
Abstract
Ewing sarcoma is a bone-associated malignancy arising primarily in childhood and adolescence. It is an aggressive cancer harbouring a characteristic translocation, t(11;22)(q24.3;q12.2). This rearrangement fuses the genes EWSR1 and FLI1, producing a fusion protein (EWS/FLI) that initiates an oncogenic transcription programme. Other rearrangements between similar genes have also been found to be drivers of Ewing sarcoma in a minority of cases. Understanding the molecular processes governed by these rearrangements promises to generate immediately actionable therapeutic strategies. This chapter discusses the defining role that translocations and their after-effects play in Ewing sarcoma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Ewing sarcoma was first described by James Ewing (Fig. 15.1) in 1921 as a tumour composed of distinctive sheets of cells with “small hyperchromatic nuclei” [1]. He noted that these tumours were distinguishable from osteogenic sarcoma by their histopathological morphology. Indeed, Ewing sarcoma continues to be characterized by its appearance as a small, round blue cell tumour, and modern molecular biology techniques have enabled scientists to elucidate many mechanistic details important for development of this tumour [2]. One particularly important discovery – made roughly 60 years after Ewing’s first description of the disease – was that Ewing sarcoma harbours a recurrent set of chromosomal translocations that drive oncogenesis [3–5]. Further study of this key translocation event and its consequences have led to greater understanding of the disease and promises to provide improved therapies for those who fall victim to this malignancy. In this chapter, we discuss the biology of Ewing sarcoma with a focus on its associated translocations, including the two most common rearrangements t(11;22)(q24.3;q12.2) and t(21;22)(q22.2;q12.2) (which generate the fusion proteins EWS/FLI and EWS/ERG, respectively) as well as other, less common translocations.

James Ewing (ca. 1890; Source: Images from the History of Medicine, National Library of Medicine; record UI: 101414702)
2 Clinical Overview
Ewing sarcoma is a relatively broad term for a group of tumours collectively known as the Ewing sarcoma family of tumours. (Previously referred to as “Ewing’s” sarcoma, the WHO has opted to avoid possessive nomenclature; hence, “Ewing” sarcoma is the current WHO-accepted term that will be utilized in this chapter.) This family is predominantly composed of classic Ewing sarcoma, which is a bone-associated tumour that harbours one of a set of oncogenic translocations (discussed hereafter), but also includes tumours such as Askin’s tumour, primitive neuroectodermal tumours (PNETs), and Ewing tumours arising in soft-tissue, known as extraosseous Ewing sarcoma [6–9]. Despite the nuances distinguishing these different members of the Ewing sarcoma family of tumours, chromosomal rearrangements are a common feature of Ewing family tumours, and are the focus of this chapter [8–11].
Ewing sarcoma is a disease of young people, occurring most commonly in children and adolescents. The mean age at diagnosis is 15 years, and ~80 % of all cases occur in patients under the age of 25 [12, 13]. For reasons that are not understood, the disease occurs at a modestly higher rate in males than females (male-to-female ratio of 1.2) (Fig. 15.2) [13, 14]. Although the disease is relatively rare, with an incidence of ~3 per million per year in the United States, Ewing sarcoma is the second most common childhood bone tumour, after osteosarcoma [15, 16]. It is most commonly encountered in patients of European ancestry, and is exceedingly uncommon in populations of African or East Asian ancestry [17–21].

Incidence of Ewing Sarcoma per year per million grouped by age at diagnosis (SEER data, 1973–2010) [13]
Ewing sarcoma is an aggressive cancer with a high propensity for metastasis. In fact, up to 25 % of patients already have metastatic disease at the time of diagnosis [22]. This may well be an underestimation, as it is thought that many patients have undetectable micrometastatic disease at diagnosis as well. Indeed, the relapse rate is ~90 % for patients who undergo surgical resection of their primary tumours without adjuvant chemotherapy [23–25]. As Ewing first observed, these tumours are often highly sensitive to radiation therapy, which was thus was a mainstay of treatment for much of the twentieth century [26, 27]. The refinement of chemotherapeutic strategies and improved surgical techniques have led to great improvements in patient survival, and current conventional treatment modalities have achieved 5-year disease-free survival rates of 60–70 % for non-metastatic disease. However, prognosis for metastatic disease remains dismal with a 5-year disease-free survival of only 10–30 % [28–30]. Moreover, survivors frequently must endure morbidities resulting from conventional anti-cancer therapy, such as severe deformities and amputations due to radical surgical resections of their tumour, and increased risk of future malignancy resulting from radiation and chemotherapy [31, 32]. Better treatments are clearly needed to provide greater survival and higher quality of life. To this end, studies continue to seek a better understanding of the molecular processes underlying Ewing sarcoma oncogenesis, including the molecular consequences of its associated chromosomal translocations.
3 Translocations in Ewing Sarcoma
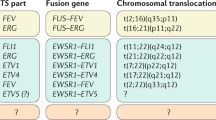
In 1983, scientists at the Curie Institute in France identified a balanced reciprocal translocation between chromosomes 11 and 22 in patient samples and cell lines of Ewing sarcoma [3, 4, 33]. This rearrangement, t(11;22)(q24.3;q12.2), was successfully cloned several years later, and the translocation breakpoint was characterized [34]. It was revealed that this translocation resulted in an in-frame fusion of two genes: Ewing Sarcoma Breakpoint Region 1 (EWSR1) on chromosome 22 and Friend Leukaemia Virus Integration Site 1 (FLI1) on chromosome 11 [34]. The fusion protein encoded by the joining of these two genes is known as EWS/FLI. Approximately 85 % of Ewing sarcoma tumours carry this hallmark cytogenetic abnormality [9, 11, 28, 33]. The remaining 15 % of tumours carry other chromosomal rearrangements resulting in similar fusions of other genes in the same families as EWSR1 and FLI1 [35–39]. A list of these chromosomal rearrangements found in Ewing sarcoma is provided in Table 15.1. Details regarding each of these translocations will be discussed in the following sections.
4 EWS/FLI
4.1 Wild-Type EWS and the FET Family of Proteins
Prior to its cloning as part of EWSR1/FLI1 in Ewing sarcoma, the EWSR1 gene had not been identified and hence bears the name of the disease. EWSR1 encodes a 656-amino acid protein called EWS. EWS is part of the FET (FUS, EWS, TAF15) family of proteins, which are involved in diverse cellular functions including gene expression and RNA processing (Fig. 15.3) [34, 40, 41].

Diagrammatic representation of FET-family proteins and their functional domains. TAD Transcriptional activation domain, RGG arginine-glycine-glycine motif, RRM RNA recognition motif, ZF zinc finger
It is ubiquitously expressed and is principally found in the nucleus, although it can be cytoplasmic or localized to the cell membrane [42–44]. The amino terminus of EWS contains a transcriptional activation region comprised of multiple pseudorepeats rich in serine, tyrosine, glycine, and glutamine (SYGQ) (Fig. 15.3) [45–48]. This SYGQ transactivation domain is critical for interaction between EWS and RNA polymerase II; indeed, wild-type EWS has also been shown to interact with other members of the transcriptional machinery including TFIID and CREBBP/CBP/p300 [45, 46, 49]. The C-terminus of EWS contains arginine-glycine-glycine (RGG) motifs and an RNA recognition motif (RRM), possibly implicating full-length EWS in RNA binding, processing and transcription [41, 50]. The two other members of the FET family of proteins, FUS (also known as TLS) and TAF15, can also be involved in the development of other non-Ewing sarcoma cancers (Table 15.2) [51–58]. These proteins bear striking similarities to EWS, particularly with respect to the domain organization found in the N-termini of EWS and FUS [59–61].
As will be discussed in the following sections, translocations between FET genes and various partners can result in fusion proteins that alter transcriptional programmes and drive oncogenic transformation. Thus, the aforementioned interactions between FET proteins and members of the transcriptional machinery have important implications for molecular mechanisms underlying Ewing sarcoma tumourigenesis, as well as other cancers driven by translocations of FET genes. Recent experiments have elegantly demonstrated that FUS and EWS are able to form both homotypic and heterotypic “amyloid-like” polymers via interactions between disordered regions of polypeptides with little diversity in amino acid sequence, termed low complexity domains [49, 62]. Such aggregates could form a platform for intermolecular binding similar to molecular “velcro”, leading to alteration of various cellular processes. Indeed, these polymers have been shown to bind to the C-terminal domain (CTD) of RNA polymerase II and induce transcription [49]. Accordingly, improper localization of FET proteins and their corresponding low complexity domains could disrupt gene expression at multiple loci, potentially contributing to an oncogenic phenotype. Such a model remains unproven, but is currently being actively tested.
4.2 Wild-Type FLI and the ETS Family of Transcription Factors
The FLI1 gene encodes the 452 amino acid FLI protein, which is a member of the ETS (E26 transformation-specific) family of transcription factors. ETS transcription factors share a highly conserved DNA binding domain. This binding domain is known as the ETS domain, and is a winged helix-turn-helix that binds to DNA, most avidly at DNA motifs containing a core sequence of GGAA or GGAT [63, 64]. Full-length murine Fli1 is capable of oncogenic function; indeed, the Fli1 gene was first characterized as an integration site for the Friend murine leukaemia virus, a function from which the gene derives its name (Friend Leukaemia Virus Integration Site 1) [65]. Integration of the virus at the murine Fli1 locus results in overexpression of Fli1 and produces erythroleukaemia in mice [66]. Wild-type FLI appears to play important roles in haematopoiesis, particularly in megakaryocyte development [67]. Deletion of Fli1 in mice results in dysfunctional megakaryocyte differentiation, and overexpression of Fli1 in erythroleukaemia cells pushes them toward a megakaryocytic programme of differentiation [68, 69].
4.3 The EWS/FLI Fusion
To form EWS/FLI, the 5′ portion of the EWSR1 gene and the 3′ region of the FLI1 gene are joined together, allowing transcription of in-frame fusion transcripts and ultimately synthesis of the EWS/FLI fusion protein. The reciprocal fusion of the 5′ end of FLI1 and the 3′ end of EWSR1 is not expressed, and the reciprocal derivative chromosome is sometimes lost [9, 70]. Interestingly, the oncogenic EWSR1/FLI1 fusion can result from several distinct translocation breakpoints occuring within introns of EWSR1 and FLI1 [71–73]. Classic splicing processes then generate fusion transcripts joining 5′ exons of EWSR1 with 3′ exons of FLI1. EWS/FLI can thus be categorized into subtypes based upon the location of the translocation breakpoint and which exons are fused together [34]. For instance, the most commonly observed translocation in Ewing sarcoma joins exons 1–7 of EWSR1 to exons 6–10 of FLI1. This rearrangement is sometimes termed a “Type I” fusion, but it is more commonly referred to simply as a “7/6” EWS/FLI fusion. Likewise, other fusions of EWS/FLI can be referred to by the exons that are fused, and a partial list of observed EWS/FLI fusions is illustrated in Fig. 15.4.

Diagrammatic representation of EWSR1 and FLI1 exons. Known translocation breakpoints are indicated
The functional significance of these subtly different EWS/FLI fusion products remains largely unknown. However, some data exist that suggest that the “7/6” EWS/FLI fusion (“Type I”) is more weakly transactivating compared to other EWS/FLI fusion subtypes [74]. This distinction was thought to be potentially useful as a prognostic variable, and retrospective analyses of patient cohorts suggested that patients with “7/6” EWS/FLI fusions had better survival rates compared to patients whose tumours harboured EWS/FLI from other translocation breakpoints [75, 76]. However, recent studies have revealed that prognostic differences no longer exist within current treatment protocols [77, 78]. Hence, the functional significance of different breakpoints, if any exists at all, remains unknown.
4.4 Oncogenic Function of EWS/FLI
EWSR1 and FLI1 genes are fused in-frame, encoding the EWS/FLI oncoprotein (Fig. 15.5). This translocation-derived oncoprotein contains the N-terminal transactivation domain of EWS fused with the DNA-binding domain of FLI, forming an oncogenic transcription factor that is indispensible for tumourigenesis [34, 42, 79–81]. The first studies implicating EWS/FLI as a driver in Ewing sarcoma observed that overexpression of EWS/FLI in NIH3T3 murine fibroblasts induced oncogenic transformation, measured by anchorage-independent growth in soft agar. This was later confirmed by experiments demonstrating the ability of EWS/FLI-expressing NIH3T3 cells to form tumours in mouse xenografts [80, 82, 83]. Furthermore, studies utilizing patient-derived Ewing sarcoma cell lines have shown that disruption of EWS/FLI expression by RNA interference (RNAi) and other means results in loss of transformation [70, 84–92]. Together, these findings clearly indicate that EWS/FLI is the driver mutation underlying Ewing sarcoma oncogenesis.

Illustration of the EWS/FLI fusion protein, joining the N-terminal portion of EWS with the C-terminal portion of FLI. PTD pointed domain, DBD DNA binding domain, Pro proline-rich activation domain, TAD Transcriptional activation domain, RGG arginine-glycine-glycine motif, RRM RNA recognition motif, ZF zinc finger
This loss of transformation is accompanied by changes in gene expression, including activation and repression of numerous EWS/FLI target genes [70, 89, 92–94]. Importantly, when EWS/FLI is reintroduced after being silenced by RNAi, the oncogenic expression profile and transformed phenotype of Ewing sarcoma are restored, indicating that EWS/FLI is at the head of an oncogenic programme of gene expression [70, 92, 94]. Studies show that thousands of genes are either upregulated or downregulated by EWS/FLI, leading to “transcriptional mayhem” [70, 81, 95]. This dysregulation of EWS/FLI target gene expression has been the focus of investigations into the mechanisms by which EWS/FLI drives tumourigenesis, and studies have revealed several EWS/FLI-regulated genes that are also required for tumourigenesis, including NR0B1, NKX2.2 and GLI1 [70, 92, 94, 96, 97].
The exact mechanisms by which EWS/FLI causes up-regulation of target genes is an area of active study. It is known that EWS/FLI alters expression of some genes in a direct manner, while it dysregulates other genes indirectly [98, 99]. Nevertheless, it has been definitively shown that the ability of EWS/FLI to bind DNA is essential for Ewing sarcoma oncogenesis [79]. Chromatin immunoprecipitation experiments followed by microarray analysis (ChIP-chip) and deep sequencing (ChIP-seq) have clearly demonstrated that EWS/FLI binds to high-affinity ETS sequences (ACCGGAAGTG) [63, 64, 100, 101]. Interestingly, it was also revealed that EWS/FLI binds to microsatellite repeats of the sequence GGAA [102, 103]. In fact, binding of EWS/FLI to microsatellites is required for upregulation of NR0B1, CAV1, and GSTM4; genes that are critical downstream effectors of EWS/FLI-driven tumourigenesis [102, 103].
Furthermore, as previously mentioned, it has been shown that wild-type EWS is capable of forming a molecular “velcro”-like polymer that facilitates protein-protein interactions between EWS and other proteins, including RNA polymerase II [49, 62]. The low complexity domain in the N-terminal region of wild-type EWS is retained in the EWS/FLI fusion protein, fused to the DNA-binding ETS domain of FLI. It is tempting to speculate, therefore, that the DNA-binding domain of FLI acts to re-direct the molecular “velcro” of EWS to different loci throughout the genome, leading to disruption of regulatory protein complexes and transcriptional activation of EWS/FLI target genes. For instance, GGAA microsatellite repeats could facilitate EWS/FLI polymerization as multiple DNA sequence repeats could permit EWS/FLI to bind in series, forming a scaffold of EWS low complexity domains to which coactivator complexes and transcriptional machinery (e.g., RNA polymerase II) could bind, thus upregulating that locus. Similarly, such a phenomenon could allow EWS/FLI to recruit repressive regulatory complexes to various loci, resulting in down-regulation of target genes. This model, while intriguing, remains unproven, and further testing will shed light on the true mechanisms underlying EWS/FLI-mediated transcriptional dysregulation.
EWS/FLI also down-regulates thousands of genes in Ewing sarcoma. This is particularly interesting considering the presence of the N-terminal transactivation domain of EWS in the EWS/FLI oncoprotein. The mechanisms by which such a transactivator-containing transcription factor causes direct repression of genes remains another active area of study, and several mechanistic insights have been revealed. For instance, it has been demonstrated that a corepressor complex called the Nucleosome Remodelling and Deacetylase (NuRD) complex plays an important role in repression of EWS/FLI targets. Interestingly, disruption of NuRD complex function by vorinostat treatment (a histone deacetylase inhibitor) or RNAi-mediated silencing of CHD4 (a core NuRD component) resulted in de-repression of EWS/FLI-repressed target genes [104]. Additionally, inhibition of lysine-specific demethylase 1 (LSD1) resulted in de-repression of EWS/FLI-regulated target genes. This effect was lost upon silencing of EWS/FLI, implicating EWS/FLI-mediated disruption of associated epigenetic factors in Ewing sarcoma oncogenesis [104, 105]. Continued investigation of these phenomena is likely to generate a clearer mechanistic understanding of EWS/FLI-driven up- and down-regulation of target genes, potentially providing targets for new and better therapeutics.
5 EWS/ERG
In 1993 it was found that a distinct translocation event between the EWSR1 gene and another ETS family member, ERG (ETS-Related Gene), also generated a fusion protein, termed EWS/ERG [38]. The t(21;22)(q22.2;q12.2) rearrangement producing this alternate fusion oncoprotein is present in approximately 10 % of Ewing tumours, making it the most common alternate translocation in Ewing sarcoma [38, 72]. Tumours carrying the EWS/ERG mutation do not carry the EWS/FLI fusion, indicating that EWS/ERG likely drives Ewing sarcoma oncogenesis in ways very similar to EWS/FLI. Indeed, the DNA-binding ETS domain of ERG is shares 98 % amino acid identity with the ETS domain of FLI, and the full-length proteins are 68 % similar [38, 106]. Furthermore, EWS/ERG-harbouring Ewing sarcoma tumours were no different compared to cases of EWS/FLI-containing tumours with respect to age at diagnosis, primary site, metastasis, as well as overall and event-free survival [107].
Like EWS/FLI, EWS/ERG induces oncogenic transformation when it is expressed in NIH3T3 cells [83]. Functionally, EWS/ERG is presumed to bind similar, if not identical, sets of loci as EWS/FLI, likely dysregulating expression of target genes in similar ways. This presumption is supported by evidence indicating that EWS/FLI and EWS/ERG dysregulate the same core subset of genes when introduced into NIH 3T3 cells, although these results must be interpreted cautiously considering the inaccuracies of this model [70, 108].
6 EWS/ETV1, EWS/ETV4, EWS/FEV
In addition to EWS/FLI and EWS/ERG, other EWS/ETS translocations have also been described in Ewing sarcoma. These alternate rearrangements result in the fusion of the EWSR1 gene with ETV1 (ETS variant gene 1), ETV4 (ETS variant gene 4) and FEV (fifth Ewing sarcoma variant) (Table 15.1) [35–37, 39]. Each of these additional fusion proteins occurs in <1 % of all Ewing sarcoma cases, making them exceptionally rare. Being members of the same family of transcription factors, ERG, ETV1, ETV4 and FEV are all highly similar, particularly in their ETS DNA-binding domains. In fact, ETS domains of FLI, ERG and FEV are 98 % similar. ETV1 and ETV4 are also similar to other ETS proteins, but are more similar to each other because they have identical DNA-binding domains.
These rare alternate fusions have been less well studied than EWS/FLI. However, their structural similarities suggest that they share much of the same oncogenic functions required for Ewing sarcoma tumourigenesis. Indeed, the mutually exclusive nature of these different types of EWS/ETS fusions suggests that they may be largely interchangeable. Notwithstanding the relative paucity of data regarding these uncommon rearrangements, some functional differences have been observed in experiments utilizing NIH3T3 cells. Using this model, it was shown that EWS/FLI, EWS/ERG and EWS/FEV were capable of inducing anchorage-independent growth in soft agar assays, whereas EWS/ETV1 and EWS/ETV4 were incapable of inducing such transformation [108]. Interestingly, each fusion protein enabled tumour formation by NIH3T3 cells in murine xenografts. The mechanism and relevance of these differences remain unknown. It has also been suggested that EWS/FEV, EWS/ETV1 and EWS/ETV4 exist predominantly in extraosseous Ewing sarcoma [109]. However, insufficient data exists at the present time to draw any definitive conclusions about this potential correlation. It is also unknown whether these different fusion proteins have any significance with regard to outcome.
7 FUS/ERG and FUS/FEV
EWSR1 is the founding member of the FET (FUS, EWSR1, TAF15) family of RNA-binding proteins involved in Ewing sarcoma translocations. However, in rare instances, other members of the family are involved. Chromosomal rearrangements between FUS (also known as TLS) and ERG or FEV, both ETS family member genes, have been identified in rare cases of Ewing sarcoma [110, 111].
The FUS protein has a similar domain structure to that of EWS, containing an N-terminal transactivation domain with SYGQ repeats, and C-terminal RGG and RRM motifs (Fig. 15.5). Considering these shared structural features, it is likely that FUS/ETS fusions drive oncogenesis via mechanisms similar to those utilized by EWS/FLI. However, this hypothesis has not been thoroughly tested, in large part due to the relative scarcity of these alternate chromosomal rearrangements. Nevertheless, some functional similarities have been observed. For instance, both EWS/FLI and FUS/ERG have been shown to disrupt RNA splicing by similar mechanisms [112]. Expression of insulin-like growth factor 1 (IGF1) is also induced by several FET/ETS fusion proteins, including FUS/ERG [113]. However, these data must be interpreted with some caution as they are based largely on murine cells, which may lack some features important for EWS/FLI function [114].
Currently, only FUS/ERG and FUS/FEV fusions have been described, but it is possible that other FET/ETS fusions could exist in Ewing sarcoma. However, such instances would be exceedingly rare. The uncommon nature of such alternate fusions makes it difficult to elucidate whether specific rearrangements have important implications for prognosis, probability of relapse, or other factors. As mentioned before, these alternate translocations do pose a potential complication for molecular diagnosis of the disease, as a tumour that appears negative for all known translocations may harbour an oncogenic FET/ETS rearrangement that has not yet been characterized and thus evades detection. These fusions are so scarce, however, that only a small minority of patients would be impacted by such a scenario.
8 “Ewing-Like Sarcomas” and Their Translocations
The existence of multiple alternate chromosomal rearrangements in Ewing sarcoma raises the question of how best to molecularly define the disease. In general, histopathological features and patient presentation give good pre-test probability for diagnosis, and definitive diagnosis commonly given by detection of CD99, a cell surface marker found on most Ewing sarcoma cells [115]. Biopsies are often subjected to molecular tests detecting the presence of the t(11;22)(q24.3;q12.2) translocation. Presence of EWS/FLI transcript are detected with RT-PCR, and translocations involving EWSR1 are detected via breakapart FISH assays. These methods will detect almost all known FET/ETS chromosomal rearrangements in Ewing sarcoma. However, a family of tumours exists in which non-FET/ETS fusions are present (Table 15.3). These cancers are termed “Ewing-like sarcomas”.
One such “Ewing-like” tumour was first reported in 2009 as a new t(20;22)(q13;q12) rearrangement between EWSR1 and NFATC2 (nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2) [116]. The wild-type NFATC2 protein is a member of the NFAT family of transcription factors and is a key player in T-cell and neuronal development. NFATC2 binds DNA cooperatively with Fos and Jun, members of the activator protein 1 (AP1) family of regulatory transcription factors [117–120]. Interestingly, ETS proteins and the EWS/FLI fusion protein are also capable of cooperative DNA binding with AP1 proteins [121–123]. Also, NFAT proteins, like ETS proteins, recognize DNA sequences with a core motif of GGAA/T [116]. Together these findings suggest possible shared mechanisms of oncogenesis between EWS/ETS and EWS/NFATC2 fusions.
EWSR1 can fuse to a number of other non-ETS proteins to drive formation of “Ewing-like” tumours. Another such fusion is EWS/POU5F1 [124]. POU5F1 (POU class 5 homeobox 1) is also known as OCT4 (octamer-binding transcription factor 4), and is a transcription factor important for regulating pluripotency of stem cells [125–127]. It is thought that this fusion protein functions as an aberrant transcription factor in these tumours, transcriptionally reprogramming cells and generating an oncogenic phenotype.
Fusions between EWSR1 and PATZ1 (POZ (BTB) and AT Hook Containing Zinc Finger 1, also known as ZSG) or SP3 are also found in some “Ewing-like” tumours [109, 128]. Both ZSG and SP3 are zinc finger-containing transcription factor proteins and, therefore, potentially function by binding DNA and allowing the EWS portion of the fusion to dysregulate gene expression profiles, similar to EWS/FLI and other Ewing sarcoma rearrangements [109, 129]. Wild-type SP3 also contains an inhibitory domain that is lost in the translocation event generating EWS/SP3, potentially contributing to its oncogenic function.
EWSR1 can also fuse with SMARCA5 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 5), an ATPase found in various chromatin remodelling complexes [130–135]. While the EWS/SMARCA5 fusion protein does not directly bind DNA in a sequence-specific manner, it alters expression of key target genes perhaps by altering a chromatin remodelling function. Interestingly, SMARCA5 can function as part of the NuRD complex, which plays an important role in EWS/FLI-mediated repression of target genes (discussed previously) [104]. Whether any relationship exists between EWS/SMARCA5 and NuRD has not been tested.
CIC/DUX4 and BCOR/CCNB3 fusions have also been described [136, 137]. However, it has not been fully determined whether these tumours represent Ewing sarcoma, “Ewing-like” sarcoma, or a distinct type of bone sarcoma. More in-depth study of the molecular mechanisms underlying these oncogenic chromosomal rearrangements must be undertaken to answer this question. Indeed, a clear molecular-based definition of Ewing sarcoma and its variations may hinge upon achieving a clearer picture of how these fusions generate an oncogenic phenotype.
9 Molecular Definitions of Ewing Sarcoma and Diagnostic Challenges
9.1 Defining the Disease
The classic diagnostic definition of Ewing sarcoma relies largely upon histopathological features of these tumours, assessed by light microscopy and/or immunohistochemistry [138, 139]. This cancer appears as a small, round cell cancer with hyperchromatic nuclei when viewed by light microscopy after H&E staining [140]. Immunohistochemical staining often reveals high levels of CD99 at the cell membrane, and is used as another diagnostic marker of Ewing sarcoma cells [115, 138].
Additionally, the presence of a balanced translocation involving EWSR1 and one of the ETS family of transcription factors are considered pathognomonic for the disease [138]. However, as discussed in this chapter, a number of different translocations involving FET family members other than EWSR1 (e.g., FUS) also exist. Additionally, several “Ewing-like” cancers have been found with fusions of EWSR1 to non-ETS proteins. These alternate molecular lesions, rare as they may be, add complexity to the question of how to properly define this disease and its variations.
Generally, Ewing sarcoma can be broadly subdivided into three groups, based on the type of translocation present in the tumour cells: (1) EWSR1/FLI1 and functionally similar translocations (i.e., FET/ETS fusions), (2) non-FET/ETS fusions (e.g., EWS/SMARCA5), and (3) totally distinct translocations (e.g., CIC/DUX4). Furthermore, tumours of EWS/FLI and other FET/ETS fusions (e.g., EWS/ERG) can be considered classic Ewing sarcoma, while rarer, non-FET/ETS fusions and distinct translocations can be generally termed “Ewing-like” sarcomas. These definitions provide a useful categorical structure for the various molecular lesions driving oncogenesis in these tumours, but definitions will be continuously refined as our understanding of the molecular mechanisms of this disease expands.
Accurate and useful definitions are important insomuch as they may assist in grouping patients in clinically useful ways, such as into groups receiving different treatments or with different prognoses. These goals are especially challenging considering the rarity of non-EWS/FLI fusion variants, and little variation currently exists in the clinical management of different types of fusions.
9.2 Challenges of Molecular Diagnosis
The existence of alternate chromosomal rearrangements has clear implications for the diagnosis of Ewing sarcoma. Current diagnostic methods utilized to identify the EWS/FLI fusion may not identify the less common translocations. For instance, breakapart FISH (fluorescence in situ hybridization) probes for EWSR1 are commonly utilized to determine that a translocation involving EWSR1 exists and are, thus, useful for detecting the most common rearrangements in Ewing sarcoma (i.e., EWS/FLI and EWS/ERG) [138, 139]. This method, however, is unable to detect Ewing sarcoma driven by non-EWSR1 rearrangements, such as the rarer FUS/ERG and FUS/FEV fusions (Fig. 15.6).

Diagrammatic representation of a breakapart fluorescence in situ hybridization (FISH) assay for EWSR1. Fluorescent red and green probes flank the EWSR1 gene. Intact EWSR1 with both probes appears yellow. A translocation splits the gene, resulting in split red and green signals. In diploid cells, separate red and green signals result from the split chromosome, and the normal second allele appears yellow
Reverse-transcriptase (RT)-PCR assays have also been utilized to detect the fusion transcript [139]. Such an approach suffers from the same weakness as the EWSR1 breakapart FISH assay in that it is unable to detect transcripts of all possible gene fusions. For instance, primers designed to amplify specific EWSR1/FLI1 fusions will not anneal to EWSR1/ERG or other alternate transcripts. Despite this weakness, one potential benefit to using a PCR-based assay is the ability to detect specific breakpoints, although this may not be clinically useful, as discussed earlier [75, 76, 78].
Hence, the rare cases of Ewing sarcoma driven by alternate translocations may theoretically result in delayed or incorrect diagnosis in uncommon cases. Clearly, the correct diagnosis of Ewing sarcoma must not rely on one single test but rather on a collection of various criteria, including patient presentation, imaging studies (e.g., X-ray, CT, MRI), histopathology, and pathognomonic molecular lesions such as EWS/FLI. Such a practice of integrating distinct pieces of data to come to a definitive diagnosis is the current practice, allowing for prompt and accurate diagnosis in almost all cases.
10 Conclusions
Although it is rare compared to other malignancies, Ewing sarcoma is a devastating disease affecting many young people, resulting in many years of life lost to morbidity and mortality. Over the past 30 years, scientists have made great strides in understanding the molecular mechanisms underlying this cancer. Nevertheless, the increased knowledge gained through studying the cellular and molecular biology of this disease has not yet led to improvements in clinical management. Current standards of care rely on conventional therapies like surgery and chemotherapy, and improved usage of these treatment modalities have achieved remarkable success in overall survival. No molecularly targeted therapy has been found to be efficacious against Ewing sarcoma, despite increased understanding of the molecular biology of the disease.
The EWS/FLI fusion protein, and the other fusions found in Ewing sarcoma, clearly offer a unique pathogenic feature of this disease that could be targeted. However, transcription factors have proven to be extraordinarily challenging targets for inhibition, often earning them the epithet “undruggable”. Thus most efforts have focused on developing deeper understanding of the functions of critical effectors of EWS/FLI-driven oncogenesis. Although progress has been slow, a few promising targets have recently emerged [141]. Future work will continue to pursue a clearer understanding of the oncogenic consequences of the chromosomal rearrangements discussed in this chapter. Understanding why these translocations drive oncogenesis will assist in developing new therapies, likely increasing the odds of survival and bettering post-survival quality of life in these patients.
References
Ewing J (1921) Diffuse endothelioma of bone. Proc N Y Phytopathol Soc 21:17–24
Stiller CA, Bielack SS, Jundt G, Steliarova-Foucher E (2006) Bone tumours in European children and adolescents, 1978–1997. Report from the Automated Childhood Cancer Information System project. Eur J Cancer 42:2124–2135
Aurias A, Rimbaut C, Buffe D, Dubousset J, Mazabraud A (1983) Translocation of chromosome 22 in Ewing’s sarcoma. C R Seances Acad Sci III Sci Vie 296:1105–1107
Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir G (1983) Chromosomal translocation (11; 22) in cell lines of Ewing’s sarcoma. C R Seances Acad Sci III Sci Vie 296:1101–1103
Janknecht R (2005) EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene 363:1–14
Horowitz ME, Malawer MM, Woo SY, Hicks MJ (1997) In: Pizzo PA, Poplack DG (eds) Principles and practice of pediatric oncology. Lippincott-Raven Publishers, Philadelphia, pp 831–863
Kimber C, Michalski A, Spitz L, Pierro A (1998) Primitive neuroectodermal tumours: anatomic location, extent of surgery, and outcome. J Pediatr Surg 33:39–41
Grier HE (1997) The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin N Am 44:991–1004
Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir GM (1984) Chromosome study of Ewing’s sarcoma (ES) cell lines. Consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet Cytogenet 12:1–19
Kovar H (1998) Ewing’s sarcoma and peripheral primitive neuroectodermal tumors after their genetic union. Curr Opin Oncol 10:334–342
Whang-Peng J et al (1984) Chromosome translocation in peripheral neuroepithelioma. N Engl J Med 311:584–585
Ries LAG et al (2001) SEER cancer statistics review, 1973–1998. National Cancer Institute, Bethesda
Surveillance, E., and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence – SEER 9 Regs Research Data, Nov 2012 Sub (1973–2010) <Katrina/Rita Population Adjustment> – Linked to county attributes – total U.S., 1969–2011 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2013, based on the November 2012 submission. Accessed February 2013
Stiller CA et al (2006) Cancer incidence and survival in European adolescents (1978–1997). Report from the Automated Childhood Cancer Information System project. Eur J Cancer 42:2006–2018
Denny CT (1998) Ewing’s sarcoma – a clinical enigma coming into focus. J Pediatr Hematol Oncol 20:421–425
Paulussen M, Frohlich B, Jurgens H (2001) Ewing tumour: incidence, prognosis and treatment options. Pediatr Drugs 3:899–913
Parkin DM, Stiller CA, Nectoux J (1993) International variations in the incidence of childhood bone tumours. Int J Cancer 53:371–376
Polednak AP (1985) Primary bone cancer incidence in black and white residents of New York State. Cancer 55:2883–2888
Bahebeck J et al (2003) Bone tumours in Cameroon: incidence, demography and histopathology. Int Orthop 27:315–317
Settakorn J et al (2006) Spectrum of bone tumors in Chiang Mai University Hospital, Thailand according to WHO classification 2002: a study of 1,001 cases. J Med Assoc Thai 89:780–787
Guo W, Xu W, Huvos AG, Healey JH, Feng C (1999) Comparative frequency of bone sarcomas among different racial groups. Chin Med J (Engl) 112:1101–1104
Terrier P, Llombart-Bosch A, Contesso G (1996) Small round blue cell tumors in bone: prognostic factors correlated to Ewing’s sarcoma and neuroectodermal tumors. Semin Diagn Pathol 13:250–257
Wang CC, Schulz MD (1953) Ewing’s sarcoma; a study of fifty cases treated at the Massachusetts General Hospital, 1930–1952 inclusive. N Engl J Med 248:571–576
Dahlin DC, Coventry MB, Scanlon PW (1961) Ewing’s sarcoma. A critical analysis of 165 cases. J Bone Joint Surg Am 43-A:185–192
Lahl M, Fisher VL, Laschinger K (2008) Ewing’s sarcoma family of tumors: an overview from diagnosis to survivorship. Clin J Oncol Nurs 12:89–97. doi:10.1188/08.CJON.89-97
Lee ES (1971) Treatment of bone sarcoma. Proc R Soc Med 64:1179–1180
Rosen G et al (1978) Curability of Ewing’s sarcoma and considerations for future therapeutic trials. Cancer 41:888–899
Sankar S, Lessnick SL (2011) Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet 204:351–365. doi:10.1016/j.cancergen.2011.07.008
Linabery AM, Ross JA (2008) Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975–1999. Cancer 113:2575–2596
Randall RL et al (2010) Is there a predisposition gene for Ewing’s sarcoma? J Oncol 2010:397632. doi:10.1155/2010/397632
May WA et al (2013) Characterization and drug resistance patterns of Ewing’s sarcoma family tumor cell lines. PLoS One 8:e80060. doi:10.1371/journal.pone.0080060
Potratz J, Dirksen U, Jurgens H, Craft A (2012) Ewing sarcoma: clinical state-of-the-art. Pediatr Hematol Oncol 29:1–11. doi:10.3109/08880018.2011.622034
Turc-Carel C et al (1988) Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet 32:229–238
Delattre O et al (1992) Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359:162–165
Jeon IS et al (1995) A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 10:1229–1234
Kaneko Y et al (1996) Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Gene Chromosome Cancer 15:115–121
Peter M et al (1997) A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 14:1159–1164
Sorensen PH et al (1994) A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 6:146–151. doi:10.1038/ng0294-146
Urano F, Umezawa A, Hong W, Kikuchi H, Hata J (1996) A novel chimera gene between EWS and E1A-F, encoding the adenovirus E1A enhancer-binding protein, in extraosseous Ewing’s sarcoma. Biochem Biophys Res Commun 219:608–612
Aman P et al (1996) Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37:1–8
Ohno T et al (1994) The EWS gene, involved in Ewing family of tumors, malignant melanoma of soft parts and desmoplastic small round cell tumors, codes for an RNA binding protein with novel regulatory domains. Oncogene 9:3087–3097
May WA et al (1993) The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol 13:7393–7398
Sutherland HG et al (2001) Large-scale identification of mammalian proteins localized to nuclear sub-compartments. Hum Mol Genet 10:1995–2011
Zakaryan RP, Gehring H (2006) Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J Mol Biol 363:27–38
Araya N et al (2003) Cooperative interaction of EWS with CREB-binding protein selectively activates hepatocyte nuclear factor 4-mediated transcription. J Biol Chem 278:5427–5432
Bertolotti A et al (1998) EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol 18:1489–1497
Petermann R et al (1998) Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 17:603–610
Rossow KL, Janknecht R (2001) The Ewing’s sarcoma gene product functions as a transcriptional activator. Cancer Res 61:2690–2695
Kwon I et al (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155:1049–1060. doi:10.1016/j.cell.2013.10.033
Deloulme JC, Prichard L, Delattre O, Storm DR (1997) The prooncoprotein EWS binds calmodulin and is phosphorylated by protein kinase C through an IQ domain. J Biol Chem 272:27369–27377
Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363:640–644
Panagopoulos I et al (1999) Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11). Oncogene 18:7594–7598
Zucman J et al (1993) EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 4:341–345
Ladanyi M, Gerald W (1994) Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res 54:2837–2840
Martini A et al (2002) Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res 62:5408–5412
Gill S et al (1995) Fusion of the EWS gene to a DNA segment from 9q22-31 in a human myxoid chondrosarcoma. Gene Chromosome Cancer 12:307–310
Sjogren H et al (2000) Fusion of the NH2-terminal domain of the basic helix-loop-helix protein TCF12 to TEC in extraskeletal myxoid chondrosarcoma with translocation t(9;15)(q22;q21). Cancer Res 60:6832–6835
Panagopoulos I et al (1996) Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 12:489–494
Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L (1996) hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J 15:5022–5031
Morohoshi F, Arai K, Takahashi EI, Tanigami A, Ohki M (1996) Cloning and mapping of a human RBP56 gene encoding a putative RNA binding protein similar to FUS/TLS and EWS proteins. Genomics 38:51–57
Stolow DT, Haynes SR (1995) Cabeza, a Drosophila gene encoding a novel RNA binding protein, shares homology with EWS and TLS, two genes involved in human sarcoma formation. Nucleic Acids Res 23:835–843
Kato M et al (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149:753–767. doi:10.1016/j.cell.2012.04.017
Seth A, Watson DK (2005) ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 41:2462–2478
Sharrocks AD (2001) The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2:827–837
Ben-David Y, Giddens EB, Bernstein A (1990) Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc Natl Acad Sci U S A 87:1332–1336
Ben-David Y, Giddens EB, Letwin K, Bernstein A (1991) Erythroleukemia induction by Friend murine leukemia virus: insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1. Genes Dev 5:908–918
Bastian LS, Kwiatkowski BA, Breininger J, Danner S, Roth G (1999) Regulation of the megakaryocytic glycoprotein IX promoter by the oncogenic Ets transcription factor Fli-1. Blood 93:2637–2644
Athanasiou M et al (1996) Increased expression of the ETS-related transcription factor FLI-1/ERGB correlates with and can induce the megakaryocytic phenotype. Cell Growth Differ 7:1525–1534
Hart A et al (2000) Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity 13:167–177
Smith R et al (2006) Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 9:405–416. doi:10.1016/j.ccr.2006.04.004
Zucman J et al (1992) Cloning and characterization of the Ewing’s sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Gene Chromosome Cancer 5:271–277
Zucman J et al (1993) Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 12:4481–4487
Zucman-Rossi J, Legoix P, Victor JM, Lopez B, Thomas G (1998) Chromosome translocation based on illegitimate recombination in human tumors. Proc Natl Acad Sci U S A 95:11786–11791
Lin PP et al (1999) Differential transactivation by alternative EWS-FLI1 fusion proteins correlates with clinical heterogeneity in Ewing’s sarcoma. Cancer Res 59:1428–1432
de Alava E et al (1998) EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol 16:1248–1255
Zoubek A et al (1996) Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol 14:1245–1251
Le Deley MC et al (2010) Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol Off J Am Soc Clin Oncol 28:1982–1988. doi:10.1200/JCO.2009.23.3585
van Doorninck JA et al (2010) Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol Off J Am Soc Clin Oncol 28:1989–1994. doi:10.1200/JCO.2009.24.5845
Lessnick SL, Braun BS, Denny CT, May WA (1995) Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI- 1 fusion gene. Oncogene 10:423–431
May WA et al (1993) Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A 90:5752–5756
Hancock JD, Lessnick SL (2008) A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle 7:250–256
Teitell MA et al (1999) EWS/ETS fusion genes induce epithelial and neuroectodermal differentiation in NIH 3T3 fibroblasts. Lab Invest 79:1535–1543
Thompson AD, Teitell MA, Arvand A, Denny CT (1999) Divergent Ewing’s sarcoma EWS/ETS fusions confer a common tumorigenic phenotype on NIH3T3 cells. Oncogene 18:5506–5513. doi:10.1038/sj.onc.1202928
Chan D et al (2003) Transformation induced by Ewing’s sarcoma associated EWS/FLI-1 is suppressed by KRAB/FLI-1. Br J Cancer 88:137–145
Chansky HA et al (2004) Targeting of EWS/FLI-1 by RNA interference attenuates the tumor phenotype of Ewing’s sarcoma cells in vitro. J Orthop Res 22:910–917
Kovar H et al (1996) EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ 7:429–437
Matsumoto Y et al (2001) Downregulation and forced expression of EWS-Fli1 fusion gene results in changes in the expression of G(1)regulatory genes. Br J Cancer 84:768–775
Ouchida M, Ohno T, Fujimura Y, Rao VN, Reddy ES (1995) Loss of tumorigenicity of Ewing’s sarcoma cells expressing antisense RNA to EWS-fusion transcripts. Oncogene 11:1049–1054
Prieur A, Tirode F, Cohen P, Delattre O (2004) EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol Cell Biol 24:7275–7283
Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y (1997) EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J Clin Invest 99:239–247
Toretsky JA, Connell Y, Neckers L, Bhat NK (1997) Inhibition of EWS-FLI-1 fusion protein with antisense oligodeoxynucleotides. J Neuro Oncol 31:9–16
Kinsey M, Smith R, Lessnick SL (2006) NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res 4:851–859
Kauer M et al (2009) A molecular function map of Ewing’s sarcoma. PLoS One 4:e5415
Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL (2009) EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res 69:9047–9055. doi:10.1158/0008-5472.CAN-09-1540
Baer C et al (2004) Profiling and functional annotation of mRNA gene expression in pediatric rhabdomyosarcoma and Ewing’s sarcoma. Int J Cancer 110:687–694
Beauchamp E et al (2009) GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem 284:9074–9082
Zwerner JP et al (2008) The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 27:3282–3291. doi:10.1038/sj.onc.1210991, 1210991 [pii]
Jaishankar S, Zhang J, Roussel MF, Baker SJ (1999) Transforming activity of EWS/FLI is not strictly dependent upon DNA-binding activity. Oncogene 18:5592–5597
Welford SM, Hebert SP, Deneen B, Arvand A, Denny CT (2001) DNA binding domain-independent pathways are involved in EWS/FLI1-mediated oncogenesis. J Biol Chem 276:41977–41984
Gangwal K et al (2008) Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc Natl Acad Sci U S A 105:10149–10154. doi:10.1073/pnas.0801073105
Mao X, Miesfeldt S, Yang H, Leiden JM, Thompson CB (1994) The FLI-1 and chimeric EWS-FLI-1 oncoproteins display similar DNA binding specificities. J Biol Chem 269:18216–18222
Gangwal K, Lessnick SL (2008) Microsatellites are EWS/FLI response elements: genomic “junk” is EWS/FLI’s treasure. Cell Cycle 7:3127–3132
Guillon N et al (2009) The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS One 4:e4932
Sankar S et al (2013) Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 32(42):5089–5100
Sorna V et al (2013) High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J Med Chem 56:9496–9508. doi:10.1021/jm400870h
Prasad DD, Rao VN, Lee L, Reddy ES (1994) Differentially spliced erg-3 product functions as a transcriptional activator. Oncogene 9:669–673
Ginsberg JP et al (1999) EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcoma. J Clin Oncol 17:1809–1814
Braunreiter CL, Hancock JD, Coffin CM, Boucher KM, Lessnick SL (2006) Expression of EWS-ETS fusions in NIH3T3 cells reveals significant differences to Ewing’s sarcoma. Cell Cycle 5:2753–2759
Wang L et al (2007) Undifferentiated small round cell sarcomas with rare EWS gene fusions: identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn 9:498–509
Ng TL et al (2007) Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn 9:459–463
Shing DC et al (2003) FUS/ERG gene fusions in Ewing’s tumors. Cancer Res 63:4568–4576
Chansky HA, Hu M, Hickstein DD, Yang L (2001) Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res 61:3586–3590
Cironi L et al (2008) IGF1 is a common target gene of Ewing’s sarcoma fusion proteins in mesenchymal progenitor cells. PLoS One 3:e2634
Rhodes M et al (1998) A high-resolution microsatellite map of the mouse genome. Genome Res 8:531–542
Kovar H et al (1990) Overexpression of the pseudoautosomal gene MIC2 in Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. Oncogene 5:1067–1070
Szuhai K et al (2009) The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 15:2259–2268
Chen L, Glover JN, Hogan PG, Rao A, Harrison SC (1998) Structure of the DNA-binding domains from NFAT. Fos and Jun bound specifically to DNA. Nature 392:42–48. doi:10.1038/32100
Macian F (2005) NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5:472–484. doi:10.1038/nri1632
Macian F, Lopez-Rodriguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20:2476–2489. doi:10.1038/sj.onc.1204386
Rao A, Luo C, Hogan PG (1997) Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15:707–747. doi:10.1146/annurev.immunol.15.1.707
Kim S, Denny CT, Wisdom R (2006) Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-ETS fusion proteins. Mol Cell Biol 26:2467–2478
Thomas RS et al (1997) ETS1, NFkappaB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene 14:2845–2855. doi:10.1038/sj.onc.1201125
Verger A et al (2001) Identification of amino acid residues in the ETS transcription factor Erg that mediate Erg-Jun/Fos-DNA ternary complex formation. J Biol Chem 276:17181–17189
Yamaguchi S et al (2005) EWSR1 is fused to POU5F1 in a bone tumor with translocation t(6;22)(p21;q12). Gene Chromosome Cancer 43:217–222
Nichols J et al (1998) Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95:379–391
Okamoto K et al (1990) A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell 60:461–472
Rosner MH et al (1990) A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature 345:686–692. doi:10.1038/345686a0
Mastrangelo T et al (2000) A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene 19:3799–3804
Bossone SA, Asselin C, Patel AJ, Marcu KB (1992) MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc Natl Acad Sci U S A 89:7452–7456
Bochar DA et al (2000) A family of chromatin remodeling factors related to Williams syndrome transcription factor. Proc Natl Acad Sci U S A 97:1038–1043
Bozhenok L, Wade PA, Varga-Weisz P (2002) WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J 21:2231–2241. doi:10.1093/emboj/21.9.2231
LeRoy G, Loyola A, Lane WS, Reinberg D (2000) Purification and characterization of a human factor that assembles and remodels chromatin. J Biol Chem 275:14787–14790. doi:10.1074/jbc.C000093200
Percipalle P, Farrants AK (2006) Chromatin remodelling and transcription: be-WICHed by nuclear myosin 1. Curr Opin Cell Biol 18:267–274. doi:10.1016/j.ceb.2006.03.001
Strohner R et al (2001) NoRC – a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J 20:4892–4900. doi:10.1093/emboj/20.17.4892
Sumegi J et al (2011) A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol Off J U S Can Acad Pathol 24:333–342. doi:10.1038/modpathol.2010.201
Machado I et al (2013) Superficial EWSR1-negative undifferentiated small round cell sarcoma with CIC/DUX4 gene fusion: a new variant of Ewing-like tumors with locoregional lymph node metastasis. Virchows Arch Int J Pathol 463:837–842. doi:10.1007/s00428-013-1499-9
Pierron G et al (2012) A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 44:461–466. doi:10.1038/ng.1107
de Alava E, Lessnick SL, Sorensen PH (2013) In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of tumours of soft tissue and bone. International Agency for Research on Cancer (IARC), Lyons, France, pp 305–309, Ch. 19
Carpentieri DF et al (2005) Protocol for the examination of specimens from pediatric and adult patients with osseous and extraosseous ewing sarcoma family of tumors, including peripheral primitive neuroectodermal tumor and ewing sarcoma. Arch Pathol Lab Med 129:866–873
Folpe AL et al (2005) Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol 29:1025–1033
Bennani-Baiti IM, Machado I, Llombart-Bosch A, Kovar H (2012) Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol 43:1300–1307. doi:10.1016/j.humpath.2011.10.010
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tanner, J.M., Lessnick, S.L. (2015). Translocations in Ewing Sarcoma. In: Rowley, J., Le Beau, M., Rabbitts, T. (eds) Chromosomal Translocations and Genome Rearrangements in Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-19983-2_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-19983-2_15
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19982-5
Online ISBN: 978-3-319-19983-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)