hydrolysis: the role of amorphogenesis. Biotechnol Biofuels 3:4
Zhang Y-HP, Lynd LR (2005) Determination of the number-average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromolecules 6(3):1510–1515
Schwald W, Chart M, Breuil C, Saddler JN (1988) Applied microbiology biotechnology comparison of HPLC and colorimetric methods for measuring cellulolytic activity, pp 398–403
Khan AW, Tremblay D, LeDuy A (1986) Assay of xylanase and xylosidase activities in bacterial and fungal cultures. Enzyme Microb Technol 8(6):373–377
Obst U (1985) Test instructions for measuring the microbial metabolic activity in water samples. Fresenius’ Zeitschrift fuer Anal Chemie 321(2):166–168
Gasch C, Hildebrandt I, Rebbe F, Röske I (2013) Enzymatic monitoring and control of a two-phase batch digester leaching system with integrated anaerobic filter. Energy Sustain Soc 3(1):10
Mshandete AM, Björnsson L, Kivaisi AK, Steven M, Rubindamayugi T, Mattiasson B (2008) Two-stage anaerobic digestion of aerobic pre-treated sisal leaf decortications residues: hydrolases activities and biogas production profile. Afr J Biochem Res 2(5):211–218
Holzapfel-Pschorn A, Obst U, Haberer K (1987) Sensitive methods for the determination of microbial activities in water samples using fluorigenic substrates. Fresenius’ Zeitschrift fuer Anal Chemie 327(5–6):521–523
Regueiro L, Veiga P, Figueroa M, Alonso-Gutierrez J, Stams AJM, Lema JM, Carballa M (2012) Relationship between microbial activity and microbial community structure in six full-scale anaerobic digesters. Microbiol Res 167(10):581–589
Parawira W (2012) Enzyme research and applications in biotechnological intensification of biogas production. Crit Rev Biotechnol 32:172–186
Ahuja SK, Ferreira GM, Moreira AR (2004) Utilization of enzymes for environmental applications. Crit Rev Biotechnol 24(2–3):125–154
Warthmann R, Baier U, Meier U, Hersener J (2013) Optimisation of anaerobic digestion by pre-treatment, additives and process engineering. In: Biogas process optimisation workshop, 2013
Bruni E, Jensen AP, Angelidaki I (2010) Comparative study of mechanical, hydrothermal, chemical and enzymatic treatments of digested biofibers to improve biogas production. Bioresour Technol 101(22):8713–8717
Shen S, Nges IA, Yun J, Liu J (2014) Pre-treatments for enhanced biochemical methane potential of bamboo waste. Chem Eng J 240:253–259
Yu S, Zhang G, Li J, Zhao Z, Kang X (2013) Effect of endogenous hydrolytic enzymes pretreatment on the anaerobic digestion of sludge. Bioresour Technol 146:758–761
Diak J, Örmeci B, Kennedy KJ (2012) Effect of enzymes on anaerobic digestion of primary sludge and septic tank performance. Bioprocess Biosyst Eng 35(9):1577–1589
Plöchl M, Hilse A, Heiermann M, Suarez Quinones T, Budde J, Prochnow A (2009) Application of hydrolytic enzymes for improving biogas feedstock fluidity. CIGR E J IX:1–16
Burgess J, Pletschke B (2008) Hydrolytic enzymes in sewage sludge treatment: a mini-review. Water Sa 34(3):343–350
Davidsson Å, Wawrzynczyk O Norrlöw, Jansen J la C (2007) Strategies for enzyme dosing to enhance anaerobic digestion of sewage sludge. J Residuals Sci Technol 4(1):1–7
Vavilin VA, Rytov SV, Lokshina LY (1996) A description of hydrolysis kinetics in anaerobic degradation of particulate organic matter. Bioresour Technol 56(2–3):229–237
Gokhale A, Lu J, Lee I (2013) Immobilization of cellulase on magnetoresponsive graphene nano-supports. J Mol Catal B Enzym 90:76–86
Weiss S, Lebuhn M, Andrade D, Zankel A, Cardinale M, Birner-Gruenberger R, Somitsch W, Ueberbacher BJ, Guebitz GM (2013) Activated zeolite—Suitable carriers for microorganisms in anaerobic digestion processes? Appl Microbiol Biotechnol 97(7):3225–3238
Montalvo S, Guerrero L, Borja R, Sánchez E, Milán Z, Cortés I, Angeles de la la Rubia M (2012) Application of natural zeolites in anaerobic digestion processes: a review. Appl Clay Sci 58:125–133
Jeganathan J, Nakhla G, Bassi A (2007) Hydrolytic pretreatment of oily wastewater by immobilized lipase. J Hazard Mater 145(1–2):127–135
Kavitha S, Adish Kumar S, Yogalakshmi KN, Kaliappan S, Rajesh Banu J (2013) Effect of enzyme secreting bacterial pretreatment on enhancement of aerobic digestion potential of waste activated sludge interceded through EDTA. Bioresour Technol 150:210–219
Wang W, Yan L, Cui Z, Gao Y, Wang Y, Jing R (2011) Characterization of a microbial consortium capable of degrading lignocellulose. Bioresour Technol 102(19):9321–9324
Yan L, Gao Y, Wang Y, Liu Q, Sun Z, Fu B, Wen X, Cui Z, Wang W (2012) Diversity of a mesophilic lignocellulolytic microbial consortium which is useful for enhancement of biogas production. Bioresour Technol 111:49–54
Weiss S, Tauber M, Somitsch W, Meincke R, Müller H, Berg G, Guebitz GM (2010) Enhancement of biogas production by addition of hemicellulolytic bacteria immobilised on activated zeolite. Water Res 44(6):1970–1980
Kovács KL, Ács N, Kovács E, Wirth R, Rákhely G, Strang O, Herbel Z, Bagi Z (2013) Improvement of biogas production by bioaugmentation. Biomed Res Int 2013:482653
Bagi Z, Acs N, Bálint B, Horváth L, Dobó K, Perei KR, Rákhely G, Kovács KL (2007) Biotechnological intensification of biogas production. Appl Microbiol Biotechnol 76(2):473–482
Peng X, Börner RA, Nges IA, Liu J (2014) Impact of bioaugmentation on biochemical methane potential for wheat straw with addition of Clostridium cellulolyticum. Bioresour Technol 152:567–571
Levine SE, Fox JM, Blanch HW, Clark DS (2010) A mechanistic model of the enzymatic hydrolysis of cellulose. Biotechnol Bioeng 107(1):37–51
Christ O, Wilderer PA, Angerhöfer R, Faulstich M (2000) Mathematical modeling of the hydrolysis of anaerobic processes. Water Sci Technol 41(3):61–65
Rajendran K, Kankanala HR, Lundin M, Taherzadeh MJ (2014) A novel process simulation model (PSM) for anaerobic digestion using Aspen Plus. Bioresour Technol 168:7–13
Koch K, Lübken M, Gehring T, Wichern M, Horn H (2010) Biogas from grass silage—Measurements and modeling with ADM1. Bioresour Technol 101(21):8158–8165
Ramirez I, Mottet A, Carrère H, Déléris S, Vedrenne F, Steyer J-P (2009) Modified ADM1 disintegration/hydrolysis structures for modeling batch thermophilic anaerobic digestion of thermally pretreated waste activated sludge. Water Res 43(14):3479–3492
Batstone D (2001) The IWA anaerobic digestion model no 1, vol 1, no. 1, pp 1–68
Narihiro T, Kamagata Y (2013) Cultivating Yet-to-be Cultivated Microbes: The Challenge Continues. Microbes Environ 28(2):163–165
Hugenholtz P (2002) Exploring prokaryotic diversity in the genomic era. Genome Biol 3(2):0003.1–0003.8
Westerholm M, Levén L, Schnürer A (2012) Bioaugmentation of syntrophic acetate-oxidizing culture in biogas reactors exposed to increasing levels of ammonia. Appl Environ Microbiol 78(21):7619–7625
Munk B, Lebuhn M (2014) Process diagnosis using methanogenic Archaea in maize-fed, trace element depleted fermenters. Anaerobe 29:22–28
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11(1):31–46
Fontaine M, Guillot E (2003) Study of 18S rRNA and rDNA stability by real-time RT-PCR in heat-inactivated Cryptosporidium parvum oocysts. FEMS Microbiol Lett 226(2):237–243
Keer JT, Birch L (2003) Molecular methods for the assessment of bacterial viability. J Microbiol Methods 53(2):175–183
Lebuhn M, Effenberger M, Gronauer A, Wilderer PA, Wuertz S (2003) Using quantitative real-time PCR to determine the hygienic status of cattle manure. Water Sci Technol 48(4):97–103
Garcés G (2005) Quantitative real-time PCR for detecting Cryptosporidium parvum in cattle manure and anaerobic digester samples—Methodological advances in {DNA} extraction. In: Proceedings of the 8th Latin American workshop and symposium on anaerobic digestion, pp 68–73
Garcés-Sanchez G, Wilderer PA, Munch JC, Horn H, Lebuhn M (2009) Evaluation of two methods for quantification of hsp70 mRNA from the waterborne pathogen Cryptosporidium parvum by reverse transcription real-time PCR in environmental samples. Water Res 43(10):2669–2678
Schmedes S, Marshall P, King JL, Budowle B (2013) Effective removal of co-purified inhibitors from extracted DNA samples using synchronous coefficient of drag alteration (SCODA) technology. Int J Legal Med 127(4):749–755
Lebuhn M, Effenberger M, Garcés G, Gronauer A, Wilderer PA (2004) Evaluating real-time PCR for the quantification of distinct pathogens and indicator organisms in environmental samples. Water Sci Technol 50(1):263–270
Dorak MT (ed) (2007) Real-time PCR, 1st edn. Taylor & Francis, New York
Lech M, Anders H-J (2014) Expression profiling by real-time quantitative polymerase chain reaction (RT-qPCR). Methods Mol Biol 1169:133–142
Kim J, Lim J, Lee C (2013) Quantitative real-time PCR approaches for microbial community studies in wastewater treatment systems: Applications and considerations. Biotechnol Adv 31(8):1358–1373
Navarro E, Serrano-Heras G, Castaño MJ, Solera J (2015) Real-time PCR detection chemistry. Clin Chim Acta 439:231–250
Terpe K (2013) Overview of thermostable DNA polymerases for classical PCR applications: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 97(24):10243–10254
McInerney P, Adams P, Hadi MZ (2014) Error rate comparison during polymerase chain reaction by DNA Polymerase. Mol Biol Int 2014:8
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ (2011) Human microbiome consortium. In: F. Petrosino J, Knight R, Birren BW (eds) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21(3):494–504
Sieber JR, McInerney MJ, Gunsalus RP (2012) Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol 66(1):429–452
Bauer C, Korthals M, Gronauer A, Lebuhn M (2008) Methanogens in biogas production from renewable resources—a novel molecular population analysis approach. Water Sci Technol 58(7):1433–1439
Widmer G, Orbacz EA, Tzipori S (1999) beta-tubulin mRNA as a marker of Cryptosporidium parvum oocyst viability. Appl Environ Microbiol 65(4):1584–1588
McKillip JL, Jaykus LA, Drake M (1999) Nucleic acid persistence in heat-killed Escherichia coli O157:H7 from contaminated skim milk. J Food Prot 62(8):839–844
Ho DP, Jensen PD, Batstone DJ (2013) Methanosarcinaceae and acetate-oxidizing pathways dominate in high-rate thermophilic anaerobic digestion of waste-activated sludge. Appl Environ Microbiol 79(20):6491–6500
Nettmann E, Bergmann I, Pramschüfer S, Mundt K, Plogsties V, Herrmann C, Klocke M (2010) Polyphasic analyses of methanogenic archaeal communities in agricultural biogas plants. Appl Environ Microbiol 76(8):2540–2548
Li J, Rui J, Pei Z, Sun X, Zhang S, Yan Z, Wang Y, Liu X, Zheng T, Li X (2014) Straw- and slurry-associated prokaryotic communities differ during co-fermentation of straw and swine manure. Appl Microbiol Biotechnol 98(10):4771–4780
Steinberg LM, Regan JM (2011) Response of lab-scale methanogenic reactors inoculated from different sources to organic loading rate shocks. Bioresour Technol 102:8790–8798
Zhang C, Yuan Q, Lu Y (2014) Inhibitory effects of ammonia on methanogen mcrA transcripts in anaerobic digester sludge. FEMS Microbiol Ecol 87(2):368–377
Lv Z, Leite AF, Harms H, Richnow HH, Liebetrau J, Nikolausz M (2014) Influences of the substrate feeding regime on methanogenic activity in biogas reactors approached by molecular and stable isotope methods. Anaerobe 29:91–99
Freitag TE, Prosser JI (2009) Correlation of methane production and functional gene transcriptional activity in a peat soil. Appl Environ Microbiol 75(21):6679–6687
Watanabe T, Kimura M, Asakawa S (2009) Distinct members of a stable methanogenic archaeal community transcribe mcrA genes under flooded and drained conditions in Japanese paddy field soil. Soil Biol Biochem 41(2):276–285
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59(1):143–169
Talbot G, Topp E, Palin MF, Massé DI (2008) Evaluation of molecular methods used for establishing the interactions and functions of microorganisms in anaerobic bioreactors. Water Res 42(3):513–537
Rolfs A, Schuller I, Finckh U, Weber-Rolfs I (1992) Detection of single base changes using PCR. In: Rolfs A, Schuller I, Finckh U, Weber-Rolfs I (eds) PCR: clinical diagnostics and research. Springer, Berlin, pp 149–167
Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51:263–273
Frigon D, Oerthe DB, Morgenroth E, Raskin L (2002) Oligonucleotide probe hybridization and modeling results suggest that populations consuming readily degradable substrate have high cellular RNA levels. Water Sci Technol 45(6):115–126
Candrian U (1995) Polymerase chain reaction in food microbiology. J Microbiol Methods 23(1):89–103
Roose-Amsaleg C, Garnier-Sillam E, Harry M (2001) Extraction and purification of microbial DNA from soil and sediment samples. Appl. Soil Ecol 18(1):47–60
Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ (2000) Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66(12):5488–5491
Maukonen J, Mättö J, Wirtanen G, Raaska L, Mattila-Sandholm T, Saarela M (2003) Methodologies for the characterization of microbes in industrial environments: a review. J Ind Microbiol Biotechnol 30(6):327–356
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414
Janssen P, Bleeker M, Kersters K (1996) Evaluation of the DNA fingerprinting method AFLP as a new tool in bacterial taxonomy, pp 1881–1893
Sklarz MY, Angel R, Gillor O, Soares MIM (2009) Evaluating amplified rDNA restriction analysis assay for identification of bacterial communities. Antonie Van Leeuwenhoek 96(4):659–664
Fisher MM, Triplett EW (1999) Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol 65(10):4630–4636
Welsh J, McClelland M (1990) Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res 18(24):7213–7218
Power EG (1996) RAPD typing in microbiology—a technical review. J Hosp Infect 34(4):247–265
Versalovic J, Schneider M, de Bruijn FJ, Lupski JR (1994) Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol Cell Biol 5(25):25–40
Mills DK, Entry JA, Gillevet PM, Mathee K (2007) Assessing microbial community diversity using amplicon length heterogeneity polymerase chain reaction. Soil Sci Soc Am J 71(2):572
Schwartz DC, Cantor CR (1984) Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37(1):67–75
Liu WT, Marsh TL, Cheng H, Forney LJ (1997) Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63(11):4516–4522
Nocker A, Burr M, Camper AK (2007) Genotypic microbial community profiling: a critical technical review. Microb Ecol 54(2):276–289
Fischer SG, Lerman LS (1983) DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc Natl Acad Sci USA 80(6):1579–1583
Muyzer G, Smalla K (1998) “Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoeck Int J Gen Mol Microbiol 73:127–141
Rosenbaum V, Riesner D (1987) Temperature-gradient gel electrophoresis. Thermodynamic analysis of nucleic acids and proteins in purified form and in cellular extracts. Biophys Chem 26(2–3):235–246
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T (1989) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 86(8):2766–2770
Schwieger F, Tebbe CC (1998) A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl Environ Microbiol 64(12):4870–4876
Su C, Lei L, Duan Y, Zhang K-Q, Yang J (2012) Culture-independent methods for studying environmental microorganisms: methods, application, and perspective. Appl Microbiol Biotechnol 93(3):993–1003
Wagner R (1994) The regulation of ribosomal RNA synthesis and bacterial cell growth. Arch Microbiol 161(2):100–109
Wintzingerode FV, Göbel UB, Stackebrandt E (2006) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21(3):213–229
García-Martínez J, Acinas SG, Antón AI, Rodríguez-Valera F (1999) Use of the 16S-23S ribosomal genes spacer region in studies of prokaryotic diversity. J Microbiol Methods 36(1–2):55–64
Gürtler V, Stanisich A (1996) New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142 (Pt 1):3–16
Schmid M, Schmitz-Esser S, Jetten M, Wagner M (2001) 16S-23S rDNA intergenic spacer and 23S rDNA of anaerobic ammonium-oxidizing bacteria: implications for phylogeny and in situ detection. Environ Microbiol 3(7):450–459
Ziganshina EE, Bagmanova AR, Khilyas IV, Ziganshin AM (2014) Assessment of a biogas-generating microbial community in a pilot-scale anaerobic reactor. J Biosci Bioeng 117(6):730–736
Mills DK, Fitzgerald K, Litchfield CD, Gillevet PM (2003) A comparison of DNA profiling techniques for monitoring nutrient impact on microbial community composition during bioremediation of petroleum-contaminated soils. J Microbiol Methods 54(1):57–74
Stahl DA, Capman WC (1994) Applications of molecular genetics to the study of microbial communities. NATO ASI Ser 35:193–206
Muyzer G (2000) Genetic fingerprinting of microbial communities—present status and future perspectives. In: Bell CR, Brylinski M, Johnson-Green P (eds) Microbial biosystems: new frontiers. proceedings of the 8th international symposium on microbial ecology. Atlantic Canada Society for Microbial Ecology, Halifax, pp 503–572
Collins G, Woods A, McHugh S, Carton MW, O’Flaherty V (2003) Microbial community structure and methanogenic activity during start-up of psychrophilic anaerobic digesters treating synthetic industrial wastewaters. FEMS Microbiol Ecol 46(2):159–170
Padmasiri SI, Zhang J, Fitch M, Norddahl B, Morgenroth E, Raskin L (2007) Methanogenic population dynamics and performance of an anaerobic membrane bioreactor (AnMBR) treating swine manure under high shear conditions. Water Res 41(1):134–144
McHugh S, Collins G, O’Flaherty V (2006) Long-term, high-rate anaerobic biological treatment of whey wastewaters at psychrophilic temperatures. Bioresour Technol 97(14):1669–1678
Scully C, Collins G, O’Flaherty V (2005) Assessment of anaerobic wastewater treatment failure using terminal restriction fragment length polymorphism analysis. J Appl Microbiol 99(6):1463–1471
Liesack W, Dunfield PF (2002) Biodiversity in soils: use of molecular methods for its characterization. In: Bitton G (ed) Encyclopedia of environmental microbiology. Wiley, New York, pp 4516–4522
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180(18):4765–4774
Schmalenberger A, Tebbe CC (2014) Profiling the diversity of microbial communities with single-strand conformation polymorphism (SSCP). Methods Mol Biol 1096:71–83
Smalla K, Oros-Sichler M, Milling A, Heuer H, Baumgarte S, Becker R, Neuber G, Kropf S, Ulrich A, Tebbe CC (2007) Bacterial diversity of soils assessed by DGGE, T-RFLP and SSCP fingerprints of PCR-amplified 16S rRNA gene fragments: do the different methods provide similar results? J Microbiol Methods 69(3):470–479
Chachkhiani M, Dabert P, Abzianidze T, Partskhaladze G, Tsiklauri L, Dudauri T, Godon JJ (2004) 16S rDNA characterisation of bacterial and archaeal communities during start-up of anaerobic thermophilic digestion of cattle manure. Bioresour Technol 93(3):227–232
Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K, Cummings TA, Beers AR, Knight R, Angenent LT (2011) Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc Natl Acad Sci USA 108(10):4158–4163
Hill TCJ, Walsh KA, Harris JA, Moffett BF (2003) Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol 43(1):1–11
Yu D, Kurola JM, Kymäläinen M, Sinkkonen A, Romantschuk M (2014) Biogas production and methanogenic archaeal community in mesophilic and thermophilic anaerobic co-digestion processes. J Environ Manage 143:54–60
De Vrieze J, Verstraete W, Boon N (2013) Repeated pulse feeding induces functional stability in anaerobic digestion. Microb Biotechnol 6(4):414–424
Franke-Whittle IH, Goberna M, Pfister V, Insam H (2009) Design and development of the ANAEROCHIP microarray for investigation of methanogenic communities. J Microbiol Methods 79(3):279–288
Franke-Whittle IH, Knapp BA, Fuchs J, Kaufmann R, Insam H (2009) Application of COMPOCHIP microarray to investigate the bacterial communities of different composts. Microb Ecol 57(3):510–521
Goberna M, Gadermaier M, Schoen MA, Sperl D, Franke-Whittle IH, Wett B, Insam H (2012) Fingerprinting the microbial communities in organic wastes using oligonucleotide microarrays and Real-Time PCR. In: Trasar-Cepeda C, Hernandez T, Garcia C, Rad C, Gonzales-Carcedo S (eds) Soil enzymology in the recycling of organic wastes and environmental restoration, Burgos, Spain. Springer, Berlin, pp 285–298
Mutz K-O, Heilkenbrinker A, Lönne M, Walter J-G, Stahl F (2013) Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol 24(1):22–30
Carvalhais LC, Dennis PG, Tyson GW, Schenk PM (2012) Application of metatranscriptomics to soil environments. J Microbiol Methods 91(2):246–251
Solomon KV, Haitjema CH, Thompson DA, O’Malley MA (2014) Extracting data from the muck: deriving biological insight from complex microbial communities and non-model organisms with next generation sequencing. Curr Opin Biotechnol 28:103–110
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom 13(1):341
Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C (2013) Computational meta’omics for microbial community studies. Mol. Syst. Biol. 9(1):666
Koren S, Phillippy AM (2015) One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol 23:110–120
Pinto AJ, Raskin L (2012) PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 7(8):e43093
Lebuhn M, Hanreich A, Klocke M, Schlüter A, Bauer C, Pérez CM (2014) Towards molecular biomarkers for biogas production from lignocellulose-rich substrates. Anaerobe 29:10–21
Zakrzewski M, Goesmann A, Jaenicke S, Jünemann S, Eikmeyer F, Szczepanowski R, Al-Soud WA, Sørensen S, Pühler A, Schlüter A (2012) Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J Biotechnol 158(4):248–258
Wagner M, Assmus B, Hartmann A, Hutzler P, Amann R (1994) In situ analysis of microbial consortia in activated sludge using fluorescently labelled, rRNA-targeted oligonucleotide probes and confocal scanning laser microscopy. J Microsc 176(3):181–187
Ploem JS (1967) The use of a vertical illuminator with interchangeable dichroic mirrors for fluorescence microscopy with incident light. Z Wiss Mikrosk 68:129–142
Caldwell DE, Korber DR, Lawrence JR (1992) Confocal laser microscopy and digital image analysis in microbial ecology. Adv Microb Ecol 12:1–67
Lawrence JR, Korber DR, Hoyle BD, Costerton JW, Caldwell DE (1991) Optical sectioning of microbial biofilms. J Bacteriol 173(20):6558–6567
Taylor DL, Salmon ED (1989) Basic fluorescence microscopy. In: Wang Y-L, Taylor DL (eds) Fluorescence microscopy of living cells in culture. Academic Press, San Diego, pp 207–237
Behnam F, Vilcinskas A, Wagner M, Stoecker K (2012) A straightforward DOPE (double labeling of oligonucleotide probes)-FISH (fluorescence in situ hybridization) method for simultaneous multicolor detection of six microbial populations. Appl Environ Microbiol 78(15):5138–5142
Amann R, Fuchs BM (2008) Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat Rev Microbiol 6(5):339–348
Amann R, Kühl M (1998) In situ methods for assessment of microorganisms and their activities. Curr Opin Microbiol 1:352–358
Raskin L, Poulsen LK, Noguera DR, Rittmann BE, Stahl DA (1994) Quantification of methanogenic groups in anaerobic biological reactors by oligonucleotide probe hybridization. Appl Environ Microbiol 60(4):1241–1248
Loy A, Horn M, Wagner M (2003) probeBase: an online resource for rRNA-targeted oligonucleotide probes. Nucleic Acids Res 31(1):514–516
Kepner RL, Pratt JR (1994) Use of fluorochromes for direct enumeration of total bacteria in environmental samples: past and present. Microbiol Rev 58(4):603–615
Sanz JL, Köchling T (2007) Molecular biology techniques used in wastewater treatment: An overview. Process Biochem 42(2):119–133
Diaper JP, Tither K, Edwards C (1992) Rapid assessment of bacterial viability by flow cytometry. Appl Microbiol Biotechnol 38(2)
Crosland-Taylor PJ (1953) A device for counting small particles suspended in a fluid through a Tube. Nature 171:37–38
Koch C, Harnisch F, Schröder U, Müller S (2014) Cytometric fingerprints: evaluation of new tools for analyzing microbial community dynamics. Front Microbiol 5:273
Wagner M, Ivleva NP, Haisch C, Niessner R, Horn H (2009) Combined use of confocal laser scanning microscopy (CLSM) and Raman microscopy (RM): investigations on EPS-Matrix. Water Res 43(1):63–76
Hoff K (1988) Rapid and simple method for double staining of bacteria with 4′,6-diamino-2-phenylindole and fluorescein isothiocyanate labelled antibodies. Appl Environ Microbiol 54(12):2949–2952
Lukinavičius G, Reymond L, D’Este E, Masharina A, Göttfert F, Ta H, Güther A, Fournier M, Rizzo S, Waldmann H, Blaukopf C, Sommer C, Gerlich DW, Arndt H-D, Hell SW, Johnsson K (2014) Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods 11(7):731–733
Grimm JB, Sung AJ, Legant WR, Hulamm P, Matlosz SM, Betzig E, Lavis LD (2013) Carbofluoresceins and carborhodamines as scaffolds for high-contrast fluorogenic probes. ACS Chem Biol 8(6):1303–1310
Hunter RC, Beveridge TJ (2005) Application of a pH-sensitive fluoroprobe (C-SNARF-4) for pH microenvironment analysis in Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71(5):2501–2510
Zebulun O (2013) Molecular and biological techniques used in landfill investigations: A mini-review. Biotechnol Mol Biol Rev 8(2):35–42
Lee N, Nielsen PH, Andreasen KH, Juretschko S, Nielsen JL, Schleifer KH, Wagner M (1999) Combination of fluorescent in situ hybridization and microautoradiography-a new tool for structure-function analyses in microbial ecology. Appl Environ Microbiol 65(3):1289–1297
Nielsen JL, Nielsen PH (2010) Combined microautoradiography and fluorescence in situ hybridization (MAR-FISH) for the identification of metabolically active microorganisms. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 4093–4102
Collins G, Kavanagh S, McHugh S, Connaughton S, Kearney A, Rice O, Carrigg C, Scully C, Bhreathnach N, Mahony T, Madden P, Enright A-M, O’flaherty V (2006) Accessing the black box of microbial diversity and ecophysiology: recent advances through polyphasic experiments. J Environ Sci Health A Tox Hazard Subst Environ Eng 41(5):897–922
Okabe S, Kindaichi T, Ito T (2004) MAR-FISH—An ecophysiological approach to link phylogenetic affiliation and in situ metabolic activity of microorganisms at a single-cell resolution. Microbes Environ 19(2):83–98
Ariesyady HD, Ito T, Yoshiguchi K, Okabe S (2007) Phylogenetic and functional diversity of propionate-oxidizing bacteria in an anaerobic digester sludge. Appl Microbiol Biotechnol 75(3):673–683
Ariesyady HD, Ito T, Okabe S (2007) Functional bacterial and archaeal community structures of major trophic groups in a full-scale anaerobic sludge digester. Water Res 41(7):1554–1568
Ito T, Yoshiguchi K, Ariesyady HD, Okabe S (2011) Identification of a novel acetate-utilizing bacterium belonging to Synergistes group 4 in anaerobic digester sludge. ISME J 5(12):1844–1856
Ito T, Yoshiguchi K, Ariesyady HD, Okabe S (2012) Identification and quantification of key microbial trophic groups of methanogenic glucose degradation in an anaerobic digester sludge. Bioresour Technol 123:599–607
Song H, Clarke WP, Blackall LL (2005) Concurrent microscopic observations and activity measurements of cellulose hydrolyzing and methanogenic populations during the batch anaerobic digestion of crystalline cellulose. Biotechnol Bioeng 91(3):369–378
Fernández N, Montalvo S, Fernández-Polanco F, Guerrero L, Cortés I, Borja R, Sánchez E, Travieso L (2007) Real evidence about zeolite as microorganisms immobilizer in anaerobic fluidized bed reactors. Process Biochem 42:721–728
Weiss S, Zankel A, Lebuhn M, Petrak S, Somitsch W, Guebitz GM (2011) Investigation of mircroorganisms colonising activated zeolites during anaerobic biogas production from grass silage. Bioresour Technol 102(6):4353–4359
Krakat N, Westphal A, Schmidt S, Scherer P (2010) Anaerobic digestion of renewable biomass: thermophilic temperature governs methanogen population dynamics. Appl Environ Microbiol 76(6):1842–1850
Cresson R, Dabert P, Bernet N (2009) Microbiology and performance of a methanogenic biofilm reactor during the start-up period. J Appl Microbiol 106(3):863–876
Karakashev D, Batstone DJ, Angelidaki I (2005) Influence of environmental conditions on methanogenic compositions in anaerobic biogas reactors. Appl Environ Microbiol 71(1):331–338
Crocetti G, Murto M, Björnsson L (2006) An update and optimisation of oligonucleotide probes targeting methanogenic Archaea for use in fluorescence in situ hybridisation (FISH). J Microbiol Methods 65:194–201
Hoshino T, Yilmaz LS, Noguera DR, Daims H, Wagner M (2008) Quantification of target molecules needed to detect microorganisms by fluorescence in situ hybridization (FISH) and catalyzed reporter deposition-FISH. Appl Environ Microbiol 74(16):5068–5077
Stoecker K, Dorninger C, Daims H, Wagner M (2010) Double labeling of oligonucleotide probes for fluorescence in situ hybridization (DOPE-FISH) improves signal intensity and increases rRNA accessibility. Appl Environ Microbiol 76(3):922–926
Kumar RK, Chapple CC, Hunter N (1999) Improved double immunofluorescence for confocal laser scanning microscopy. J Histochem Cytochem 47:1213–1218
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, Lai T, Steppi S, Jobb G, Förster WI, Brettske S, Gerber A, Ginhart W, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer K-H (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32(4):1363–1371
Rotaru A-E, Shrestha PM, Liu F, Shrestha M, Shrestha D, Embree M, Zengler K, Wardman C, Nevin KP, Lovley DR (2014) A new model for electron flow during anaerobic digestion: direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ Sci 7(1):408–415
Rotaru A-E, Shrestha PM, Liu F, Markovaite B, Chen S, Nevin K, Lovley D (2014) Direct interspecies electron transfer between Geobacter metallireducens and Methanosarcina barkeri. Appl Environ Microbiol 80(24):AEM.00895
Dubé CD, Guiot SR (2014) Direct interspecies electron transfer in granular sludge. In: International conference biogas science 2014, pp 85–86
Teng Z, Hua J, Wang C, Lu X (2014) Design and optimization principles of biogas reactors in large scale applications: research progress in lab—Immobilization. In: Shi F (ed) Reactor and process design in sustainable energy technology. Springer, Amsterdam, pp 125–127
Lalov IG, a. Krysteva M, Phelouzat JL (2001) Improvement of biogas production from vinasse via covalently immobilized methanogens. Bioresour Technol 79:83–85
Gong W, Liang H, Li W, Wang Z (2011) Selection and evaluation of biofilm carrier in anaerobic digestion treatment of cattle manure. Energy 36(5):3572–3578
Ahammad SZ, Davenport RJ, Read LF, Gomes J, Sreekrishnan TR, Dolfing J (2013) Rational immobilization of methanogens in high cell density bioreactors. RSC Adv 3:774
Bauer A, Theuretzbacher F, Rincón M, Enguidanos R (2014) Steam explosion pretreatment production of Alpine hay for enhancing biogas. In: Proceedings international conference of agricultural engineering, 2014, pp 4–7
Bharathiraja B, Sudharsanaa T, Bharghavi A, Sowmeya GS, Balaram G (2014) Insights on lignocellulosic pretreatments for biofuel production- SEM and reduction of lignin analysis, vol 6, no 9
Serrano-Lotina A, Daza L (2013) Highly stable and active catalyst for hydrogen production from biogas. J Power Sources 238:81–86
Knott G, Marchman H, Wall D, Lich B (2008) Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J Neurosci 28(12):2959–2964
Ishitani T, Yaguchi T (1996) Cross-sectional sample preparation by focused ion beam: a review of ion-sample interaction. Microsc Res Tech 35(4):320–333
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Lebuhn, M., Weiß, S., Munk, B., Guebitz, G.M. (2015). Microbiology and Molecular Biology Tools for Biogas Process Analysis, Diagnosis and Control. In: Guebitz, G., Bauer, A., Bochmann, G., Gronauer, A., Weiss, S. (eds) Biogas Science and Technology. Advances in Biochemical Engineering/Biotechnology, vol 151. Springer, Cham. https://doi.org/10.1007/978-3-319-21993-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-21993-6_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21992-9
Online ISBN: 978-3-319-21993-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)