
Abstract
Metastasis is a multistep process that implies genetic modifications and is strongly influenced by the interactions between host and tumor cells, and by tumor microenvironment. Before tumor cells colonize distant organs, they can prepare foreign soil by remotely coordinating a “premetastatic niche” from the primary tumor. The premetastatic niche provides an array of cells, cytokines, growth factors, and adhesion molecules to support metastatic cells on their arrival and to guide metastases to specific organs. Factors secreted by tumor cells, such as VEGF, LOX, IL-6, IL-10, and exosomes, participate in the premetastatic niche formation. Also extracellular matrix (ECM) molecules, namely periostin, tenascin and osteopontin can supply the necessary resources for successful metastatic colonization. One of the key underlying hypotheses of the cancer stem cell (CSC) model proposes that CSCs are the basis of metastases. CSCs in situ may transform to metastatic stem cells (MetSCs) by epithelial-mesenchymal transition (EMT) and subsequently disseminate and form metastatic colonies. Alternatively, MetSCs may derive from disseminated tumor cells that reacquire the competence to initiate tumor growth after a period of indolence. CSCs exhibit properties that are beneficial to metastasize and adapt in the foreign microenvironment, such as mesenchymal characteristics, increased capacity for DNA repair, resistance to apoptosis and to antitumor therapy. Circulating tumor cells (CTCs) are linked to tumor progression in a variety of solid tumors. CTCs are therefore assumed as precursors of distant metastasis. Potentially, a fraction of CTCs have CSC activity; stem-like CTCs may be a critical subset of CTCs with the capacity to form distant metastases. Many therapeutic strategies against CSCs strategies have been investigated. Among them, therapies directed at CSC niche and pre-metastatic niche are of particular interest. These therapies are aimed at targeting vasculature, extrinsic signals and tumor associated macrophages.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Tumor Progression and Metastasis
Metastasis is a multistep process that allows primary tumor cells to invade the surrounding tissue, intravasate through blood vessels to enter the circulatory or lymphatic system, survive environmental changes, extravasate into new tissue, proliferate at secondary sites and develop a vascular system to support growth (Giaccia and Erler 2008). Different tumor types have the ability to colonize the same or different organ sites (Fidler 2003). Research in this field is identifying genes that support metastasis to particular organs (Yin et al. 1999; Minn et al. 2005a, b; Kang et al. 2003). Another important variable is the temporal course of metastasis. Breast and lung adenocarcinomas typically relapse within a similar range of organs, including bone, lung, liver and brain (Hess et al. 2006). However, breast cancer recurrences are often detected following years or decades of remission (Schmidt-Kittler et al. 2003), whereas lung cancers establish distant macrometastases within months of diagnosis (Hoffman et al. 2000). The temporal gap between organ infiltration and colonization produces a period of metastatic latency (Nguyen et al. 2009).
1.1 Genetic Driven Metastasisation
The genes and activities that underlie the general steps of metastasis can be grouped into several classes, which have been defined as metastasis initiation, metastasis progression and metastasis virulence genes (Chiang and Massagué 2008; Nguyen and Massagué 2007). Metastasis initiation genes allow transformed cells to invade the surrounding tissue, attract a supportive stroma and facilitate the dispersion of cancer cells. These genes could promote cell motility, epithelial to mesenchymal transition (EMT ), extracellular matrix degradation, bone marrow progenitor mobilization, angiogenesis or evasion of the immune system (Guo et al. 2008; Tavazoie et al. 2008).
Metastasis progression genes allow cancer cell passage through capillary walls and survival in the newly invaded parenchyma. Metastasis progression genes could have different functions at the primary site and in distant organs. As the structure and composition of capillary walls and the subjacent parenchyma vary in different organs, the functions required for metastatic infiltration, survival and colonization might also differ depending on the target organ. Metastasis virulence genes confer activities that are essential for the metastatic colonization of a certain organ and for which expression becomes detectable only in cancer cells that metastasize to those tissues. For example, osteoclast mobilizing factors, such as parathyroid hormone-related protein (pTHRp) and interleukin (IL)-11 do not provide an advantage to breast cancer cells in primary tumors but enable them to establish osteolytic metastases in bone (Yin et al. 1999; Kang et al. 2003; Mundy 2002). The hypothetical classes of metastasis genes are summarized in Fig. 2.1.

Principal steps of metastasis and hypothetical classes of metastasis genes. Tumor initiation genes include oncogenes as ERBB2, KRAS, PI3K, EGFR , MYC and tumor suppressor genes as APC, TP53, PTEN , BRCA1, BRCA2; metastasis initiation genes include TWIST1, SNAI1, SNAI2, MET , miR-126, miR-335; metastasis progression genes include MMP -1, LOX, ANGPTL4; metastases virulence genes include GM-CSF, IL6, TNF-α
1.2 Interactions Between Host and Tumor Cells
Metastasis is strongly influenced by the interactions between host and tumor cells, and by tumor microenvironment. Tumor cells must overcome a different barriers to metastasize, including physical barriers such as extracellular matrix (ECM ) and basement membranes, and physiological barriers such as hypoxia and the immune system (Gupta and Massagué 2006). Cells respond to external microenvironmental influences by altering gene expression such as they are able to adapt and survive. The tumor microenvironment thus exerts a selection pressure for cells capable of overcoming these barriers, driving tumor progression and acquisition of metastasis functions.
During preinvasive tumor growth, oxygen and glucose typically can only diffuse 100–150 μm, resulting in portions of the expanding mass becoming hypoxic. Hypoxia selects for cells with low apoptotic potential (Graeber et al. 1996; Erler et al. 2004) and increases genomic instability (Reynolds et al. 1996). Hypoxia also increases the expression of genes involved in glucose transportation, angiogenesis, anaerobic metabolism, cell survival, invasion and metastasis (Knowles and Harris 2001; Le et al. 2004).
In particular, hypoxia-inducible factors HIF-1α and HIF-2α induce the transcription of over 100 target genes involved in angiogenesis, glycolysis and invasion. Up-regulated angiogenesis genes include vascular endothelial growth factor (VEGF ) and platelet-derived growth factor (PDGF) that induce blood vessels remodeling. In addition, HIF-α up-regulates matrix metalloproteinase (MMP )-1 and -2, lysyl oxidase (LOX), and the chemokine receptor CXCR4 . Degradation of the basement membrane by MMP2 and alteration of the extracellular matrix (ECM ) by MMP1 and LOX clears away a barrier to migration. The activation of CXCR4 stimulates cancer cells to migrate to regions of angiogenesis (Bergers and Benjamin 2003; Gatenby and Gillies 2004).
Cancer cells are often surrounded by activated fibroblasts and bone marrow-derived cells (BMDCs). The presence of an inflammatory response in cancer would apply significant selective pressure on the tumor to evade immune-mediated attack. Progressing tumors orchestrate an immunosuppressive environment, a process known as immunoediting (Dunn et al. 2006). Cells involved in chronic inflammation can facilitate tumor formation and progression, mostly mediated by nuclear factor-kB (NF-kB) and cyclooxygenase 2 (COX-2) (Karin 2006; Dannenberg and Subbaramaiah 2003). Tumor-associated macrophages (TAMs ) may have tumor-suppressing and tumor-promoting roles. TAMs are stimulated by hypoxia and secrete angiogenesis inducers (including VEGF ) and proteases (including MMPs) (Lewis and Pollard 2006; Murdoch and Lewis 2005); TAMs express high levels of HIF-2 transcription factor that is needed for myeloid cell infiltration and activation (Knowles et al. 2004; Cramer et al. 2003). Furthermore, TAMs release growth factors such as PDGF, epidermal growth factor (EGF ), hepatocyte growth factor (HGF), which enhance proliferation, survival and invasion (Lewis and Pollard 2006).
TAMs are the main population of inflammatory cells in solid tumors and the cytokines released from them possess diversified significance in tumor development (Lewis and Pollard 2006). TAMs are derived from circulating monocytes and differentiate within the tumor microenvironment (Sica and Bronte 2007; Biswas et al. 2008). The majority of TAMs are M2-like macrophages, with properties that differ from the M1 macrophages, which are usually present in tissue areas with acute inflammation (Lewis and Pollard 2006; Biswas et al. 2008). TAMs generally fail to express pro-inflammatory cytokines for T helper type 1 (Th1) responses but are excellent producers of immunosuppressive cytokines for T helper type 2 responses (Allavena et al. 2008). As TAMs generally exhibit low antigen-presenting and co-stimulating capacity, they ordinarily fail to activate T-cell-mediated adaptive immunity. Therefore the M2- like TAMs are immunosuppressive and facilitate tumor progression (Allavena et al. 2008; Solinas et al. 2009).
Rather than simply suppress the inflammatory response, cancer cells develop mechanisms to both co-opt and perpetuate it. For example, myeloid-derived suppressor cells (MDSCs) are contributing to immunosuppression, but they also facilitate tumor invasion by residing at the invasive front and secreting MMPs. TAMs are often found at points of basement membrane breakdown and at the invasive front. Growth factors secreted by the TAMs activate fibroblasts; activated fibroblasts become carcinoma-associated fibroblasts (CAFs ) and promote primary tumor growth by secreting CXCL12 (chemokine stromal cell-derived growth factor 1, SDF-1 ), that binds CXCR4 on tumor cells. Angiogenesis is also aided by the action of CAFs through recruitment of endothelial progenitor cells by CXCL12 and by the action of TAMs that are recruited to areas of hypoxia to produce VEGF . In addition to exerting selection for general metastasis-supporting traits, the primary tumor stroma can also select for organ-specific seeding traits. This specificity was recently shown in the case of bone metastatic breast cancer (Zhang et al. 2013). A CAF -rich stroma in breast tumors produces CXCL12/SDF1 and insulin-like growth factor-1 (IGF1), which select for Src hyperactive cancer clones that are superior at responding to these signals with activation of the phosphoinositide-3 kinase (PI3K)/PI3K/protein kinase B (Akt) survival pathway. Src-high clones are thereby primed for seeding the bone marrow where local sources of CXCL12 and IGF1 provide them with a higher chance of survival. As a corollary to these findings, CAF content, CXCL12/IGF1 signaling, and high Src activity in breast tumors all predict an increased likelihood of bone relapse in breast cancer patients (Zhang et al. 2009, 2013).
Cancer cells may leave a primary tumor early and evolve separately from the tumor. It has been proposed that the parallel evolution of early disseminated cancer cells over a period of indolence affords these cells a superior adaptation to their metastatic microenvironment and a leading role in metastatic relapse (Klein 2009). Cancer cell entry into the circulation and lodging in distant organs can certainly occur after minimal genetic changes (Podsypanina et al. 2008; Schardt et al. 2005). However, large-scale genome sequencing studies have shown more similarities than differences between primary tumors and their metastases, suggesting that most of the genetic changes required for metastasis accumulate in primary tumors (Yachida et al. 2010). Actively growing cancer cells in primary tumors may be more likely to undergo variation for the selection of metastatic traits than their precociously dispersed, indolent comrades.
1.3 Epithelial to Mesenchymal Transition and Invasion
Changes in cell-cell and cell-matrix adhesion interactions are necessary to dissociate cancer cells from the tumor (Cavallaro and Christofori 2004). Cell-cell adhesion is mediated primarily by E-cadherin proteins expressed at junctions between cells. Reduced expression of E-cadherin is often observed in aggressive cancers (Friedl and Wolf 2003) and the loss of this protein is highly associated with EMT (Lee et al. 2006). The acquisition of the invasive phenotype has many similarities with EMT, including loss of cell-cell adhesion and increase in cell mobility. During EMT, there is a switch from E-cadherin expression to N-cadherin expression (a mesenchymal cell marker), which promotes cell-matrix adhesion (Lee et al. 2006).
EMT can confer invasive migration capacity to enter circulatory or lymphatic system. Invasive migration involves changes in cell-matrix adhesion and cytoskeleton; cell-matrix adhesion is largely regulated by integrins that bind to specific components of ECM (Guo and Giancotti 2004). Integrins are activated by the contact with specific ECM substrates or through growth factor stimulated signalling (Mitra et al. 2005; Playford and Schaller 2004). Integrin stimulation promotes formation of focal adhesion contacts, focal adhesion kinase (FAK) activation and formation of FAK-Src complexes (Playford and Schaller 2004). Intracellular signaling mediated by FAK leads to actin-myosin contraction and recruitment of MMPs to focal adhesion sites where they degrade ECM (Mitra et al. 2005; Friedl and Wolf 2003).
The basement membrane provides a physical barrier between stroma and epithelial cells. Glycoproteins and proteoglycans provide ligands for integrins, permitting cell orientation and signalling. Tumor cells can overcome the basement membrane by altering their surface receptors such that they can adhere to basement membrane components; for example, tumor cells can increase expression of integrins (that bind laminin and collagen) and CD44 (that permits cell binding to proteoglycans) (Friedl and Wolf 2003; Behrens 1994). In addition, tumor cell can modify the basement membrane composition to facilitate penetration, for example reducing laminin expression. Besides, they can proteolytically disrupt the basement membrane by altering the balance between ECM proteases and their inhibitory proteins; for example, elevated MMP expression is associated with collagen degradation (Morikawa et al. 1988). MMP degradation of ECM generates bioactive peptides, growth factors and cytokines (Egeblad and Werb 2002; Andres et al. 1991; Chakrabarty et al. 1990).
Tumor blood vessels are malformed and irregular and often present breaks that permit the easy access of tumor cells into the circulation; this abnormal vasculature is the result of dysregulated expression of proangiogenic growth factors, inhibition of antiangiogenic pathways, and recruitment of vascular progenitor cells from bone marrow. Tumors do not possess abundant lymphatic vessels. Tumors secrete lymphangiogenic factors such as VEGF -C, but the development of lymphatics is abnormal.
Knowledge of genetic determinants involved in intravasation is limited. Chemo-attractant proteins such as chemokines have been proposed to guide cells toward the circulatory system. Tumor cells also move along collagen fibers, a process facilitated by host macrophages (Condeelis and Segall 2003).
EMT genes can be essential for metastasis. In a breast cancer model, inhibition of Twist potently reduced the number of metastatic lesions in the lung. Consistently, inhibiting Twist in either hypoxic cells or in cells overexpressing hypoxia-induced factor (HIF)-1α reversed both EMT and metastasis, and inhibiting Snail decreased metastasis induced by inflammatory signals (Yang et al. 2008). It has been shown a role for Twist in establishing high levels of circulating tumor cells through enhancing intravasation and/or survival in the circulation (Yang et al. 2004). The ability of cells undergoing EMT to intravasate is consistent with observations that EMT occurs at the invasive front of tumors whereby cells lose E-cadherin , detach, invade, and break down the basement membrane. Accordingly, experiments that directly analyzed EMT and non-EMT cells showed that only the EMT cells were able to penetrate surrounding stroma and intravasate (Tsuji et al. 2009).
A high proportion of distant metastases are differentiated and in some cases metastases can show a greater degree of cellular differentiation than the primary tumors. For example, increased E-cadherin expression in metastases compared to the primary tumors has been reported in human patient specimens (Oka et al. 1993; Kowalski et al. 2003; Chao et al. 2010). Furthermore, the importance of epithelial phenotype in the formation of secondary tumors has been demonstrated in different metastasis models, including bladder cancer (Chaffer et al. 2005, 2006, 2007), prostate cancer (Oltean et al. 2006; Yates et al. 2007), colorectal cancer (Vincan et al. 2007), and breast cancer (Tsuji et al. 2008, 2009; Chao et al. 2010). Hence, both clinical and experimental evidence points to the necessity of disseminated cancer cells undergoing a mesenchymal-to-epithelial reverting transition (MET ) in the secondary microenvironment to form macrometastases (Nieto 2013). Consequently, it has been proposed that metastatic cancer cells possess the phenotypic plasticity and acquired EMT -like phenotype for disseminating from the primary tumor, and subsequently a second transition from the EMT-like to MET-like state occurs to facilitate the formation of metastatic tumors at target organs (Brabletz 2012). MET can take part of metastatic formation with tumor cells regaining their epithelial properties at their secondary homing sites (Hugo et al. 2007; Yao et al. 2011). This hypothesis is in accord with the observation that metastatic lesions generally share epithelial features of the primary tumor (e.g., E-cadherin expression) (Chao et al. 2010; Imai et al. 2004).
Tumor cells in the circulatory system are subjected to immune attack, circulatory forces and apoptosis induced by loss of adhesion (anoikis) (Gupta and Massagué 2006). Circulating tumor cell s (CTCs ) bind platelets that protect them from dangers and increase their chances of survival (Nash et al. 2002; Gasic 1984). Tumor cells also bind thrombin, fibrinogen, tissue factor, fibrin, thus creating emboli (Zhan et al. 2004). These tumor emboli are more resistant to circulatory forces and immune attack (Nash et al. 2002). In the circulation, aggregates of tumor cells associated with platelets are defined as heterotypic clumps. Both CTCs and platelets can express the αvβ3 integrin to promote aggregation of these cells to form tumor emoboli (Guo and Giancotti 2004). This aggregation facilitates arrest and can protect against shear forces and natural killer (NK) cell-mediated killing. Activation of αvβ3 integrin can result from CXCL12 /CXCR4 signaling and has been shown to be required for formation of tumor emboli and metastasis (Sun et al. 2007; Felding-Habermann et al. 2001).
Platelets have been implicated to actively induce an EMT in circulating tumor cells, either through direct cell–cell contact or secretion of transforming growth factor (TGF)-beta, which is supposed to act in combination with other factors (Labelle et al. 2011). A transient exposure to platelets was shown to be enough for tumor cells to adopt a more mesenchymal state resulting in enhanced invasive and metastatic behavior (Fig. 2.1) (Labelle et al. 2011). One possible implication of platelet-induced EMT in disseminated cancer cells is thus the conversion of these cells to a more stem-like state, which enables them to seed metastasis (see below).
The ability to resist apoptosis is also very important. Loss of cell adhesion can induce anoikis; a variety of receptor tyrosine kinases can confer resistance to anoikis; also tumor emboli formation can promote resistance to anoikis as well (Zhan et al. 2004). Antiapoptosis genes such as BCL2 or BCL -XL, or the loss of proapoptotic genes and genes downstream of the tumor necrosis factor (TNF)-related receptor family, can result in increased metastasis (Martin et al. 2004; Stupack et al. 2006). Part of this may be the result of survival both in the circulation and shortly after extravasation.
Endothelial cells can guard against wandering tumor cells through expression of DARC, a Duffy blood group glycoprotein (Bandyopadhyay et al. 2006). DARC interacts with KAI1 expressed on circulating tumor cells causing them to undergo senescence. KAI1 was originally identified as a metastasis suppressor gene. The immune system can also actively attack circulating tumor cells (Mehlen and Puisieux 2006). For example, NK cells can engage and kill cancer cells via TNF-related molecules such as TNF related apoptosis-inducing ligand (TRAIL) or CD95L. In total, mechanical and cell-mediated stresses can result in a short half-life for CTCs , so their half-life is often as short as a few hours (Meng et al. 2004).
Tumor cell arrest can occur passively through mechanical lodging or can be allowed by cell surface molecules (Arap et al. 1998; Pasqualini et al. 2000). The vasculature of normal tissues where tumor cells extravasate is intact. In normal vessels, endothelial cells are constantly shed from the vessel walls, so creating temporary gaps where tumor cells can attach, as basement membrane components are exposed (el-Sabban ME and Pauli 1994). Vessel wall damage also attracts platelets and tumor cells associated to platelets (Karpatkin and Pearlstein 1981; Karpatkin et al. 1988). Fibrin clots at the site of tumor cell arrest can further attract platelets and circulating tumor cells (Dvorak et al. 1983). Tumor cell arrest can be allowed by P and E-selectin that are expressed by endothelial cells and bind to tumor cells (Kim et al. 1998); tumor glycosylation patterns and cell-cell adhesion molecules such as integrins and CD44 may also have a role (Wang et al. 2004; Ruoslahti 1994; Friedrichs et al. 1995).
VEGF expression by the tumor can also lead to disruptions in endothelial cell junctions and facilitate extravasation of cancer cells through enhanced vascular permeability. This is likely mediated by the activation of SRC family kinases in the endothelial cells (Criscuoli et al. 2005). Expression of hypoxia-induced CXCR4 on CTCs allows for the selective extravasation into certain organs. This selectivity is due to the expression of its ligand CXCL12 by certain organs that include the lung, liver, bone, and lymph nodes (Müller et al. 2001). Also, tumor clump formation facilitates tumor cell arrest by increasing adhesive interactions. ECM components such as fibronectin and laminin enhance tumor cell arrest (Terranova et al. 1984); tumor cells may reside and growth in the intravascular space until they physically break through the vessel. Tumor cells may also extravasate by inducing endothelial cell retraction that permits cell attachment to ECM (Al-Mehdi et al. 2000).
Resumption of cell proliferation at the secondary site needs angiogenesis to supply oxygen and nutrients. The host tissue can influence tumor growth through autocrine, paracrine and endocrine signals, and the balance between positive and negative signals determines metastatic proliferation. This can partially explain organ specificity of metastases, as only certain cells can respond to specific proliferation signals (Fidler 2001). For example, IGF -1, hepatocyte growth factor (HGF) and TGF-α are highly expressed in the liver (Zarrilli et al. 1994; Radinsky 1991; Khatib et al. 2005), and cancer cells from colon and breast cancers (that often metastasize to liver) overexpress receptors for these ligands, e.g. epidermal growth factor receptor (EGFR ) and c-met receptor (Gross et al. 1991; Radinsky et al. 1995; Bottaro et al. 1991).
The angiogenic “switch” occurs when the ratio of inducers to inhibitors is increased. Inhibitors of angiogenesis include ECM proteins thrombospondin and endostatin (Dameron et al. 1994; O’Reilly et al. 1997); inducers include VEGF , PDGF, basic fibroblast growth factors (bFGF), TGF-β , and ephrin (Steeg 2006). VEGF stimulates endothelial cells, mobilizes endothelial progenitor cells, stimulates outgrowth of pericytes, increases vascular permeability (Senger et al. 1983; Leung et al. 1989); in addition, VEGF is thought to be a key molecule for the homing of VEGFR-positive bone marrow-derived progenitor cells involved in premetastatic niche formation (Kaplan et al. 2005) and for homing of VEGFR-positive tumor cells to metastatic sites (Price et al. 2001).
Tumor cells that have colonized secondary organs are capable of further colonization of other organs. Cells within the metastatic tumor are subjected to similar microenvironmental pressure as the primary tumor, and adapt to overcome the external barriers and seed new terrain. These cells from metastases are able to constantly reseed both primary and secondary tumor (Norton and Massagué 2006). Tumor cells can move multidirectionally, seeding not only distant sites but also their tumors of origin (Comen and Norton 2012). At least in theory, it would seem that compared with uncharted foreign environments or even premetastatic niches, the primary tumor would impose the least resistance to colonization (Karnoub et al. 2007). Support for this concept of tumor self-seeding has recently been provided using mouse model systems and a variety of different cancer types by demonstrating that CTCs can seed the primary tumor and contribute to its mass (Kim et al. 2009). The ability to self-seed is promoted by IL-6 and IL-8, common prometastatic cytokines found in the tumor microenvironment.
2 The Premetastatic Niche: Factors Secreted by Tumor Cells That Affect the Formation of the Premetastatic Niche
Before tumor cells colonize distant organs, they can prepare foreign soil for the subsequent arrival of disseminated tumor cells (DTCs) by remotely coordinating a “premetastatic niche” from the primary tumor (Psaila and Lyden 2009). These niches are often located within distant organs around terminal veins and are characterized by newly recruited hematopoietic progenitor cells of the myeloid lineage and by stromal cells. The premetastatic niche provides an array of cytokines, growth factors, and adhesion molecules to help support metastatic cells on their arrival, so it is essential for the growth of extravasated tumor cells. An additional function is to guide metastases to specific organs (Kaplan et al. 2005).
Factors secreted by primary tumor cells stimulate mobilization of BMDCs that enter the circulation and reside in sites of future metastases. BMDCs express VEGFR-1 and several other hematopoietic markers including CD34 , CD11b, c-kit, and Sca-1, defining them as early hematopoietic progenitors cells engaged with the parenchyma of the distant organ (Kaplan et al. 2005, 2006, 2007; Wels et al. 2008). The key tumor-secreted factors that determine metastatic sites and mediate pre-metastatic niche formation have to be fully identified, although a role of TNF-α , TGF-β and VEGF -A has been demonstrated (Hiratsuka et al. 2006). These factors induce the expression of chemoattractants such as S100A8 and S100A9 by myeloid and endothelial cells, and promote the homing of tumor cells to the premetastatic sites as well as the invasion of circulating tumor cells through a p38-mediated activation of invadopodia (Hiratsuka et al. 2008).
Homing of VEGFR1+VLA4+ BMDCs is mediated via the induction of fibronectin, which is a ligand of VLA-4. BMDCs express the VLA-4 (α4β1), thus priming them to sites rich in fibronectin to establish the clusters in preparation for metastasis (Kaplan et al. 2005). It is thought that these cells may become educated within tumors to hunt out and lay the foundations for distant metastasis. Alternatively, the cells may become activated locally or in the circulation due to chemokines secreted by tumor. Secretion of placental growth factor (PlGF), a ligand for VEGFR1 may activate resident organ fibroblasts to synthesize fibronectin, which facilitates the binding of VLA-4 expressing hematopoietic progenitor cells (Peinado et al. 2012).
Tumor cells secrete LOX, an amine oxidase that plays a role in crosslinking collagens and elastins in the ECM , to provoke systemic alterations and induce the formation of the premetastatic niche (Erler et al. 2009). Under hypoxic conditions, breast cancer tumors secrete LOX, which accumulates in premetastatic sites. This favours the recruitment of CD11b+ myeloid cells that adhere to cross-linked collagen IV and produce MMP -2, which cleaves collagen and makes it easier for BMDCs and tumor cells to invade the area.
Cancer cells secrete factors such as IL-6 and IL-10 that activate the S1PR1–STAT3 pathway in myeloid cells. This in turn promotes activation of fibroblasts and up-regulation of fibronectin (Deng et al. 2012). Targeting the pro-invasive S1PR1-STAT3 pathway in Cd11b+ myeloid cells eliminates de novo formation of premetastatic niches and metastasis, and reduces preformed metastatic niches. In addition, the expression of tissue factor by tumor cells induces the formation in the future metastatic sites of platelet clots, which recruit myeloid cells (Gil-Bernabé et al. 2012.).
Exosomes are another class of tumor derived products which may prime metastases. Exosomes are small secreted vesicles derived from the endocytic pathway. It has been shown that melanoma cells use exosomes to deliver signals that prime the future metastatic sites. These exosomes instruct BMDCs to contribute to tumor growth and metastatic colonization through the transfer of different molecules such as the Met oncoprotein (Hood et al. 2011; Peinado et al. 2012). In renal carcinoma, microvesicles released from CD105+ CSCs , but not from CD105− tumor cells, were able to trigger angiogenesis and significantly enhanced the capacity of renal carcinoma cells to metastasize to the lungs (Grange et al. 2011).
MMPs may also play an important role in this process. VEGFR1 signalling is necessary for pre-metastatic induction of MMP -9 expression in endothelial cells and macrophages of the lungs by distant primary tumors (Hiratsuka et al. 2002). Furthermore, stromal derived MMP-2 and MMP-9 have also been shown to contribute to establishment and growth of metastases (Masson et al. 2005). Periostin, tenascin and osteopontin have been previously linked to the induction of angiogenesis in different systems; these ECM proteins are able to regulate VEGF and its receptors and induce angiogenesis. Thus, ECM molecules can supply the necessary resources for successful metastatic colonization and secondary tumor growth (Chakraborty et al. 2008; Tanaka et al. 2004; Shao et al. 2004; Tokes et al. 1999).
3 Characteristics of Cancer Stem Cells That Are Linked to Metastasis
One of the key underlying hypotheses of the CSC model proposes that CSCs are the basis of metastases. To study the role of CSCs in the process of tumor metastasis, Brabletz et al. (2005) suggested the migrating cancer stem (MetCS)-cell concept. They proposed that CSCs in situ can transform to MetCS cells by EMT . Subsequently, the MetCS cells disseminate and form metastatic colonies. MetSC is any DTC that is capable of reinitiating macroscopic tumor growth in a distant tissue. Metastatic stem cell s (MetSCs) may already exist in the primary tumor with the necessary traits to overcome the bottlenecks of the metastatic process, or, alternatively, may derive from DTCs that reacquire the competence to initiate tumor growth after a period of indolence (Fig. 2.2).

Hypotheses on the origin of cancer stem cells and metastases. A. Only CSCs can metastasize to distant tissues; B. In animal models of some tumor types, all the cancer cells demonstrate no significant differences in the ability to generate tumors or establish metastases distant sites; C. Cancer cells migrate as CTCs from primary tumors to distant tissues, where become CSCs through dedifferentiation
CSCs exhibit properties that are beneficial to adapt in the foreign microenvironment and eventually form metastasis. Several unique properties necessary for ensuring long life span of normal stem cells may contribute to protection of CSCs in the adverse microenvironment.
3.1 EMT
EMT is characterized by epithelial cells loosening their cell-cell adhesion, losing cell polarity, and gaining the ability to invade and migrate. EMT regulators include Notch and Wnt/β-catenin pathway s, TGF-β family members and FGF proteins that serve to set up regulatory networks involving EMT transcription factors such as Snail and Twist. These networks drive morphogenetic movements by repression of the cell-cell adhesion protein E-cadherin , promoting cytoskeletal rearrangement, and increasing MMP activity. After cells complete EMT-mediated morphogenetic migration, they can then differentiate into epithelial structures by repressing Snail and undergoing a MET .
CSCs express EMT markers, and induction of EMT in transformed epithelial cells promotes the generation of CSCs (Yang et al. 2004; Mani et al. 2008; Floor et al. 2011; Jordan et al. 2011; Wu 2011; Wu and yang 2011; Krantz et al. 2012). In colon cancer, nuclear accumulation of β-catenin, the feature of Wnt signaling activation and stem cell signaling, is found at the invasive front of the primary tumor (Fodde and Brabletz 2007). Stem-like cells isolated from normal mammary glands and breast tumors also express EMT markers (Damonte et al. 2007; Mani et al. 2008). Overexpression of EMT-inducing transcription factors such as Snail or Twist in transformed mammary epithelial cells increased tumor-initiating frequency in immune-deficient mice (Mani et al. 2008). The mesenchymal phenotype marker Zeb1 may facilitate the acquisition of stem cell-like properties (Peter 2010). Untransformed immortalized human mammary epithelial cells are capable of undergoing an EMT-like state by expressing FoxC2, Zeb factors, and N-cadherin , all of which have been linked to a CSC state (Morel et al. 2008). Likewise, by over-expressing Ras or Her2/neu, a stem-like subpopulation of CD44high/CD24low cells with an enhanced EMT phenotype has been identified (Radisky and LaBarge 2008).
The acquisition of an EMT phenotype may be regulated by signals from the microenvironment or niche. Tumor associated fibroblasts have been shown to enhance the metastatic potential of tumors by promoting migration and extravasation through an EMT process as well as the establishment of a CSC -like state (Aktas et al. 2009; Armstrong et al. 2011; Gregory et al. 2008; Kalikin et al. 2003; Martin et al. 2010).
3.2 Increased Capacity for DNA Repair and Resistance to Apoptosis
Normal stem cell s have increased capacity for DNA repair and express higher levels of anti-apoptotic proteins than differentiated cells (Cairns 2002; Potten et al. 2002; Park and Gerson 2005). The enhanced anti-apoptotic and DNA repair capability of CSCs could increase the survival of CSCs for a long period of time under metabolic and/or other environmental stress (e.g., hypoxia) in the target organ and allow them to find adaptive solutions. Autocrine production of cytokines such as IL-4 has been shown to increase anti-apoptotic proteins and induces resistance to therapy-induced cytotoxicity in different cancer types (Conticello et al. 2004).
3.3 Resistance to Anti-tumor Therapy
Many studies have shown that CSCs have increased drug resistance capacity. For example, it has been shown that stem-like subpopulation of cancer cells express high levels of ATP-binding cassette (ABC) transporters that can actively efflux drugs and shield them from the adverse effects of chemotherapeutic insult (Pardal et al. 2003; Lou and Dean 2007; Dean 2009; Donnenberg et al. 2009; Ding et al. 2010; Moitra et al. 2011). There is also growing evidence that CSCs are inherently resistant to radiation (Rich 2007; Debeb et al. 2009; Pajonk et al. 2010; Croker and Allan 2012; D’Andrea 2012). For example, the effectiveness of radiotherapy is mediated by the induction of reactive oxygen species (ROS ) in cancer cells. However, it has been found that both human and mouse mammary CSCs contain lower ROS levels than more differentiated tumor cells and accumulate less DNA damage upon radiation (Diehn et al. 2009). Lower ROS levels in CSCs appear to result from increased expression of free radicals scavenging systems (Diehn et al. 2009; Kobayashi and Suda 2012; Shi et al. 2012). The inherent feature of drug resistance in CSCs could activate stress responses to protect them from growth-suppressing conditions in the target organ microenvironment and allow them to persist in foreign tissues for a long period of time.
3.4 Genetic Signatures
Genetic signatures in CSCs are thought to predict tumor recurrence and metastases, providing some support for the concept that CSCs may be metastatic precursors. For example, expression of the CSC marker CD133 in glioblastoma and lung adenocarcinoma is correlated with both the proliferation marker Ki67 and poorer clinical outcomes (Pallini et al. 2008). CD133 antigen expression has also been shown to correlate with patient survival in high-grade oligodendroglial tumors (Beier et al. 2008), rectal cancer (Wang et al. 2009), gastric adenocarcinoma (Zhao et al. 2010), and non-small cell lung cancer (Shien et al. 2012). In patients with colorectal carcinoma, the combination of CD133, CD44 , and CD166 can identify patients at low-, intermediate-, and high-risk of recurrence and metastasis (Horst et al. 2009). Methylation of Wnt-target-gene promoters is also a strong predictor for recurrence in colorectal cancer (de Sousa et al. 2011). Finally, the vast majority of disseminated breast cancer cells in the bone marrow displays a CSC phenotype based on CD44 and CD24 expression (CD44+CD24−/low) (Balic et al. 2006).
3.5 Experimental Observations on Metastatic Potential of CSCs
The expression of markers such as CD44 or CD24−/low by tumor cells alone does not prove that these cells can generate metastases or that they are necessarily CSCs . Moreover, identification of CSCs within metastatic lesions or in circulating or disseminated tumor cell populations (Balic et al. 2006) does not necessarily mean these cells are capable of establishing disseminated lesions.
Some of the most direct evidence that CSCs establish metastases comes from the demonstration that breast cancer CSCs isolated based upon the putative stem cell markers CD44 + and CD24−/low are able to generate primary tumors in an orthotopic site and subsequently produce lung metastases (Liu et al. 2010). In pancreatic cancer models, it has been shown that a distinct subpopulation of CD133 +/CXCR4 + cells localizes to the invasive edge of tumors and is more migratory than CD133+/CXCR4− cells. Although both populations were equally capable of initiating primary tumor growth, only the CD133+/CXCR4+ cells could metastasize to the liver (Hermann et al. 2007). The authors could identify a subpopulation of CSCs that were positive for CD133 and for the stromal-derived factor-1 (SDF1) receptor CXCR4, which showed highly increased migratory abilities. Ablation of these migrating CSCs abolished the capability of pancreatic cancer cells to form metastases (Hermann et al. 2007). In colon cancer different subtypes of CSCs could be identified, one displaying metastases formation abilities (Dieter et al. 2011). Likewise, in inflammatory breast cancer (IBC) a subpopulation of cancer cells displaying stem cell properties was identified as being relevant for metastatic spread (Charafe-Jauffret et al. 2010). Furthermore, the presence of these aldehyde dehydrogenase-positive cells was suggested to be an independent prognostic factor for early metastasis in patients with IBC (Charafe-Jauffret et al. 2010).
Different populations of CSCs may be responsible for primary vs. secondary tumor sites, implicating CSC heterogeneity as a critical component of this model. Dieter et al. (2011) have demonstrated this heterogeneity within the CSC compartments, reporting at least three phenotypically distinct CSCs in a human colon cancer animal model. To track the contribution of tumor-initiating cell clones, the group generated tumorigenic cells from cancer specimens, marked them with lentiviral vectors, and then sequenced the integration sites in serially transplanted tumors. A population of CSCs was identified as tumor transient amplifying cells (T-TACs) which had limited self-renewal capacity but did form tumors in primary transplants. A second population of CSCs exhibiting extensive self-renewing long-term tumor initiating cells (LTTICs) were able to generate tumors in serial xenotransplants. A third population described as rare delayed contributing TICs (DC-TICs) were exclusively active in secondary or tertiary mice. The marrow could serve as a major source of LT-TICs; however, metastasis formation was predominantly driven by self-renewing LT-TICs (Dieter et al. 2011).
4 The Stem Cell Niche
Similar to normal stem cells, CSCs are thought to reside in a relative stable microenvironment, or niche, in order to retain an undifferentiated state and give rise to more differentiated progenitor cells (Calabrese et al. 2007). The stem cell niche is critical for stem cell self-renewal, survival, function and for maintaining CSC properties (Sneddon 2007).
4.1 The Normal Stem Cell Niche
Normal stem cell s in adult tissues reside in specific sites or “niches,” the cellular and molecular components of which regulate the self-renewal potential of stem cells and their access to differentiation cues. The location and constitution of stem cell niches have been defined in various tissues, including the intestinal epithelium, hematopoietic bone marrow, epidermis, and brain (Clevers 2013; Hsu and Fuchs 2012; Moore and Lemischka 2006; Morrison and Spradling 2008).
In normal tissues, self-renewal and differentiation of stem cells are tightly regulated, a function fulfilled by the stem cell niche (Morrison and Spradling 2008). For example, the intestinal stem cell resides at the crypt base in close proximity to a secretory non-goblet-like cell type (Sato et al. 2011). These so-called Paneth cells were shown to express components of the various morphogenetic signaling pathways demonstrated to be essential for stem cell maintenance (Sato et al. 2011). Paneth cells support the in vitro outgrowth of LGR5+ cells that is one of the cells thought to be the intestinal stem cell, to organoids. Also the stromal myofibroblasts residing at the crypt bottom provide essential signals for stem cell maintenance (Clevers 2006) and therewith contribute to the stem cell niche. In primary tumors, cancer cells may interact with these native stem cell niches.
4.2 The Cancer Stem Cell Niche
As pathways regulating normal intestinal stem cell biology significantly overlap with those influencing colorectal CSCs , it was hypothesized that the essential stem cell features in tumors are affected by niche cells in equal measure (Medema and Vermeulen 2011), a hypothesis being confirmed by recent studies on colon, pancreatic and brain cancers.
Activin/Nodal signaling molecules, that are essential for sustaining pancreatic CSCs , were shown to not only be provided by the CSCs themselves, but also by stromal pancreatic stellate cells in a paracrine fashion (Lonardo et al. 2011). Similarly, endothelial cells in brain cancers support the CSC state (Borovski et al. 2009). In analogy to normal neural stem cells, Nestin+/CD133 + brain CSCs could be located in direct vicinity of endothelial cells (Louissaint et al. 2002). Soluble factors derived from endothelial cells were sufficient to increase the selfrenewing capacity of brain CSCs (Calabrese et al. 2007). In a PDGF-induced glioma mouse model, soluble nitric oxide was identified as the paracrine mediator secreted by endothelial cells and to activate the Notch signaling pathway in a paracrine fashion in glioma CSCs, leading to enhanced tumorigenesis in mice (Charles et al. 2010). Myofibroblasts residing in the tumor microenvironment of colorectal cancer can maintain and even induce a cancer stem-like state through the secretion of HGF, which leads to a boost of the Wnt signaling pathway in adjacent cancer cells (Vermeulen et al. 2010). Interleukin 6 (IL-6), IL-8, and IL-1b directly promote breast CSC self-renewal and survival (Korkaya et al. 2011; Coussens and Werb 2002). The activation of STAT3 by IL-6 through the IL-6 receptor/GP130 complex has been shown to induce breast CSC expansion (Iliopoulos et al. 2009). IL-6 also stimulates the recruitment of mesenchymal stem cells, which produce CXC chemokine ligand (CXCL)-7 to increase the number of breast CSCs in the tumor (Liu et al. 2011b). In addition, breast CSCs express high levels of IL-8 receptor CXCR1, which prevents CSC apoptosis (Ginestier et al. 2010). Also receptor activator of NF-kB ligand (RANKL) has been found to be an important stem cell-stimulating cytokine in the breast (Asselin-Labat et al. 2010; Joshi et al. 2010). The activation of the RANKL-RANK pathway induces EMT and increases the population of CD44high/CD24low CSCs (Palafox et al. 2012). Finally, multiple stromal cell types including carcinoma associated fibroblasts and mesenchymal stem cells can produce prostaglandin (PG)-E2 (Li et al. 2012; Rudnick et al. 2011), that increases the number of aldehyde dehydrogenase (ALDH)-high CSCs through the activation of Wnt/β-catenin signaling (Li et al. 2012). Stem cell niche s are sources of developmental and self-renewal signals including Wnt, Notch, the TGF-β family, CXCL12 /SDF1, and hedgehog (Clevers 2013; Hsu and Fuchs 2012; Morrison and Spradling 2008). A source of these signals in the bone marrow are mesenchymal cells that produce CXCL12/SDF1 for hematopoietic stem cell maintenance. The cognate chemokine receptor CXCR4 is frequently overexpressed in bone metastatic cells and provides these cells with chemotaxis and PI3K-mediated survival signals (Müller et al. 2001; Zlotnik et al. 2011).
5 Relationships among Cancer Stem Cells and Pre-metastatic and Metastatic Niches
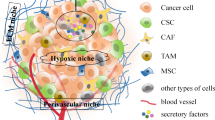
Establishing CSC niches at distant sites is crucial for the survival of CSCs and is required for the activation of their self-renewal ability for metastatic colonization (Giancotti 2013). There are three distinct sources of metastatic niche functionality: (1) native stem cell niches that metastatic cells may occupy in the host tissues; (2) niche functions provided by stromal cells not belonging to stem cell niches; (3) stem cell stem cell niche components components that the cancer cells themselves may produce (Fig. 2.3).

The metastatic niche. Disseminated cancer cells can occupy native stem cell niches (including perivascular and hematopoietic sites) and recruit stromal cells that produce stem cell niche-like components; they also can produce niche components themselves. The supportive niche stimulates Wnt and Notch signalling pathways to increase viability and stem cell expansion. Periostin (POSTN) presents Wnt ligands to Lrp and Frz receptors; tenascin C (TNC) promotes Wnt and Notch signalling. L1CAM L1 cell adhesion molecule
5.1 Hematopoietic and Perivascular Niche
Cancer cells that infiltrate distant organs may lodge in random locations of the invaded parenchyma. However, recent research provides evidence that cancer cells can occupy native stem cell niches of the host tissue. For example, prostate cancer cells showed affinity for the hematopoietic stem cell niche within the bone marrow, where they may benefit from cues that enhance stem cell properties and deter differentiation (Shiozawa et al. 2011). Using an in vivo micrometasasis model in which DTCs were introduced into immunodeficient mice, it was shown that DTCs target and displace hematopoietic stem cells (HSCs) out of their niche, and establish metastatic foci within the niche space (Shiozawa et al. 2011; Havens et al. 2008).
Another location where cancer cells initiate metastatic outgrowth is around blood capillaries, that is the perivascular niche. This niche has been studied as a preferred residence for glioma stem cells that supplies these cells with hedgehog-, Notch-, and PI3K-activating signals (Charles and Holland 2010; Hambardzumyan et al. 2008). Breast cancer , lung cancer, and melanoma cells that infiltrate the brain conspicuously place themselves around capillaries (Carbonell et al. 2009; Kienast et al. 2010). Perivascular niche s may support MetSCs by supplying not only attachment, oxygen, and nutrients but also paracrine factors from the activated endothelium, in what is called “angiocrine” stimulation (Butler et al. 2010). Endothelial cells also express various extracellular matrix (ECM ) components that promote metastatic functions in tissue culture (Ghajar et al. 2013). As metastatic lesions grow, the cancer cells recruit TAMs , myeloid precursors, and mesenchymal cells that establish paracrine loops feeding back to the cancer cells with various survival and self-renewal factors (Acharyya et al. 2012; Calon et al. 2012; Joyce and Pollard 2009). A recent work showed that brain metastasis-initiating cells express L1 cell adhesion molecule (L1CAM) and use it to stretch over the perivascular basal lamina (Valiente et al. 2014). L1CAM expression in many types of cancer is associated with poor prognosis (Doberstein et al. 2011; Schröder et al. 2009), raising the possibility of a role for L1CAM in metastasis to other organs besides the brain.
5.2 Periostin, Tenascin and VCAM1
Periostin (POSTN) is an extracellular matrix (ECM ) molecule highly expressed not only in normal stem cell niches but also in the stroma of the primary tumor and in newly forming metastases. Periostin expression is down-regulated in the adult, except in mesenchymal niches in close contact with tissue-specific stem cells. It was shown that only CD90+CD24+ breast CSCs (derived from mouse breast tumors) are able to seed metastases in the lung after tail-vein injection (Malanchi et al. 2011). CSCs arriving in the lung strongly depended on periostin (POSTN) expression. Infiltrating tumor cells –via secretion of TGFb3– induced POSTN expression in the stromal compartment of the lung. POSTN binds Wnt ligands that signal to the CSCs and maintain their stem-like state. Breast tumors arising in Postn−/− mice led to significantly reduced metastatic burden. Cancer cells stimulate the expression of periostin by stromal fibroblasts; in fact, in the stromal compartment of breast tumors (both human and mouse), POSTN is widely expressed by aSMA+VIM+ fibroblasts (Malanchi et al. 2011).
Breast cancer cells also contribute to their own CSC niche by secreting tenascin C (TNC) at metastatic sites; TNC is a hexameric glycoprotein that is found in stem cell niches and supports stem cell functions (von Holst 2008). TNC expression in breast tumors is associated with increased risk of lung metastasis (Minn et al. 2005b). In xenotransplantation models, breast cancer cells that express high levels of TNC have a distinct advantage at initiating metastases after extravasating in the lungs (Oskarsson et al. 2011). TNC enhances Notch and Wnt signaling in the cancer cells. Each of these pathways have been shown to be critical for metastasis and in stem cell biology (de Sousa et al. 2011; Duncan et al. 2005; Malanchi et al. 2011; Reya and Clevers 2005). By expressing their own TNC, breast cancer cells have a higher probability of surviving during micrometastatic outgrowth. Myofibroblasts and S100A4+ fibroblasts eventually migrate into the growing lesion to provide additional sources of TNC (O’Connell et al. 2011; Oskarsson et al. 2011).
TNC and periostin thus enhance Wnt and Notch signaling to promote the fitness of MetSCs during the initiation of metastatic colonization. TNC and periostin bind to cell surface integrins and bind tightly to each other (Kii et al. 2010). The physical interaction of TNC and periostin in the ECM may underlie a functional cooperation of these two proteins in stem cell niches (Oskarsson and Massagué 2012). Periostin and TNC in the case of Wnt and Notch signaling, like Src and vascular cell adhesion molecule-1 (VCAM1) in the case of PI3K-AKT signaling, act as amplifiers of the ability of MetSCs to respond to limiting levels of stromal Wnt and Notch ligands for activation of vital self-renewal pathways. The expression of VCAM-1 on cancer cells allows them to interact with macrophages and monocytic osteoclast progenitors via integrin a4b1. This interaction activates PI3K/Akt-mediated survival signals in cancer cells and promotes their osteolytic expansion (Lu et al. 2011; Chen et al. 2011).
5.3 Tumor Dormancy and CSCs
A major limiting step in metastasis is acquiring the ability to sustain growth within a distant site after extravasation. Many cancers such as breast and prostate will not give rise to metastasis until years or even decades after eradication of the primary tumor. Experimentally, it has been shown that the vast majority of extravasated cancer cells do not form macrometastasis (Chambers et al. 2002). These observations of latency are referred to as metastatic dormancy.
Dormant cells are frequently observed in prostate, melanoma and breast cancer (Crowley and Seigler 1992; Demicheli et al. 1996; Van Moorselaar and Voest 2002) and often reside in the lungs, liver and bone marrow. These micrometastases represent a minimal residual disease that results from the inefficiency of metastasizing tumor cells to colonize organs properly following extravasation (Luzzi et al. 1998). Incompatibilities between tumor cells and their tissue soil as well as inability of tumor cells to generate sufficient angiogenesis may result in cell cycle arrest and dormancy (Townson and Chambers 2006). Genes and pathways controlling metastatic dormancy are largely unknown and are important to identify, as they represent a metastatic tumor suppressor mechanism.
Most DTCs detected in bone marrow are proliferatively quiescent, or “dormant” (Müller et al. 2005). Although entry into G0 has been regarded as a failure of cancer cells to proceed with their tumor-propagating potential, it may represent a defense under adverse conditions (Barkan et al. 2010; Klein 2011). Isolation and re-implantation of dormant cells can generate primary tumors, demonstrating that these cells are viable (Luzzi et al. 1998; Naumov et al. 2002; Goodison et al. 2003). Growth of these cells can be activated by angiogenesis or removal of primary tumor, suggesting that limited levels of growth factors or cytokines may induce this dormant state (Holmgren et al. 1995). Unlike the active stroma in primary tumors, the distant tissue where disseminated tumor cells arrive tends to have a more quiescent microenvironment and these quiescent signals may force DTCs into dormancy. For example, abundant bone morphogenetic protein (BMP) ligands in the lung parenchyma inhibit CSC self-renewal, thereby causing metastatic dormancy. Expression of a BMP antagonist, Coco, promotes tumor-initiation ability and allows DTCs to reactivate and colonize (Gao et al. 2012).
A single dormant cancer cell or a dormant micrometastasis can turn into clinically detectable metastasis through an increased secretion of angiogenic factors in the metastatic niche to promote the recruitment and formation of new blood vessels (Takahashi and Mai 2005; Gao et al. 2008; Garcia and Kandel 2012). It has been reported that CSCs promote tumor angiogenesis by actively secreting angiogenic factors such as vascular endothelial growth factor (VEGF ) (Bao et al. 2006b; Seton-Rogers 2011). Dormant tumor cells were found to reside in microvasculatures, where quiescent endothelial cell-derived thrombospondin-1 induces tumor dormancy. Upon the induction of neoangiogenesis, the sprouting vasculatures produce active TGF-b1 and POSTN, two important CSC niche signals, to promote metastasis outgrowth (Ghajar et al. 2013).
Mechanisms that contribute to cellular dormancy may relate to the balance between the RAF/MAP kinase kinase (MEK)/mitogen-activated protein kinase (ERK) pathway and the p38-mitogen-activated protein kinase (MAPK ) pathway. Inhibition of the former and activation of the latter is associated with cellular quiescence in a G0-G1 state, and the exact balance between the two may depend on cross-talk between the tumor and the microenvironment. Genes that may be important in blocking productive cross-talk between dormant metastasis and its microenvironment include metastasis suppressor genes such as NME23, MKK4, and RKIP (Aguirre-Ghiso 2007; Dangi-Garimella et al. 2009).
6 Relationships between Circulating Tumor Cells and Cancer Stem Cells
A number of studies have linked circulating tumor cells (CTCs ) to tumor progression in a variety of solid tumors, and CTC enumeration has begun to be utilized as a prognostic tool in patients with metastatic breast (Cristofanilli et al. 2004), colon (Cohen et al. 2008) and prostate cancer (Danila et al. 2007). These cells are therefore assumed to be a surrogate marker of minimal residual disease and precursors of distant metastasis.
Despite the prognostic relevance of tumor cell dissemination, detection of tumor cells in blood is not necessarily followed by relapse of disease. While most of these cells are already apoptotic or dead and others will successfully be eliminated by shear forces of the bloodstream, only a small group of CTCs possesses the ability to extravasate and migrate through the endothelial cell layer (Frisch and Screaton 2001; Cameron et al. 2000; Sleeman et al. 2011). Merely a fraction of those is able to survive at secondary sites and cause tumor growth “metastatic inefficiency” (Méhes et al. 2001; Larson et al. 2004).
6.1 CTCs with CSC /EMT Phenotype
Whether CTCs are simply associated with disease worsening or whether they directly contribute to metastatic progression remains to be determined. Potentially, a fraction of CTCs have CSC activity, and it is hypothesized that CSCs in a primary tumor which enter the circulation become circulating CSCs and remain so until they lodge or home to a target organ. If true, then stem-like CTCs may be a critical subset of CTCs with the capacity to form distant metastases. If the spread of CSCs leads to metastasis, then it would be expected that some CTCs would express stem cell markers (Aktas et al. 2009; Kasimir-Bauer et al. 2012). Markers useful in the isolation and characterization of CTCs are shown in Table 2.1.
A study identified CSCs in a CTC population among breast cancer patient peripheral blood samples. This study showed that among a total of 1439 CTCs , 66.7 % of patients showed a putative stem cell/progenitor phenotype (35.2 % CD44 +/CD24−/low or 17.7 % ALDH1high/CD24−/low) in CTCs; 35 % of the CTCs in 20 out of 30 patients exhibited the BCSC CD44+/CD24−/low phenotype, whereas 17.7 % of the CTCs identified in seven patients were ADLH1high/CD24−/low (Theodoropoulos et al. 2010).
Metastasis initiating cells containing CTC populations originating from primary human luminal breast cancer expressing epithelial cell adhesion molecule (EpCAM ), CD44 , CD47, and MET caused lung, liver, and bone metastasis in mice. In a small patient cohort exhibiting tumor metastasis, the population of EpCAM+CD44+CD47+MET+ correlated with increased metastasis and low overall survival (Baccelli et al. 2013). CTCs obtained from patients with Dukes’ B and C colon cancers were shown to express CD133 , carcinoembryonic antigen (CEA) and cytokeratin. Prognosis among these patients is significantly poorer due to metastasis than those individuals who were found not to express these markers in their CTCs (Pantel and Alix-Panabières 2007). Circulating CSCs were detected in the blood of patients positive for colonic adenocarcinomas. Authors isolated a relatively pure population of CSCs (CD45−/CK19+), free of red blood cells and largely free of contaminating CD45+ white blood cells. Enriched circulating CSCs from patients with colon adenocarcinomas had a malignant phenotype and co-expressed CSC markers (DCLK1/LGR5) with CD44/Annexin A2. CSCs were not found in the blood of non-cancer patients, free of colonic growths. Enriched circulating CSCs from colon cancer patients grew primary spheroids, suggesting the presence of tumor-initiating cells in the blood of these patients (Kantara et al. 2015).
In a human-to-mouse xenotransplantation experimental model, viable tumorigenic melanoma CTCs were isolated and it was demonstrated that they were capable of metastasis formation. The detection of melanoma CTC in human-to-mouse s.c. tumor xenotransplantation models correlated significantly with pulmonary metastasis formation. Moreover CTCs isolated from murine recipients of s.c. melanoma xenografts were capable of primary tumor initiation and caused metastasis formation upon xenotransplantation to secondary murine NOD -scid IL2Rγ (null) recipients. These results provide initial evidence that melanoma CTC are tumorigenic and demonstrate that CTC are capable of causing metastatic tumor progression (Ma et al. 2010).
It has been recently postulated that EMT plays a key role in the process of tumor cell dissemination (Kasimir-Bauer et al. 2012; Giordano et al. 2012; Barrière et al. 2012; Aktas et al. 2009). Tumor cells undergoing EMT may migrate into peripheral blood as CTCs ; due to their mesenchymal stemness features, these cells might be able to reach distant sites of the body and initiate metastases. Loss of E-cadherin , overexpression of N-cadherin , and cytoskeletal alterations (e.g., expression of vimentin) are the hallmarks of EMT. So far, defining the CSCs in a population of CTCs has proven extremely challenging given current limitations in the capture of CTCs (Monteiro and Fodde 2010). CTCs seem to represent a highly heterogeneous cell population with regard to their morphology, molecular characteristics, implantation efficiency after dissemination and their metastatic potential (Lianidou et al. 2013; Fehm et al. 2010).
6.2 Stemness and EMT Identification in CTCs
The challenge in identifying and detecting CTCs is based on their rare number as well as the lack of a universal marker. The majority of methods are based on the detection of epithelial markers, and cells undergoing EMT or with a mesenchymal phenotype might thus be missed. Only a few markers useful in the isolation of CTCs with a mesenchymal phenotype have been evaluated.
In the past 10 years, the number of assays to detect and characterize has increased. Due to the low frequency of the isolated tumor cells, all techniques have to be extremely sensitive. In several cases the first step is the enrichment of tumor cells (Ross et al. 1993). The choice of enrichment and characterization steps depending on the markers analyzed (especially EpCAM ) is crucial to allow as well as to limit the detection of cells undergoing EMT or not. One way to enrich disseminated tumor cells is density gradient centrifugation. Due to the lack of a general marker, tumor cells are characterized as epithelial cells which are positive for EpCAM or cytokeratins (Fehm et al. 2002). Another way to enrich CTCs is to label the cells with specific antibodies which are conjugated with magnetic particles. Several tests are based on the immunomagnetic enrichment of epithelial markers, especially EpCAM (Cristofanilli et al. 2004; Fehm et al. 2009), therefore limiting the possibilities to detect mesenchymal tumor cells which have undergone EMT. Tests differ in the subsequent characterization of the CTCs.
The semiautomatic CellSearch system (Janssen Diagnostics, Raritan, NJ, USA) which has been approved by the FDA is based on an immunomagnetic enrichment of epithelial cells using EpCAM -specific antibodies coated with magnetic beads. CTCs are quantified and further characterized by immunofluorescence detecting cytokeratins (CKs) 8, 18, and 19 and CD45 to exclude leucocytes as well as staining of the nuclei (DAPI) (Cristofanilli et al. 2004, 2005). Additional staining of the CSC marker CD44 can be made (Lowes et al. 2012).
In the AdnaTest Breast Cancer (AdnaGen GmbH, Langenhagen, Germany) this enrichment step is performed using magnetic beads which are coated with EpCAM - and mucin-1 (MUC1) specific antibodies. The additional characterization of the CTCs is made by detection of the EMT and stem cell markers TWIST, Akt2, PI3K, and ALDH1, respectively (Kasimir-Bauer et al. 2012; Aktas et al. 2009).
Several approaches to enrich CTCs use special chips combining microfluidics and immobilization of CTCs by binding of specific antibodies (e.g., CTC -chip, Herringbone Chip) (Nagrath et al. 2007; Stott et al. 2010). The latter chip was used by Yu et al. (2013) to establish an RNA in situ hybridization assay to detect and quantify CTCs with either an epithelial or mesenchymal phenotype or with a phenotype in between (partial EMT ). The expression levels of seven epithelial transcripts (EpCAM ; CK 5, 7, 8, 18, and 19 and cadherin 1) and three mesenchymal transcripts (SERPINE1/PAI1, cadherin 2, and fibronectin 1) were analyzed to characterize CTCs which were detected by binding at least one of the following antibodies: EpCAM, HER2 or epidermal growth factor receptor 2 (EGFR ). Flow cytometry is another technique which allows an individual characterization of rare cells like CTCs. Using flow cytometry, Giordano et al. (2012) could detect a subpopulation of cancer stem cells expressing either ALDH1, CD44 , or low amounts of CD24 or ALDH1 and CD133 . Although the majority of assays use EpCAM as detection marker, different markers are currently used to detect and enrich CTC. As CTCs change their phenotype during EMT and MET , false negative results can be obtained depending on which detection marker was used. EpCAM-based assays involve the risk that CTC showing a mesenchymal phenotype might be missed.
The hypothesis that EMT markers can be detected among the CTCs of breast cancer patients has been confirmed by various studies in both metastatic and early breast cancer (Giordano et al. 2012; Barrière et al. 2012; Aktas et al. 2009; Mego et al. 2012a, b; Armstrong et al. 2011; Kallergi et al. 2011; Raimondi et al. 2011). EMT markers positive CTCs can be detected in up to 26 % of metastatic breast cancer patients. Moreover, a high expression of EMT markers predicted shorter progression free survival in these patients (Mego et al. 2012b). Aktas et al. (2009) showed in 39 metastatic breast cancer patients that EMT markers, such as Twist1, Akt2, and PI3Kα, can be co-detected in up to 62 % of CTC positive blood samples; EMT markers were more likely to be found in patients resistant to therapy, suggesting increased invasiveness of tumor cells undergoing this process. In primary breast cancer EMT markers could be detected in 72 % of CTC positive and 18 % of CTC negative patients, respectively (Kasimir-Bauer et al. 2012). Expression of EMT markers (e.g., vimentin, fibronectin) was found in up to 38 % of all stage breast cancer patients tested by the standard definition as CTC negative (Raimondi et al. 2011).
These findings suggest that, in addition to CTCs expressing epithelial antigens, a fraction of CTCs with exclusively mesenchymal phenotype could exist and thus remain undetectable for assays based on epithelial character of these cells. However, due to the methodology, morphological features of the cells were not evaluated in these trials (Aktas et al. 2009; Kasimir-Bauer et al. 2012). In this regard, CTCs coexpressing mesenchymal and epithelial markers have been visualized in three other studies in breast cancer patients confirming that both kinds of markers can be expressed in the same cell (Yu et al. 2013; Armstrong et al. 2011; Kallergi et al. 2011). Additionally, Vimentin positive CTCs were detected in peripheral blood of metastatic breast cancer patients while paired metastases from the same patients were shown to be negative for this marker (Armstrong et al. 2011). This suggests a reversibility of the EMT process once tumor cells reach their destination resembling the phenomenon of epithelial plasticity known from embryonic development (Nieto 2013).
In a recent study by Kasimir-Bauer et al. (2012) on 502 primary breast cancer patients 46 % of CTC positive and 5 % of CTC negative blood samples were positive for ALDH1, a common stem cell marker. Similar findings have been shown by Aktas et al. (2009) in the metastatic situation. Moreover, a presence of stem cell-like CTCs in peripheral blood of breast cancer patients was shown to be associated with therapy resistance; stem cell markers or EMT factors or both were detected in 74 % (25/34) of nonresponders and in 10 % (2/21) of patients who responded to systemic treatment. In the trial by Raimondi et al. (2011), an overexpression of stem markers in CTCs was correlated with advanced stage of disease.
7 Principal Therapeutic Strategies Against Cancer Stem Cells: Focusing in Particular on Metastatic Niche and Microenvironment as Potential Therapeutic Targets
To prevent disease relapse and achieve permanent cure, the CSCs that sustain tumor growth must be eradicated in addition to killing the bulk cells of the tumor. However, properties of CSCs, such as quiescence or expression of drug-resistance transporters, may make them difficult to eliminate using conventional cytotoxic drugs that kill the bulk tumor cells. It will be crucial to understand the unique biology of CSCs in order to develop novel treatments that effectively target these cells.
There are several obstacles to be overcome in the development of effective CSC-targeted therapies. Such treatments must be selective for CSCs and spare normal stem cells. There is recent evidence in acute myeloid leukemia that the pathways that regulate self-renewal in normal stem cells are not completely abolished in leukemia stem cells (Hope et al. 2004). Thus, drugs that target critical processes in CSCs, such as survival or self-renewal, may prove intolerably harmful to their normal counterparts. Furthermore, CSCs will likely have acquired genetic or epigenetic changes that allow them to bypass normal tumor-suppressing processes such as senescence or apoptosis in response to DNA damage, and CSCs are believed to be more resistant to chemotherapy than more differentiated cancer cells. Treatment with agents that normally induce senescence or apoptosis may actually provide a growth advantage to CSCs (Bao et al. 2006a). Ideally, effective therapies will target pathways that are necessary for CSC survival but not for the survival of normal stem cells. Here the principal therapeutic strategies against CSCs are summarized (Fig. 2.4).

Therapeutic target s potentially useful in anti- CSCs therapy. ECM extracellular matrix, TAMs tumor-associated macrophages
7.1 Directly Targeting CSCs via Surface Markers
CD133 is a well characterized marker for putative cancer stem cells (Wu and Wu 2009). Blockage of CD133 reduced the capacity of the melanoma to metastasize (Rappa et al. 2008), suggesting that CD133 might be a potential therapeutic target for CSCs in melanoma and other cancer types (Wu and Wu 2009).
CD44 is a marker of CSCs and also an adhesion receptor involved in metastasis and drug-resistance. Inhibition of CD44 using a siRNA decreases cancer cell adhesion to bone marrow endothelial cells in prostate and breast cancer cell lines (Draffin et al. 2004). A CD44v6-targeting immuno-conjugate, bivatuzumab mertansine, has been evaluated in phase I clinical trial in the case of head and neck squamous cell carcinoma (Riechelmann et al. 2008). Targeting CD44 by an A6 peptide (acetyl-KPSSPPEE-amino) blocked the migration and metastasis of CD44-expressing cells (Piotrowicz et al. 2011). In hepatocellular carcinoma, neutralizing CD44 can also inhibit CD90+ cell-mediated tumor formation and metastasis in vivo, suggesting an therapeutic strategy against CD90+ CSCs by targeting CD44 (Lee et al. 2013).
In an acute myeloid leukemia mouse model, in vivo administration of an activating monoclonal antibody directed at CD44 resulted in significant reduction in the levels of leukemic repopulation (Jin et al. 2006). CD44 is a regulator of several miRNAs known to maintain CSCs . When CD44 expression in PCa cells is down-regulated, miR-34a levels increase leading to reduced tumor regeneration and metastasis in xenografts (Liu et al. 2011a).
7.2 Targeting Self-Renewal and Differentiation Pathways
Signaling pathway s , such as Wnt, Notch, and Hedgehog (Hh), are essential for both regulation of EMT /metastasis and self-renewal of CSCs in several cancers. Development of agents that target critical steps in these pathways will be complicated due to signaling cross-talk (Takebe et al. 2011). Several novel agents targeting Wnt/β-catenin have been developed. Some of these agents have been shown to selectively target the cancer stem cell subpopulation in vivo, inhibit tumor growth and inhibit metastasis (Takebe et al. 2011). Inhibition of Notch1 can significantly decrease the CD44 +CD24−/low subpopulation and inhibited the development of brain metastases from breast cancer (McGowan et al. 2011). Pharmacologic blockage of aberrant Hedgehog signaling might be an effective therapeutic strategy for inhibiting metastases in human cancers through targeting CSCs. A small-molecule Hedgehog inhibitor, IPI-269609, has been proved to profoundly inhibit systemic metastases in orthotopic xenografts derived from human pancreatic cancer cell lines, accompanied with a significant reduction in the population of ALDH-positive cells in pancreatic cancer (Feldmann et al. 2008).
Salinomycin, a wnt/β-catenin inhibitor, inhibits tumor growth, induces epithelial differentiation of tumor cells, and down-regulates CSC genes in tumor cells (Gupta et al. 2009).
As stem cells are often dependent on bone morphogenetic protein (BMP) signaling, a number of therapeutic strategies are being sought to target this pathway (Joseph et al. 2012; Zhang et al. 2003; Zhu and Emerson 2004). Piccirillo et al. (2006) reported that the BMP-BMPR signaling system – which controls the activity of normal brain stem cells – may also act as a key inhibitory regulator of tumor-initiating, stem-like cells from glioblastoma by a reduction in proliferation and increased expression of markers of neural differentiation. The reduction in the size of the CD133 + population and the growth kinetics of the glioblastoma cells suggest that targeted BMP-pathway therapeutics are worth pursuing (Massard et al. 2006; Piccirillo et al. 2006).
The histone deacetylase inhibitor valproic acid, commonly used to treat epilepsy, has recently been found to have anti-cancer activity that may target CSCs (Blaheta et al. 2005). Valproic acid induces the terminal differentiation of cancer cells by increasing the DNA binding of activating protein-1 transcription factor, decreasing protein kinase C (PKC) activity, inhibiting the Wnt signaling pathways, and activating the peroxisome proliferator-activated receptors, in addition to blocking histone deacetylase (Blaheta et al. 2005).
Histone deacetylase (HDAC) inhibitors inhibit growth, induce differentiation and apoptosis of neurosphere derived from glioblastoma (GBM). GBM neurospheres contain cancer stem cell like that propagate tumor and resist cytotoxic therapeutics. Using MS-275, a specific gene product induced by HDAC inhibition, Delta/Notch like epidermal growth factor (DNER), inhibited the growth of GBM derived neurospheres, induced their differentiation and inhibited their engraftment and growth as tumor xenografts (Sun et al. 2009). The HDAC inhibitor entinostat reverses EMT in xenografts (Tate et al. 2012).
7.3 Differentiation Therapies
Another proposed therapeutic approach is to stimulate differentiation of CSCs such that they lose their capacity for self-renewal and resistance to chemotherapeutic agents (Nguyen et al. 2012). This is particularly critical where CSCs are widely distributed at low density, making conventional interventions challenging. Thus far, the most well developed therapeutic agent is vitamin A and its analogues (retinoid acid) for the treatment of acute promyelocytic leukemia (APL). These agents enhance tumor differentiation and reverse malignant progression by modulation of signal transduction networks regulated by nuclear retinoid receptors. In patients with APL, a 90 % remission rate and a 70 % cure rate with all-trans retinoic acid therapy followed by chemotherapy has been observed (Burnett et al. 2010). In vitro, retinoid acid can also induce differentiation in embryonic cells, teratocarcinomas and melanomas (Rohwedel et al. 1999).
7.4 Therapies Directed at CSC Niche and Pre-metastatic Niche
The challenges of targeting disseminated CSCs may be even more pronounced, as the distant microenvironment may help protect these cells from therapeutic insults (Hovinga et al. 2010).
7.4.1 Targeting Vasculature
In particular, the vasculature likely plays an important role in forming stem and progenitor cell niches and has been suggested to regulate many tumor microenvironments (Bautch 2011). Therefore, damaging the CSC niche environment may impact the survival and tumor-initiating properties of CSCs (Folkins et al. 2007). The impact of angiogenesis inhibitors such as bevacizumab, thalidomide, sorafenib, sunitinib, pazopanib may be in part related to their effects on the vascular niche and disruption of the CSC microenvironment (Tonini et al. 2003).
The VEGF -specific antibody bevacizumab has direct and rapid anti-vascular effects and seem to be useful in targeting CSCs by disturbing niche (Willett et al. 2004). On the other hand, hypoxic tumor microenvironment promotes tumor progression, regulates CSCs and increases their metastatic potential (Hill et al. 2009). Inhibition of hypoxia eliminates metastasis in mice without effect on the primary tumor, suggesting that hypoxia is an important process in the formation of pre-metastatic niche (Sceneay et al. 2013).
Preclinical models suggest that antiangiogenic agents actually increase invasive and metastatic properties of breast cancer cells (Ebos et al. 2009; Pàez-Ribes et al. 2009). Hypoxia induced by administration of antiangiogenic agents might accelerate tumor growth and metastasis by increasing the CSC population. Conley et al. (2012) demonstrated that administration of sunitinib and bevacizumab increased CSC population in breast cancer xenografts as a consequence of tumor hypoxia, and this effect was mediated by HIF-1α through the activation of Wnt pathway via Akt/β-catenin signalling. Authors concluded that antiangiogenic agents might have to be combined with CSC targeting drugs.
These results differ from those obtained in glioblastoma. Glioblastomas express high levels of vascular endothelial growth factor (VEGF ) (Bao et al. 2006b). A functional interaction between brain CSCs and endothelial cells is supported by the close association of CD133 + brain cancer cells with vascular endothelial cells in vitro and in vivo, and more importantly by the demonstration that coinjection of primary human endothelial cells enhances tumor formation by CD133+ medulloblastoma cells in immune-deficient mice (Calabrese et al. 2007). Tumors initiated in mice by CD133+ cells from either primary glioblastoma biopsy specimens or xenograft cell lines are highly vascular (Bao et al. 2006b) Treatment of xenograft tumors with bevacizumab not only potently inhibits tumor growth in mice (Calabrese et al. 2007; Bao et al. 2006b) but also results in depletion of cells coexpressing CD133 and nestin, a marker of primitive neural cells, without directly affecting bulk tumor cell proliferation or death. Together, these results suggest that inhibition of brain tumor growth by antiangiogenic agents is mediated at least in part by disruption of a vascular niche required for maintenance of CSCs.
7.4.2 Targeting the Extrinsic Signals at the CSC Niche
The CXCL12 /CXCR4 plays a central role in cancer cell proliferation, invasion, and dissemination in the majority of malignant diseases. Although the signals generated by the metastatic niche that regulate CSCs are not yet fully understood, accumulating evidence suggests a key role of the CXCL12/CXCR4 axis (Cojoc et al. 2013). Strategies aimed at modulating the CXCL12/CXCR4 axis may have important clinical applications to inhibit CSC growth (Gil et al. 2014; Barone et al. 2014). In a phase I study evaluating LY2510925, a peptide agonist blocking stromal cell derived factor-1 (SDF1) from CXCR4 binding, in 45 advanced cancer patients, stable disease was obtained in nine (20 %) of them (Galsky et al. 2014).
Multiple agents are currently being developed to target CXCL12 /CXCR4 signaling in cancer. The anti-CXCR4 drug AMD3100 (plerixafor) is approved for stem cell mobilization in patients with non-Hodgkin’s lymphoma and multiple myeloma; the CXCL12 analog CTCE-9908 is approved for clinical use in patients with osteosarcoma. Novel CXCR4 antagonists are currently in clinical trials for multiple myeloma, leukemia, and lymphoma. CXCR4 inhibitor MSX-122 is in Phase I trials for advanced malignant disease resistant to standard therapy. NOX-A12 neutralizes CXCL12 and is in clinical trial for the treatment of chronic lymphocytic leukemia and multiple myeloma (Ramsey and McAlpine 2013; De Nigris et al. 2012). AMD3100 has been shown to decrease metastatic potential in animal models for different types of tumors (Smith et al. 2004; Kim et al. 2010; D’Alterio et al. 2012; Kajiyama et al. 2008; Matsusue et al. 2009). Similarly, blocking CXCR4 receptor function by a monoclonal antibody or polypeptide inhibits cancer cell proliferation, motility, and invasion in multiple preclinical models both in vitro and in vivo (Zeng et al. 2006; O’Boyle et al. 2013). Recent data suggest that inactivation of the CXCL12/CXCR4 axis by neutralizing antibody or by the CXCR4-specific small molecule antagonist AMD3100 inhibits glioma, renal, colon, pancreas, and prostate cancer progenitors as well as tumor initiating population within gefitinib-resistant lung cancer and tamoxifen-resistant breast cancer cells in vitro and in animal models (Gassenmaier et al. 2013; Dubrovska et al. 2012; Redjal et al. 2006). Low oxygen tension is a critical microenvironmental factor in regulating tumor initiating axis in cancer cells; hypoxia promotes expansion of CSCs and converts non-stem cancer cells into CSC populations with increased self-renewal capacity (Heddleston et al. 2009; Soeda et al. 2009). The effects of reduced oxygen tension on CSCs are mediated at least in part through the activation of the HIF signaling pathway (Li and Rich 2010). CXCR4 expression is induced under hypoxic stress via activation of the HIF pathway (Ishikawa et al. 2009). As tumor cells can be protected from the effect of ionizing radiation by hypoxia, pharmacologic inhibition of the CXCL12/CXCR4 interaction by AMD3100 or neutralizing antibody prevents the recurrence of glioblastoma after irradiation in mice by inhibition of vasculogenesis (Kioi et al. 2010). Activation of CXCR4-mediated STAT3 signaling in non-small cell lung cancer cells is functionally crucial for the maintenance of stemness and resistance to radiotherapy (Jung et al. 2013). Recent prostate tumor xenograft studies in mice showed that a combination of AMD3100, which targets prostate cancer stem-like cells, and the conventional chemotherapeutic drug docetaxel, which targets the bulk tumor, is significantly more effective in eradicating tumors as compared to monotherapy (Dubrovska et al. 2010, 2012; Domanska et al. 2012).
Human breast cancer cells expressing CXCR1, a receptor that binds the proinflammatory chemokine IL-8, are present almost exclusively within the CSC-containing ALDH1+ population (Ginestier et al. 2010). IL-8 has been implicated in tumor invasion, metastasis, and self-renewal. Treatment of orthotopically transplanted tumors in NOD /SCID mice with the CXCR1/2 inhibitor repertaxin, the standard chemotherapeutic agent docetaxel, or a combination of both drugs all resulted in impaired tumor growth. However, tumors treated with docetaxel alone showed either unchanged or increased percentage of ALDH1+ cells compared with untreated controls, whereas repertaxin treatment alone or in combination with docetaxel significantly reduced the ALDH1+ population. Upon serial transplantation, tumor cells derived from control or docetaxel-treated primary animals were able to generate tumors in secondary mice with similar efficiency, while cells from repertaxin-treated animals showed a two- to fivefold reduction in tumor growth and were only able to generate tumors at the highest injected cell dose (Ginestier et al. 2010).
STAT3 signalling has an important role in self-renewal and differentiation of stem cells. IL-6 is implicated in promoting STAT3 mediated CSC expansion in several other types of tumors. Expression levels of IL-6 and its receptor are highly elevated in prostate CSCs , and a crucial role of JAK-STAT3 in mediating IL-6-induced stem cell maintenance in prostate cancer has been shown. Furthermore, IL-6–JAK2–STAT3 signalling is required for the maintenance of breast CSCs and tumor growth (Yu et al. 2014). In addition, IL-6 induces the recruitment of mesenchymal stem cells (MSCs), into the tumor microenvironment. IL-6 increases STAT3 activation in MSCs, which contributes to MSC survival and MSC-mediated tumor progression (Rattigan et al. 2010).
Given the central role of STAT3 in the promotion and maintenance of a stem cell phenotype, controlling STAT3 activity in this population should inhibit tumor progression. In stem cells, STAT3 activity can be regulated by EZH2-mediated protein methylation (Kim et al. 2013). STAT3 protein phosphorylation affects the regulation of genes driving stemness, EZH2-induced STAT3 protein methylation, and possibly also STAT3 acetylation induced by p300 acetyltransferase, and is thus crucial for regulating the formation of a transcription complex bound to the promoters of genes with a propensity to promote stem cell characteristics. Therefore, functional disruption of STAT3 modification enzymes such as EZH2 and p300 may serve as a promising therapeutic strategy for human cancers. However, targeting these modification enzymes may generate broad biological effects that lead to unwanted toxicity (Yu et al. 2014).
7.4.3 Targeting Tumor Associated Macrophages
Tumor associated macrophage s (TAMs ) play an important role in tumor growth, angiogenesis, metastasis, matrix remodelling and immune evasion in various human and animal tumors (Sica and Bronte 2007; Biswas et al. 2008; Solinas et al. 2009; Sica et al. 2008). In mouse tumor models, an increased density of TAMs is associated with poor efficacy of chemotherapy and radiotherapy (Zhang et al. 2010; Meng et al. 2010). The density, activation and histological location of TAMs can predict patients’ survival in different types of cancer (Zhu et al. 2008; Kurahara et al. 2011; Hanada et al. 2000). Therefore, TAMs are now considered as a promising target for tumor therapy. Some tumor-released and stroma-released cytokines and chemokines facilitate the recruitment of macrophages to tumor tissues, and possible therapeutic strategies are aimed at inhibiting macrophage recruitment. For example, overexpression of C-C motif chemokine ligand 2 (CCL2) was correlated with macrophage infiltration and poor prognosis in human cancers (Roca et al. 2009); macrophage infiltration and the growth of tumors were reduced when CCL2 was inhibited (Mizutani et al. 2009; Qian et al. 2011; Zhu et al. 2010). A CCL2-targeting agent, trabectedin, used in clinic to treat human ovarian cancer and myxoid liposarcoma, could suppress the recruitment of monocytes to tumor sites and inhibit their differentiation to mature TAMs (Allavena et al. 2005; Germano et al. 2010).
In a phase II clinical study, siltuximab, anti-interleukin- 6 (IL-6) antibody, reduced macrophage infiltration in tumor tissue by decreasing the plasma level of some chemoattractants such as CCL2, vascular endothelial growth factor (VEGF ) and C-X-C motif chemokine ligand-12 (CXCL-12) (Coward et al. 2011). An alternative way to suppress the chemoattractive activity of CCL2 is neutralizing its receptor, C-C motif chemokine receptor 2 (CCR2). A CCR2 inhibitor, RS102895, has exhibited negative effects on macrophage migration (Jin et al. 2010). Another important chemoattractant for macrophages is macrophage colony-stimulating factor (M-CSF). In human hepatocellular carcinoma, there is a significant association between high M-CSF expression and high macrophage density, each relates to poor overall survival of patients (Zhu et al. 2008). Treatment with M-CSF antibody suppressed tumor growth by 40 % in human MCF-7 breast cancer xenografts (Paulus et al. 2006). Two M-CSF receptor inhibitors (JNJ-28312141 andGW2580) were found to decrease TAM count and suppress tumor growth, angiogenesis and metastasis (Manthey et al. 2009; Kubota et al. 2009).
Other chemoattractants for macrophages, such as VEGF , CXCL-12 and CCL5, also seem to be potential targets for TAM depletion. Selectively inhibiting VEGFR-2 reduced macrophage density and prevented tumor growth and angiogenesis in orthotropic pancreatic and breast tumors (Dineen et al. 2008; Roland et al. 2009). Repressing either the CXCL12 /CXCR4 or the placental growth factor (PIGF)/VEGFR-1 pathway reduced macrophage count (Welford et al. 2011). The tumor microenvironment is usually hypoxic and hypoxia-inducible factors are transcriptional activators for VEGF and CXCR4 genes (Fang et al. 2009); HIF-1a deficiency reduced macrophage density, tumor angiogenesis and invasion in murine glioblastoma via blocking the matrix metalloproteinase 9 (MMP9)/VEGF pathway (Du et al. 2008). HIF-2a mediates macrophage migration to the tumor microenvironment partly through regulating M-CSFR and CXCR4 (Imtiyaz et al. 2010).
Some drugs commonly used in clinical practice can directly suppress TAMs survival. Clodronate has a selective cytotoxicity to macrophages and this clodronate-induced depletion of macrophages can result in the regression of tumor growth, angiogenesis and metastasis (Zeisberger et al. 2006; Hiraoka et al. 2008). Zoledronic acid selectively depletes MMP9-expressing TAMs (Tsagozis et al. 2008), impairs the differentiation of myeloid cells to TAMs and induces the tumoricidal activity of macrophages (Tsagozis et al. 2008; Veltman et al. 2010; Coscia et al. 2010). Dasatinib, a Src kinase inhibitor and a preclinical drug for chronic-phase chronic myeloid leukemia, could reduce MMP9+ macrophage density and inhibit MMP9 expression in the tumor microenvironment (Liang et al. 2010).
Another approach is to deplete TAMs by targeting their surface molecules with immunotoxin-conjugated agents. In ovarian cancer, alemtuzumab (anti CD52) induced lysis of myeloid cells in vitro and ex vivo, supporting the use of lemtuzumab in clinical trials to test its efficacy as an anti-myeloid cell antiangiogenic therapeutic (Pulaski et al. 2009). Folate receptor b (FRb) is another surface protein over-expressed in M2-like TAMs (Nagai et al. 2009; Puig-Kröger et al. 2009), and the existence of FRb+ macrophages positively associates with high vessel density, high incidence of haematogenous metastasis and a poor prognosis in patients with pancreatic cancer (Kurahara et al. 2012). A recombinant immunotoxin to folate receptor beta affects tumor growth, accomplished with the depletion of TAMs (Nagai et al. 2009). In this approach while pro-tumoral M2 TAMs could be depleted, the M1 tumoricidal ones are not affected.
Inhibiting the signals essential for M2 differentiation so impairing the pro-tumoral and immunosuppressive profile of TAMs is another strategy in development. STAT3 pathway is consistently active in many tumors and acts as a negative regulator for macrophage activation and the host’s inflammatory responses. When the activation of STAT3 was blocked, either with a dominant negative variant or an antisense oligonucleotide, macrophages could increase the release of IL-12 and RANTES and reverse the systemic immune tolerance (Cheng et al. 2003). Two tyrosine kinase inhibitors (sunitinib and sorafenib) have shown their inhibitory effects on STAT3 in macrophages in vitro (Xin et al. 2009; Edwards and Emens 2010). Sorafenib can restore IL-12 production but suppress IL-10 expression in prostaglandin E2 conditioned macrophages, indicating its effects on reversing the immunosuppressive cytokine profile of TAMs (Edwards and Emens 2010). Another STAT family member important for TAM biology is STAT6. In one study, STAT6–/– mice produced predominantly M1-like tumoricidal TAMs and > 60 % of STAT6–/– mice rejected tumor metastasis (Sinha et al. 2005). Several up-/down-stream mediators of STAT6 could act as modulators of TAM function. These modulators include phosphatidylinositol 3-kinase (PI3K), Src homology 2-containing inositol-5′-phosphatase (SHIP), Kruppel-like factor 4 (KLF4) and c-Myc.
It has been reported that the expression of KLF4 was induced in M2 macrophages and reduced in M1 macrophages. A study indicated that KLF4 cooperated with STAT6 to induce an M2 pattern. Levels of KLF4 can be manipulated by diverse agonists such as statins, resveratrol, bortezomib and dietary compounds (Liao et al. 2011). Other proteins and signalling pathways are known to promote M2-like properties of macrophages and are also the potential targets for tumor therapy. Peroxisome proliferator-activated receptor (PPAR)-c can promote M2 type differentiation of human macrophages by acting as a transcriptional inhibitor of NF-jB.132 PPAR-a plays a role in macrophages by antagonizing M1 polarization and supporting M2 polarization (Van Ginderachter et al. 2008). As synthetic inhibitors of PPAR-a/c have now been identified, the evaluation of their role in TAM targeted therapy is essential.
HIFs are a possible target because of their over-expression in TAMs residing in the hypoxic tumor microenvironment and their ability to induce the production of angiogenic factors, including VEGF , platelet-derived growth factor-b, NOS2, fibroblast growth factor 2, IL-8 and cyclooxygenase-2. Macrophage-targeted depletion of HIF-1a reduced tumor growth in mice (Doedens et al. 2010).
Among anti-tumor drugs, cisplatin promotes macrophages to produce large amounts of NO, a reactive oxygen intermediate and proinflammatory cytokines, leading to enhanced tumoricidal activity (Chauhan et al. 2009). Silibinin inhibits the production of angiogenic cytokines and interleukins in macrophages, leading to angiogenesis regression (Tyagi et al. 2009). Finally, pantoprazole enhances TAM recruitment but increases TAMs to an M1-like tumoricidal state (Vishvakarma and Singh 2010).
8 Future Prospects
Metastatic cancer remains an incurable disease and targeting CSCs is a novel promising approach. Many drugs active on CSCs or metastatic niche are already used in clinical practice; other approaches are under clinical trials or still in a preclinical phase (Ferrari and Nicolini 2012; Ferrari et al. 2013). It is likely that drugs targeting CSCs or their microenvironment would be mostly useful in an early phase of cancer history, when there is a micrometastatic spread not clinically evident. In this context, such drugs, eventually combined with standard anticancer drugs, could theoretically eradicate the minimal residual disease.
Targeting only CSCs may not be enough to prevent metastasis or relapse due to metastasis. Continued development of combination therapies with multiple targets (e.g. targeting CSCs, combination of chemotherapy, differentiation therapy, and targeting microenvironment) will be essential. A study demonstrated that a neutralizing antibody against a membrane-associated NOTCH ligand inhibits tumor growth and CSC self-renewal in human colon cancer implanted mice (Hoey et al. 2009). Another study showed that the administration of an anti-CD123 antibody prevents the engraftment of serially transplanted acute myeloid leukemia into the animals, suggesting this antibody impedes the stem-like characteristics of leukemia cells (Jin et al. 2009). In both cases, the inhibitory abilities of the antibodies were enhanced in combination with chemotherapy. Releasing CSCs from their niche could enhance their susceptibility to chemotherapy. Indeed, when acute myeloid leukemia cells (Nervi et al. 2009) and multiple myeloma cells (Azab et al. 2009) are treated prior to chemotherapy, with a CXCR4 inhibitor, that prevents the lodging of cancer cells into select microenvironments, the chemosensitivity of these cells was strongly enhanced. In part, release from the protection of the microenvironmental niche could sensitize CSCs to chemotherapeutics. It is also possible that disruption of CXCL12 /CXCR4 signaling activates CSC cycling which in turn could sensitize CSCs to agents targeting proliferating cells. This strategy could be helpful for targeting potentially dormant DTCs in patients with no clinically apparent distant disease (Shiozawa et al. 2013).
Tumor dormancy is another important issue. In fact, as most neoplasms are identified after they have reached a critical mass, the ability to block the reactivation of dormant CSCs at distant sites of metastases is a critical area of research. Dormancy of disseminated tumor cells (DTCs) may not be a process exclusive to metastatic cells that arise from established primary tumors. In fact, pre-invasive lesions also contain epithelial cells that can undergo epithelial–mesenchymal transition and disseminate; these cells are referred to as early DTCs. Early DTCs can develop metastatic growth capacity that manifests after long periods of dormancy. By disseminating at early stages, DTCs that survive may evolve divergently from the primary tumor. This may generate metastases with different characteristics from the primary lesion and may explain the lack of success of treating metastasis with therapies designed on the basis of primary tumor characteristics. Moreover, the vast majority of early DTCs in mouse models seem to be dormant, and clinical evidence supports this hypothesis. This suggests that persistence in a dormant state may protect these DTCs from treatment, contributing to late recurrence of disease (Sosa et al. 2014). Possible therapeutic strategies mimic the dormancy programme to sustain dormant DTCs and thereby prevent relapse. Cancer therapy may force surviving residual tumor cells into dormancy by activating stress signalling (Schewe and Aguirre-Ghiso 2009; Kobayashi et al. 2011). An example of this may be tumor cells that are known as drug-tolerant persisters that survive targeted therapies by altering epigenetic mechanisms (Sharma et al. 2010). Drugs commonly used in clinic may be useful to induce dormancy in DTCs. One study showed that, in both primary cells and breast cancer and leukemia cell lines, the DNA methylation inhibitor 5‑azacytidine alone caused decreased expression of G0 to G1 exit genes (Tsai et al. 2012). Also, HDAC inhibitors or DNA demethylating agents might represent alternative adjuvant therapies to induce prolonged dormancy of uveal melanoma or other types of DTCs (Sosa et al. 2014).
Among new therapeutic targets, ECM has gained importance in the recent years. Recent advances in knowledge about the role of TNC and POSTN in the metastatic microenvironment suggest that these ECM components as new therapeutic targets (Malanchi et al. 2011; Oskarsson et al. 2011).
As pro-tumoral activity of TAMs largely depends on their recruitment and activation, therefore therapeutic strategies against TAMs should be aimed at inhibiting macrophage recruitment, suppressing TAM survival, enhancing M1 tumoricidal activity of TAMs and blocking M2 tumor-promoting activity of TAMs. So far, many agents have been identified as candidate drugs, either as inhibitors of macrophage accumulation or as modulators of TAM properties. Using immune system to treat cancer is a promising approach. As TAMs contribute to chemoresistance and radio-protective effects, TAM-targeted strategies may also improve the efficacy of conventional therapies in some cases (Tang et al. 2013).
Finally, an important step will be the identification of additional markers that provide even more specific isolation and characterization of CSCs , particularly in solid tissues, particularly markers that can be used for localization and visualization of CSCs in situ, to facilitate anatomical localization of the niche as well. However, it will likely be a significant task given the complexity of the niche, comprising fibroblastic cells, myeloid and other inflammatory cells, endothelial and perivascular cells (or their progenitors), and ECM components (Sneddon 2007). Functional studies will be crucial for understanding the contribution of defined molecular constituents of metastatic niche to CSC physiology.
References
Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG et al (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150(1):165–178
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7:834–846
Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S (2009) Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res 11(4):R46
Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F et al (2005) Anti-inflammatory properties of the novel anti-tumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res 65(7):2964–2971
Allavena P, Sica A, Solinas G, Porta C, Mantovani A (2008) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 66(1):1–9
Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ (2000) Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med 6(1):100–102
Andres JL, Rönnstrand L, Cheifetz S, Massagué J (1991) Purification of the transforming growth factor-beta (TGF-beta) binding proteoglycan betaglycan. J Biol Chem 266(34):23282–23287
Arap W, Pasqualini R, Ruoslahti E (1998) Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279(5349):377–380
Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD et al (2011) Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res 9(8):997–1007
Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER et al (2010) Control of mammary stem cell function by steroid hormone signalling. Nature 465(7299):798–802
Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X et al (2009) CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 113(18):4341–4351
Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V et al (2013) Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol 31(6):539–544
Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G et al (2006) Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res 2(19):5615–5621
Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S et al (2006) Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med 12(8):933–938
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006a) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760
Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB et al (2006b) Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66(16):7843–7848
Barkan D, Green JE, Chambers AF (2010) Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer 46(7):1181–1188
Barone A, Sengupta R, Warrington NM, Smith E, Wen PY, Brekken RA et al (2014) Combined VEGF and CXCR4 antagonism targets the GBM stem cell population and synergistically improves survival in an intracranial mouse model of glioblastoma. Oncotarget
Barrière G, Riouallon A, Renaudie J, Tartary M, Rigaud M (2012) Mesenchymal and stemness circulating tumor cells in early breast cancer diagnosis. BMC Cancer 12:114
Bautch VL (2011) Stem cells and the vasculature. Nat Med 17(11):1437–1443
Behrens J (1994–1995) Cell contacts, differentiation, and invasiveness of epithelial cells. Invasion Metastasis 14(1–6):61–70
Beier D, Wischhusen J, Dietmaier W, Hau P, Proescholdt M, Brawanski A et al (2008) CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathol 18(3):370–377
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Biswas SK, Sica A, Lewis CE (2008) Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol 180(4):2011–2017
Blaheta RA, Michaelis M, Driever PH, Cinatl J Jr (2005) Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev 25(4):383–397
Borovski T, Verhoeff JJ, Cate R, Cameron K, de Vries NA, van Tellingen O et al (2009) Tumor microvasculature supports proliferation and expansion of glioma-propagating cells. Int J Cancer 125(5):1222–1230
Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF et al (1991) Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251(4995):802–804
Brabletz T (2012) To differentiate or not–routes towards metastasis. Nat Rev Cancer 12(6):425–436
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T (2005) Opinion: migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat Rev Cancer 5(9):744–749
Burnett AK, Hills RK, Green C, Jenkinson S, Koo K, Patel Y et al (2010) The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood 115(5):948–956
Butler JM, Kobayashi H, Rafii S (2010) Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 10(2):138–146
Cairns J (2002) Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci U S A 99(16):10567–10570
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11(1):69–82
Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV et al (2012) Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 22(5):571–584
Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC et al (2000) Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60(9):2541–2546
Carbonell WS, Ansorge O, Sibson N, Muschel R (2009) The vascular basement membrane as “soil” in brain metastasis. PLoS One 4(6), e5857
Cavallaro U, Christofori G (2004) Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 4(2):118–132
Chaffer CL, Dopheide B, McCulloch DR, Lee AB, Moseley JM, Thompson EW et al (2005) Upregulated MT1-MMP/TIMP-2 axis in the TSU-Pr1-B1/B2 model of metastatic progression in transitional cell carcinoma of the bladder. Clin Exp Metastasis 22(2):115–125
Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED (2006) Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res 66(23):11271–11278
Chaffer CL, Thompson EW, Williams ED (2007) Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 185(1-3):7–19
Chakrabarty S, Fan D, Varani J (1990) Modulation of differentiation and proliferation in human colon carcinoma cells by transforming growth factor beta 1 and beta 2. Int J Cancer 46(3):493–499
Chakraborty G, Jain S, Kundu GC (2008) Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res 68(1):152–161
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2(8):563–572
Chao YL, Shepard CR, Wells A (2010) Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 9:179
Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B et al (2010) Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res 16(1):45–55
Charles N, Holland EC (2010) The perivascular niche microenvironment in brain tumor progression. Cell Cycle 9(15):3012–3021 (B)
Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D et al (2010) Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6(2):141–152
Chauhan P, Sodhi A, Shrivastava A (2009) Cisplatin primes murine peritoneal macrophages for enhanced expression of nitric oxide, proinflammatory cytokines, TLRs, transcription factors and activation of MAP kinases upon co-incubation with L929 cells. Immunobiology 214:197–209
Chen Q, Zhang XH, Massagué J (2011) Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20(4):538–549
Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J et al (2003) A critical role for Stat3 signaling in immune tolerance. Immunity 19(3):425–436
Chiang AC, Massagué J (2008) Molecular basis of metastasis. N Engl J Med 359(26):2814–2823
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127(3):469–480
Clevers H (2013) The intestinal crypt, a prototype stem cell compartment. Cell 154(2):274–284
Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY et al (2008) Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 26(19):3213–3221
Cojoc M, Peitzsch C, Trautmann F, Polishchuk L, Telegeev GD, Dubrovska A (2013) Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther 6:1347–1361
Comen E, Norton L (2012) Self-seeding in cancer. Recent Results Cancer Res 195:13–23
Condeelis J, Segall JE (2003) Intravital imaging of cell movement in tumours. Nat Rev Cancer 3(12):921–930
Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN et al (2012) Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A 109(8):2784–2789
Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G et al (2004) IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J Immunol 172(9):5467–5477
Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C et al (2010) Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med 14(12):2803–2815
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867
Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA et al (2011) Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res 17(18):6083–6096
Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N et al (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112(5):645–657
Criscuoli ML, Nguyen M, Eliceiri BP (2005) Tumor metastasis but not tumor growth is dependent on Src-mediated vascular permeability. Blood 105:1508
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC et al (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351(8):781–791
Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM et al (2005) Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol 23(7):1420–1430
Croker AK, Allan AL (2012) Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat 133(1):75–87
Crowley NJ, Seigler HF (1992) Relationship between disease-free interval and survival in patients with recurrent melanoma. Arch Surg 127(11):1303–1308
D’Alterio C, Barbieri A, Portella L, Palma G, Polimeno M, Riccio A et al (2012) Inhibition of stromal CXCR4 impairs development of lung metastases. Cancer Immunol Immunother 61(10):1713–1720
D’Andrea FP (2012) Intrinsic radiation resistance of mesenchymal cancer stem cells and implications for treatment response in a murine sarcoma model. Dan Med J 59(2):B4388
Dameron KM, Volpert OV, Tainsky MA, Bouck N (1994) Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 265(5178):1582–1584
Damonte P, Gregg JP, Borowsky AD, Keister BA, Cardiff RD (2007) EMT tumorigenesis in the mouse mammary gland. Lab Invest 87(12):1218–1226
Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM et al (2009) Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J 28(4):347–358
Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E et al (2007) Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res 13(23):7053–7058
Dannenberg AJ, Subbaramaiah K (2003) Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell 4(6):431–436
De Nigris F, Schiano C, Infante T, Napoli C (2012) CXCR4 inhibitors: tumor vasculature and therapeutic challenges. Recent Pat Anticancer Drug Discov 7(3):251–264
de Sousa EM, Vermeulen L, Richel D, Medema JP (2011) Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res 17(4):647–653
Dean M (2009) ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 14(1):3–9
Debeb BG, Xu W, Woodward WA (2009) Radiation resistance of breast cancer stem cells: understanding the clinical framework. J Mammary Gland Biol Neoplasia 14(1):11–17
Demicheli R, Abbattista A, Miceli R, Valagussa P, Bonadonna G (1996) Time distribution of the recurrence risk for breast cancer patients undergoing mastectomy: further support about the concept of tumor dormancy. Breast Cancer Res Treat 41(2):177–185
Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C et al (2012) S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 21(5):642–654
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN et al (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458(7239):780–783
Dieter SM, Ball CR, Hoffmann CM, Nowrouzi A, Herbst F, Zavidij O et al (2011) Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 9(4):357–365
Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS et al (2008) Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res 68(11):4340–4346
Ding XW, Wu JH, Jiang CP (2010) ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci 86(17-18):631–637
Doberstein K, Wieland A, Lee SB, Blaheta RA, Wedel S, Moch H et al (2011) L1-CAM expression in ccRCC correlates with shorter patients survival times and confers chemoresistance in renal cell carcinoma cells. Carcinogenesis 32(3):262–270
Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG et al (2010) Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res 70(19):7465–7475
Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, Ananias HJ et al (2012) CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 14(8):709–718
Donnenberg VS, Meyer EM, Donnenberg AD (2009) Measurement of multiple drug resistance transporter activity in putative cancer stem/progenitor cells. Methods Mol Biol 568:261–279
Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ (2004) CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res 64:5702–5711
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E et al (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 3(3):206–220
Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR et al (2010) Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res 16(23):5692–56702
Dubrovska A, Hartung A, Bouchez LC, Walker JR, Reddy VA, Cho CY et al (2012) CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br J Cancer 107(1):43–52
Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C et al (2005) Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol 6(3):314–322
Dunn GP, Koebel CM, Schreiber RD (2006) Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6(11):836–848
Dvorak HF, Senger DR, Dvorak AM (1983) Fibrin as a component of the tumor stroma: origins and biological significance. Cancer Metastasis Rev 2(1):41–73
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15:232–239
Edwards JP, Emens LA (2010) The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE2 in murine macrophages. Int Immunopharmacol 10:1220–1228
Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2(3):161–174
el-Sabban ME, Pauli BU (1994–1995) Adhesion-mediated gap junctional communication between lung-metastatic cancer cells and endothelium. Invasion Metastasis 14(1–6):164–176
Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C et al (2004) Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol 24(7):2875–2889
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A et al (2009) Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15(1):35–44
Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL et al (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114(4):844–859
Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D et al (2002) Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res 8(7):2073–2084
Fehm T, Hoffmann O, Aktas B, Becker S, Solomayer EF, Wallwiener D et al (2009) Detection and characterization of circulating tumor cells in blood of primary breast cancer patients by RT-PCR and comparison to status of bone marrow disseminated cells. Breast Cancer Res 11(4):R59
Fehm T, Müller V, Aktas B, Janni W, Schneeweiss A, Stickeler E et al (2010) HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Res Treat 124(2):403–412
Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH et al (2001) Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A 98(4):1853–1858
Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H et al (2008) An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther 7:2725–2735
Ferrari P, Nicolini A (2012) Breast cancer stem cells: new therapeutic approaches. Breast Cancer Manag 1(4):277–294
Ferrari P, Nicolini A, Carpi A (2013) Targeted therapies of metastatic breast cancer: relationships with cancer stem cells. Biomed Pharmacother 67(6):543–555
Fidler IJ (2001) Seed and soil revisited: contribution of the organ microenvironment to cancer metastasis. Surg Oncol Clin N Am 10(2):257–269
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3:453–458
Floor S, van Staveren WC, Larsimont D, Dumont JE, Maenhaut C (2011) Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: distinct, overlapping or same populations. Oncogene 30(46):4609–4621
Fodde R, Brabletz T (2007) Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 19(2):150–158
Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS (2007) Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res 67:3560–3564
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3(5):362–374
Friedrichs K, Franke F, Lisboa BW, Kügler G, Gille I, Terpe HJ et al (1995) CD44 isoforms correlate with cellular differentiation but not with prognosis in human breast cancer. Cancer Res 55(22):5424–5433
Frisch SM, Screaton RA (2001) Anoikis mechanisms. Curr Opin Cell Biol 13(5):555–562
Galsky MD, Vogelzang NJ, Conkling P, Raddad E, Polzer J, Roberson S et al (2014) A phase I trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin Cancer Res 20(13):3581–3588
Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V (2008) Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319(5860):195–198
Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A et al (2012) The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150(4):764–779
Garcia A, Kandel JJ (2012) Notch: a key regulator of tumor angiogenesis and metastasis. Histol Histopathol 27(2):151–156
Gasic GJ (1984) Role of plasma, platelets, and endothelial cells in tumor metastasis. Cancer Metastasis Rev 3(2):99–114
Gassenmaier M, Chen D, Buchner A, Henkel L, Schiemann M, Mack B et al (2013) CXC chemokine receptor 4 is essential for maintenance of renal cell carcinoma-initiating cells and predicts metastasis. Stem Cells 31(8):1467–1476
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4(11):891–899
Germano G, Frapolli R, Simone M, Tavecchio M, Erba E, Pesce S et al (2010) Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res 70(6):2235–2244
Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H et al (2013) The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15(7):807–817
Giaccia AJ, Erler JT (2008) The cellular microenvironment and metastases. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG (eds) Clinical oncology, 4th edn. Churchill Livingstone-Elsevier, Philadelphia, pp 1177–1228 (Chapter 3)
Giancotti FG (2013) Mechanisms governing metastatic dormancy and reactivation. Cell 155(4):750–764
Gil M, Komorowski MP, Seshadri M, Rokita H, McGray AJ, Opyrchal M et al (2014) CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol
Gil-Bernabé AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH et al (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 119(13):3164–3175
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M et al (2010) CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 120(2):485–497
Giordano A, Gao H, Anfossi S, Cohen E, Mego M, Lee BN et al (2012) Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol Cancer Ther 11(11):2526–2534
Goodison S, Kawai K, Hihara J, Jiang P, Yang M, Urquidi V et al (2003) Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin Cancer Res 9(10, Pt 1):3808–3814
Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW et al (1996) Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379(6560):88–91
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC et al (2011) Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71(15):5346–5356
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10(5):593–601
Gross ME, Zorbas MA, Danels YJ, Garcia R, Gallick GE, Olive M et al (1991) Cellular growth response to epidermal growth factor in colon carcinoma cells with an amplified epidermal growth factor receptor derived from a familial adenomatous polyposis patient. Cancer Res 51(5):1452–1459
Guo W, Giancotti FG (2004) Integrin signalling during tumor progression. Nat Rev Mol Cell Biol 5:816
Guo C, Sah JF, Beard L, Willson JK, Markowitz SD, Guda K (2008) The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer 47(11):939–946
Gupta GP, Massagué J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA et al (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening 1. Cell 138:645–659
Hambardzumyan D, Becher OJ, Holland EC (2008) Cancer stem cells and survival pathways. Cell Cycle 7(10):1371–1378
Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y (2000) Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol 7:263–269
Havens AM, Pedersen EA, Shiozawa Y, Ying C, Jung Y, Sun Y et al (2008) An in vivo mouse model for human prostate cancer metastasis. Neoplasia 10(4):371–380
Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN (2009) The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8(20):3274–3284
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M et al (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1(3):313–323
Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R et al (2006) Metastatic patterns in adenocarcinoma. Cancer 106(7):1624–1633
Hill RP, Marie-Egyptienne DT, Hedley DW (2009) Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol 19:106–111
Hiraoka K, Zenmyo M, Watari K, Iguchi H, Fotovati A, Kimura YN et al (2008) Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci 99(8):1595–1602
Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H et al (2002) MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2(4):289–300
Hiratsuka S, Watanabe A, Aburatani H, Maru Y (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8(12):1369–1375
Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K et al (2008) The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 10(11):1349–1355
Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S et al (2009) DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 5:168–177
Hoffman PC, Mauer AM, Vokes EE (2000) Lung cancer. Lancet 355:479–485
Holmgren L, O’Reilly MS, Folkman J (1995) Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1:149–153
Hood JL, San RS, Wickline SA (2011) Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res 71:3792–3801
Hope KJ, Jin L, Dick JE (2004) Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 5(7):738–743
Horst D, Kriegl L, Engel J, Kirchner T, Jung A (2009) Prognostic significance of the cancer stem cell markers CD133, CD44, and CD166 in colorectal cancer. Cancer Invest 27(8):844–850
Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H et al (2010) Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 28(6):1019–1029
Hsu YC, Fuchs E (2012) A family business: stem cell progeny join the niche to regulate homeostasis. Nat Rev Mol Cell Biol 13(2):103–114
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED et al (2007) Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol 213(2):374–383
Iliopoulos D, Hirsch HA, Struhl K (2009) An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139(4):693–706
Imai T, Horiuchi A, Shiozawa T, Osada R, Kikuchi N, Ohira S et al (2004) Elevated expression of E-cadherin and alpha-, beta-, and gamma-catenins in metastatic lesions compared with primary epithelial ovarian carcinomas. Hum Pathol 35(12):1469–1476
Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ et al (2010) Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120(8):2699–2714
Ishikawa T, Nakashiro K, Klosek SK, Goda H, Hara S, Uchida D et al (2009) Hypoxia enhances CXCR4 expression by activating HIF-1 in oral squamous cell carcinoma. Oncol Rep 21(3):707–712
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 12:1167–1174
Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L et al (2009) Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 5:31–42
Jin G, Kawsar HI, Hirsch SA, Zeng C, Jia X, Feng Z et al (2010) An antimicrobial peptide regulates tumor-associated macrophage trafficking via the chemokine receptor CCR2, a model for tumorigenesis. PLoS One 5(6), e10993
Jordan NV, Johnson GL, Abell AN (2011) Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 10(17):2865–2873
Joseph J, Shiozawa Y, Jung Y, Kim JK, Pedersen E, Mishra A et al (2012) Disseminated prostate cancer cells can instruct hematopoietic stem and progenitor cells to regulate bone phenotype. Mol Cancer Res 10:282–292
Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL et al (2010) Progesterone induces adult mammary stem cell expansion. Nature 465(7299):803–807
Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9(4):239–252
Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, Ko YG et al (2013) Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 32(2):209–221
Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F (2008) Involvement of SDF-1alpha/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int J Cancer 122(1):91–99
Kalikin LM, Schneider A, Thakur MA, Fridman Y, Griffin LB, Dunn RL et al (2003) In vivo visualization of metastatic prostate cancer and quantitation of disease progression in immunocompromised mice. Cancer Biol Ther 2(6):656–660
Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S (2011) Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res 13(3):R59
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C et al (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3(6):537–549
Kantara C, O’Connell MR, Luthra G, Gajjar A, Sarkar S, Ullrich RL et al (2015) Methods for detecting circulating cancer stem cells (CCSCs) as a novel approach for diagnosis of colon cancer relapse/metastasis. Lab Invest 95:100–112
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438(7069):820–827
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66(23):11089–11093
Kaplan RN, Psaila B, Lyden D (2007) Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med 13(2):72–81
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441(7092):431–436
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW et al (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449(7162):557–563
Karpatkin S, Pearlstein E (1981) Role of platelets in tumor cell metastases. Ann Intern Med 95(5):636–641
Karpatkin S, Pearlstein E, Ambrogio C, Coller BS (1988) Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest 81(4):1012–1019
Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T (2012) Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res 14(1):R15
Khatib AM, Auguste P, Fallavollita L, Wang N, Samani A, Kontogiannea M et al (2005) Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am J Pathol 167(3):749–759
Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J et al (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16(1):116–122
Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N et al (2010) Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem 285(3):2028–2039
Kim YJ, Borsig L, Varki NM, Varki A (1998) P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A 95(16):9325–9330
Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L et al (2009) Tumor self-seeding by circulating cancer cells. Cell 139(7):1315–1326
Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K et al (2010) CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res 70(24):10411–10421
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N et al (2013) Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23(6):839–852
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120(3):694–705
Klein CA (2009) Parallel progression of primary tumours and metastases. Nat Rev Cancer 9(4):302–312
Klein CA (2011) Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev 21(1):42–49
Knowles HJ, Harris AL (2001) Hypoxia and oxidative stress in breast cancer. Hypoxia and tumourigenesis. Breast Cancer Res 3(5):318–322
Knowles H, Leek R, Harris AL (2004) Macrophage infiltration and angiogenesis in human malignancy. Novartis Found Symp 256:189–200
Kobayashi CI, Suda T (2012) Regulation of reactive oxygen species in stem cells and cancer stem cells. J Cell Physiol 227(2):421–430
Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S et al (2011) Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 208(13):2641–2655
Korkaya H, Liu S, Wicha MS (2011) Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 121(10):3804–3809
Kowalski PJ, Rubin MA, Kleer CG (2003) E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 5(6):R217–R222
Krantz SB, Shields MA, Dangi-Garimella S, Munshi HG, Bentrem DJ (2012) Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J Surg Res 173(1):105–112
Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M et al (2009) MCSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med 206:1089–1102
Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F et al (2011) Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res 167(2):e211–e219
Kurahara H, Takao S, Kuwahata T, Nagai T, Ding Q, Maeda K et al (2012) Clinical significance of folate receptor β-expressing tumor-associated macrophages in pancreatic cancer. Ann Surg Oncol 19(7):2264–2271
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20(5):576–590
Larson CJ, Moreno JG, Pienta KJ, Gross S, Repollet M, O’hara SM et al (2004) Apoptosis of circulating tumor cells in prostate cancer patients. Cytometry A 62(1):46–53
Le QT, Denko NC, Giaccia AJ (2004) Hypoxic gene expression and metastasis. Cancer Metastasis Rev 23(3-4):293–310
Lee JM, Dedhar S, Kalluri R, Thompson EW (2006) The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172(7):973–981
Lee TK, Cheung VC, Ng IO (2013) Liver tumor-initiating cells as a therapeutic target for hepatocellular carcinoma. Cancer Lett 338:101–109
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309
Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66:605–612
Li Z, Rich JN (2010) Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr Top Microbiol Immunol 345:21–30
Li HJ, Reinhardt F, Herschman HR, Weinberg RA (2012) Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov 2(9):840–855
Liang W, Kujawski M, Wu J, Lu J, Herrmann A, Loera S et al (2010) Antitumor activity of targeting SRC kinases in endothelial and myeloid cell compartments of the tumor microenvironment. Clin Cancer Res 16(3):924–935
Lianidou ES, Mavroudis D, Georgoulias V (2013) Clinical challenges in the molecular characterization of circulating tumour cells in breast cancer. Br J Cancer 108(12):2426–2432
Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H et al (2011) Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest 121(7):2736–2749
Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J et al (2010) Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A 107(42):18115–18120
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H et al (2011a) The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 17:211–215
Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F et al (2011b) Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 71(2):614–624
Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I et al (2011) Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 9(5):433–446
Lou H, Dean M (2007) Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene 26(9):1357–1360
Louissaint A Jr, Rao S, Leventhal C, Goldman SA (2002) Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 34(6):945–960
Lowes LE, Hedley BD, Keeney M, Allan AL (2012) User-defined protein marker assay development for characterization of circulating tumor cells using the Cell Search® system. Cytometry A 81(11):983–995
Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M et al (2011) VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 20(6):701–714
Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF et al (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153(3):865–873
Ma J, Lin JY, Alloo A, Wilson BJ, Schatton T, Zhan Q et al (2010) Isolation of tumorigenic circulating melanoma cells. Biochem Biophys Res Commun 402(4):711–717
Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF et al (2011) Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481(7379):85–89
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715
Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF et al (2009) JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther 8(11):3151–3161
Martin SS, Ridgeway AG, Pinkas J, Lu Y, Reginato MJ, Koh EY et al (2004) A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 23(26):4641–4645
Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C et al (2010) Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res Treat 2:317–326
Massard C, Deutsch E, Soria JC (2006) Tumour stem cell-targeted treatment: elimination or differentiation. Ann Oncol 17:1620–1624
Masson V, de la Ballina LR, Munaut C, Wielockx B, Jost M, Maillard C et al (2005) Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J 19(2):234–236
Matsusue R, Kubo H, Hisamori S, Okoshi K, Takagi H, Hida K et al (2009) Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Ann Surg Oncol 16(9):2645–2653
McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS et al (2011) Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res 9:834–844
Medema JP, Vermeulen L (2011) Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 474(7351):318–326
Mego M, Mani SA, Lee BN, Li C, Evans KW, Cohen EN et al (2012a) Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int J Cancer 130(4):808–816
Mego M, Gao H, Lee BN, Cohen EN, Tin S, Giordano A et al (2012b) Prognostic value of EMT-circulating tumor cells in metastatic breast cancer patients undergoing high-dose chemotherapy with autologous hematopoietic stem cell transplantation. J Cancer Educ 3:369–380
Méhes G, Witt A, Kubista E, Ambros PF (2001) Circulating breast cancer cells are frequently apoptotic. Am J Pathol 159(1):17–20
Mehlen P, Puisieux A (2006) Metastasis: a question of life or death. Nat Rev Cancer 6(6):449–458
Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF et al (2004) Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 10(24):8152–8162
Meng Y, Beckett MA, Liang H, Mauceri HJ, Nv R, Cohen KS et al (2010) Blockade of tumor necrosis factor a signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res 70:1534–1543
Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M et al (2005a) Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 115(1):44–55
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD et al (2005b) Genes that mediate breast cancer metastasis to lung. Nature 436(7050):518–524
Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6(1):56–68
Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ et al (2009) The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia 11(11):1235–1242
Moitra K, Lou H, Dean M (2011) Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther 89(4):491–502
Monteiro J, Fodde R (2010) Cancer stemness and metastasis: therapeutic consequences and perspectives. Eur J Cancer 46(7):1198–1203
Moore KA, Lemischka IR (2006) Stem cells and their niches. Science 311(5769):1880–1885
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3(8):e2888
Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ (1988) Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res 48(23):6863–6871
Morrison SJ, Spradling AC (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132(4):598–611
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410(6824):50–56
Müller V, Stahmann N, Riethdorf S, Rau T, Zabel T, Goetz A et al (2005) Circulating tumor cells in breast cancer: correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin Cancer Res 11(10):3678–3685
Mundy GR (2002) Metastasis to bone: causes, consequences therapeutic opportunities. Nat Rev Cancer 2:584–593
Murdoch C, Lewis CE (2005) Macrophage migration and gene expression in response to tumor hypoxia. Int J Cancer 117(5):701–708
Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K et al (2009) Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother 58(10):1577–1586
Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L et al (2007) Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450(7173):1235–1239
Nash GF, Turner LF, Scully MF, Kakkar AK (2002) Platelets and cancer. Lancet Oncol 3(7):425–430
Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM et al (2002) Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res 62(7):2162–2218
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK et al (2009) Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113:6206–6214
Nguyen DX, Massagué J (2007) Genetic determinants of cancer metastasis. Nat Rev Genet 8:341–352
Nguyen DX, Bos PD, Massagué J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9(4):274–284
Nguyen LV, Vanner R, Dirks P, Eaves CJ (2012) Cancer stem cells: an evolving concept. Nat Rev Cancer 12:133–143
Nieto MA (2013) Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342(6159):1234850
Norton L, Massagué J (2006) Is cancer a disease of self-seeding? Nat Med 12(8):875–878
O’Boyle G, Swidenbank I, Marshall H, Barker CE, Armstrong J, White SA et al (2013) Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by AMD11070. Br J Cancer 108(8):1634–1640
O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS et al (2011) VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci U S A 108(38):16002–16007
O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS et al (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88(2):277–285
Oka H, Shiozaki H, Kobayashi K, Inoue M, Tahara H, Kobayashi T et al (1993) Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res 53(7):1696–1701
Oltean S, Sorg BS, Albrecht T, Bonano VI, Brazas RM, Dewhirst MW et al (2006) Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci U S A 141103(38):14116–14121
Oskarsson T, Massagué J (2012) Extracellular matrix players in metastatic niches. EMBO J 31(2):254–256
Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG et al (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17(7):867–874
Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F et al (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15(3):220–231
Pajonk F, Vlashi E, McBride WH (2010) Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells 28(4):639–648
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A et al (2012) RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res 72(11):2879–2888
Pallini R, Ricci-Vitiani L, Banna GL, Signore M, Lombardi D, Todaro M et al (2008) Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res 14(24):8205–8212
Pantel K, Alix-Panabières C (2007) The clinical significance of circulating tumor cells. Nat Clin Pract Oncol 4(2):62–63
Pardal R, Clarke MF, Morrison SJ (2003) Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 3(12):895–902
Park Y, Gerson SL (2005) DNA repair defects in stem cell function and aging. Annu Rev Med 56:495–508
Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A et al (2000) Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 60(3):722–727
Paulus P, Stanley ER, Schafer R, Abraham D, Aharinejad S (2006) Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res 66:4349–4356
Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G et al (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18(6):883–891
Peter ME (2010) Regulating cancer stem cells the miR way. Cell Stem Cell 6(1):4–6
Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G et al (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444:761–765
Piotrowicz RS, Damaj BB, Hachicha M, Incardona F, Howell SB, Finlayson M (2011) A6 peptide activates CD44 adhesive activity, induces FAK and MEK phosphorylation, and inhibits the migration and metastasis of CD44-expressing cells. Mol Cancer Ther 10:2072–2082
Playford MP, Schaller MD (2004) The interplay between Src and integrins in normal and tumor biology. Oncogene 48:7928–7946
Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H (2008) Seeding and propagation of untransformed mouse mammary cells in the lung. Science 321(5897):1841–1844
Potten CS, Owen G, Booth D (2002) Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci 115(11):2381–2388
Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H (2001) Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ 12(3):129–135
Psaila B, Lyden D (2009) The metastatic niche: adapting the foreign soil. Nat Rev Cancer 9(4):285–293
Puig-Kröger A, Sierra-Filardi E, Domínguez-Soto A, Samaniego R, Corcuera MT, Gómez-Aguado F et al (2009) Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res 69(24):9395–9403
Pulaski HL, Spahlinger G, Silva IA, McLean K, Kueck AS, Reynolds RK et al (2009) Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J Transl Med 7:49
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR et al (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475(7355):222–225
Radinsky R (1991) Growth factors and their receptors in metastasis. Semin Cancer Biol 2(3):169–177
Radinsky R, Risin S, Fan D, Dong Z, Bielenberg D, Bucana CD et al (1995) Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin Cancer Res 1(1):19–31
Radisky DC, LaBarge MA (2008) Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2(6):511–512
Raimondi C, Gradilone A, Naso G, Vincenzi B, Petracca A, Nicolazzo C et al (2011) Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res Treat 130(2):449–455
Ramsey DM, McAlpine SR (2013) Halting metastasis through CXCR4 inhibition. Bioorg Med Chem Lett 23(1):20–25
Rappa G, Fodstad O, Lorico A (2008) The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 26:3008–3017
Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D (2010) Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res 316(20):3417–3424
Redjal N, Chan JA, Segal RA, Kung AL (2006) CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin Cancer Res 12(22):6765–6771
Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434(7035):843–850
Reynolds TY, Rockwell S, Glazer PM (1996) Genetic instability induced by the tumor microenvironment. Cancer Res 56(24):5754–5757
Rich JN (2007) Cancer stem cells in radiation resistance. Cancer Res 67(19):8980–8984
Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K et al (2008) Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol 44:823–829
Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ (2009) CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem 284:34342–34354
Rohwedel J, Guan K, Wobus AM (1999) Induction of cellular differentiation by retinoic acid in vitro. Cells Tissues Organs 165:190–202
Roland CL, Dineen SP, Lynn KD, Sullivan LA, Dellinger MT, Sadegh L et al (2009) Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol Cancer Ther 8(7):1761–1771
Ross AA, Cooper BW, Lazarus HM, Mackay W, Moss TJ, Ciobanu N et al (1993) Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 82(9):2605–2610
Rudnick JA, Arendt LM, Klebba I, Hinds JW, Iyer V, Gupta PB et al (2011) Functional heterogeneity of breast fibroblasts is defined by a prostaglandin secretory phenotype that promotes expansion of cancer-stem like cells. PLoS One 6(9), e24605
Ruoslahti E (1994–1995) Fibronectin and its alpha 5 beta 1 integrin receptor in malignancy. Invasion Metastasis 14(1–6):87–97
Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M et al (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469(7330):415–418
Sceneay J, Smyth MJ, Moller A (2013) The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 32:449–464
Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C et al (2005) Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell 8(3):227–239
Schewe DM, Aguirre-Ghiso JA (2009) Inhibition of eIF2α dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res 69:1545–1552
Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ et al (2003) From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A 100(13):7737–7742
Schröder C, Schumacher U, Fogel M, Feuerhake F, Müller V, Wirtz RM et al (2009) Expression and prognostic value of L1-CAM in breast cancer. Oncol Rep 22(5):1109–1117
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985
Seton-Rogers S (2011) Cancer stem cells. VEGF promotes stemness. Nat Rev Cancer 11(12):831
Shao R, Bao S, Bai X, Blanchette C, Anderson RM, Dang T et al (2004) Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol 9:3992–4003
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S et al (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141(1):69–80
Shi X, Zhang Y, Zheng J, Pan J (2012) Reactive oxygen species in cancer stem cells. Antioxid Redox Signal 16(11):1215–1228
Shien K, Toyooka S, Ichimura K, Soh J, Furukawa M, Maki Y et al (2012) Prognostic impact of cancer stem cell-related markers in non-small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer 77(1):162–167
Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J et al (2011) Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 121(4):1298–1312
Shiozawa Y, Nie B, Pienta KJ, Morgan TM, Taichman RS (2013) Cancer stem cells and their role in metastasis. Pharmacol Ther 138(2):285–293
Sica A, Bronte V (2007) Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 117:1155–1166
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG et al (2008) Macrophage polarization in tumour progression. Semin Cancer Biol 18(5):349–355
Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174:636–645
Sleeman JP, Nazarenko I, Thiele W (2011) Do all roads lead to Rome? Routes to metastasis development. Int J Cancer 128(11):2511–2526
Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D et al (2004) CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res 64(23):8604–8612
Sneddon JB (2007) The cancer stem cell niche. Cell Stem Cell 1:607–661
Soeda A, Park M, Lee D, Lee D, Mintz A, Androutsellis-Theotokis A et al (2009) Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28(45):3949–3959
Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86:1065–1073
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14(9):611–622
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12(8):895–904
Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA et al (2010) Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A 107(43):18392–18397
Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ et al (2006) Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 439(7072):95–99
Sun YX, Fang M, Wang J, Cooper CR, Pienta KJ, Taichman RS (2007) Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 67(1):61–73
Sun P, Xia S, Lal B, Eberhart CG, Quinones-Hinojosa A, Maciaczyk J et al (2009) DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells 27(7):1473–1486
Takahashi Y, Mai M (2005) Antibody against vascular endothelial growth factor (VEGF) inhibits angiogenic switch and liver metastasis in orthotopic xenograft model with site-dependent expression of VEGF. J Exp Clin Cancer Res 24(2):237–243
Takebe N, Harris PJ, Warren RQ, Ivy SP (2011) Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 8:97–106
Tanaka K, Hiraiwa N, Hashimoto H, Yamazaki Y, Kusakabe M (2004) Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int J Cancer 108:31–40
Tang X, Mo C, Wang Y, Wei D, Xiao H (2013) Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 138(2):93–104
Tate CR, Rhodes LV, Segar HC, Driver JL, Pounder FN, Burow ME (2012) Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res 14:R79
Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD et al (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451(7175):147–152
Terranova VP, Williams JE, Liotta LA, Martin GR (1984) Modulation of the metastatic activity of melanoma cells by laminin and fibronectin. Science 226(4677):982–985
Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D et al (2010) Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett 288:99–106
Tokes AM, Hortovanyi E, Kulka J, Jackel M, Kerenyi T, Kadar A (1999) Tenascin expression and angiogenesis in breast cancers. Pathol Res Pract 195:821–828
Tonini T, Rossi F, Claudio PP (2003) Molecular basis of angiogenesis and cancer. Oncogene 22:6549–6556
Townson JL, Chambers AF (2006) Dormancy of solitary metastatic cells. Cell Cycle 5(16):1744–1750
Tsagozis P, Eriksson F, Pisa P (2008) Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol Immunother 57:1451–1459
Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV et al (2012) Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21(3):430–446
Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A et al (2008) Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 68(24):10377–10386
Tsuji T, Ibaragi S, Hu GF (2009) Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res 69(18):7135–7139
Tyagi A, Singh RP, Ramasamy K, Raina K, Redente EF, Dwyer-Nield LD et al (2009) Growth inhibition and regression of lung tumors by silibinin: modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev Res 2(1):74–83
Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ et al (2014) Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156(5):1002–1016
Van Ginderachter JA, Movahedi K, Van den Bossche J, De Baetselier P (2008) Macrophages, PPARs, and Cancer. PPAR Res 2008:169414
van Moorselaar RJ, Voest EE (2002) Angiogenesis in prostate cancer: its role in disease progression and possible therapeutic approaches. Mol Cell Endocrinol 197(1-2):239–250
Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans JP et al (2010) Zoledronic acid impairs myeloid differentiation to tumour associated macrophages in mesothelioma. Br J Cancer 103:629–641
Vermeulen L, de Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH et al (2010) Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 12(5):468–476
Vincan E, Brabletz T, Faux MC, Ramsay RG (2007) A human three-dimensional cell line model allows the study of dynamic and reversible epithelial-mesenchymal and mesenchymal-epithelial transition that underpins colorectal carcinogenesis. Cells Tissues Organs 185(1-3):20–28
Vishvakarma NK, Singh SM (2010) Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: implication in antitumor activation of tumor-associated macrophages. Immunol Lett 134:83–92
Von Holst A (2008) Tenascin C in stem cell niches: redundant, permissive or instructive? Cells Tissues Organs 188(1-2):170–177
Wang H, Fu W, Im JH, Zhou Z, Santoro SA, Iyer V et al (2004) Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol 164(6):935–941
Wang Q, Chen ZG, Du CZ, Wang HW, Yan L, Gu J (2009) Cancer stem cell marker CD133+ tumour cells and clinical outcome in rectal cancer. Histopathology 55(3):284–293
Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F et al (2011) TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest 121(5):1969–1973
Wels J, Kaplan RN, Rafii S, Lyden D (2008) Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev 22(5):559–574
Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT et al (2004) Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 10:145–147
Wu KJ (2011) Direct activation of Bmi1 by Twist1: implications in cancer stemness, epithelial-mesenchymal transition, and clinical significance. Chang Gung Med J 34(3):229–238
Wu Y, Wu PY (2009) CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev 18:1127–1134
Wu KJ, Yang MH (2011) Epithelial-mesenchymal transition and cancer stemness: the Twist1-Bmi1 connection. Biosci Rep 31(6):449–455
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H (2009) Sunitinib inhibition of STAT3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 69:2506–2513
Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B et al (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467(7319):1114–1117
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117(7):927–939
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ et al (2008) Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 10(3):295–305
Yao D, Dai C, Peng S (2011) Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res 9(12):1608–1620
Yates CC, Shepard CR, Stolz DB, Wells A (2007) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96(8):1246–1252
Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R et al (1999) TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 103(2):197–206
Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT et al (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339(6119):580–584
Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14(11):736–746
Zarrilli R, Bruni CB, Riccio A (1994) Multiple levels of control of insulin-like growth factor gene expression. Mol Cell Endocrinol 101(1-2):R1–R14
Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AHM, Ballmer-Hofer K, Schwendener RA (2006) Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer 95:272–281
Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E et al (2006) Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther 5(12):3113–3121
Zhan M, Zhao H, Han ZC (2004) Signalling mechanisms of anoikis. Histol Histopathol 19(3):973–983
Zhang JW, Niu C, Ye L, Huang HY, He X, Tong WG et al (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425:836–841
Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M et al (2009) Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16(1):67–78
Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX et al (2010) Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 16(13):3420–3430
Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E et al (2013) Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154(5):1060–1073
Zhao P, Li Y, Lu Y (2010) Aberrant expression of CD133 protein correlates with Ki-67 expression and is a prognostic marker in gastric adenocarcinoma. BMC Cancer 10:218
Zhu J, Emerson SG (2004) A new bone to pick: osteoblasts and the haematopoietic stem-cell niche. Bioessays 26(6):595–599
Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ et al (2008) High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol 26(16):2707–2716
Zhu X, Fujita M, Snyder L, Okada H (2010) Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol 104:83–92
Zlotnik A, Burkhardt AM, Homey B (2011) Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol 11(9):597–606
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ferrari, P., Nicolini, A. (2015). Cellular Plasticity, Cancer Stem Cells and Metastasis. In: Babashah, S. (eds) Cancer Stem Cells: Emerging Concepts and Future Perspectives in Translational Oncology. Springer, Cham. https://doi.org/10.1007/978-3-319-21030-8_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-21030-8_2
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21029-2
Online ISBN: 978-3-319-21030-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)