
Abstract
It is increasingly apparent that cancer stem cells (CSCs) play a substantial role in the response of human cancers to therapy. Indeed, the failure of mainstream chemotherapies to reduce the CSC burden may explain the high rates of tumor recurrence and metastasis. The development of new, anti-CSC agents is thus of great importance to reduce cancer-related mortality. One strategy to target CSCs focuses on their dependence on cell-signaling pathways, which differ from the majority of the tumor cells; these pathways include the embryonic Notch, Wingless-related (Wnt), and Hedgehog (Hh) pathways. Recently, there has been a surge in the development and clinical evaluation of targeted anti-Notch, anti-Wnt, and anti-Hh agents. Herein, we discuss the signaling paradigm for each of these pathways, identify druggable targets, and discuss selected pre-clinical and clinical findings with agents targeting each pathway. A number of natural molecules have shown some efficacy in inhibiting these stemness pathways. Importantly, we consider other disease-specific targeted agents to discuss roadblocks to the success of these anti-stemness agents – including financial considerations, the development of resistance, and on-target adverse effects. Novel clinical trial elements are required to adequately assess the success of these agents; however, the future for anti-CSC therapy is promising.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cancer stem cells
- Stemness pathway s
- Notch signaling
- Wnt signaling
- Hedgehog signaling
- Druggable target s
- Targeted therapy
1 Introduction
1.1 Cancer Stem Cells and Stemness Pathways
There is mounting evidence that, regardless of the cell-of-origin, the dysregulated proliferation and differentiation observed in many cancer types represents a return to an earlier developmental stage. The dependence of cancer cells and cancer stem cells (CSCs ) in particular, on self-renewal and multipotency make them reliant on a select few signaling pathways governing these characteristics. Indeed, the difference between cancerous and normal tissues has been characterized as dependent on the loss of stem-cell regulated homeostatic mechanisms which contribute to the maintenance of normal cell numbers (Tan et al. 2006). We will briefly discuss the reliance of CSCs on Notch, Wingless-related (Wnt), and Hedgehog (Hh) signaling before discussing drug targets to modulate these pathways.
1.2 Signaling Paradigm
A few pathways govern the development of entire organisms, including Notch, Wnt, Hh, receptor tyrosine kinase (RTK), Janus kinase/signal transducer and activator of transcription (Jak/STAT), and transforming growth factor beta (TGF-β ) pathways. As a result, they must be highly specific and well organized. Barolo and Posakony (2002) identified important characteristics which define the signaling paradigm of these developmental pathways. First, these select pathways must be able to activate different or overlapping subsets of genes in various contexts. To facilitate this, pathways demonstrate activator insufficiency. Activation of the pathway is insufficient to activate transcription of all target genes with the same response element. This can be mediated by active repression of target genes in inappropriate signaling contexts. This requires the presence of cis-regulatory elements which bind repressors or additional activators. Alterations often exist in negative regulators of these signaling pathways in various types of cancer (Pece et al. 2004; Westhoff et al. 2009). Second, developmental pathways require the cooperation of tissue-specific or cell-type-specific activators (Barolo and Posakony 2002). Binding sites for these local activators are often located near the signal-activated promoters and are signal-independent. For example, transcription activation in the Notch pathway requires the “CBF-1, Suppressor of Hairless, Lag-2” (CSL) complex and the mastermind-like proteins (MAML1-3 in humans). An alternatively spliced form of CSL (CSL-TREX) was identified in acute myeloid leukemia (AML) and was associated with improved outcomes (Mansour et al. 2008). Alterations in the co-activator MAML have been identified in mucoepidermoid carcinomas via a chromosomal translocation disrupting the Notch pathway (Tonon et al. 2003). In human-papillomavirus (HPV)-induced cervical cancer, preliminary data has suggested that the E6 protein interacts with and interferes with MAML as a transcriptional co-activator in Notch signaling . This provides a possible mechanism for the inhibition of epithelial differentiation in HPV-induced cervical cancer (Wu and Griffin 2004).
The final characteristic identified by Barolo and Posakony is default repression (Barolo and Posakony 2002). In the absence of signaling through these developmental pathways, transcription is repressed. Each pathway has unique DNA-binding co-repressors; however, they often share non-DNA-binding co-repressors [such as the silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) and nuclear receptor corepressor (N-Cor)]. A number of alterations in co-repressors have been described in various cancer types (Bosserhoff et al. 2001; Sheng et al. 2004; Tostar et al. 2005; Fernández-Majada et al. 2007; Scales and de Sauvage 2009; Phelps et al. 2009), suggesting that these co-repressors play a not-insignificant role in modulating the self-renewal and cell-fate decisions of malignant cells. The signaling paradigm described by Barolo and Posakony (2002) is important to understand how alterations in developmental signaling pathways contribute to the pathogenesis of cancer. Additionally, the three characteristics they have identified contribute to the selection of appropriate targets in the pharmacological modulation of signaling pathways.
2 Targeting Stem Cell Signaling Pathways
2.1 Identifying Druggable Targets in Signaling Pathways
The convoluted nature and extensive cross-talk between the Wnt, Hh and Notch pathways makes identifying appropriate druggable targets difficult. Gashaw et al. of Bayer Health set out a list of five characteristics to define actionable drug targets (Gashaw et al. 2011). These include ensuring that: (1) target has a role in disease; (2) the target is disease-specific; (3) the target is not uniformly expressed throughout the body; (4) there is a target- or disease-specific biomarker to monitor efficacy; and (5) prediction of side effects is minimal. Finally, targets are more favorable for drug development if they, or corresponding biomarkers, are easily assayed.
The stem cell signaling pathways culminate in transcriptional responses, often characterized by the transcriptional activation of target genes. Targeting these transcriptional responses can be difficult as drugs must pass through the nuclear membrane, and only small molecules which can diffuse through the membrane, or proteins which can be chaperoned, will enter the nucleus (Lusk et al. 2007). The transcriptional co-factors involved in these responses also have convoluted structures and lack deep binding sites for ideal drug targeting (Grivas and Papavassiliou 2012). Targeting upstream segments of these signaling pathways, such as ligand:receptor interactions or kinases usually lack sufficient specificity. The potential of these targets is further limited by the redundancy between pathways and general cross-talk.
In many cases, targeting stem cell signaling pathways will not be disease-specific, which leads to a number of on-target side effects. These adverse effects are sometimes dose-limiting and have led to the pursuit of alternate druggable targets. One potential solution to this issue is the use of naturally-occurring molecules, as discussed in Sect. 2.2.
2.2 Targeted Molecules or Naturally Occurring Molecules?
Several important issues should be reflected upon when considering the costs and benefits of targeted therapies compared to naturally occurring molecules. The cost of targeted therapy development is often astronomical when considering the number of patients who will benefit (Kantarjian et al. 2013). Many of these drugs are tested in cancer patients who have exhausted all other means of treatment, resulting in minimal benefits to overall survival.
Targeted therapies will always be of benefit to cancers which display consistent and widespread oncogene addiction (such as Her2-amplified breast cancers and MET -overexpressing liver tumors). Gleevec (imatinib), the tyrosine kinase inhibitor, is one of the major successes of targeted therapy development and is used to treat chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors. However, many drugs under development are beginning to focus on smaller and smaller subsets of patients, and many have idealized this narrowing focus as the future of personalized medicine. At an average cost of $1 billion USD for FDA-approved clinical drugs (Goozner 2004), it will confer an enormous, perhaps unsustainable, burden to those patients who are being targeted and their health insurance providers.
Since 2007, at least 12 natural products or derivatives have been approved for cancer therapy (Basmadjian et al. 2014). This is an indication of the reemergence of naturally occurring molecules in the pharmaceutical field. It is important to consider why natural molecules have been historically successful as anti-cancer therapeutics (e.g. etoposide, campothecin, paclitaxel, and rapamycin). Natural molecules have been described to occupy a different “area” of biochemical space than synthetic compounds (Ganesan 2008). They are subject to different restrictions in structure and are made up of different building blocks than synthetic molecules. The structural complexity of these molecules contributes to their specific interactions with targets, decreasing the possibility of dose-limiting side effects (Basmadjian et al. 2014). Notably, as the evolutionary purpose of these natural molecules is not as disease-modifying drugs, iterative alterations to their structures can improve their profile as pharmaceutical agents, such as the semi-synthetic paclitaxel analog, docetaxel (Ganesan 2008).
Drug development in the area of embryonic signaling pathways provides an opportunity to look at the benefits of both targeted therapies and natural molecules. Importantly, many cancers display aberrant signaling through the Notch, Wnt and Hh pathways; this suggests a possible benefit to many patients via treatment with signaling antagonists. A variety of targeted agents have been developed to each of these pathways, and are discussed in the following sections. Additionally, many existing medicinal agents (such as non-steroidal anti-inflammatory drugs) and natural molecules (such as resveratrol and curcumin) have been investigated for their modulation of Notch, Wnt, or Hh signaling. These agents will also be discussed.
3 Notch Signaling Pathway
3.1 The Notch Pathway and Druggable Targets
The Notch pathway is an intercellular communication pathway which is highly conserved among multicellular organisms (Egan et al. 1997). Notch facilitates the maintenance of an undifferentiated state in stem cells, participates in cell fate decisions, and can induce terminal differentiation.
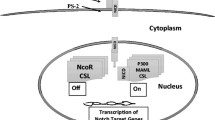
The four Notch receptors (NOTCH1-4) are single-pass transmembrane proteins; the extracellular portion interacts with Delta-like ligands (DLLs) or Jagged ligands (JAGs) on nearby cells (Fig. 15.1). Upon receiving a signal via DLL or JAG, tumor necrosis factor-alpha-converting enzyme (TACE) or another ADAM protease (that containing a disintegrin and a metalloprotease domain) cleaves the extracellular domain. This allows recognition of the Notch intracellular domain (NICD) by the y-secretase complex. The γ-secretase complex, consisting of nicastrin (NCSTN), presenilin (PSEN), presenilin enhancer 2 (PEN2), and anterior pharynx-defective 1 (APH1), releases the NICD from the transmembrane portion of the protein. NICD translocates to the nucleus, where it binds with the CSL complex to release co-repressors and recruit MAML and other co-activators. This activates transcription of Notch target genes, such as the Hes and Hey families of transcription factors.

Notch signaling results in transcriptional activation at target genes. (a) In its inactive state, DLL or Jagged ligands on signaling cells undergo endocytosis and degradation which is mediated by Neuralized (NEURL) and Mindbomb (MIB) ubiquitin ligases. Signaling from Notch intracellular domain (NICD) is inhibited by Numb and Deltex, and Notch-target genes are repressed by a combination of histone deacetylases (HDAC), other co-repressors (coR) and the CSL complex. (b) When Notch ligands bind to the Notch receptor, Notch undergoes a conformational change allowing cleavage of the extracellular domain by ADAM/TACE and subsequent cleavage of NICD by the γ-secretase complex. Following release, NICD translocates to the nucleus where it activates transcription in cooperation with Mastermind-like (MAML) and other co-activators (coA).
The role of Notch signaling in oncogenesis is most clearly illustrated by T-cell acute lymphoblastic leukemia/lymphoma (T-ALL). Initially, Notch signaling was implicated in approximately 1 % of T-ALLs via the t(7;9)(q34;q34.3) chromosomal translocation. This translocation fuses the intracellular domain of Notch1 to the TCRβ promoter/enhancer, coupling T-cell development to constitutively activated Notch signaling (Reynolds et al. 1987). Two additional activating mutations were identified in Notch1, which occur in up to 60 % of T-ALL patients. The first of these leads to ligand-independent metalloproteases (ADAM/TACE) cleavage and release of the intracellular domain. The second stabilizes the intracellular domain and prevents its degradation.
While Notch-activating mutations are frequent in T-ALL, they have not been observed in other solid cancer types; this indicates that ligand-dependent activation predominates in activating aberrant Notch signaling (Roy et al. 2006). This activation of Notch signaling can be oncogenic in many contexts, resulting in increased invasion, migration, and proliferation. Oncogenic Notch signaling has been described in breast cancer, pancreatic cancer, glioblastoma, colon cancer, lymphoma and multiple myeloma (Stylianou et al. 2006; Wang et al. 2009; Li et al. 2011; Ylivinkka et al. 2013; Dai et al. 2014). Interestingly, there may be a specific role for Notch signaling in chemotherapeutic resistance and hypoxia-induced epithelial-to-mesenchymal transition (EMT ) (Sahlgren et al. 2008; Wang et al. 2009).
Despite the multitude of evidence regarding the oncogenic role of Notch signaling , a number of groups have identified Notch as a tumor suppressor in several models (Sriuranpong et al. 2001; Nicolas et al. 2003; Proweller et al. 2006). Interestingly, Notch has been described as a tumor suppressor within the hematopoietic system, suggesting that the role of Notch is context specific, even within the hematopoietic system (Klinakis et al. 2011).
Identifying druggable targets in the Notch pathway is best done sequentially from extra-cellular-ligand binding through to activation of transcription at target genes (Fig. 15.1). First, preventing ligand:receptor interactions involves targeting the Notch receptor or the JAG/DLL ligands. Next, release of NICD, the intracellular molecule required for signaling activation, involves cleavage by ADAM/TACE and γ-secretase enzymes. Finally, transcription of target genes requires the CSL complex and MAML. A number of agents directed at these targets have been developed, and are in various stages of pre-clinical and clinical evaluation (Fig. 15.2). The most advanced agents are γ-secretase inhibitors, owing to overlap between Alzheimer’s drug discovery and cancer therapy.

Clinical trial s of targeted anti-Notch agents have shown varying degrees of efficacy. A number of anti-DLL4 antibodies (e.g. demcizumab) and anti-Notch antibodies (e.g. OMP-59R5) have demonstrated promise in numerous cancer types. While GSIs are the most advanced in clinical development (e.g. RO4929091), they have not been as successful as those therapeutics inhibiting the Notch:ligand interaction.
3.2 Targeted Anti-notch Agents
3.2.1 DLL4 Monoclonal Antibodies
DLL4 is a Notch ligand which is also important for tumor angiogenesis. It is expressed by the tumor vasculature, and not often by the tumor cells. The expression of DLL4 in the vessels supplying the tumor seems to be regulated by VEGF , and expression levels of both DLL4 and VEGF correlate in tumors. The expression of DLL4 is low in the vasculature in normal tissues (Mailhos et al. 2002; Patel et al. 2006; Li et al. 2007; Jubb et al. 2009). Inhibition of DLL4-Notch signaling has led to increased vasculature; however, this is in general non-productive. This is due to hypersprouting of immature vessels, which are not able to perfuse the tissue efficiently (Thurston et al. 2007; Kuhnert et al. 2011). In fact, this non-productive angiogenesis inhibits tumor growth (Noguera-Troise et al. 2006). While DLL4 has a function in angiogenesis, DLL4-Notch signaling also plays an important role in CSC maintenance. Inhibition of DLL4 reduced CSC populations (Hoey et al. 2009). In colon cancer, inhibition of DLL4 leads to more differentiated colon cells (Hoey et al. 2009). However, targeting DLL4 is not without safety concerns. A study of chronic anti-DLL4 therapy identified changes in the livers of mice, rats, and cynomolgus monkeys; as well, skin lesions with features of vascular neoplasms were identified (Yan et al. 2010).
3.2.1.1 Demcizumab
In 2014, FDA granted Orphan Drug status for demcizumab (OMP-21M18, Fig. 15.2) in the treatment of pancreatic cancer. Early preclinical studies demonstrated that demcizumab inhibited expression of Notch target genes (Hoey et al. 2009). In combination with irinotecan, demcizumab decreased tumor growth and CSC frequency in a colorectal tumor model. A similar effect was seen when paclitaxel was combined with demcizumab in a breast tumor xenograft (Hoey et al. 2009). Preclinical studies in ovarian cancer xenografts demonstrated that demcizumab inhibited tumor growth and reduced CSC frequency (Yen et al. 2012). Treatment of pancreatic tumor xenografts with demcizumab also demonstrated the anti-tumor effects; interestingly, these effects were stronger when both human and mouse DLL4 were targeted (Yen et al. 2012). The most dangerous side effect observed in clinical studies (phase I) of demcizumab has been grade III asymptomatic hypertension in 28 % of patients. If anti-DLL4 treatment is to be combined with anti-VEGF therapy, patients must be carefully monitored (Ranpura et al. 2010; Twardowski et al. 2010).
3.2.1.2 Enoticumab (REGN421)
Enoticumab, a monoclonal anti-DLL4 antibody, is in phase I of development for advanced malignancies, led by Regeneron and Sanofi (Fig. 15.2). Preclinical treatment of ovarian tumor xenografts demonstrated an inhibition of tumor growth; accompanied by an increase in tumor vascularization but reduced tumor perfusion (Kuhnert et al. 2013). These effects are consistent with those of other anti-DLL4 treatments. In a phase I study of patients with advanced solid tumors, several patients demonstrated prolonged stable disease or partial response (Jimeno et al. 2013a).
3.2.1.3 MEDI0639
The monoclonal antibody, MEDI0639 was identified by AstraZeneca as a specific, anti-DLL4 modulator of Notch signaling (Jenkins et al. 2012). Results of a safety study in cynomolgus monkeys identified a starting dose for a first-in-human phase 1 clinical trial; however, serious adverse events included reversible effects associated with gastrointestinal bleeding and heart failure (Ryan et al. 2013).
3.2.2 Notch-Targeted Antibodies
3.2.2.1 OMP-59R5 (Tarextumab)
Led by OncoMed Pharmaceuticals and GlaxoSmithKline, OMP-59R5 is an anti-Notch2/3 antibody in clinical testing (Fig. 15.2). Limited results are available from clinical studies. Phase I trials revealed dosages which were well-tolerated, and preliminary evidence of efficacy was observed (Spigel et al. 2014). Phase Ib and phase II proof-of-concept trials are ongoing in pancreatic cancer (with Abraxane® and gemcitabine) and in small cell lung cancer (with cisplatin and etopside).
3.2.2.2 OMP-52M51
OMP-52M51 is a humanized monoclonal anti-NOTCH1 antibody developed by OncoMed Pharmaceuticals (Fig. 15.2). Preclinical testing of OMP-52M51 in T-ALL demonstrated delayed tumorigenicity in samples from poor responders or relapsed patients (Agnusdei et al. 2013), and decreased CSC frequency in a xenograft model of breast cancer (Cancilla et al. 2013). Phase 1 single-agent trials are ongoing in hematologic and solid malignancies where NOTCH1 activation is implicated. Preliminary data from those with solid tumors demonstrates treatment was well tolerated (Davis et al. 2013).
3.2.3 γ-Secretase Inhibitors
Inhibitors of the γ-secretase complex, or GSIs, were initially developed to target the cleavage of the amyloid beta-protein precursor (AβPP) in Alzheimer’s disease. Cleavage of AβPP by β- and γ-secretases generate the amyloid beta-peptide (Aβ) implicated in Alzheimer’s disease. Treatment with GSIs in Alzheimer’s clinical trials identified a number of significant and serious side effects which have been attributed to the role of γ-secretases in Notch signaling throughout the body. These include an effect on the thymus, spleen and intestines (Wong et al. 2004; van Es et al. 2005; Demehri et al. 2009). A number of pre-clinical and clinical trials identified dose-limiting gastrointestinal side effects (Milano et al. 2004; van Es et al. 2005); however, combining GSIs with steroids, such as glucocorticoid or dexamethasone, has contributed to a decrease in these side effects (Real et al. 2008). These ‘off-target’ effects in the treatment of Alzheimer’s disease lead to the investigation of these as ‘on-target’ effects in cancer therapy. Alarmingly, however, treatment with GSIs may increase the risk of skin cancer (Xia et al. 2001; Li et al. 2007; Demehri et al. 2009), suggesting that further characterization of patient tumors is required to determine the contexts in which Notch signaling is oncogenic or tumor-suppressive. A number of theoretical risks have also been suggested when considering GSIs as a cancer therapeutic, including damage to normal stem cells leading to goblet cell metaplasia (Searfoss et al. 2003; Wong et al. 2004). Drug discovery for Alzheimer’s disease now focuses on modulators of γ-secretase activity, or Notch-sparing inhibitors; thus, there is no longer significant overlap between the cancer field and Alzheimer’s field.
3.2.3.1 DAPT
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), is a dipeptide non-transition state analog, specific γ-secretase inhibitor (Dovey et al. 2001). DAPT targets presenilin and prevents γ-secretase activity at a site distinct from the catalytic and substrate binding sites (Morohashi et al. 2006). In vitro, DAPT has been shown to deplete or inhibit CSC populations in nasopharyngeal carcinoma, lung carcinoma, metastatic breast cancer, and ovarian carcinoma (Jiang et al. 2011; McGowan et al. 2011; Yu et al. 2012; Liu et al. 2014). A number of other GSIs were developed from DAPT which are significantly more effective (e.g. RO-4929097, discussed below). It is thus not surprising that there are no clinical studies using DAPT.
3.2.3.2 L-685,458
An aspartyl protease transition state mimic, L-685,458 was identified in 2000 as a AβPP y-secretase inhibitor (Shearman et al. 2000). This GSI is not Notch-sparing and was demonstrated to block the colony forming ability of lymphoma CSCs by inhibiting the Notch pathway (Wang et al. 2011). In addition, inhibition of Notch by L-685,458 inhibited the growth of human tongue squamous cell carcinoma cells, accompanied by cell cycle arrest and apoptosis (Yao et al. 2007). L-685,458 has been observed to inhibit the activity of signal peptide peptidases (SPPs), a family of aspartyl proteases that is closely related to the γ-secretase complex; as such, any observations about the anti-tumor efficacy of L-685,458 cannot be assumed to be γ-secretase dependent (Weihofen et al. 2003).
3.2.3.3 RO4929097
Preclinical profiling of RO4929097 (Hoffman-La Roche, Fig. 15.2) demonstrated it was a very selective and potent inhibitor of γ-secretase activity and inhibited Notch signaling in vitro and in vivo (Luistro et al. 2009). RO4929097 was effective in reducing tumor growth of a number of xenograft models including pediatric models and melanomas; this was accompanied by a decrease in tumor initiating potential of melanoma (Huynh et al. 2011). Preclinical studies suggested intermittent dosing in clinical studies (Luistro et al. 2009). Interestingly, preclinical studies in inflammatory breast cancer (IBC) indicated that RO4929097 sensitized IBC to radiotherapy; however, mammosphere formation efficiency increased, contradicting previous evidence from the melanoma xenograft study (Debeb et al. 2012). Characterization of clinical CSC frequency will be required to determine the effects of RO4929097 on tumorigenicity and CSC number. Data from a phase I study with RO4929097 and cediranib in patients with advanced solid tumors suggested the combination was well tolerated and some evidence of antitumor efficacy was observed (Sahebjam et al. 2013). Similar results were observed by Diaz-Padilla et al. in advanced solid tumors and Tolcher et al. in refractory metastatic or locally advanced solid tumors (Tolcher et al. 2012; Diaz-Padilla et al. 2013). A phase II trial in refractory metastatic colorectal cancer revealed no antitumor efficacy and suggested it not be pursued as a monotherapy for this patient population (Strosberg et al. 2012). A phase II study in previously treated metastatic pancreatic adenocarcinoma was well tolerated and stable disease was achieved in 25 % of patients. Enrollment was halted after development of RO4929097 was discontinued (De Jesus-Acosta et al. 2014). A number of clinical trials with RO4949097 are in progress (Fig. 15.2); however, the majority of these trials are no longer recruiting patients. Ultimately, while RO4929097 may have some synergistic effects with existing chemotherapies, it is unlikely it will achieve success as a single agent.
3.2.3.4 MRK003 and MK0752
Merck and Co., Inc. have developed two sulfonamide-containing non-transition-state analog GSIs, MRK003 and its human analog MK0752 (Fig. 15.2). MRK003 has been tested in pre-clinical settings, and informed the use of MK0752 in clinical trials. In a mouse model of Her2-amplified breast cancer, where tumors contain a larger percentage of CSCs , treatment with MRK003 eliminated CSCs and initiated tumor regression. MRK003 also inhibited the survival and tumor-initiating capabilities of CSCs (Kondratyev et al. 2011). In a xenograft model of pancreatic cancer, MRK003 enhanced the anti-tumor effects of gemcitabine; up-regulation of B-cell receptor signaling and nuclear factor erythroid-derived 2-like 2 pathway correlated with the response of xenografts to the MRK003/gemcitabine regimen (Mizuma et al. 2012). In a patient-derived xenograft of uterine serous carcinoma, MRK003 enhanced the effect of paclitaxel and carboplatin therapy (Groeneweg et al. 2014b). Using platinum-resistant patient-derived xenografts of ovarian cancers, MRK003 in combination with paclitaxel and carboplatin demonstrated anti-tumor effects greater than that of paclitaxel and carboplatin alone (Groeneweg et al. 2014a). Preclinical testing of MRK003 demonstrated a reduction of CSCs in breast cancer tumor xenograft models and an enhanced effect of docetaxel. Although several studies did not observe a strong effect of MRK003 (Watters et al. 2009; Efferson et al. 2010), it is likely that enhanced profiling of those cancers which do benefit will determine a previously-unidentified factor affecting the response of these tumors to MRK003 – and possibly to other GSIs. Clinically, the human analog, MK0752, in combination with docetaxel, resulted in a decrease of CSCs in patient tumors. Preliminary evidence of efficacy was observed, suggesting further clinical trials are warranted (Schott et al. 2013). Results from a phase I trial in pediatric patients with refractory central nervous system (CNS) tumors determined that MK0752 was well tolerated; however, no objective responses were observed. Interestingly, dose-limiting GI symptoms were not observed in this pediatric study (Fouladi et al. 2011). Results from a phase I trial in adult patients with advanced solid tumors suggested a clinical benefit to patients with high-grade gliomas (Krop et al. 2012). The range of effects seen following treatment with MK0752 demonstrates that further stratification of patients is warranted to isolate only those who will benefit.
3.2.3.5 PF-03084014
Pfizer has developed PF-03084014, a selective tetralin amino imidazole GSI (Fig. 15.2). A 2010 pre-clinical study determined that PF-03084014 reduced NICD levels and down-regulated the transcription of Notch target genes. The same study identified a dosing schedule which reduced gastrointestinal toxicity (Wei et al. 2010). In T-cell acute lymphoblastic leukemia (T-ALL), the combination of PF-03084014 with glucocorticoids contributed to a reduction of leukemic burden in a xenograft model (Samon et al. 2012). A pre-clinical study in breast cancer used docetaxel to activate the Notch pathway; subsequent treatment with PF-03084014 reversed these effects and synergistically induced tumor regression in a xenograft model (Zhang et al. 2013a). A combination of PF-03084014 and gemcitabine was effective at inducing tumor regression in a xenograft model of pancreatic ductal adenocarcinoma (PDAC ) (Yabuuchi et al. 2013) and also reduced CSC (CD24−/CD44 + and Aldefluor+) burden. PF-03084014 also demonstrated efficacy in colorectal xenografts with high activation of the Notch and Wnt pathways (Arcaroli et al. 2013); however, demonstrated limited efficacy as a single agent in pediatric xenograft models of solid and T-ALL tumors (Carol et al. 2014). We await the results of ongoing clinical trials to evaluate the efficacy of PF-03084014.
3.2.3.6 MPC-7869
The use of γ-secretase modulators (GSMs), such as MPC-7869 (tarenflurbil, Flurizan™), was intended to reduce the off-target effects of GSIs and minimize their dose-limiting toxicities. GSMs do not affect the rate of enzyme processing or cause a build-up of substrates. MPC-7869 is based on the non-steroidal anti-inflammatory drug (NSAID) scaffold. Ultimately, MPC-7869 did not affect the γ-secretase cleavage of Notch, allowing signal transduction through the Notch pathway (Kukar and Golde 2008). After a double-blind, placebo-controlled clinical trial in prostate cancer failed to meet its efficacy endpoints (NCT00045123), Myriad Genetics Inc. discontinued its development as a cancer therapeutic (Fig. 15.2).
3.2.3.7 Conclusion
Current clinical trials of several GSIs are addressing the toxicity and efficacy of these drugs. Unfortunately, numerous mechanisms of resistance have been identified which will affect the success of GSIs in cancer therapy. One example is PTEN loss, which commonly occurs in T-ALL and contributes to GSI resistance (Palomero et al. 2008). Overexpression of MYC also contributes to GSI resistance (Rao et al. 2009). Cells which are resistant to GSIs demonstrate distinct signaling and transcriptional profiles, which have been attributed to a modified epigenetic status (Knoechel et al. 2014). Other mechanisms for GSI resistance have also been described (Watters et al. 2009; Wang et al. 2011; Miyamoto et al. 2013). Several of these mechanisms may be bypassed if GSIs are included with other classes of agents such as histone deacetylases (HDACs) or proteasome inhibitors, which have enhanced the effects of GSIs in T-All (Sanda et al. 2009). Complete pre-clinical testing is essential to rationalize the use of GSIs in various disease states (Tejada et al. 2014).
3.2.4 Other Agents
3.2.4.1 MAML-Stapled Peptide
MAML proteins are critical coactivators for the transcription of Notch-target genes, and have been implicated in the cross-talk with other signaling pathways such as Wnt/β-catenin (Alves-Guerra et al. 2007). As mentioned earlier, targeting nuclear proteins presents a significant difficulty for drug delivery. A 2006 study identified that a dominant-negative (dn) form of MAML functioned as a pan-Notch inhibitor (Proweller et al. 2006), and further investigations led to the development of a stapled fragment of dnMAML to prevent binding of its full-length, functional counterpart of the CSL complex. This prevents transcriptional activation of Notch-target genes. Preclinical testing of this model in GSI-sensitive T-ALL cell lines reduced the proliferation and leukemia-initiating capabilities of these cells (Moellering et al. 2009).
3.2.4.2 Anti-nicastrin Agents
In a pre-clinical study, silencing of nicastrin (a component of the γ-secretase complex) resulted in a decrease of breast cancer cell motility and invasion. Similar findings were observed with anti-nicastrin antibodies in vitro. The authors suggest that a nicastrin-blocking antibody may be an effective therapy against metastasis of breast cancer (Filipović et al. 2014). Further in vitro testing as well as investigations in clinical settings will determine the efficacy of this strategy in other cancers.
3.3 Conclusion
GSIs remain the most advanced drugs targeting the Notch pathway. While GSIs have been associated with a number of side-effects including dose-limiting gastrointestinal toxicity and an increased risk of skin cancer, it is unclear whether the other Notch-targeting agents will have these same side effects. Further clinical testing will identify the consequences of chronic treatment using anti-DLL4 or anti-Notch antibodies.
4 Wnt Signaling Pathway
4.1 Wnt Signaling and Druggable Targets
The canonical Wnt signaling pathway functions in embryonic development and carcinogenesis by regulation of gene transcription. Wnt signaling is activated by the binding of a WNT ligand to the frizzled (FZD) receptor and low-density lipoprotein receptor-related protein (LRP) 5 or LRP6 on the cell surface. Dishevelled (DVL), adenomatous polyposis coli (APC), and axin are recruited to FZD, where they inhibit the activity of glycogen synthase kinase 3β (GSK3β) (Fig. 15.3). This promotes the stabilization of β-catenin, which enters the nucleus, binds to TCF /LEF transcription factors and activates the transcription of β-catenin target genes (e.g. c-myc, cyclin D, c-Jun, CTLA4). In the absence of WNT ligands, GSK3β phosphorylates β-catenin which leads to its degradation in the proteasome. The T-cell factor /lymphoid enhancer factor (TCF/LEF) transcription factor is bound to Groucho and HDACs, preventing the transcription of target genes.

Wnt signaling is dependent on the accumulation of β-catenin and its translocation to the nucleus. (a) In the absence of the WNT ligand, a “destruction complex” consisting of APC, GSK3β, and axin cooperate to phosphorylate β-catenin. This allows its ubiquitination, mediated by β-TRCP, and leads to proteasomal degradation. Wnt-target genes are inhibited by Groucho and HDAC binding to the TCF /LEF transcription factors. (b) When WNT ligands bind to the Frizzled receptor and LRP5/6, the “destruction complex” is recruited to Disheveled (DSH) at the membrane, inhibiting GSK3β. β-catenin is not phosphorylated and thus accumulates in the cytoplasm. The increasing levels of β-catenin drive it into the nucleus, where it can bind to TCF/LEF and activate transcription of target genes.
Wnt signaling is a major contributor to oncogenesis of colorectal cancers. Mutations in APC and β-catenin frequently occur, leading to constitutive activation of the signaling pathway. In other cancers, dysfunctional Wnt signaling is often a result of irregular activation. Breast CSCs have displayed increased nuclear localization of β-catenin, suggesting highly active Wnt signaling in this population, and a number of agents which inhibit Wnt signaling also selectively inhibit the growth and tumorigenicity of CSCs (Gupta et al. 2009; Khramtsov et al. 2010). Wnt signaling is essential for the initiation of pancreatic cancer, and β-catenin is highly expressed in cisplatin-resistant lung cancer cells (Zhang et al. 2013b, Wang et al. 2014).
Inhibiting Wnt signaling can be done at many levels. First, it may be possible to prevent the secretion of Wnt ligands. Next, the interaction between WNT and FZD or LRP5/6 can prevent activation of downstream signaling. Finally, transcription of Wnt/β-catenin target genes can be prevented by antagonizing the binding of β-catenin to the TCF /LEF transcription factors or the CREB-binding protein (CBP) co-activator.
4.2 Targeted Anti-Wnt Agents
4.2.1 Porcupine Inhibitors
Porcupine (PORCN) is a membrane-bound O-acetyltransferase required for proper Wnt ligand secretion. Blocking Wnt ligand secretion by inhibiting porcupine activity may prevent full activation of the Wnt signaling pathway.
4.2.1.1 LGK974
A small-molecule screen led to the identification of LGK974 as a specific PORCN inhibitor by Liu et al. (Novartis, Fig. 15.4). They demonstrated its efficacy in murine models of Wnt-dependent breast cancer and human head-and-neck squamous cell carcinoma. Additionally, when used in combination with paclitaxel, it inhibited the growth of a human breast tumor xenograft (Liu et al. 2013). The results from an ongoing Phase I clinical trial will inform further use of this agent.

Targeted anti-Wnt agents are early in clinical development. These clinical agents target a range of interactions in Wnt signaling. Molecules targeting WNT secretion (LGK974, PORCN inhibitor) or acting as Frizzled decoy receptors are in early phases of clinical testing, allowing minimal conclusions about their efficacy. PRI724, a CBP-inhibitor which antagonizes transcription of target genes, has a surprisingly low toxicity profile.
4.2.1.2 IWP Compounds
A cell-based synthetic-chemical screen identified several inhibitors of Wnt production (IWPs) as well as a number of inhibitors of Wnt response (IWRs). The IWP compounds, all sharing the same core chemical structure, specifically inhibited PORCN and subsequent secretion of Wnt ligands (Chen et al. 2009a). While IWP-2 has been tested pre-clinically in a number of models (Covey et al. 2012; Mo et al. 2013), its use as a clinical agent has not yet been determined.
4.2.2 Anti-frizzled Molecules
4.2.2.1 FZD8-Fc (Ipafricept)
The decoy receptor, FZD8-Fc (OMP-54F28, Fig. 15.4), consists of an immunoglobulin fragment-crystallizable (Fc) region fused to the cysteine-rich domain of FZD8 by a series of 8 amino acids. The minimal Fzd8 protein contains residues 1–155 and possible protease cleavage sites have been removed (DeAlmeida et al. 2007). This molecule binds Wnt ligands and prevents their signaling through native FZD receptors. Preclinical testing in an MMTV-Wnt1 tumor model as well as teratoma cell lines demonstrated significant anti-tumor activity accompanied by a decrease in expression of WNT-target genes (DeAlmeida et al. 2007). The FDA placed a partial clinical hold on ipafricept for 2 months (July–August 2014) due to observed on-target bone-related adverse events. Amendments have been incorporated into the ongoing Phase Ib clinical trial.
4.2.2.2 OMP-18R5 (Vantictumab)
Preclinical analysis of OMP-18R5, a monoclonal antibody (Fig. 15.4) which binds to five FZD receptors (FZD1, FZD2, FZD5, FZD7, FZD8), revealed anti-tumor effects on a range of tumor types including breast, NSCLC, pancreatic, colon, and teratocarcinoma; a decrease in tumorigenicity lowered to a decrease in CSC frequency (Gurney et al. 2012). Treatment of a mouse model of Kras-dependent pancreatic cancer with OMP-18R5 inhibited Wnt signaling and fewer pancreatic lesions were observed (Zhang et al. 2013b). Samples from patients enrolled in a phase Ia study of OMP-18R5 revealed that Wnt pathway target genes were regulated by vantictumab. There were dose-dependent effects on bone turnover markers (Smith et al. 2013). Increased bone turnover was observed, and more stringent exclusion criteria were developed in combination with prophylactic use of vitamin D and calcium, and use of zoledronic acid if required. Similar to the hold placed on ipafricept, the FDA placed a hold on vantictumab until amendments were made to phase Ib trials.
4.2.3 CREB-Binding Protein Targeted Agents
4.2.3.1 ICG-001
The small molecule ICG-001 binds CREB-binding protein (CBP) to disrupt its interaction with β-catenin and inhibit CBP function as a co-activator of Wnt/β-catenin-mediated transcription; however, its growth-inhibiting effects in PDAC cells were not due to inhibition of β-catenin-mediated transcription. Instead, microarray gene expression analyses implicated the potential disruption of DNA replication and cell cycle progression induced by CBP inhibition. Importantly, treatment prolonged survival of PDAC-bearing mice, indicating the potential for CBP inhibition in PDAC treatment (Arensman et al. 2014).
4.2.3.2 PRI-724
Improvements to the ICG-001 structure led to the development of PRI-724. PRI724 is a specific CBP/beta-catenin antagonist with an extremely low toxicity profile (Fig. 15.4) (El-Khoueiry et al. 2013). This is somewhat surprising as CBP interacts with as many as 500 other cellular entities, including a large number of transcription factors (Lenz and Kahn 2014). Nevertheless, ongoing clinical trials will determine its efficacy as an anti-cancer agent.
4.3 Anti-Wnt Activity of Existing Medicinal Agents
4.3.1 Non-steroidal Anti-inflammatory Drugs
Non-steroidal anti-inflammatory drugs (NSAIDS) exert their anti-inflammatory, analgesic, and antipyretic effects by inhibiting cyclooxygenase (COX)-1 and COX2. An acetic-acid derivative NSAID, sulindac, and the COX2 inhibitor, celecoxib, have been shown to reduce ademonas in patients with familial adenomatous polyposis (FAP) (Huls et al. 2003). Patients with FAP commonly have inactivating mutations in APC, a negative regulator of Wnt signaling (Fig. 15.4). When NSAIDs are used in APC-mutant colorectal cells, Wnt signaling appears to be modulated (Stolfi et al. 2013); however, the precise mechanism of Wnt inhibition by NSAIDs is not fully understood. Some studies attribute the effects of NSAIDs to COX-dependent regulation of prostaglandin E2, which can suppress β-catenin degradation, while other studies have reported COX-independent mechanisms (Castellone et al. 2005; Buchanan and DuBois 2006). Understanding the mechanisms by which NSAIDs regulate Wnt signaling may lead to the derivation of new inhibitors which may have increased effectiveness as anti-cancer agents.
4.3.1.1 Acetaminophen
Wnt signaling is implicated in acetaminophen-induced liver injury (North et al. 2010), suggesting that acetaminophen may be able to modulate Wnt signaling at alternative dosages. Treatment of breast cancer cells in vitro with acetaminophen caused a decrease in β-catenin. The growth of subsequent engraftments of acetaminophen-treated cells was significantly impaired (Takehara et al. 2011).
4.3.1.2 Sulindac and Phosphosulindac
Sulindac binds to the PDZ domain (an interaction domain often found in scaffolding proteins) of DVL and blocks Wnt signaling (Lee et al. 2009). In patients treated with sulindac, nuclear β-catenin expression decreased from pre-treatment levels, suggesting a modulation of Wnt signaling (Boon et al. 2004). Sulindac treatment of colon cancer xenografts inhibited metastasis (Stein et al. 2011). Concomitant with a decrease in β-catenin levels, sulindac treatment inhibited proliferation of colon, lung, breast and prostate cancer cells (Han et al. 2008; Lu et al. 2008; Stein et al. 2011). Phosphosulindac, a safer and more effective derivative of sulindac, has been shown to inhibit the growth of breast and pancreatic cancer xenografts via inhibition of Wnt signaling and EMT in breast CSCs (Mackenzie et al. 2010, 2011; Zhu et al. 2012; Murray et al. 2013).
4.3.1.3 Celecoxib
The COX2 inhibitor, celecoxib, was approved by the FDA in 1999 for the treatment of FAP; however, this approval was withdrawn in 2011 as a decrease in colorectal cancer incidence upon treatment with celecoxib was not demonstrated. Treatment of colorectal cancer cells with celecoxib increases GSK3β kinase activity and phosphorylation of β-catenin. This was accompanied by a reduction of β-catenin/TCF dependent transcription (Sakoguchi-Okada et al. 2007; Tuynman et al. 2008). These effects have been attributed to the prostaglandin-E2 bioactive component of celecoxib (Castellone et al. 2005; Buchanan and DuBois 2006). However, a phase II trial of celecoxib in combination with gemcitabine and cisplatin in pancreatic cancer did not appear to have any benefit over the gemcitabine and cisplatin combination (El-Rayes et al. 2005). Selective targeting of tumors with high activation of Wnt signaling may be required to see any clinical benefit from celecoxib.
4.3.2 Antimicrobials
4.3.2.1 Streptonigrin
An antibiotic with anticancer activity, streptonigrin was investigated as early as 1967 (Smith et al. 1967). Treatment with streptonigrin has been demonstrated to inhibit proliferation of cancer cells with activated β-catenin/Wnt signaling. Streptonigrin treatment decreased nuclear β-catenin and β-catenin/TCF transcriptional activity. It is unclear whether this effect on transcription is a direct activity or whether it is due to suppression of upstream components such as GSK3β (Park and Chun 2011). Interestingly, a natural product screen determined that while streptonigrin was cytotoxic against melanoma cells, it was not effective against a CML cell line. Streptonigrin treatment also left a side-population of slow-cycling putative CSCs unaffected (Sztiller-Sikorska et al. 2014).
4.3.2.2 Salinomycin
The anti-CSC properties of salinomycin, an antibiotic potassium ionophore used in veterinary medicine, were first described in 2009 (Gupta et al. 2009). Salinomycin was isolated from Streptomyces albus in a soil sample from Japan (Naujokat and Steinhart 2012). Salinomycin has been demonstrated to down-regulate Wnt target genes in endometrial cancer cells (Kusunoki et al. 2013). This may be due to inhibition of phosphorylation of LRP6 (Lu et al. 2011a) or by activation of GSK3β and subsequent degradation of β-catenin (Tang et al. 2011; He et al. 2012; Wang et al. 2012). Evidence from breast cancer suggests that salinomycin is 100-fold more efficacious than paclitaxel at reducing the CSC frequency (Gupta et al. 2009). Unfortunately, salinomycin treatment has been associated with severe toxicity; a recent report attributes this to elevated cytosolic sodium levels, which subsequently increase cytosolic calcium levels, activating caspase 9 and 3 to reduce cell viability (Boehmerle and Endres 2011). Evidence from chronic lymphocytic leukemia suggests, however, that the effects of salinomycin on cell viability were specific to leukemic lymphocytes (Lu et al. 2011a). Safety evaluations and further pre-clinical testing will clarify the risk-to-benefit ratio of salinomycin.
4.3.2.3 Nigericin
Another potassium ionophore with a similar structure to that of salinomycin, nigericin, was observed to have anti-CSC characteristics (Gupta et al. 2009; Deng et al. 2013). Evidence has suggested that nigericin can inhibit the Wnt pathway, though the mechanism for this interaction is unclear (Lu et al. 2011a; Zhou et al. 2012).
4.3.2.4 Quinacrine
Wnt signaling can be inhibited by quinacrine, which up-regulates APC. This is followed by a subsequent decrease in activated GSK3β, and increased degradation of β-catenin (Preet et al. 2012). These effects have contributed to an inhibition of growth in breast cancer cells, while sparing normal breast epithelial cells (Preet et al. 2012).
4.3.2.5 Niclosamide
As an anti-helminthic, nicolasmide is used primarily in the treatment of tapeworms. Niclosamide blocks Wnt signaling in cancer cells via LRP6 degradation (Lu et al. 2011b). This induced apoptosis and inhibited proliferation of breast and prostate cancer cells. However, alternate evidence suggests that niclosamide antagonizes upstream Wnt signaling by promoting the endocytosis of FZD1 and down-regulating the DVL2 ligand (Chen et al. 2009b).
4.3.3 Other Agents
4.3.3.1 Tetrandrine
The calcium channel inhibitor, tentrandrine is a bis-benzylisoquinoline alkaloid purified from the root of Stephania tetrandra. In preclinical tests, tetrandrine exhibited better anticancer effects than 5-fluorouracil and carboplatin. In treated tumors, there was a decrease in β-catenin levels, suggesting that the anticancer activity of tetrandrine may be due to a modulation of Wnt signaling (He et al. 2010). The addition of tetrandrine enhanced the effects of cisplatin in cell line and xenograft models (Zhang et al. 2011b). One study suggested that tetrandrine specifically targets CSCs in breast cancer (Xu et al. 2012). In clinical testing, the addition of tetrandrine to a gemcitabine/cisplatin combination regimen in patients with advanced NSCLC improved short-term efficacy (Liu et al. 2012).
4.3.3.2 Trifluoperazine
The antipsychotic, trifluoperazine, inhibited the formation of tumorospheres in lung cancer models, which was accompanied by an inhibition of Wnt signaling. These effects enhanced the activity of gefitinib in animal models of lung cancer (Yeh et al. 2012). A network-based analysis suggests that these effects may also be observed when using other phenothiazine drugs such as chlorpromazine and fluphenazine (Qi and Ding 2013).
4.4 Conclusions
Of the three stemness pathways discussed in this chapter, it is intriguing that Wnt has been the focus of few targeted therapies. Instead, research has primarily focused on the use of natural products or existing medicinal agents in modulating Wnt signaling. It is unclear why this balance is different for Notch (Sect. 3) or Hh (Sect. 5). To date, some of the most successful pre-clinical findings in Wnt inhibition have been derived from natural molecules. While targeted therapies such as anti-FZD antibodies may reach an endpoint in their efficacy, developmental iterations of natural molecules will likely improve their efficacy.
5 Hedgehog Signaling Pathway
5.1 Hedgehog Pathway and Druggable Targets
The Hh signaling pathway functions in embryonic development and carcinogenesis by regulating gene transcription. The binding of a hedgehog ligand (Desert hedgehog DHH, Sonic hedgehog SHH, or Indian hedgehog IHH) to a 12-pass transmembrane patched (PTCH) protein triggers the reversal of suppressor-of-fused (SUFU) inhibition of activating GLI proteins. The GLI proteins are effectors of Hh signaling and enter the nucleus, initiating a transcriptional response with CBP/p300 at Hh target genes (Fig. 15.5).

Hedgehog signaling requires a balance between repressive and activating GLI proteins. (a) Endogenous Patched inhibits Smoothened, preventing its interactions with Sufu. Active Sufu inhibits activating GLI proteins, allowing repressive GLI to bind to Hh-target genes. When HH ligands bind to PTCH, the inhibition of Sufu is relieved, allowing activating GLI proteins to bind to CBP at target genes, activating transcription.
Hh signaling has been unambiguously linked to a particular subtype of medulloblastoma. Hh signaling regulates cerebellar patterning, linking mutations in pathway components such as PTCH or SUFU to the development of malignant brain tumors such as medulloblastoma. Approximately 30 % of medulloblastomas can be characterized by dysregulated Hh signaling (Northcott et al. 2012). Other cancers display activated Hh signaling, though to a lesser extent. For example, breast CSCs have higher expression of PTCH and GLI proteins compared to the non-CSCs (Liu et al. 2006; Shipitsin et al. 2007).
Important druggable interactions in the signaling pathway are the binding of HH ligands to PTCH, the PTCH: SMO interaction, and the GLI-mediated transcriptional response. In some cases, activation of Hh is downstream from SMO and these drug candidates will not be effective (Nagao-Kitamoto et al. 2014). Thus, it is important to target downstream interactions such as GLI-mediated transcription.
5.2 Targeted Anti-Hedgehog Agents
5.2.1 Hedgehog: PTCH Inhibitors
5.2.1.1 5E1
This Hh pathway antagonist has been used in vitro and in vivo to study Hh signaling. 5E1, a monoclonal antibody, blocks binding of the Hh ligands to PTCH. In hepatocellular carcinoma cells with activated Hh signaling, 5E1 decreased expression of Hh target genes, inhibited cell growth and resulted in apoptosis (Huang et al. 2006). Xenograft growth of colorectal cancer cells and pancreatic was significantly decreased upon treatment with 5E1 (Yauch et al. 2008; Bailey et al. 2009). It has not progressed to clinical trials.
5.2.1.2 Robotnikinin
A high-throughput screen of aminoalcohol-derived macrocycles identified robotnikinin as a small molecule which binds the SHH ligand and prevents its interactions with PTCH (Stanton et al. 2009; Peng et al. 2009). A number of analogues were identified in a 2012 publication; however, none of these molecules have progressed to clinical trials (Dockendorff et al. 2012).
5.2.2 Smoothened Inhibitors
5.2.2.1 Cyclopamine
Sheep grazing on corn lily (Veratrum californicum) on a farm in Idaho gave birth to lambs with cyclopia, or one-eyed lambs. Cyclopamine and jervine were finally identified as the teratogenic components of the corn lily. It was not until the 1990s that the defects observed in these lambs were associated with dysregulated Hh signaling (Chiang et al. 1996; Cooper et al. 1998). Cyclopamine is a steroidal jervetraum alkaloid which binds SMO to inhibit Hh signaling (Chen et al. 2002). The mechanism of action of cyclopamine is not fully understood; however, it likely influences the balance between the active and inactive forms of SMO (Taipale et al. 2000; Chen et al. 2002). Cyclopamine, however, exhibits poor solubility, acid sensitivity, and weak potency when compared to other small-molecule antagonists. As such, derivatives of cyclopamine have been identified which have increased bioavailability and are more potent against human cancers (Zhang et al. 2008; Tremblay et al. 2008). One such derivative, IPI-926, is discussed below.
5.2.2.2 GDC-0449 (Vismodegib)
Development of GDC-0449, a small molecule of the 2-arylpyridine class (Genentech Inc. and Curis Inc., Fig. 15.6), was approved in 2012 by the FDA for treatment of metastatic or locally advanced basal cell carcinoma (BCC). Locally advanced BCC includes those patients with post-surgical recurrent tumors, and patients who are not candidates for surgery or radiation. While vismodegib is an important addition to the treatment options for those with locally advanced BCC, phase II evidence leading to the approval of vismodegib for locally advanced BCC consisted of a small number of patients in a single-arm study (Lyons et al. 2014). A 2012 report identified a novel phenomenon of BCC tumor regrowth in or near to the original vismodegib-sensitive tumor bed while therapy is ongoing. The mechanism for this is not clear and may be due to heterogeneity of the original tumor (Chang and Oro 2012). Further evidence and long-term follow-up data will be essentially to fully evaluate the efficacy of vismodegib in BCC and the benefit to patient survival.

Drug development against Hedgehog signaling focuses on Smoothened inhibition. The numerous anti-Hh agents in clinical testing almost exclusively target SMO. Vismodegib (anti-SMO) has already been approved by the FDA for the treatment of basal cell carcinoma. While resistant variants have been described, it appears that vismodegib resistance does not confer resistance to all SMO inhibitors. Pre-clinical testing of downstream targets suggests that the next line of anti-Hh therapeutics will have different modes of action than those already in clinical use.
Vismodegib is also under investigation in tumors of other origins. A phase I study determined that vismodegib was well tolerated in pediatric medulloblastoma patients (Gajjar et al. 2013). A phase II trial in metastatic colorectal cancer identified no benefit from vismodegib, and actually described lower treatment intensity for the other standard-of-care components. The authors suggest that toxicity may have contributed to this decreased efficacy (Berlin et al. 2012). A phase II trial in patients with ovarian cancer in second or third complete remission did not meet expectations for increased progression-free survival (Kaye et al. 2012).
5.2.2.3 BMS-833923
Bristol-Myers Squibb Co. and Exelixis Inc. have developed BMS-833923 (XL-139, Fig. 15.6). Treatment with BMS-833923 inhibited transcription of Hh target genes in esophageal adenocarcinoma cells and induced apoptosis (Zaidi et al. 2013). A phase I study of BMS-833923 demonstrated a partial response in one patient with basal cell nevoid syndrome with a known mutation in PTCH1 (Siu et al. 2009). Treatment was well-tolerated. The results from ongoing clinical trials will define its use as an anti-cancer agent.
5.2.2.4 PF-04449913
The identification of PF-04449913 was described by Munchhof et al. (Munchhof et al. 2011). Treating with PF-04449913 decreased tumorigenicity and leukemia-initiating potential of AML cells (Fukushima et al. 2013). The numerous clinical trials ongoing with PF-04449913 will instruct its future use in various cancer types (Fig 15.6).
5.2.2.5 TAK-441
Takeda Pharmaceutical Company, Ltd. modified a previous molecule to generate TAK-441 with an improved pharmacological profile including increased potency and bioavailability (Ohashi et al. 2012a, b). TAK-441 binds to SMO and blocks Hh signal transduction (Ishii et al. 2013). Preclinical profiling revealed anti-tumor effects in a murine model of medulloblastoma and in castration-resistant prostate xenografts (Ohashi et al. 2012a; Ibuki et al. 2013). It may be possible to use GLI1 mRNA expression (a target of Hh transcriptional response) as a biomarker to predict the effect of TAK-441 in clinical trials (Fig. 15.6) (Kogame et al. 2013).
5.2.2.6 LEQ506
Novartis has led the development of a SMO inhibitor, LEQ506 (Fig. 15.6). When compared to sonidegib (LDE225, another Novartis-lead pharmaceutical), LEQ506 has improved aqueous solubility, increased potency against a mouse model of medulloblastoma, and increased inhibition of GLI-dependent transcription. LEQ506 was effective against a SMO-mutant and vismodegib-resistant cell line. LEQ506, however, has a shorter half-life than sonidegib and requires a higher dosage (Peukert et al. 2013).
5.2.2.7 LY 2940680 (Taladegib)
Taladegib inhibits the Hh pathway by directly binding to Smo (Wang et al. 2013; Bai et al. 2014). This was observed in human xenograft and murine models of medulloblastoma. It was effective against the D473H-mutant cell line which is resistant to vismodegib (Bender et al. 2011).
5.2.2.8 SANT1-4
A small-molecule compound screen identified four molecules (SANT1-4) which modulate SMO activity. SANT1 and SANT2 have been demonstrated to lock SMO into an inactive state, preventing its engagement of downstream Hh signaling (Rohatgi et al. 2009).
5.2.2.9 IPI-926
Developed by Infinity Pharmacetuticals Inc., IPI-926 (saridegib, Fig. 15.6) is a semisynthetic analogue of cyclopamine. Preclinical profiling revealed improved potency and pharmacokinetic profile relative to cyclopamine. IPI926 induced complete tumor regression in a Hh-dependent medulloblastoma allograft model (Tremblay et al. 2009). Treatment prolonged overall survival in a similar model and was active against the D473H point mutation (Lee et al. 2012). In phase I study, IPI-926 was well tolerated and a response was observed in one third of patients (Jimeno et al. 2013b).
5.2.2.10 LDE225
In phase I testing, LDE225 (sonidegib or erismodegib, Fig. 15.6) exhibited activity in advanced basal-cell carcinoma and relapsed medulloblastoma. Side effects were relatively mild, with the exception of elevated serum creatine kinase in 18 % of patients. Reduction of GLI1 mRNA was observed in a dose-dependent manner (Rodon et al. 2014). Further clinical testing will identify if the effects of LDE225 can be translated to other cancer types.
5.2.3 GLI-Mediated Transcription Inhibitors
5.2.3.1 GANT58 and GANT61
GANT (GLI ANTagonist)-58 and GANT61 were identified in a small-molecule screen described by Lauth et al. (2007). GANT58 has a thiophene core with four pyridine rings. Inhibition of GLI-mediated transcription by GANT58 in acute T-cell leukemia showed anti-cancer activity and demonstrated reduced viability of T-ALL cells (Hou et al. 2014). Treatment of prostate cancer xenografts with GANT58 contributed to the development of stable disease in mice; however, GANT61 was more potent in initial testing. The vast majority of pre-clinical studies have thus focused on GANT61. GANT61 is a hexahydropyrimidine derivative shown to inhibit Hh signaling and reduce tumor growth of prostate cancer cells (Lauth et al. 2007). It is suggested that GANT61 alters the conformation of GLI1 and as a result compromises DNA binding of GLI1 (Lauth et al. 2007). Treatment with GANT61 has been effective against Eweing Sarcoma cells, biliary tract carcinoma, lung squamous carcinoma, and PDAC (Xu et al. 2013; Huang et al. 2014; Matsumoto et al. 2014).
5.2.3.2 HPI1 and HPI4
Four HPI (Hedgehog Pathway Inhibitor) molecules were identified in a small-molecule screen conducted by Hyman et al. They describe two of these compounds, HPI1 and HPI4, as modulators of GLI-dependent transcription. Both HPI1 and HPI4 affect the stability and processing of GLI1 and GLI2 (Hyman et al. 2009). Most recently, HPI1 has been packaged in a polymeric nanoparticle (NanoHHI) and shown to inhibit the growth of pancreatic and hepatocellular carcinoma xenografts (Chenna et al. 2011). NanoHHI treatment inhibited the expression of CD133 , which marks a subpopulation of hepatocellular carcinoma CSCs (Xu et al. 2011).
5.2.4 Conclusions
Most of the side-effects of anti-Hh therapy have been mild (Amakye et al. 2013). The agents which have progressed into clinical testing almost exclusively target SMO. While several of them are effective against cancers which are resistant to first-line SMO-inhibitor vismodegib, further resistance will require agents which target other aspects of the pathway.
6 Cross-Talk Between Signaling Pathways
The development of an entire organism through several signaling pathways requires extensive cooperation, or cross-talk, between them. These interactions represent additional layers of complexity in targeting stem cell signaling in cancer, as inhibition of signaling through one pathway may lead to compensation via the remaining pathways.
Crosstalk between stemness pathways has been described and can occur by several mechanisms (Guo and Wang 2008; Javelaud et al. 2012). First, there may be physical interactions between components of two pathways (e.g. Wnt effector, DVL inhibits Notch) (Axelrod et al. 1996). The GLI3 repressor protein can interact with β-catenin and prevent transactivation (Fig. 15.7) (Ulloa et al. 2007).

Stemness pathway s exhibit numerous points of “cross-talk”. A few interactions between the Notch, Wnt, and Hh pathways are depicted here. (a) First, both Sufu and repressive GLI proteins (Hh signaling) can inhibit the activation of transcription by β-catenin (Wnt signaling). (b) Next, transcriptional targets of Hh and Wnt signaling act as ligands for the Notch receptors. (c) Wnt ligands are also transcriptional targets of Hh signaling, suggesting that Hh can activate Wnt signaling. (d) Finally, Dishevelled (DVL) can inhibit the function of NICD. These interactions demonstrate that stem cell signaling is a convoluted network of multiple pathways.
Next, one component may be an enzymatic or transcriptional target of another pathway. Both Hh and Wnt signaling result in transcription of genes which are Notch-receptor ligands. One transcriptional target of Hh signaling is JAG2, while a target of TCF /LEF transcription is JAG1 (Fig. 15.7) (He et al. 2006). Wnt signaling also results in the transcription of the Hh repressor protein, GLI3 (Alvarez-Medina et al. 2007). Alternatively, GLI proteins allow Hh to induce Wnt signaling as the WNT proteins are targets of GLI-mediated transcription (Mullor et al. 2001; Yang et al. 2009). This Hh-induced Wnt signaling has been observed in pancreatic cancer models (Pasca di Magliano et al. 2007).
Finally, one pathway may compete with or modulate a mediator of the other pathway. For example, SUFU can inhibit both activating GLI proteins (Hh signaling) and β-catenin (Wnt signaling). Hh signaling has been reported to up-regulate a Wnt antagonist, secreted frizzled-related protein 1 (SFRP1), resulting in inhibition of Wnt signaling (Fig. 15.7) (He et al. 2006).
A number of publications have identified additive growth suppression when more than one stem-cell pathway is inhibited. For example, simultaneous inhibition of Hh and Notch in leukemia, pancreatic and prostate cancer suggests these pathways cooperate in cancer progression as additive suppressive effects are observed (Ristorcelli and Lombardo 2010; Okuhashi et al. 2011). Similarly, inhibition of the TGF-β and Notch pathway suggests that these pathways cooperate in EMT (Guo and Wang 2008).
7 Molecules with Pan-inhibitory Effects
7.1 Genistein
Genistein (4,5,7-trihydroxyisoflavone) is an isoflavone phytoestrogen, derived from Genista tinctoria. A variety of evidence indicates that genistein can inhibit Notch signaling (Wang et al. 2005; Pan et al. 2012; Dandawate et al. 2013). The precise mechanism is unknown; however, it may be due to miR-34a up-regulation (Xia et al. 2012a). In phase I testing, isoflavone supplementation in prostate cancer patients revealed no toxicity (Miltyk et al. 2003; Takimoto et al. 2003; Fischer et al. 2004). An analog of genistein, phenoxodiol, inhibited breast cancer development in a rat model (Constantinou et al. 2003). Interestingly, it has also been demonstrated to enhance the activity of conventional chemotherapy drugs (Alvero et al. 2006). Further efficacy testing is necessary before any conclusions can be made about the use of genistein or its derivatives in human cancers.
7.2 Curcumin
Curcumin is a diarylheptanoid and a natural phenol. It is the principle curcuminoid of turmeric. It has poor bioavailability as it is insoluble in water. Inhibition of Wnt signaling has been described in osteosarcoma, liver, breast, and colon cancers, resulting in potent growth inhibition (Jaiswal et al. 2002; Prasad et al. 2009; Leow et al. 2009; Kim et al. 2013a). Natural analogs of curcumin down-regulated p300, an essential positive regulator of Wnt signaling (Ryu et al. 2008). Intriguingly, activation of Wnt by curcumin has also been described in neuroblastoma cells and in adipocytes (Ahn et al. 2010; Zhang et al. 2011a), suggesting that further characterization is required to determine in which contexts curcumin can be used to inhibit Wnt signaling. Evidence suggests that curcumin may also modulate Notch signaling by down-regulating Notch1 (Subramaniam et al. 2012; Li et al. 2012). The growth-inhibitory effects observed may be due to crosstalk with the NFκβ pathway (Wang et al. 2006). The preventative effects of curcumin have also been investigated in a phase IIa trial of patients at high risk for developing colorectal cancers. Patients receiving curcumin had a lower number of aberrant crypt foci, suggesting that high-risk patients may benefit from curcumin as a preventative treatment (Carroll et al. 2011). Curcumin has also been observed to inhibit Hh signaling (Elamin et al. 2009; Slusarz et al. 2010; Sun et al. 2013). These pan-inhibitory effects of curcumin make it a particularly appealing natural molecule for cancer therapy. Modifications to the structure of curcumin may increase its bioavailability and potency, thus enhancing its anti-cancer effects.
7.3 Resveratrol
Resveratrol (trans-3,5,4′-trihydroxystilbene) is a natural phenol and a member of the phyoalexin family. It is found in red grapes, wine, nuts, and several plants. A number of its anti-cancer effects have been attributed to inhibition of topoisomerase activity, or its estrogen-antagonizing structure (Bowers et al. 2000; Leone et al. 2012; Basso et al. 2013).
Interestingly, several studies have described activation of Notch signaling by resveratrol in carcinoid, medullary thyroid cancer, and glioblastoma cells, inducing apoptosis (Pinchot et al. 2010; Truong et al. 2010; Lin et al. 2011). A separate study, however, observed resveratrol-mediated inhibition of Notch signaling in T-ALL, which induced apoptosis (Cecchinato et al. 2007). Similar effects were seen in cervical cancer cells; however, selective Notch inhibition did not achieve the same result (Zhang et al. 2014). The authors suggest that concurrent inhibition of Notch, Wnt, and STAT3 signaling resulted in the observed apoptotic effects of resveratrol. Additional studies have demonstrated obstruction of Wnt signaling by resveratrol (Hope et al. 2008; Vanamala et al. 2010). Many of these have focused on colon cancer, likely due to the importance of APC and Wnt signaling in FAP. A 2012 study determined that resveratrol inhibits the formation of the β-catenin/TCF complex, thus modulating transcription initiation at target genes (Chen et al. 2012a). Phase I trials of resveratrol have demonstrated inhibition of Wnt signaling in normal colonic mucosa; and, using a micronized formulation, increased apoptosis of hepatic metastases (Nguyen et al. 2009; Howells et al. 2011). In human trials, the major dose-limiting side effect of resveratrol has been gastrointestinal toxicity (la Porte et al. 2010; Brown et al. 2010). Resveratrol may also inhibit Hh signaling. While the mechanisms range from decreased nuclear translocation of GLI and decreased transcription of target genes to down-regulation of PTCH and SMO, resveratrol has been described to modulate Hh signaling in AML, prostate cancer, and pancreatic cancer (Slusarz et al. 2010; Su et al. 2013; Qin et al. 2014).
A major limiting factor in the clinical use of resveratrol is its poor bioavailability (Walle 2011). While resveratrol is easily absorbed, it is extensively metabolized in the intestine and liver resulting in limited efficacy. The use of methylated derivatives of resveratrol may decrease clearance of resveratrol by increasing metabolic stability and result in improved anti-cancer effects of resveratrol (Walle et al. 2007; Cai et al. 2010).
7.4 Celastrol
Celastrol (tripterene) is a triterpenoid, isolated from the root extracts of Tripterygium wilfordii (Thunder god vine) and Celastrus regelii. It has been described to have anti-oxidant, anti-inflammatory, and anti-cancer activity (Allison et al. 2001). Some of its anti-cancer effects may be a result of its modulation of Notch signaling , as treatment of leukemia cells resulted in a down-regulation of Notch1 (Wang et al. 2010). Interestingly, celastrol has been described to induce apoptosis via the activation of Wnt signaling. In colorectal cancer cells, celastrol increased nuclear beta-catenin levels (Lu et al. 2012).
7.5 Honokiol
Honokiol is a small-molecule polyphenol, isolated from various components of trees belonging to the genus Magnolia. It has been shown to have anti-inflammatory, anti-angiogenic, and anti-cancer properties (Fried and Arbiser 2009). Treatment with honokiol in preclinical models can modulate Wnt signaling, and may have CSC -specific effects. In oral squamous cell carcinoma CSCs , honokiol decreased β-catenin and a down-regulation of downstream targets was observed (Yao et al. 2013). Similar effects were seen in non-small cell lung cancer cells. Antagonism of the Notch pathway has also been observed following honokiol treatment. In a colon cancer model, honokiol sensitized CSCs to ionizing radiation. The expression of components of the γ-secretase complex as well as downstream target genes were reduced (Ponnurangam et al. 2012). The effects of honokiol could be reversed by the addition of NICD, suggesting that Notch signaling is vital for this response. A similar decrease in γ-secretase components was observed when melanoma cells were treated with honokiol (Kaushik et al. 2012).
7.6 Arsenic Trioxide
Arsenic has been used as a medicinal agent for thousands of years. Currently, arsenic trioxide (ATO) is used in combination with all-trans retinoic acid in the treatment of acute promyelocytic leukemia (APL). ATO promotes cellular differentiation, induces apoptosis in malignant and normal cells, and induces an accumulation of reactive oxygen species (Rojewski et al. 2002; List et al. 2003; Park et al. 2005). These effects may be mediated by inhibition of the Notch pathway. In gliomas, treatment with ATO resulted in decreased transcription of Notch-dependent genes. This was accompanied by a depletion of the CSC population (Zhen et al. 2009). Similar results have been observed in breast cancer and glioblastoma (Xia et al. 2012b; Wu et al. 2013). ATO may also antagonize Hh signaling (Raju 2010; Kim et al. 2013b). In a mouse model of Hh-dependent medulloblastoma, ATO treatment improved survival (Beauchamp et al. 2010). It is suggested that ATO binds GLI1 and inhibits its transcriptional activity; however, a separate study observed an ATO-induced reduction of GLI2 (Kim et al. 2010a). It is likely that the effects of ATO on the Hh pathway are mediated by the GLI proteins, and further experimentation will elucidate the precise mechanisms.
8 Conclusion and Future Perspectives
8.1 Roadblocks to Success
8.1.1 Preclinical/Clinical Failures
Drug development for stemness pathways closely follows that for many other targets. The vast majority of therapeutic agents remain in preclinical studies, and a number of agents which show promise in preclinical models fail in clinical trials. These disappointments may be due to any number of differences between preclinical and clinical testing. Cell line models lack the inherent heterogeneity of human cancers, and the use of xenograft models requires immunocompromised hosts. Neither of these popular preclinical paradigms properly recapitulates the complexity of treating patients.
It will be important to require the same success in preclinical models as we require in clinical settings – if clinical success is defined as inducing tumor regression or stable disease, then slowing tumor growth in preclinical tests is insufficient. The interesting concept of co-clinical trials presents an opportunity to hasten the progress of targeted therapies (Nardella et al. 2011; Chen et al. 2012b). In principle, co-clinical trials encompass a genetically-engineered murine model paralleling a human clinical trial. This allows real-time feedback on treatment failures and successes, and simultaneous integration of preclinical and clinical data.
8.1.2 Strategies to Overcome Resistance
This approach to clinical testing of targeted therapies will allow rapid redeployment of alternate therapies when resistance develops. While targeting stemness pathways is a relatively young field of anti-cancer therapy, it is not surprising that resistance to a number of these therapeutic agents has already been described. Indeed, it is most surprising that the emergence of resistance has not altered the strategies being used to target stemness pathways. The success of imatinib (Novartis) in treating BCR-ABL CML was followed quickly by the emergence of resistant variants (Valent 2007). This necessitated the development of second-generation tyrosine kinase inhibitors (dasatinib, nilotinib, and bosutinib) and third-generation ponatinib (Golas et al. 2003; Lombardo et al. 2004; Weisberg et al. 2005). Finally, a novel treatment for CML (omacetaxine), which acts independently of BCR-ABL tyrosine kinase inhibition, was developed; it has shown promise in treating patients who have failed first- and second-generation tyrosine-kinase-inhibitor therapy and was approved by the FDA in 2012 (Pérez-Galán et al. 2007).
The development of anti-SMO therapies to inhibit Hh signaling mimics the BCR-ABL story. Mutations have already been described which confer resistance to the first-line vismodegib (Metcalfe and de Sauvage 2011; Chang and Oro 2012), and while other SMO-antagonists may still be effective, it is likely only a matter of time before resistance to second- and third-line antagonists emerges. It will be essential to hurry the development of therapies which target other aspects of the Hh signaling pathway as the SMO-antagonists move into wider clinical use (Metcalfe and de Sauvage 2011). In the Hh pathway, it may be essential to use a therapeutic such as GANT61 to target GLI-mediated transcription once resistance emerges at the SMO-level (Fig. 15.6) (Matsumoto et al. 2014). Therapeutic agents which target different aspects of the Notch, Wnt, and Hh pathways are in various stages of development – while some classes of drugs, such as the Notch-targeted GSIs or the Hh-targeted SMO antagonists, are further ahead, the emergence of resistance will place a selective pressure on those less-developed agents. Alternatively, resistance to these targeted therapies may be addressed by combining anti-stemness agents with other specific agents. In SMO-antagonist-resistant tumors, this may mean the addition of a PI3K-inhibitor (Kim et al. 2010b, 2013b).
8.1.3 Dealing with On-Target Side Effects
It is important to recognize that even targeted therapies have serious on- and off-target side effects. For example, a number of CML patients treated with imatinib developed congestive heart failure (Kerkelä et al. 2006). This was caused by a build-up of misfolded proteins in the endoplasmic reticulum, activating apoptosis. Inhibiting the BCR-ABL fusion protein also systemically inhibits the function of the ABL tyrosine kinase, leading to imatinib’s particular effects on cardiac function.
The clinical use of GSIs for leukemia patients and those with solid tumors exposed the importance of considering on-target side effects of anti-stemness agents. While the Notch, Wnt, and Hh pathways are vital for embryonic patterning and development, they are also active in many adult stem cell populations. Treatment with GSIs led to gastrointestinal toxicity due to the involvement of Notch in the intestinal tract (Searfoss et al. 2003; Milano et al. 2004; Wei et al. 2010). The resulting dose-limiting goblet cell hyperplasia has curtailed the use of GSIs in clinical settings and modified dosing schedules have been investigated (Krop et al. 2012). The use of steroidal agents in combination with GSIs has also been investigated and seems promising (Real et al. 2008).
Similarly, on-target side effects have been observed in patients treated with Wnt signaling antagonists. Wnt and Hh signaling cooperate extensively in regulating bone turnover; thus, the use of targeted therapies in these pathways has resulted in abnormal bone mass. The FDA halted clinical testing of two anti-Wnt agents (ipafricept and vantictumab) until the on-target bone side effects were addressed. The use of zoledronic acid in these patients appears to mediate these effects.
Targeting stemness pathways will not be without consequence until a tumor-specific delivery platform can be mobilized. Ado-trastuzumab emtansine (Kadcycla or T-DM1, Genentech) consists of the Her2 monoclonal antibody, Herceptin, conjugated to a cyctotoxic agent, mertansine (Verma et al. 2012). Approved by the FDA in 2013 for the treatment of metastatic Her2-positive breast cancer, T-DM1 exhibited a better safety profile and improved efficacy over trastuzumab alone. This is an important harbinger of the potential for tumor-specific delivery.
8.2 Evidence for Success
8.2.1 Immediate Clinical Successes
Several agents discussed in this chapter have already demonstrated clinical success, leading to FDA approval. Vismodegib (a SMO antagonist, Fig. 15.6) is approved for the treatment of locally-advanced or metastatic basal cell carcinoma, and demicizumab (anti-DLL4 agent, Fig. 15.2) received an orphan-drug designation for the treatment of pancreatic cancer. This demonstrates that targeted anti-stemness therapy is an active and successful field of drug development. Other agents in advanced stages of clinical testing, such as the Hh antagonist LDE225, the PF-03084014 GSI, and PRI-724, a CBP inhibitor, demonstrate benefits to patient outcomes.
Just as relevant, however, are those agents which have exhibited little-to-no clinical success. RO4929097, a GSI, has little benefit as a monotherapy, though it may still yet exhibit synergistic effects with conventional chemotherapies or even other targeted anti-Notch agents (Strosberg et al. 2012; De Jesus-Acosta et al. 2014). While some may call this a failure of the drug-development pipeline, it is important to consider how the success of anti-stemness agents is measured.
8.2.2 Measuring Long-Term Effects
It is difficult to evaluate the long-term efficacy of the targeted anti-Notch, Wnt, or Hh therapeutics discussed in this chapter, as many of them are fairly recent developments. An additional factor confounding the assessment of these agents is the rarity of the cell populations they target. CSCs often exhibit high signaling via these pathways when compared to the non-CSC component of the tumor. Importantly, however, the frequency of CSCs in many cancers is less than 1 %. Thus, targeting stemness pathways in human cancers may show little immediate success over conventional chemotherapy, as increased toxicity to 1 % of cells in a tumor is difficult to quantify. From another perspective, however, the hypothesized role for CSCs in cancer recurrence suggests that targeting CSCs may reduce recurrence rate and increase overall survival (Beck and Blanpain 2013).
Clinical testing of these targeted agents should include long-term follow-up as well as a determination of CSC frequency before, during, and after treatment. This data will allow us to determine if the overall efficacy of the agent can be attributed to anti-CSC effects.
8.3 The Future of Targeting Stemness Pathways
With the hypothesized importance of CSCs in tumorigenesis, metastasis, chemotherapy resistance, and recurrence gaining increasing credence (Bonnet and Dick 1997; Singh et al. 2004; Ginestier et al. 2007), there has been a major thrust to identify novel therapies that target CSCs. The intrinsic linkage of stem cell signaling pathways with CSC maintenance and tumorigenicity provides an avenue for therapeutic development and a more thorough study of CSCs in human cancers. The number of pre-clinical investigations and clinical trials examining the potential use of anti-stemness drugs has grown exponentially in recent years. The success of future trials will likely depend on extensive consideration of the cross-talk between stemness pathways. Future therapies may include dual-purpose agents such as the recently-described NL-103, a Hh and HDAC inhibitor (Zhao et al. 2014). Additionally, it is becoming increasingly apparent that the end result of signaling through these stemness pathways depends heavily on the cellular context – signaling may be oncogenic or tumor-suppressive. Even the use of a single agent can activate or inhibit signaling (e.g. resveratrol). The identification of patients who may benefit from these therapies or combinations of anti-stemness therapeutics will necessitate an evaluation of stemness pathway cross-talk in patient tumors. Additionally, altered clinical paradigms should be considered, such as co-clinical trials and outcome measures that incorporate CSC frequency measurements.
Targeted therapies have outpaced natural product research in terms of resources spent by pharmaceutical companies on the development of novel anti-stemness pathway drugs for cancer. We will learn in the coming years if this strategy was effective, or if a new shift in research focus may occur. It has been suggested that natural molecules with novel mechanisms are more likely to be successful than many small molecules targeted at the same interaction (Ganesan 2008). Major advances may come from identifying the targets of natural molecules with proven anti-stemness/cancer activity and utilizing this information to generate semi-synthetic natural compounds with enhanced activity or developing novel strategies for targeted therapy (Pucheault 2007).
References
Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S et al (2013) Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia 28:278–288
Ahn J, Lee H, Kim S, Ha T (2010) Curcumin-induced suppression of adipogenic differentiation is accompanied by activation of Wnt/beta-catenin signaling. Am J Physiol Cell Physiol 298:C1510–C1516
Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C (2001) Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 25:1341–1357
Alvarez-Medina R, Cayuso J, Okubo T, Takada S, Martí E (2007) Wnt canonical pathway restricts graded Shh/Gli patterning activity through the regulation of Gli3 expression. Development 135:237–247
Alvero AB, O’Malley D, Brown D, Kelly G, Garg M, Chen W et al (2006) Molecular mechanism of phenoxodiol-induced apoptosis in ovarian carcinoma cells. Cancer 106:599–608
Alves-Guerra M, Ronchini C, Capobianco AJ (2007) Mastermind-like 1 is a specific coactivator of beta-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res 67:8690–8698
Amakye D, Jagani Z, Dorsch M (2013) Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat Med 19:1410–1422
Arcaroli JJ, Quackenbush KS, Purkey A, Powell RW, Pitts TM, Bagby S et al (2013) Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF-03084014, a γ-secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer 109:667–675
Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, Dawson DW (2014) The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther 13:2303–2314
Axelrod JD, Matsuno K, Artavanis-Tsakonas S, Perrimon N (1996) Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science 271:1826–1832
Bai Q, Shen Y, Jin N, Liu H, Yao X (2014) Molecular modeling study on the dynamical structural features of human smoothened receptor and binding mechanism of antagonist LY2940680 by metadynamics simulation and free energy calculation. Biochim Biophys Acta 1840:2128–2138
Bailey JM, Mohr AM, Hollingsworth MA (2009) Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 28:3513–3525
Barolo S, Posakony JW (2002) Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev 16:1167–1181
Basmadjian C, Zhao Q, Bentouhami E, Djehal A, Nebigil CG, Johnson RA et al (2014) Cancer wars: natural products strike back. Front Chem 2:20
Basso E, Fiore M, Leone S, Degrassi F, Cozzi R (2013) Effects of resveratrol on topoisomerase II-α activity: induction of micronuclei and inhibition of chromosome segregation in CHO-K1 cells. Mutagenesis 28:243–248
Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC et al (2010) Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest 121:148–160
Beck B, Blanpain C (2013) Unravelling cancer stem cell potential. Nat Rev Cancer 13:727–738
Bender MH, Hipskind PA, Capen AR, Cockman M, Credille KM, Gao H et al (2011) Abstract 2819: identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res 71(8):A2819
Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC et al (2012) A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res 19:258–267
Boehmerle W, Endres M (2011) Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis 2, e168
Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737
Boon EMJ, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, Pais ST (2004) Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer 90:224–229
Bosserhoff AK, Echtenacher B, Hein R, Buettner R (2001) Functional role of melanoma inhibitory activity in regulating invasion and metastasis of malignant melanoma cells in vivo. Melanoma Res 11:417–421
Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM (2000) Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 141:3657–3667
Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD et al (2010) Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res 70:9003–9011
Buchanan FG, DuBois RN (2006) Connecting COX-2 and Wnt in cancer. Cancer Cell 9:6–8
Cai H, Sale S, Britton RG, Brown K, Steward WP, Gescher AJ (2010) Pharmacokinetics in mice and metabolism in murine and human liver fractions of the putative cancer chemopreventive agents 3′,4′,5′,5,7-pentamethoxyflavone and tricin (4′,5,7-trihydroxy-3′,5′-dimethoxyflavone). Cancer Chemother Pharmacol 67:255–263
Cancilla B, Cain J, Wang M, Beviglia L, Shah J, Gurney A et al (2013) Abstract 3728: anti-Notch1 antibody (OMP-52M51) impedes tumor growth and cancer stem cell frequency (CSC) in a chemo-refractory breast cancer xenograft model with an activating Notch1 mutation and screening for activated Notch1 across multiple solid tumor types. Cancer Res 73(8):A3728
Carol H, Maris JM, Kang MH, Reynolds CP, Kolb EA, Gorlick R et al (2014) Initial testing (stage 1) of the notch inhibitor PF-03084014, by the pediatric preclinical testing program. Pediatr Blood Cancer 61:1493–1496
Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L et al (2011) Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res 4:354–364
Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS (2005) Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310:1504–1510
Cecchinato V, Chiaramonte R, Nizzardo M, Cristofaro B, Basile A, Sherbet GV, Comi P (2007) Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem Pharmacol 74:1568–1574
Chang ALS, Oro AE (2012) Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Arch Dermatol 148:1324–1325
Chen JK, Taipale J, Cooper MK, Beachy PA (2002) Inhibition of hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 16:2743–2748
Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW et al (2009a) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5:100–107
Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK et al (2009b) The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 48:10267–10274
Chen H, Hsu L, Shia Y, Lin MW, Lin CM (2012a) The β-catenin/TCF complex as a novel target of resveratrol in the Wnt/β-catenin signaling pathway. Biochem Pharmacol 84:1143–1153
Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T et al (2012b) A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483:613–617
Chenna V, Hu C, Pramanik D, Aftab BT, Karikari C, Campbell NR et al (2011) A polymeric nanoparticle encapsulated small-molecule inhibitor of hedgehog signaling (NanoHHI) bypasses secondary mutational resistance to Smoothened antagonists. Mol Cancer Ther 11:165–173
Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383:407–413
Constantinou AI, Mehta R, Husband A (2003) Phenoxodiol, a novel isoflavone derivative, inhibits dimethylbenz[a]anthracene (DMBA)-induced mammary carcinogenesis in female Sprague-Dawley rats. Eur J Cancer 39:1012–1018
Cooper MK, Porter JA, Young KE, Beachy PA (1998) Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280:1603–1607
Covey TM, Kaur S, Ong TT, Proffitt KD, Wu Y, Tan P, Virshup DM (2012) PORCN moonlights in a Wnt-independent pathway that regulates cancer cell proliferation. PLoS One 7, e34532
Dai Y, Wilson G, Huang B, Peng M, Teng G, Zhang D et al (2014) Silencing of Jagged1 inhibits cell growth and invasion in colorectal cancer. Cell Death Dis 5, e1170
Dandawate P, Padhye S, Ahmad A, Sarkar FH (2013) Novel strategies targeting cancer stem cells through phytochemicals and their analogs. Drug Deliv Transl Res 3:165–182
Davis SL, Lorusso P, Xu L, Kapoun AM, Dupont J, Munster P et al (2013) Abstract B48: a first-in-human phase I study of the novel cancer stem cell (CSC) targeting antibody OMP-52M51 (anti-Notch1) administered intravenously to patients with certain advanced solid tumors. Mol Cancer Ther 12:B48
De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A et al (2014) A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs 32:739–745
DeAlmeida VI, Miao L, Ernst JA, Koeppen H, Polakis P, Rubinfeld B (2007) The soluble wnt receptor Frizzled8CRD-hFc inhibits the growth of teratocarcinomas in vivo. Cancer Res 67:5371–5379
Debeb BG, Cohen EN, Boley K, Freiter EM, Li L, Robertson FM et al (2012) Pre-clinical studies of Notch signaling inhibitor RO4929097 in inflammatory breast cancer cells. Breast Cancer Res Treat 134:495–510
Demehri S, Turkoz A, Kopan R (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16:55–66
Deng CC, Liang Y, Wu MS, Fent GT, Hu WR, Chen LZ et al (2013) Nigericin selectively targets cancer stem cells in nasopharyngeal carcinoma. Int J Biochem Cell Biol 45:1997–2006
Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M et al (2013) A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 31:1182–1191
Dockendorff C, Nagiec MM, Weïwer M, Buhrlage S, Ting A, Nag PP et al (2012) Macrocyclic hedgehog pathway inhibitors: optimization of cellular activity and mode of action studies. ACS Med Chem Lett 3:808–813
Dovey HF, John V, Anderson JP, Chen LZ, de Saint AP, Fang LY et al (2001) Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 76:173–181
Efferson CL, Winkelmann CT, Ware C, Sullivan T, Giampaoli S, Tammam J et al (2010) Downregulation of Notch pathway by a gamma-secretase inhibitor attenuates AKT/mammalian target of rapamycin signaling and glucose uptake in an ERBB2 transgenic breast cancer model. Cancer Res 70:2476–2484
Egan SE, St-Pierre B, Leow CC (1997) Notch receptors, partners and regulators: from conserved domains to powerful functions. Curr Top Microbiol Immunol 228:273–324
Elamin MH, Shinwari Z, Hendrayani S, Al-Hindi H, Al-Shail E, Khafaga Y et al (2009) Curcumin inhibits the Sonic hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol Carcinog 49:302–314
El-Khoueiry AB, Ning Y, Yang D, Cole S, Kahn M, Zoghbi M et al (2013) Abstract 2501: a phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol 31:A2501
El-Rayes BF, Zalupski MM, Shields AF, Ferris AM, Vaishampayan U, Heilbrun LK et al (2005) A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs 23:583–590
Fernández-Majada V, Pujadas J, Vilardell F, Capella G, Mayo MW, Bigas A, Espinosa L (2007) Aberrant cytoplasmic localization of N-CoR in colorectal tumors. Cell Cycle 6:1748–1752
Filipović A, Lombardo Y, Fronato M, Abrahams J, Aboagye E, Nguyen QD et al (2014) Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res Treat 148(2):455–462
Fischer L, Mahoney C, Jeffcoat AR, Koch MA, Thomas BE, Valentine JL et al (2004) Clinical characteristics and pharmacokinetics of purified soy isoflavones: multiple-dose administration to men with prostate neoplasia. Nutr Cancer 48:160–170
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M et al (2011) Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 29:3529–3534
Fried LE, Arbiser JL (2009) Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal 11:1139–1148
Fukushima N, Minami Y, Hayakawa F, Kiyoi H, Sadarangani A, Jamieson C, Naoe T (2013) Abstract 1649: treatment with hedgehog inhibitor, PF-04449913, attenuates leukemia-initiation potential in acute myeloid leukemia cells. Blood 122(21):A1649
Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ et al (2013) Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res 19:6305–6312
Ganesan A (2008) The impact of natural products upon modern drug discovery. Curr Opin Chem Biol 12:306–317
Gashaw I, Ellinghaus P, Sommer A, Asadullah K (2011) What makes a good drug target? Drug Discov Today 17:S24–S30
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M et al (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1:555–567
Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J et al (2003) SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res 63:375–381
Goozner M (2004) The $800 million pill: the truth behind the cost of new drugs. University of California Press, Berkley
Grivas PD, Papavassiliou AG (2012) Transcriptional corepressors in cancer: emerging targets for therapeutic intervention. Cancer 119:1120–1128
Groeneweg JW, DiGloria CM, Yuan J, Richardson WS, Growdon WB, Sathyanarayanan S et al (2014a) Inhibition of notch signaling in combination with Paclitaxel reduces platinum-resistant ovarian tumor growth. Front Oncol 4:171
Groeneweg JW, Hall TR, Zhang L, Kim M, Byron VF, Tambouret R et al (2014b) Inhibition of gamma-secretase activity impedes uterine serous carcinoma growth in a human xenograft model. Gynecol Oncol 133:607–615
Guo X, Wang X (2008) Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res 19:71–88
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138:645–659
Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L et al (2012) Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA 109:11717–11722
Han A, Song Z, Tong C, Hu D, Bi X, Augenlicht LH, Yang W (2008) Sulindac suppresses beta-catenin expression in human cancer cells. Eur J Pharmacol 583:26–31
He J, Sheng T, Stelter AA, Li C, Zhang X, Sinha M et al (2006) Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J Biol Chem 281:35598–35602
He BC, Gao JL, Zhang BQ, Luo Q, Shi Q, Kim SH et al (2010) Tetrandrine inhibits Wnt/β-catenin signaling and suppresses tumor growth of human colorectal cancer. Mol Pharmacol 79:211–219
He L, Wang F, Dai WQ, Wu D, Lin CL, Wu SM et al (2012) Mechanism of action of salinomycin on growth and migration in pancreatic cancer cell lines. Pancreatology 13:72–78
Hoey T, Yen W, Axelrod F, Basi J, Donigan L, Dylla S et al (2009) DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 5:168–177
Hope C, Planutis K, Planutiene M, Moyer MP, Johal KS, Woo J et al (2008) Low concentrations of resveratrol inhibit Wnt signal throughput in colon-derived cells: implications for colon cancer prevention. Mol Nutr Food Res 52(Suppl 1):S52–S61
Hou X, Chen X, Zhang P, Fan Y, Ma A, Pang T et al (2014) Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 101:50–59
Howells LM, Berry DP, Elliott PJ, Jacobson EW, Hoffman E, Hegarty B et al (2011) Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases–safety, pharmacokinetics, and pharmacodynamics. Cancer Prev Res 4:1419–1425
Huang S, He J, Zhang X, Bian Y, Yang L, Xie G et al (2006) Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 27:1334–1340
Huang L, Walter V, Hayes DN, Onaitis M (2014) Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res 20:1566–1575
Huls G, Koornstra JJ, Kleibeuker JH (2003) Non-steroidal anti-inflammatory drugs and molecular carcinogenesis of colorectal carcinomas. Lancet 362:230–232
Huynh C, Poliseno L, Segura MF, Medicherla R, Haimovic A, Menendez S et al (2011) The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One 6, e25264
Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K et al (2009) Small-molecule inhibitors reveal multiple strategies for hedgehog pathway blockade. Proc Natl Acad Sci USA 106:14132–14137
Ibuki N, Ghaffari M, Pandey M, Iu I, Fazli L, Kashiwagi M et al (2013) TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int J Cancer 133:1955–1966
Ishii T, Shimizu Y, Nakashima K, Kondo S, Ogawa K, Sasaki S, Matsui H (2013) Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur J Pharmacol 723:305–313
Jaiswal AS, Marlow BP, Gupta N, Narayan S (2002) Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene 21:8414–8427
Javelaud D, Pierrat M, Mauviel A (2012) Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett 586:2016–2025
Jenkins DW, Ross S, Veldman-Jones M, Foltz IN, Clavette BC, Manchulenko K et al (2012) MEDI0639: a novel therapeutic antibody targeting Dll4 modulates endothelial cell function and angiogenesis in vivo. Mol Cancer Ther 11:1650–1660
Jiang L, Zhang X, Du P, Zheng J (2011) γ-secretase inhibitor, DAPT inhibits self-renewal and stemness maintenance of ovarian cancer stem-like cells in vitro. Chin J Cancer Res 23:140–146
Jimeno A, Lorusso P, Strother RM, Diamond JR, Plato L, Younger A et al (2013a) Abstract 2502: phase I study of REGN421 (R)/SAR153192, a fully-human delta-like ligand 4 (Dll4) monoclonal antibody (mAb), in patients with advanced solid tumors. J Clin Oncol 31(15):A2502
Jimeno A, Weiss GJ, Miller WH, Gettinger S, Eigl BJ, Chang AL et al (2013b) Phase I study of the hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res 19:2766–2774
Jubb AM, Turley H, Moeller HC, Steers G, Han C, Li JL et al (2009) Expression of delta-like ligand 4 (Dll4) and markers of hypoxia in colon cancer. Br J Cancer 101:1749–1757
Kantarjian HM, Fojo T, Mathisen M, Zwelling LA (2013) Cancer drugs in the United States: justum pretium–the just price. J Clin Oncol 31:3600–3604
Kaushik G, Ramalingam S, Subramaniam D, Rangarajan P, Protti P, Rammamoorthy P et al (2012) Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am J Surg 204:868–873
Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B et al (2012) A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res 18:6509–6518
Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C et al (2006) Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 12:908–916
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH (2010) Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 176:2911–2920
Kim J, Lee JJ, Kim J, Gardner D, Beachy PA (2010a) Arsenic antagonizes the hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA 107:13432–13437
Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV et al (2010b) Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell 17:388–399
Kim HJ, Park SY, Park OJ, Kim Y (2013a) Curcumin suppresses migration and proliferation of Hep3B hepatocarcinoma cells through inhibition of the Wnt signaling pathway. Mol Med Rep 8:282–286
Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M et al (2013b) Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23:23–34
Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S et al (2011) A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 473:230–233
Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ et al (2014) An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet 46:364–370
Kogame A, Tagawa Y, Shibata S, Tojo H, Miyamoto M, Tohyama K et al (2013) Pharmacokinetic and pharmacodynamic modeling of hedgehog inhibitor TAK-441 for the inhibition of Gli1 messenger RNA expression and antitumor efficacy in xenografted tumor model mice. Drug Metab Dispos 41:727–734
Kondratyev M, Kreso A, Hallett RM, Girgis-Gabardo A, Barcelon ME, Ilieva D et al (2011) Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene 31:93–103
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Cinnaiyan P et al (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 30:2307–2313
Kuhnert F, Kirshner JR, Thurston G (2011) Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell 3:20
Kuhnert F, Chen G, Thurston G (2013) Abstract 5091: potent anti-tumor activity of blocking stromal Dll4 in ovarian xenograft models. Cancer Res 73(8):A5091
Kukar T, Golde TE (2008) Possible mechanisms of action of NSAIDs and related compounds that modulate gamma-secretase cleavage. Curr Top Med Chem 8:47–53
Kusunoki S, Kato K, Tabu K, Inagaki T, Okabe H, Kaneda H et al (2013) The inhibitory effect of salinomycin on the proliferation, migration and invasion of human endometrial cancer stem-like cells. Gynecol Oncol 129:598–605
la Porte C, Voduc N, Zhang G, Seguin I, Tardiff D, Singhal N, Cameron DW (2010) Steady-state pharmacokinetics and tolerability of trans-resveratrol 2000 mg twice daily with food, quercetin and alcohol (ethanol) in healthy human subjects. Clin Pharmacokinet 49:449–454
Lauth M, Bergström A, Shimokawa T, Toftgård R (2007) Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA 104:8455–8460
Lee H, Wang NX, Shi D, Zheng JJ (2009) Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein dishevelled. Angew Chem Int Ed Engl 48:6448–6452
Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S et al (2012) Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci USA 109:7859–7864
Lenz H, Kahn M (2014) Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci 105:1087–1092
Leone S, Basso E, Polticelli F, Cozzi R (2012) Resveratrol acts as a topoisomerase II poison in human glioma cells. Int J Cancer 131:E173–E178
Leow P, Tian Q, Ong Z, Yang Z, Ee PL (2009) Antitumor activity of natural compounds, curcumin and p KF118–310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Invest New Drugs 28:766–782
Li J, Sainson RCA, Shi W, Leek R, Harrington LS, Preusser M et al (2007) Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res 67:11244–11253
Li W, Frame LT, Hoo KA, Li Y, D’Cunha N, Cobos E (2011) Genistein inhibited proliferation and induced apoptosis in acute lymphoblastic leukemia, lymphoma and multiple myeloma cells in vitro. Leuk Lymphoma 52:2380–2390
Li Y, Zhang J, Ma D, Zhang L, Si M, Yin H, Li J (2012) Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J 279:2247–2259
Lin H, Xiong W, Zhang X, Liu B, Zhang W, Zhang Y et al (2011) Notch-1 activation-dependent p53 restoration contributes to resveratrol-induced apoptosis in glioblastoma cells. Oncol Rep 26:925–930
List A, Beran M, DiPersio J, Slack J, Vey N, Rosenfeld CS, Greenberg P (2003) Opportunities for Trisenox (arsenic trioxide) in the treatment of myelodysplastic syndromes. Leukemia 17:1499–1507
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW et al (2006) Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 66:6063–6071
Liu W, Zhang J, Ying C, Wang Q, Yan C, Jingyue Y et al (2012) Tetrandrine combined with gemcitabine and Cisplatin for patients with advanced non-small cell lung cancer improve efficacy. Int J Biomed Sci 8:28–35
Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T et al (2013) Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci USA 110:20224–20229
Liu J, Mao Z, Huang J, Xie S, Liu T, Mao Z (2014) Blocking the NOTCH pathway can inhibit the growth of CD133-positive A549 cells and sensitize to chemotherapy. Biochem Biophys Res Commun 444:670–675
Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K et al (2004) Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 47:6658–6661
Lu W, Tinsley HN, Keeton A, Qu Z, Piazza GA, Li Y (2008) Suppression of Wnt/beta-catenin signaling inhibits prostate cancer cell proliferation. Eur J Pharmacol 602:8–14
Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA (2011a) Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci USA 108:13253–13257
Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y (2011b) Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS One 6, e29290
Lu W, Jia G, Meng X, Zhao C, Zhang L, Ren Y et al (2012) Beta-catenin mediates the apoptosis induction effect of celastrol in HT29 cells. Life Sci 91:279–283
Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D et al (2009) Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 69:7672–7680
Lusk CP, Blobel G, King MC (2007) Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol 8:414–420
Lyons TG, O’Kane GM, Kelly CM (2014) Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 13:1125–1132
Mackenzie GG, Sun Y, Huang L, Xie G, Ouyang N, Gupta RC et al (2010) Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe and effective in colon cancer prevention in mice. Gastroenterology 139:1320–1332
Mackenzie GG, Ouyang N, Xie G, Vrankova K, Huang L, Sun Y et al (2011) Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prev Res 4:1052–1060
Mailhos C, Modlich U, Lewis J, Harris A, Bicknell R, Ish-Horowicz D (2002) Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation 69:135–144
Mansour MR, Gale R, Khwaja A, Pule M, Hills RK, Burnett AK, Linch D (2008) Abstract 3364: alternatively spliced isoform of CSL (RBPJ-K) predominates over the full-length isoform in many patients with acute myeloid leukaemia, can activate Notch signalling and is associated with improved outcome. Blood 112(11):A3364
Matsumoto T, Tabata K, Suzuki T (2014) The GANT61, a GLI inhibitor, induces caspase-independent apoptosis of SK-N-LO cells. Biol Pharm Bull 37:633–641
McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS et al (2011) Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res 9:834–844
Metcalfe C, de Sauvage FJ (2011) Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res 71:5057–5061
Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R et al (2004) Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci 82:341–358
Miltyk W, Craciunescu CN, Fischer L, Jeffcoat RA, Koch MA, Lopaczynski W et al (2003) Lack of significant genotoxicity of purified soy isoflavones (genistein, daidzein, and glycitein) in 20 patients with prostate cancer. Am J Clin Nutr 77:875–882
Miyamoto S, Nakanishi M, Rosenberg DW (2013) Suppression of colon carcinogenesis by targeting Notch signaling. Carcinogenesis 34:2415–2423
Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF et al (2012) The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther 11:1999–2009
Mo M, Li M, Chen Z, Liu XW, Sheng Q, Zhou HM (2013) Inhibition of the Wnt palmitoyltransferase porcupine suppresses cell growth and downregulates the Wnt/β-catenin pathway in gastric cancer. Oncol Lett 5:1719–1723
Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC et al (2009) Direct inhibition of the NOTCH transcription factor complex. Nature 462:182–188
Morohashi Y, Kan T, Tominari Y, Fuwa H, Okamura Y, Watanabe N et al (2006) C-terminal fragment of presenilin is the molecular target of a dipeptidic gamma-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J Biol Chem 281:14670–14676
Mullor JL, Dahmane N, Sun T, Altaba ARI (2001) Wnt signals are targets and mediators of Gli function. Curr Biol 11:769–773
Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS et al (2011) Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med Chem Lett 3:106–111
Murray OT, Wong CC, Vrankova K, Rigas B (2013) Phospho-sulindac inhibits pancreatic cancer growth: NFATc1 as a drug resistance candidate. Int J Oncol 44:521–529
Nagao-Kitamoto H, Nagata M, Nagano S, Kitamoto S, Ishidou Y, Yamamoto T et al (2014) GLI2 is a novel therapeutic target for metastasis of osteosarcoma. Int J Cancer 136:1276–1284
Nardella C, Lunardi A, Patnaik A, Cantley LC, Pandolfi PP (2011) The APL paradigm and the “co-clinical trial” project. Cancer Discov 1:108–116
Naujokat C, Steinhart R (2012) Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol 2012:950658
Nguyen AV, Martinez M, Stamos MJ, Moyer MP, Planutis K, Hope C, Holcombe RF (2009) Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag Res 1:25–37
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M et al (2003) Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33:416–421
Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW et al (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444:1032–1037
North TE, Babu IR, Vedder LM, Lord AM, Wishnok JS, Tannenbaum SR et al (2010) PGE2-regulated wnt signaling and N-acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci USA 107:17315–17320
Northcott PA, Korshunov A, Pfister SM, Taylor MD (2012) The clinical implications of medulloblastoma subgroups. Nat Rev Neurol 8:340–351
Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Shibata S, Sato Y et al (2012a) Discovery of pyrrolo[3,2-c]quinoline-4-one derivatives as novel hedgehog signaling inhibitors. Bioorg Med Chem 20:5496–5506
Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Tanaka Y, Shibata S et al (2012b) Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: modification of the core skeleton for improved solubility. Bioorg Med Chem 20:5507–5517
Okuhashi Y, Itoh M, Nara N, Tohda S (2011) Effects of combination of notch inhibitor plus hedgehog inhibitor or Wnt inhibitor on growth of leukemia cells. Anticancer Res 31:893–896
Palomero T, Dominguez M, Ferrando AA (2008) The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle 7:965–970
Pan H, Zhou W, He W, Liu X, Ding Q, Ling L et al (2012) Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-κB activity via the Notch-1 pathway. Int J Mol Med 30:337–343
Park S, Chun S (2011) Streptonigrin inhibits β-Catenin/Tcf signaling and shows cytotoxicity in β-catenin-activated cells. Biochim Biophys Acta 1810:1340–1345
Park M, Lee J, Kwak H, Park CM, Lee HC, Woo SH et al (2005) Arsenic trioxide (As2O3) inhibits invasion of HT1080 human fibrosarcoma cells: role of nuclear factor-kappaB and reactive oxygen species. J Cell Biochem 95:955–969
Pasca di Magliano M, Biankin AV, Heiser PW, Cano DA, Gutierrez PJ, Deramaudt T et al (2007) Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One 2, e1155
Patel NS, Dobbie MS, Rochester M, Steers G, Poulsom R, Le Monnier K et al (2006) Up-regulation of endothelial delta-like 4 expression correlates with vessel maturation in bladder cancer. Clin Cancer Res 12:4836–4844
Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V et al (2004) Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167:215–221
Peng LF, Stanton BZ, Maloof N, Wang X, Schreiber SL (2009) Syntheses of aminoalcohol-derived macrocycles leading to a small-molecule binder to and inhibitor of Sonic hedgehog. Bioorg Med Chem Lett 19:6319–6325
Pérez-Galán P, Roué G, Villamor N, Camp N, Colomer D (2007) The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood 109:4441–4449
Peukert S, He F, Dai M, Zhang R, Sun Y, Miller-Moslin K et al (2013) Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 8:1261–1265
Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K et al (2009) A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 137:623–634
Pinchot SN, Jaskula-Sztul R, Ning L, Peters NR, Cook MR, Kunnimalaiyaan M, Chen H (2010) Identification and validation of Notch pathway activating compounds through a novel high-throughput screening method. Cancer 117:1386–1398
Ponnurangam S, Mammen JMV, Ramalingam S, He Z, Zhang Y, Umar S et al (2012) Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther 11:963–972
Prasad CP, Rath G, Mathur S, Bhatnagar D, Ralhan R (2009) Potent growth suppressive activity of curcumin in human breast cancer cells: modulation of Wnt/beta-catenin signaling. Chem Biol Interact 181:263–271
Preet R, Mohapatra P, Das D, Satapathy SR, Choudhuri T, Wyatt MD, Kundu CN (2012) Lycopene synergistically enhances quinacrine action to inhibit Wnt-TCF signaling in breast cancer cells through APC. Carcinogenesis 34:277–286
Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J et al (2006) Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res 66:7438–7444
Pucheault M (2007) Natural products: chemical instruments to apprehend biological symphony. Org Biomol Chem 6:424–432
Qi L, Ding Y (2013) Potential antitumor mechanisms of phenothiazine drugs. Sci China Life Sci 56:1020–1027
Qin Y, Ma Z, Dang X, Li W, Ma Q (2014) Effect of resveratrol on proliferation and apoptosis of human pancreatic cancer MIA PaCa-2 cells may involve inhibition of the Hedgehog signaling pathway. Mol Med Rep 10:2563–2567
Raju GP (2010) Arsenic: a potentially useful poison for Hedgehog-driven cancers. J Clin Invest 121:14–16
Ranpura V, Pulipati B, Chu D, Zhu X, Wu S (2010) Increased risk of high-grade hypertension with bevacizumab in cancer patients: a meta-analysis. Am J Hypertens 23:460–468
Rao SS, O’Neil J, Liberator CD, Hardwick JS, Dai X, Zhang T et al (2009) Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res 69:3060–3068
Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E et al (2008) Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 15:50–58
Reynolds TC, Smith SD, Sklar J (1987) Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the beta T cell receptor gene in human lymphoblastic neoplasms. Cell 50:107–117
Ristorcelli E, Lombardo D (2010) Targeting Notch signaling in pancreatic cancer. Expert Opin Ther Targets 14:541–552
Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J et al (2014) A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res 20:1900–1909
Rohatgi R, Milenkovic L, Corcoran RB, Scott MP (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci U S A 106:3196–3201
Rojewski MT, Baldus C, Knauf W, Thiel E, Schrezenmeier H (2002) Dual effects of arsenic trioxide (As2O3) on non-acute promyelocytic leukaemia myeloid cell lines: induction of apoptosis and inhibition of proliferation. Br J Haematol 116:555–563
Roy M, Pear WS, Aster JC (2006) The multifaceted role of Notch in cancer. Curr Opin Genet Dev 17:52–59
Ryan PC, Huang J, Bao H, Cho S, Brohawn P, Burke P et al (2013) Abstract 4424: nonclinical safety evaluation of MEDI0639 (Anti-DLL4 Mab) to support first time in human: linking DLL4-notch signaling blockade to exaggerated pharmacology effects in cynomolgus monkeys. Cancer Res 73(8):A4424
Ryu M, Cho M, Song J, Yun YS, Choi IW, Kim DE et al (2008) Natural derivatives of curcumin attenuate the Wnt/beta-catenin pathway through down-regulation of the transcriptional coactivator p300. Biochem Biophys Res Commun 377:1304–1308
Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G et al (2013) A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer 109:943–949
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA 105:6392–6397
Sakoguchi-Okada N, Takahashi-Yanaga F, Fukada K, Shiraishi F, Taba Y et al (2007) Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem Pharmacol 73:1318–1329
Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J et al (2012) Preclinical analysis of the γ-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther 11:1565–1575
Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O’Neil J et al (2009) Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood 115:1735–1745
Scales SJ, de Sauvage FJ (2009) Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci 30:303–312
Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I et al (2013) Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res 19:1512–1524
Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR et al (2003) Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem 278:46107–46116
Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P et al (2000) L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry 39:8698–8704
Sheng T, Li C, Zhang X, Chi S, He N, Chen K et al (2004) Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer 3:29
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J et al (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11:259–273
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Siu LL, Papadopoulos KP, Alberts S, Kirchoff-Ross R, Vakkalagadda B, Lang L et al (2009) Abstract 2501: a first-in-human, phase I study of an oral hedgehog pathway antagonist, BMS-833923 (XL139), in subjects with advanced or metastatic solid tumors. J Clin Oncol 28(15 Supp):A2501
Slusarz A, Shenouda NS, Sakla MS, Drenkhahn SK, Narula AS, MacDonald RS et al (2010) Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer Res 70:3382–3390
Smith GM, Gordon JA, Sewell IA, Ellis H (1967) A trial of streptonigrin in the treatment of advanced malignant disease. Br J Cancer 21:295–301
Smith DC, Rosen L, Wang M, Zhang C, Xu L, Chugh R et al (2013) Abstract B24: biomarker analysis in the first-in-human phase 1a study for vantictumab (OMP-18R5; anti-Frizzled) demonstrates pharmacodynamic (PD) modulation of the Wnt pathway in patients with advanced solid tumors. Mol Cancer Ther 12(11 Suppl):B24
Spigel DR, Spira AI, Jotte R, Gadgeel SM, Mita AC, Hart LL et al (2014) Abstract 7601: phase 1b of anti-cancer stem cell antibody OMP-59R5 (anti-Notch2/3) in combination with etoposide and platinum therapy (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). J Clin Oncol 32(5 Suppl):A7601
Sriuranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, Ball DW (2001) Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res 61:3200–3205
Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL et al (2009) A small molecule that binds hedgehog and blocks its signaling in human cells. Nat Chem Biol 5:154–156
Stein U, Arlt F, Smith J, Sack U, Herrmann P, Walther W et al (2011) Intervening in β-catenin signaling by sulindac inhibits S100A4-dependent colon cancer metastasis. Neoplasia 13:131–144
Stolfi C, De Simone V, Pallone F, Monteleone G (2013) Mechanisms of action of non-steroidal anti-inflammatory drugs (NSAIDs) and mesalazine in the chemoprevention of colorectal cancer. Int J Mol Sci 14:17972–17985
Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G et al (2012) A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 48:997–1003
Stylianou S, Clarke RB, Brennan K (2006) Aberrant activation of notch signaling in human breast cancer. Cancer Res 66:1517–1525
Su Y, Li S, Wu Y, Wang LM, Chao KS, Liao HF (2013) Resveratrol downregulates interleukin-6-stimulated sonic hedgehog signaling in human acute myeloid leukemia. Evid Based Complement Alternat Med 2013:547430
Subramaniam D, Ponnurangam S, Ramamoorthy P, Standing D, Battafarano RJ, Anant S, Sharma P (2012) Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PLoS One 7, e30590
Sun X, Liu X, Huang D (2013) Curcumin reverses the epithelial-mesenchymal transition of pancreatic cancer cells by inhibiting the hedgehog signaling pathway. Oncol Rep 29:2401–2407
Sztiller-Sikorska M, Koprowska K, Majchrzak K, Harman M, Czyz M (2014) Natural compounds’ activity against cancer stem-like or fast-cycling melanoma cells. PLoS One 9, e90783
Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L et al (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406:1005–1009
Takehara M, Hoshino T, Namba T, Yamakawa N, Mizushima T (2011) Acetaminophen-induced differentiation of human breast cancer stem cells and inhibition of tumor xenograft growth in mice. Biochem Pharmacol 81:1124–1135
Takimoto CH, Glover K, Huang X, Hayes SA, Gallot L, Quinn M et al (2003) Phase I pharmacokinetic and pharmacodynamic analysis of unconjugated soy isoflavones administered to individuals with cancer. Cancer Epidemiol Biomarkers Prev 12:1213–1221
Tan BT, Park CY, Ailles LE, Weissman IL (2006) The cancer stem cell hypothesis: a work in progress. Lab Invest 86:1203–1207
Tang QL, Zhao ZQ, Li JC, Liang Y, Yin JQ, Zou CY et al (2011) Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett 311:113–121
Tejada FNH, Silva JRG, Zweidler-McKay PA (2014) The challenge of targeting notch in hematologic malignancies. Front Pediatr 2:54
Thurston G, Noguera-Troise I, Yancopoulos GD (2007) The delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer 7:327–331
Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG et al (2012) Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 30:2348–2353
Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T et al (2003) t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet 33:208–213
Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB (2005) Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol 208:17–25
Tremblay MR, Nevalainen M, Nair SJ, Porter JR, Castro AC, Behnke ML et al (2008) Semisynthetic cyclopamine analogues as potent and orally bioavailable hedgehog pathway antagonists. J Med Chem 51:6646–6649
Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC et al (2009) Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J Med Chem 52:4400–4418
Truong M, Cook MR, Pinchot SN, Kunnimalaiyaan M, Chen H (2010) Resveratrol induces Notch2-mediated apoptosis and suppression of neuroendocrine markers in medullary thyroid cancer. Ann Surg Oncol 18:1506–1511
Tuynman JB, Vermeulen L, Boon EM, Kemper K, Zwinderman AH, Peppelenbosch MP, Richel DJ (2008) Cyclooxygenase-2 inhibition inhibits c-Met kinase activity and Wnt activity in colon cancer. Cancer Res 68:1213–1220
Twardowski P, Stadler WM, Frankel P, Lara PN, Ruel C, Chatta G et al (2010) Phase II study of Aflibercept (VEGF-Trap) in patients with recurrent or metastatic urothelial cancer, a California Cancer Consortium Trial. Urology 76:923–926
Ulloa F, Itasaki N, Briscoe J (2007) Inhibitory Gli3 activity negatively regulates Wnt/beta-catenin signaling. Curr Biol 17:545–550
Valent P (2007) Imatinib-resistant chronic myeloid leukemia (CML): current concepts on pathogenesis and new emerging pharmacologic approaches. Biologics 1:433–448
van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H et al (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435:959–963
Vanamala J, Reddivari L, Radhakrishnan S, Tarver C (2010) Resveratrol suppresses IGF-1 induced human colon cancer cell proliferation and elevates apoptosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer 10:238
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J et al (2012) Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 367:1783–1791
Walle T (2011) Bioavailability of resveratrol. Ann N Y Acad Sci 1215:9–15
Walle T, Wen X, Walle UK (2007) Improving metabolic stability of cancer chemoprotective polyphenols. Expert Opin Drug Metab Toxicol 3:379–388
Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH (2005) Inhibition of nuclear factor kappab activity by genistein is mediated via Notch-1 signaling pathway in pancreatic cancer cells. Int J Cancer 118:1930–1936
Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH (2006) Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer 106:2503–2513
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS et al (2009) Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 69:2400–2407
Wang XN, Wu Q, Yang X, Zhang LS, Wu YP, Lu C (2010) Effects of celastrol on growth inhibition of U937 leukemia cells through the regulation of Notch1/NF-kappaB signaling pathway in vitro. Chin J Cancer 29:385–390
Wang Y, Liu Y, Malek SN, Zheng P, Liu Y (2011) Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8:399–411
Wang F, He L, Dai W, Xu YP, Wu D, Lin CL et al (2012) Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS One 7, e50638
Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W et al (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497:338–343
Wang H, Zhang G, Zhang H, Zhang F, Zhou B, Ning F et al (2014) Acquisition of epithelialmesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol 723:156–166
Watters JW, Cheng C, Majumder PK, Wang R, Yalavarthi S, Meeske C et al (2009) De novo discovery of a gamma-secretase inhibitor response signature using a novel in vivo breast tumor model. Cancer Res 69:8949–8957
Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A et al (2010) Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther 9:1618–1628
Weihofen A, Lemberg MK, Friedmann E, Rueeger H, Schmitz A, Paganetti P et al (2003) Targeting presenilin-type aspartic protease signal peptide peptidase with gamma-secretase inhibitors. J Biol Chem 278:16528–16533
Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A et al (2005) Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7:129–141
Westhoff B, Colaluca IN, D'Ario G, Donzelli M, Tosoni D, Volorio S et al (2009) Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci USA 106:22293–22298
Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T et al (2004) Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 279:12876–12882
Wu L, Griffin JD (2004) Modulation of Notch signaling by mastermind-like (MAML) transcriptional co-activators and their involvement in tumorigenesis. Semin Cancer Biol 14:348–356
Wu J, Ji Z, Liu H, Liu Y, Han D, Shi C, Shi C et al (2013) Arsenic trioxide depletes cancer stem-like cells and inhibits repopulation of neurosphere derived from glioblastoma by downregulation of Notch pathway. Toxicol Lett 220:61–69
Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ et al (2001) Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA 98:10863–10868
Xia J, Duan Q, Ahmad A, Bao B, Banerjee S, Shi Y et al (2012a) Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr Drug Targets 13:1750–1756
Xia J, Li Y, Yang Q, Mei C, Chen Z, Bao B et al (2012b) Arsenic trioxide inhibits cell growth and induces apoptosis through inactivation of Notch signaling pathway in breast cancer. Int J Mol Sci 13:9627–9641
Xu Y, Chenna V, Hu C, Sun HX, Khan M, Bai H et al (2011) Polymeric nanoparticle-encapsulated hedgehog pathway inhibitor HPI-1 (NanoHHI) inhibits systemic metastases in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res 18:1291–1302
Xu X, Gan Y, Xu G, Chen T, Zhou H, Tang JF et al (2012) Tetrandrine citrate eliminates imatinib-resistant chronic myeloid leukemia cells in vitro and in vivo by inhibiting Bcr-Abl/β-catenin axis. J Zhejiang Univ Sci B 13:867–874
Xu Y, An Y, Wang X, Zha W, Li X (2013) Inhibition of the hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol Rep 31:707–712
Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, Korangath P et al (2013) Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 335:41–51
Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G et al (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463:E6–E7
Yang L, Xie G, Fan Q, Xie J (2009) Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 29:469–481
Yao J, Duan L, Fan M, Wu X (2007) Gamma-secretase inhibitors exerts antitumor activity via down-regulation of Notch and Nuclear factor kappa B in human tongue carcinoma cells. Oral Dis 13:555–563
Yao C, Lai G, Yeh C, Lai MT, Shih PH, Chao WJ et al (2013) Honokiol eliminates human oral cancer stem-like cells accompanied with suppression of Wnt/ β -catenin signaling and apoptosis induction. Evid Based Complement Alternat Med 2013:146136
Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP et al (2008) A paracrine requirement for hedgehog signalling in cancer. Nature 455:406–410
Yeh C, Wu ATH, Chang PM, Chen KY, Yang CN, Yang SC et al (2012) Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am J Respir Crit Care Med 186:1180–1188
Yen W, Fischer MM, Hynes M, Wu J, Kim E, Beviglia L et al (2012) Anti-DLL4 has broad spectrum activity in pancreatic cancer dependent on targeting DLL4-Notch signaling in both tumor and vasculature cells. Clin Cancer Res 18:5374–5386
Ylivinkka I, Hu Y, Chen P, Rantanen V, Hautaniemi S, Nyman TA et al (2013) Netrin-1-induced activation of Notch signaling mediates glioblastoma cell invasion. J Cell Sci 126:2459–2469
Yu S, Zhang R, Liu F, Wang H, Wu J, Wang Y (2012) Notch inhibition suppresses nasopharyngeal carcinoma by depleting cancer stem-like side population cells. Oncol Rep 28:561–566
Zaidi AH, Komatsu Y, Kelly LA, Malhotra U, Rotoloni C, Kosovec JE et al (2013) Smoothened inhibition leads to decreased proliferation and induces apoptosis in esophageal adenocarcinoma cells. Cancer Invest 31:480–489
Zhang J, Garrossian M, Gardner D, Garrossian A, Chang YT, Kim YK, Chang CW (2008) Synthesis and anticancer activity studies of cyclopamine derivatives. Bioorg Med Chem Lett 18:1359–1363
Zhang X, Yin W, Shi X, Li Y (2011a) Curcumin activates Wnt/β-catenin signaling pathway through inhibiting the activity of GSK-3β in APPswe transfected SY5Y cells. Eur J Pharm Sci 42:540–546
Zhang Y, Wang C, Wang H, Wang K, Du Y, Zhang J (2011b) Combination of tetrandrine with cisplatin enhances cytotoxicity through growth suppression and apoptosis in ovarian cancer in vitro and in vivo. Cancer Lett 304:21–32
Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q et al (2013a) Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med 2:233–242
Zhang Y, Morris JP, Yan W, Schofield HK, Gurney A, Simeone DM et al (2013b) Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res 73:4909–4922
Zhang P, Li H, Yang B, Yang F, Zhang LL, Kong QY et al (2014) Biological significance and therapeutic implication of resveratrol-inhibited Wnt, Notch and STAT3 signaling in cervical cancer cells. Genes Cancer 5:154–164
Zhao J, Quan H, Xie C, Lou L (2014) NL-103, a novel dual‐targeted inhibitor of histone deacetylases and hedgehog pathway, effectively overcomes vismodegib resistance conferred by Smo mutations. Pharmacol Res Perspect 2, e00043
Zhen Y, Zhao S, Li Q, Li Y, Kawamoto K (2009) Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett 292:64–72
Zhou HM, Dong TT, Wang LL, Feng B, Zhao HC, Fan XK, Zheng MH (2012) Suppression of colorectal cancer metastasis by nigericin through inhibition of epithelial-mesenchymal transition. World J Gastroenterol 18:2640–2648
Zhu C, Cheng K, Ouyang N, Huang L, Sun Y, Constantinides P, Rigas B (2012) Phosphosulindac (OXT-328) selectively targets breast cancer stem cells in vitro and in human breast cancer xenografts. Stem Cells 30:2065–2075
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Coyle, K.M., Thomas, M.L., Sultan, M., Marcato, P. (2015). Targeting Key Stemness-Related Pathways in Human Cancers. In: Babashah, S. (eds) Cancer Stem Cells: Emerging Concepts and Future Perspectives in Translational Oncology. Springer, Cham. https://doi.org/10.1007/978-3-319-21030-8_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-21030-8_15
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21029-2
Online ISBN: 978-3-319-21030-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)