
Abstract
Amyloid fibrils are an intriguing class of protein aggregates with distinct physicochemical, structural and morphological properties. They display peculiar membrane-binding behavior, thus adding complexity to the problem of protein-lipid interactions. The consensus that emerged during the past decade is that amyloid cytotoxicity arises from a continuum of cross-β-sheet assemblies including mature fibrils. Based on literature survey and our own data, in this chapter we address several aspects of fibril-lipid interactions, including (i) the effects of amyloid assemblies on molecular organization of lipid bilayer; (ii) competition between fibrillar and monomeric membrane-associating proteins for binding to the lipid surface; and (iii) the effects of lipids on the structural morphology of fibrillar aggregates. To illustrate some of the processes occurring in fibril-lipid systems, we present and analyze fluorescence data reporting on lipid bilayer interactions with fibrillar lysozyme and with the N-terminal 83-residue fragment of amyloidogenic mutant apolipoprotein A-I, 1-83/G26R/W@8. The results help understand possible mechanisms of interaction and mutual remodeling of amyloid fibers and lipid membranes, which may contribute to amyloid cytotoxicity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Protein-lipid interactions
- Amyloid fibrils
- Lysozyme
- N-terminal fragment of apolipoprotein A-I
- Fluorescence spectroscopy
6.1 Introduction
Protein-lipid interactions have long been recognized as an effective modulator of a wide range of membrane processes, including intracellular transport, enzyme function, respiration, antimicrobial defense, signal transduction, motility, etc. (Lee 2003, 2004; Palsdottir and Hunte 2004). Along with continuous expansion, this research area currently undergoes a substantial shift in focus towards ascertaining the role of lipids in modulating the polypeptide self-association and elucidating membrane responses to aggregated proteins. Of paramount interest are amyloid fibrils, a special type of protein aggregates involved in the pathogenesis of numerous conformational disorders, such as Parkinson’s, Alzheimer’s and Huntington’s diseases, type II diabetes, systemic amyloidosis, etc. (Stefani 2004). These aggregates are assembled from proteins or peptides adopting a non-native β-structure-enriched conformation, and have distinct highly ordered structural organization with β-sheets propagating along the fibril axis (Serpell 2000; Kelly 2002). Due to fundamental similarity between intra- and interchain interactions, protein fibrillization represents an alternative folding pathway of partially unfolded or misfolded proteins that acquire a stable structure by reaching the minimum on the energy landscape (Zbilut et al. 2003). Fibril formation is nucleation-dependent and proceeds through thermodynamically unfavorable monomer association into a critical oligomeric nucleus followed by its subsequent energetically favorable elongation and the exponential fibril growth (Dima and Thirumalai 2002).
Emerging evidence indicates that lipid bilayer can substantially accelerate amyloid nucleation through accumulation of the protein species with specific aggregation-prone conformation, orientation and location at the lipid-water interface (Stefani and Dobson 2003; Gorbenko and Kinnunen 2006; Stefani 2008; Aisenbrey et al. 2008). At the same time, cell membranes are thought to be a primary target for toxic amyloid assemblies (Bucciantini et al. 2014). Membrane damage produced by the early protein oligomers is regarded as the main cause of cytotoxicity (Kinnunen 2009; Relini et al. 2009; Stefani 2010; Butterfield and Lashuel 2010). Cytotoxic action of protein oligomers can be attributed to compromised membrane integrity (Meratan et al. 2011; Huang et al. 2009), formation of non-specific ion channels (Caughey and Lansbury 2003), uptake of lipids into the fibers growing on a membrane template (Sparr et al. 2004; Engel et al. 2008), alterations in the intracellular redox status and free calcium level (Arispe et al. 1993; Squier 2001; Tabner et al. 2002), and impaired functions of membrane proteins (Stefani 2007). Several lines of evidence suggest that oligomeric species display higher membrane-binding affinity compared to mature fibrils. This was demonstrated for several proteins including the N-terminal domain of the hydrogenase maturation factor HypF-N (Relini et al. 2004; Canale et al. 2006, AC), stefin B (Anderluh et al. 2005), α-synuclein (Giannakis et al. 2008; Smith et al. 2008), lysozyme (Meratan et al. 2011) and Abeta (Williams and Serpell 2011). This increased affinity was explained in terms of hydrophobicity-based toxicity mechanism, highlighting the importance of factors such as extensive hydrophobic surfaces and high flexibility of the protein oligomers (Meratan et al. 2011). Nevertheless, it is becoming increasingly clear that, although mature amyloid fibrils bind membrane relatively weakly, they are far from being chemically inert nontoxic species. Numerous studies indicate substantial cytotoxic potential of fibrillar aggregates (Weldon et al. 1998; Forloni 1996; Gharibyan et al. 2007; Matsuzaki 2011; Bucciantini et al. 2012). In particular, Novitskaya et al. showed that mature amyloid fibrils of mammalian prion protein are as toxic as the soluble oligomers to cultured cells and primary neurons (Novitskaya et al. 2006). Different toxicity mechanisms operating through apoptotic and necrotic pathways were revealed for oligomers and fibrils of hen egg white lysozyme in a process leading to neuroblastoma cell death. Mature lysozyme fibrils can induce mitochondrial failure and increase plasma membrane permeability. Hence, cytotoxicity is inherent to a continuum of cross-β-sheet-rich structures rather than to a single uniform species (Gharibyan et al. 2007).
The relationship between the morphology of fibrillar aggregates and their toxicity was reported by Petkova and colleagues who examined the action of mature amyloid fibrils of Alzheimer’s amyloid-beta peptide, Aβ1–40, on neuronal cell cultures (Petkova et al. 2005). The absence of direct correlation between disease symptoms and total amyloid deposition was considered as the main evidence of the nontoxic nature of mature fibrils. High surface hydrophobicity and solvent accessibility of disulfides in the structure of fibrillar lysozyme were proposed to be responsible for the membrane-disruptive effect of lysozyme fibrils that brought about hemolysis of erythrocytes and their aggregation coupled with intermolecular disulfide cross-linking (Huang et al. 2009). Notably, amyloid fibrils can manifest their toxicity not only nonspecifically, but also via specific pathways involving the activation of cellular receptors, as was demonstrated for transthyretin and Aβ peptide (Bamberger et al. 2003; Sousa et al. 2001). Finally, fragmentation of mature fibrils upon mechanical stress, thermal motion or chaperone activity was reported to enhance cytotoxic potential of amyloid (Carulla et al. 2005; Smith et al. 2006; Xue et al. 2009). Together, these studies highlight the importance of further in-depth analyses of biological activities of amyloid fibrils in relation to their physicochemical, structural and morphological properties. Evidently, gaining better insights into the membrane-associating and bilayer-modifying behavior of amyloid fibrils, which represent a specific aggregation state of polypeptide chain, adds a new exciting dimension to the problem of protein-lipid interactions.
The present chapter provides a concise overview of the available information concerning structural basis for membrane damage by fibrillar aggregates and presents some of our own pertinent data. Specifically, we scrutinize such processes as (i) changes in membrane structure in response to fibril binding; (ii) competition between fibrillar and monomeric proteins for association with a lipid bilayer; and (iii) the ability of lipid membrane to remodel amyloid fibrils.
6.2 Effects of Amyloid Fibrils on the Membrane
6.2.1 Effects of Protein Fibrils and Oligomers on the Membrane Structure and Dynamics
One of the mechanisms by which amyloid fibril can exert their cytotoxicity involves changes in the structure and dynamics of the cell membrane. It is well established that protein-lipid interactions commonly involve interrelated processes of conformational changes in the protein molecule as well as structural rearrangement of the membrane lipids. However, the conceptual framework developed in this field cannot be directly extrapolated to fibril-lipid interactions. In fact, fibrillar protein aggregates of amyloid type possess unique physical properties, including exceptional rigidity largely arising from highly ordered cross-β array of hydrogen bonds, with Young’s modulus on the order of several GPa and persistence length of several tens of micrometers (Adamcik and Mezzenga 2012a). Compared to other biological filaments, amyloid fibrils are extremely stiff, approaching most rigid proteinaceous materials, such as silk, collagen and keratin (Knowles and Buehler 2011). Moreover, regular linear arrangement of the charged, hydrophobic or aromatic amino-acid side chains in fibrillar structure creates reactive surfaces uncharacteristic of natively folded proteins (Adamcik and Mezzenga 2012b). Accordingly, there may be substantial differences in membrane responses to monomeric, oligomeric and fibrillar polypeptides.
This issue has been addressed in a number of recent studies by using a variety of experimental approaches such as atomic force microscopy (AFM) (Giannakis et al. 2008), infrared reflection absorption spectroscopy (Lopes et al. 2007), surface plasmon resonance (Smith et al. 2008), small-angle neutron scattering (Dante et al. 2008), circular dichroism (Quist et al. 2005), fluorescence spectroscopy (Kremer et al. 2001), micropipette manipulation (Kim and Frangos 2008), conductivity measurements (Valincius et al. 2008), and dye release assay (van Rooijen et al. 2009).
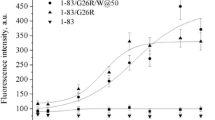
To address the problem of fibril-lipid interactions, we used two proteins, hen egg white lysozyme and 1-83/G26R/W@8, which is the N-terminal 1-83 fragment of human apolipoprotein A-I containing a common amyloidogenic mutation G26R and an engineered Trp8 as a reporter group (Adachi et al. 2013). We explored the influence of these two fibrillar proteins on the structural state of model membranes composed of phosphatidylcholine and its mixtures with cholesterol (30 mol%) or cardiolipin (10 mol%). Figure 6.1 shows transmission electron microscopy images of the examined lysozyme (A) and 1-83/G26R/W@8 (B) fibrils.

Transmission electron micrographs of negatively stained fibrils of lysozyme (a) and 1-83/G26R/W@8 (b). Scale bar is 100 nm
Lysozyme is a ubiquitous multifunctional protein displaying bactericidal, antitumor and immunomodulatory activities. The mutants of human lysozyme (I56T, F57I, W64R, D67H) undergo pathological fibrillization associated with familial non-neuropathic systemic amyloidosis, a disease affecting kidney, liver and spleen (Pepys et al. 1993). Amyloidogenicity of this protein is thought to arise from the enhanced propensity of its mutants to adopt a partially unfolded aggregation-prone conformation (Frare et al. 2004). Specific segment in hen egg white lysozyme encompassing residues 54–62 was recently proposed to serve as the amyloid core that triggers fibril formation (Tokunaga et al. 2013).
ApoA-I is the major protein component of high-density lipoproteins promoting efflux of phospholipid and cholesterol from plasma membrane (Phillips 2013). Specific naturally occurring variants of human apoA-I, the most common of which is a single substitution mutant G26R, can form amyloid fibrils associated with renal or liver failure in hereditary systemic amyloidosis (Joy et al. 2003). The N-terminal fragments 1-83–1-93 have been identified as the predominant form of apoA-I in amyloid fibril deposits (Nichols et al. 1990; Obici et al. 2006). One of such fragments is used in our research.
To monitor the changes in the molecular organization of the lipid bilayer induced by fibrillar lysozyme and 1-83/G26R/W@8 peptide, two fluorescent probes differing in their bilayer location have been employed: Laurdan that resides at the lipid-water interface and pyrene that is partitioned in the apolar core of lipid bilayer.
Fluorescent membrane probe Laurdan is characterized by a high sensitivity to variations in membrane hydration and lipid packing density (Parasassi and Gratton 1995; Parasassi et al. 1998). In a lipid bilayer, this amphiphilic fluorophore is localized at the level of the glycerol backbone, with lauric acid tail anchored in the acyl chain region. Laurdan’s emission spectrum has two clearly distinct components that are attributed to solvent-unrelaxed (shorter-wavelength band centered circa 440 nm) and solvent-relaxed states (longer-wavelength band centered circa 490 nm) (Lúcio et al. 2010; Sanchez et al. 2012). This photophysical property is thought to originate from the reorientation of water dipoles around the excited-state dipole of the probe molecule. The environment-dependent spectral changes of Laurdan are generally quantitatively described by the steady-state fluorescence parameter known as the generalized polarization (GP) (Parasassi et al. 1991).
As illustrated in Fig. 6.2, fibrillar lysozyme produced GP increase in the model membranes comprised of phosphatidylcholine (PC) and its mixture with cardiolipin (CL) or cholesterol (Chol). In contrast, amyloid fibrils of the apoA-I fragment 1-83/G26R/W@8 brought about decrease in GP value of PC and PC/Chol bilayers. These findings imply that association of fibrillar apoA-I mutant with the membrane leads to the increase in lipid bilayer hydration and decrease of lipid packing density at the level of glycerol backbone, whereas lysozyme fibrils produce opposite effects. The latter is consistent with the observation that the binding of fibrillar lysozyme to liposomes is followed by the short-wavelength shift of Trp emission maximum, from ~352 to ~343 nm, indicating the transfer Trp62 and Trp108 (which dominate the emission) to interfacial bilayer region containing bound water with restricted mobility (Gorbenko et al. 2012). Therefore, different protein fibrils can produce distinctly different effects on the properties of model liposomes.

Relative changes in the generalized polarization of Laurdan induced by fibrillar lysozyme and 1-83/G26R/W@8 in the model membranes comprised of PC, PC/CL (9:1, mol:mol) and PC/Chol (7:3, mol:mol). Unilamellar liposomes 100 nm in diameter were used in the lysozyme studies, and 50 nm in diameter in 1-83/G26R/W@8 studies
Notably, compared to PC vesicles, in similar vesicles of PC:Chol (7:3, mol:mol), both these fibrillar proteins produced much less pronounced changes in GP. Hence, the ability of fibrils to modify physical properties of the interfacial membrane region can be hampered by cholesterol. This observation is in good agreement with numerous studies suggesting that cholesterol can prevent membrane disruption by the aggregated proteins (Sponne et al. 2004; Cecchi et al. 2005; Qiu et al. 2011). In particular, cholesterol was demonstrated to protect primary cortical neurons from neurotoxic effects of soluble oligomeric Aβ by modulating the physical properties of lipid bilayer. Model membrane studies showed that increase in cholesterol content inhibited aggregation and fusion of liposomes induced by Aβ (1-40) peptide (Sponne et al. 2004). The cell membranes rich in cholesterol were found to strongly resist to the remodelling by prefibrillar aggregates of the N-terminal domain of the prokaryotic hydrogenase maturation factor HypF (Cecchi et al. 2005). The perturbations in the polar part of the model PC membrane by lysozyme oligomers were also suppressed upon cholesterol inclusion in this membrane (Gorbenko and Trusova 2011). Taken together, these studies suggest that cholesterol in lipid membranes prevents their remodeling by pre-fibrillar and fibrillar protein aggregates, perhaps because increased phospholipid headgroup packing in the presence of cholesterol precludes fibril binding to the bilayer.
Next, to monitor lipid bilayer modifications occurring at the level of acyl chains, we employed a classical fluorescent probe pyrene, a polycyclic aromatic compound that is primarily distributed at the level of carbons 4–13 in the hydrocarbon region of the bilayer (Loura et al. 2013). Emission spectrum of this probe has characteristic vibronic structure in the wavelength range 370–400 nm, with relative intensities of vibronic transitions depending on the polarity of the fluorophore microenvironment via the so-called “Ham effect” (Nakajima 1971). Specifically, the intensity of the third vibronic peak (0–2 transition) is significantly enhanced in the hydrophobic environment, while the intensity of the first vibronic peak (0-0 transition) is increased in polar media. For this reason, the intensity ratio of the first-to-third vibronic band, I 1 /I 3 , has long been employed as an indicator of polarity in the vicinity of pyrene monomers. Accordingly, in water I 1 /I 3 was reported to be 1.96, while in the solvents of lower polarity this ratio decreases, reaching the value 0.6 in n-hexane (Karpovich and Blanchard 1995).
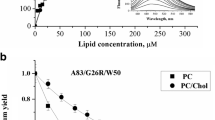
Figure 6.3 shows the emission spectra of pyrene in lipid vesicles in the absence and in the presence of fibrillar 1-83/G26R/W@8. Together with similar studies of lysozyme fibers, these results suggest that neither 1-83/G26R/W@8 nor lysozyme fibrils could induce any significant changes in relative intensity of the first-to-third vibronic bands. This suggests that the fibril contacts with the lipid surface do not affect membrane polarity at the level of the initial acyl chain carbons where pyrene monomers are thought to reside.

Pyrene emission spectra in PC and PC/Chol unilamellar liposomes in the absence or in the presence of 1-83/G26R/W@8 peptide. Lipid concentration was 16 μM, protein concentration was 1 μM, pyrene concentration was 0.1 μM; liposome diameter was 50 nm
Moreover, these fibrillar proteins did not markedly influence another pyrene spectral parameter, excimer-to-monomer intensity ratio (E/M). This parameter depends upon the rate of pyrene lateral diffusion and reflects dimerization of the excited- and ground-state probe molecules; such excimer formation manifests itself as a new fluorescent band circa 460–470 nm. Since pyrene excimerization is controlled by the frequency of collisions between the probe monomers in a lipid bilayer which, in turn, is a function of molecular packing density, this process is commonly analyzed in terms of the free volume model (Ioffe and Gorbenko 2005). This model considers pyrene diffusion in a lipid phase as a three-step process: (i) formation of dynamic defects (kinks) in the acyl chains followed by opening of the cavities in a lipid monolayer; (ii) jump of diffusing molecules into the cavities coupled with the generation of voids; (iii) sealing the voids by the movement of the packing defects along the adjacent hydrocarbon chains. The appearance of dynamic defects in the membrane interior is associated with trans-gauche isomerization of acyl chains initiated by thermal motion and packing constraints. Free volume of the membrane, which is produced by lateral displacements of hydrocarbon chains after kink formation, is defined as the difference between the effective and van der Waals volumes of lipid molecules. Accordingly, the changes in E/M ratio reflect altered rate of trans-gauche isomerization of hydrocarbon chains.
The observed invariance of I 1 /I 3 and E/M ratios, coupled with pronounced GP changes, strongly suggest that fibrillar lysozyme and 1-83/G26R/W@8 tend to perturb the interfacial bilayer region, with lipid tail order remaining virtually unaffected. This is probably a consequence of superficial fibril-lipid binding, which may involve adsorption of several liposomes onto a single fibril or wrapping of fibrillar strand around the lipid vesicle (Fig. 6.4). Our data agree with the results of Milanesi and colleagues indicating that fibrillar β2-microglobulin associates with the membrane surface, as follows from the fibril-induced clustering of liposomes (Milanesi et al. 2012). These studies suggest that protein fibers interact mainly with the surface of the lipid monolayer but cause little or no perturbation in its core.

Schematic representation of adsorption of multiple lipid vesicles along the amyloid fibril
In contrast to mature fibrils, lysozyme oligomers brought about the decrease in E/M ratio which probably reflects lower rate of trans-gauche isomerization of hydrocarbon chains (Gorbenko and Trusova 2011). Furthermore, the magnitude of bilayer perturbations induced by oligomeric lysozyme in the nonpolar region of the bilayer appeared to be independent of the membrane charge, suggesting that electrostatic forces do not play determining role in this perturbation. Consequently, hydrophobic interactions are likely to dominate the modification of the membrane structure by lysozyme pre-fibrillar aggregates. In agreement with this finding, various aggregated forms of Aβ amyloid peptide were reported to decrease bilayer fluidity in a fashion that correlated with surface hydrophobicity of the aggregated species (Kremer et al. 2000). Likewise, Aβ aggregates exerted ordering effect on the nonpolar part of the charged and neutral membranes, with little or no perturbation of the headgroup region (Kremer et al. 2001). Furthermore, the accessibility of the hydrocarbon core of the bilayer was demonstrated to modulate the membrane-disruptive effects of synuclein oligomers (van Rooijen et al. 2009). Taken together, these studies suggest that, compared to mature fibers, pre-fibrillar aggregates tend to insert more deeply into lipid bilayers, and that hydrophobic interactions dominate the ensuing structural perturbations in the hydrocarbon core of the bilayer.
6.2.2 Other Mechanisms of Fibril-Membrane Interactions
Evidently, aggregated proteins can influence natural membranes via diverse mechanisms, with lipid bilayer perturbations representing only one of the many effects. For example, lysozyme protofibrils can disintegrate erythrocyte membranes through intermolecular disulfide cross-linking of the membrane proteins (Huang et al. 2009) and induce apoptosis-like death of neurons, fibroblasts and neuroblastoma cells (Malisauskas et al. 2005; Gharibyan et al. 2007). Amyloid Aβ peptide can scramble erythrocyte membrane with subsequent cell death (Nicolay et al. 2007). Similarly to the rigidification of the model membranes (Kremer et al. 2001), Aβ aggregates can also reduce fluidity of hippocampal membranes (Eckert et al. 2000) which results from the peptide influence on the hydrocarbon core. However, other studies reported that aggregated Aβ perturbs mainly the interfacial region of the bilayer and has little effect on its hydrocarbon core (Ma et al. 2002). The main source of such discrepancies probably results from the high heterogeneity of the pre-fibrillar species as well as the structural polymorphism of mature fibrils (Stefani 2010).
Accumulating evidence substantiates the idea that disruption of cellular membrane by amyloid aggregates is one of the principal mechanisms of fibril-induced cellular dysfunction (Gorbenko and Kinnunen 2006; Bucciantini and Cecchi 2010; Bucciantini et al. 2014). The current consensus is that the mechanisms by which amyloidogenic proteins disrupt the membranes resemble those of antimicrobial peptides and include carpeting, toroidal or barrel-stave pore formation, non-specific membrane permeabilization, detergent- and raft-like membrane dissolution, etc. (Demuro et al. 2005; Lashuel and Lansbury 2006; Smith et al. 2009). These mechanisms are not mutually exclusive, so an individual peptide may cause membrane damage via different mechanisms depending on the membrane lipid composition. Changes in membrane integrity, which are initiated by mature fibrils, may result in increased lipid bilayer permeability (de Planque et al. 2007), lipid loss (Lee et al. 2012), receptor activation (Verdier et al. 2004), membrane fragmentation (Sciacca et al. 2012), and oxidation of membrane lipids (Butterfield et al. 2002).
Recent studies revealed a specific membrane distortion by fibrillar assemblies, which is different from the previously observed mechanisms of lipid bilayer disruption (Milanesi et al. 2012). Using confocal microscopy and cryoelectron tomography, Milanesi and coauthors visualized 3D membrane damage produced by fibrillar β2-microglobulin. Fibril-lipid interactions were found to result in re-shaping of lipid vesicles, interruptions to the bilayer structure, extraction of lipids from the membrane by removal or blebbing of the outer membrane leaflet, and formation of tiny vesicles from the extracted lipids. Remarkably, the largest distortions were generated by the fragmented fibrils, indicating that fibril ends possess much stronger membrane-modifying propensity than fibril shaft. This was ascribed to the enhanced hydrophobicity of fibril ends, a property shared by the prefibrillar oligomers that are currently regarded as the most toxic species of aggregated proteins (Butterfield and Lashuel 2010; Campioni et al. 2010; Winner et al. 2011; Cremades et al. 2012). This kind of membrane reorganization is thought to be distinct from the membrane breakage characteristic of pore-forming peptides (Tilley and Saibil 2006).
In general, concerted action of factors such as surface topography, hydrophobicity, charge distribution, hydrogen-bonding propensity, conformational flexibility, etc. seems to determine the reactivity of protein aggregates. On the other hand, membrane response to the aggregates of a certain type is likely to be controlled by a range of adjustable parameters of a lipid bilayer, such as surface charge, polarity, acyl chain order, fluidity, lateral pressure, curvature, etc. Therefore, while analyzing the structural basis for amyloid cytotoxicity, a membrane should be considered as a dynamic entity whose properties depend on its interactions with other components such as protein aggregates (Bucciantini et al. 2014).
6.3 Competitive Binding Behavior of Fibrillar Aggregates
The involvement of membrane structures in a wide variety of cellular processes implies multiple potential mechanisms of cell injury by aggregated proteins. In this regard, one cannot rule out competitive relationships between fibrillar species and native proteins within cellular environment. We hypothesized that in vivo amyloid fibrils can impair functionally relevant protein-membrane interactions, thereby initiating deleterious cell responses. As a first step to verify this idea, we determined whether fibrillar lysozyme can compete with the native cytochrome c (cyt c) for binding to the negatively charged model membranes composed of PC mixtures with varying proportions of anionic phospholipids, phosphatidylglycerol (PG), phosphatidylserine (PS) or CL. Biological activities of cytochrome c involve electron transport in the inner mitochondrial membrane and triggering of programmed cell death (Gray and Winkler 2010; Ascenzi et al. 2011). Monomeric lysozyme and cyt c, which are similar in their size (diameter ~3 nm) and charge (~ + 9e at physiological pH), display similar modes of membrane interaction, with relative contributions of electrostatic and hydrophobic interactions depending on the physicochemical characteristics of the membrane and the environmental conditions (Al Kayal et al. 2012).
To track membrane association of cyt c and its displacement with monomeric or polymerized lysozyme, we measured the efficiency of Förster resonance energy transfer (FRET) between anthrylvinyl-labeled PC (AV-PC) as a donor and heme group of cyt c as an acceptor. As shown by 1H-NMR-spectroscopy and AV fluorescence quenching by iodide, anthrylvinyl fluorophore resides at the level of terminal methyl groups preferentially orienting parallel to acyl chains (Molotkovsky et al. 1982). The competition between cyt c and lysozyme for the membrane binding sites manifested itself in the rise of AV quantum yield (Q r ) with increasing lysozyme concentration. Compared to its monomeric counterpart, fibrillar lysozyme caused substantially more detachment of cyt c from the membranes (Fig. 6.5). Moreover, the character of competitive adsorption to lipid bilayers was different for fibrillar and native states of lysozyme. Specifically, upon increasing the content of anionic lipid from 10 to 40 mol% (PG, PS) or from 5 to 25 mol% (CL), desorption curves changed their shape from hyperbolic to sigmoidal, suggesting that desorption of cyt c caused by lysozyme fibrils was a cooperative process. One possible reason for such a cooperativity may lie in simultaneous covering of multiple lipid headgroups in the contact areas of fibrils with liposomes and hence, exclusion of multiple cyt c molecules from these areas.

Relative quantum yield of AV-PC in protein-lipid systems as a function of cytochrome c concentration for PC/PG (3:2, mol:mol) and PC/CL (3:1, mol:mol) liposomes. Lipid concentration was 10 μM, lysozyme concentration was 0.5 μM. Liposome diameter was 100 nm
As follows from the mean-field thermodynamic analysis of surface adsorption in a binary mixture of charged model proteins, competitive behavior of a protein strongly depends upon its molecular parameters such as size, shape, and charge distribution (Fang and Szleifer 2003). The binding of lysozyme and cyt c to negatively charged membranes is largely governed by electrostatic interactions, since these proteins have high positive net charge at physiological pH. At strongly acidic fibrillization conditions (pH 2.0), the net charge on the lysozyme molecule reaches +15e. Notably, the net repulsion among the protein monomers favors fibril growth, while net attraction gives rise to precipitation, as reported by Hill and coauthors (Hill et al. 2011). Moreover, the magnitude of charge repulsion is thought to modulate the assembly pathway and morphology of fibrillar aggregates. Recent studies emphasize the importance of net charge of amyloid fibrils as one of the principal determinants of their cytotoxicity (Hirano et al. 2010, 2012). Higher bilayer-disruptive activity of lysozyme fibrils compared to monomeric protein was attributed to the enhanced electrostatic interactions due to increased charge density upon fibrillization. Likewise, effects on the membranes of the positively charged lysozyme fibrils differed from those of the negatively charged Aβ fibrils (Yoshiike et al. 2007). These observations led Hirano and colleagues to propose that electrostatic interactions dominate the membrane association of fibrillar lysozyme and concomitant bilayer disruption (Hirano et al. 2012). Accordingly, electrostatics is likely to play an essential role in determining the competitive membrane binding of lysozyme fibrils and cyt c discussed here.
Consistent with this idea are the results of Trp fluorescence quenching by acrylamide indicating that decrease in Stern-Volmer constant upon fibril-lipid binding become more pronounced with increasing membrane charge (Gorbenko et al. 2012). For electrostatically-controlled adsorption, the equilibrium association constant can be represented as a combination of an intrinsic (or non-electrostatic) term and an electrostatic component that depends on surface charge density, environmental conditions (pH, ionic strength), and the degree of surface coverage by a protein (Gorbenko et al. 2007). As the proportion of anionic lipid rises, cyt c-bilayer interaction becomes stronger and the number of membrane binding sites increases; therefore, higher lysozyme concentrations are required to reduce cyt c – lipid association constant and surface occupancy to the values at which competition between the proteins becomes significant. This effect may account for the delayed threshold effect in Q r dependencies on lysozyme concentration observed for liposomes containing 40 mol% PG or PS, or 25 mol% CL, as illustrated in Fig. 6.6 for PC/PG vesicles.

Relative quantum yield of AV-PC in protein-lipid systems as a function of lysozyme concentration for PC/PG (3:2, mol:mol) liposomes. Lipid concentration was 10 μM, cytochrome c concentration was 0.08 μM. Liposome diameter was 100 nm
However, there is no unequivocal relation between the membrane charge and the extent of protein competition, since fibrillar lysozyme induced distinct changes in Q r of the membranes with similar electrostatic surface potentials but different lipid composition. This implies that the amount of desorbed cyt c is not determined exclusively by nonspecific electrostatic factors. The possibility of specific interactions between lysozyme fibrils and phospholipid headgroups is also supported by our finding that the degree of Trp solvent exposure differs for lipid bilayers of similar charge but different chemical nature of phospholipid headgroups (Gorbenko et al. 2012).
In sum, our model studies corroborate the idea that pathological fibrillar aggregates forming in vivo can potentially impair proper membrane binding of cellular proteins. The modulating effects of membrane charge and chemical nature of anionic phospholipid headgroups observed in our studies enabled us to propose that lipid-associating and competitive binding properties of lysozyme fibrils are governed by both specific and non-specific protein-lipid interactions. These results suggest that enhanced ability of fibrillar protein aggregates to compete for membrane binding sites are among the possible determinants of amyloid cytotoxicity.
6.4 Fibril Restructuring on a Membrane Template
Specific molecular architecture of amyloid assemblies is stabilized by the enthalpic contribution from the main-chain hydrogen bonding, ionic pairing, aromatic π-π interactions and hydrogen bonds between amino-acid side chains (Nelson et al. 2005; Makin et al. 2005; Petkova et al. 2006), as well as the entropic contribution originating from the release of structured water molecules from the tightly packed amyloid core (Sawaya et al. 2007; Williams et al. 2006). Clearly, all forces stabilizing fibril structure can be modulated by the environmental factors inherent to a lipid bilayer. This notion is supported by the fact that lipids can destabilize and re-solubilize mature fibrils with formation of “reverse oligomers” similar in their cytotoxicity to the oligomers assembled from monomeric proteins. This effect was demonstrated, for instance, for Aβ peptide (Martins et al. 2008). Hence, amyloid plaques can be considered as reservoirs of toxicity in which mature fibrils can be transformed into highly toxic oligomeric species due to alterations in local physicochemical parameters controlled by lipid metabolism.
To further explore fibril transformations induced by the lipid surface, we designed a series of FRET experiments aimed at elucidating the effects of lipids on the structure and morphology of fibrillar aggregates. The aforesaid N-terminal fragment of the amyloidogenic variant of human apoA-I, 1-83/G26R/W@8, was chosen as a model protein for these experiments, while lipid vesicles were formed from PC and its mixture with cholesterol. The idea was to recruit amyloid-specific dye with a defined location within fibril structure as an energy acceptor for a membrane fluorescent probe. To this end, we employed classical amyloid marker Thioflavin T (ThT). Fibril binding of ThT is accompanied by a dramatic fluorescence enhancement arising from restricted torsional oscillations of the benzothiazole and aminobenzoyl rings and nearly planar conformation of the dye molecule incorporated in the solvent-exposed grooves spanning across consecutive β-strands parallel to the fibril axis (Sulatskaya et al. 2010; Stsiapura et al. 2007; Hawe et al. 2008). Another remarkable feature of ThT is its preferential association with the grooves lined with aromatic residues (Biancalana and Koide 2010). Due to these distinct properties, ThT can be used not only for identification of amyloid fibrils, but also for their structural characterization. Our experimental strategy involved: (i) quantitating the dye interaction with fibrillar 1-83/G26R/W@8 peptide and defining possible ThT binding sites through measuring FRET between the engineered Trp8 in this peptide and ThT; (ii) monitoring morphologic changes in 1-83/G26R/W@8 fibrils adsorbing onto the surface of lipid vesicles by examining FRET between Laurdan and ThT.
As a first step, we used the method of double fluorimetric titration to determine quantitative characteristics of ThT-fibril binding (association constant, K a , and binding stoichiometry, n, in mole of ThT per mole of protein). Scatchard plot obtained from the binding data had a concave-up shape suggesting two types of ThT binding sites. Global fitting based on simultaneous analysis of two-dimensional data arrays acquired by varying both ThT and protein concentration yielded two sets of parameters: \( {K}_{a1}=\left(6.2\pm 0.7\right)\mu {\mathrm{M}}^{-1} \), \( {n}_1=0.1\pm 0.02 \) (high-affinity sites) and \( {K}_{a2}=\left(0.14\pm 0.03\right)\mu {\mathrm{M}}^{-1} \), \( {n}_2=0.17\pm 0.03 \) (low-affinity sites), as described in detail in (Girych et al. 2014). The observed heterogeneity of ThT binding sites can stem from: (i) the structurally and compositionally distinct sites within fibrillar assemblies of a certain morphology, and/or (ii) polymorphism inherent to most amyloid preparations. Indeed, AFM shows that 1-83/G26R/W@8 fibrils have either smooth or twisted appearance suggestive of structural polymorphism. The height profiles over contour length analyzed for the two types of fibrils suggested that different polymorphs are represented by the twisted and helical ribbons (Fig. 6.7a, b). Accordingly, we proposed that high- and low-affinity ThT binding sites reside on distinct fibril polymorphs. To better understand these polymorphs, we attempted to develop a tentative structural model of fibrillar 1-83/G26R/W@8 and to ascertain what solvent-exposed grooves within the fibril structure are most likely to bind ThT.

Atomic force microscopy images of 1-83/G26R/W@8 fibrils illustrating the presence of helical and twisted ribbon polymorphs (a). Height profiles acquired over contour length for different polymorphs of 1-83/G26R/W@8 fibrils (b)
Amino acid sequence analysis of the self-associating properties of apoA-I 1-83 fragment by using AGGRESCAN (Conchillo-Sole et al. 2007), Zyggregator (Tartaglia and Vendruscolo 2008) and TANGO (Linding et al. 2004) algorithms showed that residue segments 14–23 and 50–58 have the highest aggregation tendency (also see Chap. 8 by Das and Gursky in this volume). Recent x-ray crystallographic studies revealed that the apoA-I segment 44–55 forms a native unpaired β-strand in lipid-free protein, suggesting high intrinsic β-sheet propensity by this segment (Gursky et al. 2012; Das et al. 2014). On the other hand, in our model the residues 1–13, 32–40 and 59–83 were excluded from the putative β-strand regions, based on the following considerations: (i) the presence of structure-breaking proline residues at positions 3, 4, 7, 66 and glycine at positions 35, 39, and 65; (ii) the proteolytic accessibility of E34 and F57 (Lagerstedt et al. 2007); and (iii) TANGO prediction that the sequences 14–31 and 41–58 have the highest propensity to form β-sheets in amyloid.
On the basis of these considerations and the observation that the height of 1-83/G26R/W@8 fibrils is ~5–10 nm, while the total length of this fully extended polypeptide is ~28 nm, we hypothesized that 1-83/G26R/W@8 protofilaments have β-strand-loop-β-strand structure in which β-strands from residues 14–31 to 41–58 form a self-complementary steric zipper stabilized by van der Waals and hydrophobic interactions, as well as by the putative salt bridge between R27 and D48 of the neighbouring strands (Girych et al. 2014). Figure 6.8 shows a tentative structure for fibrillar 1-83/G26R/W@8 created for four protein monomers using Rosetta and VMD software (Delano 2005). Notably, the proposed parallel in-register β-sheet structure is consistent with our recent FTIR data recorded of this fibrillar peptide (Adachi et al. 2013).

3D model structure of 1-83/G26R/W@8 in a fibrillar state derived from Rosetta calculations. Four individual peptide molecules which were used to build the model are shown
According to our structural model, surface grooves that can potentially bind ThT sites are formed by the solvent-exposed residues L14_T16_Y18_D20_L22_D24_R26_D28_V30 in the N-terminal β-strand and by the residues Q41_N43_K45_L47_N49_D51, S52_T54_T56_S58 in the C-terminal β-strand. Of those, the grooves lined with aromatic and hydrophobic residues are probably preferred for ThT binding, as demonstrated by Wu and coauthors (Wu et al. 2008, 2009, 2011; Biancalana et al. 2009). These considerations, together with the FRET-based distance estimates between fibril-bound ThT and Trp8 in 1-83/G26R/W@8 (Girych et al. 2014), suggest that the high-affinity ThT binding sites reside within the groove T16_Y18 on the helical ribbon polymorphs. The constant curvature of this region favors nearly planar motionally restricted conformation of the dye, while the low-affinity sites are lined up at the groove D20_L22 on the twisted ribbon polymorphs with varying curvature.
Next, we used this model as a structural basis to determine whether protein-lipid interactions can alter the structure and morphology of 1-83/G26R/W@8 fibrils. To this end, we employed fluorescent probe Laurdan located at the lipid-water interface as an energy donor for ThT. Fibrillar 1-83/G26R/W@8 peptide was incubated with small (50 nm) PC liposomes doped with Laurdan, followed by titration of ThT. Since in these protein-lipid systems ThT was distributed between the lipid bilayer and the binding sites in the helical and twisted ribbon fibrils, a separate series of FRET experiments was directed towards quantifying the ThT partitioning into the lipid phase and evaluating its membrane location. As illustrated in Fig. 6.9, FRET efficiency in the PC vesicles + fibrillar 1-83/G26R/W@8 system is markedly higher compared to the PC vesicles alone, implying that the number of ThT molecules serving as the energy acceptors increases upon fibril adsorption on the surface of the lipid vesicles. Notably, this effect was observed only for fibril-forming apoA-I variant, while in the case of its non-amyloidogenic counterpart lacking G26R mutation, energy transfer between Laurdan and ThT appeared to be indistinguishable from that in the protein-free PC liposomes. Hence, FRET enhancement could be attributed exclusively to fibril-bound ThT molecules.

Relative quantum yield of Laurdan, which was incorporated in PC liposomes and PC + 1-83/G26R/W@8 complexes, as a function of Thioflavin T concentration. Lipid concentration was 13 μM, protein concentration was 1.6 μM, Laurdan concentration was 0.5 μM. Liposome diameter was 50 nm. Solid line represents the simulation data yielding the best agreement between experiment and theory
Our next goal was to ascertain what kind of spatial distribution of donors and acceptors was consistent with the experimentally measured FRET profiles. In the simulation-based FRET analysis, donor and acceptor coordinates were generated in a virtual square box with edge length l b and calculating the relative quantum yield averaged over all donors as:
Here r ij is the distance between j-th donor and i-th acceptor; N D and N AC stand for the number of donors and acceptors in a box, respectively; and l b was taken as 10 R 0, where R 0 is the Förster radius for the Laurdan-ThT donor-acceptor pair estimated to be ~2.9 nm. The N AC value was determined assuming three populations of ThT molecules associated either with lipids or with helical ribbon (HR) or twisted ribbon (TR) fibrils. The respective molar concentrations of these species (B L , B HR and B TR ) were calculated for any given total concentration of ThT by solving the system of the following equations:
Here, P is the total concentration of the protein monomers, K P is the dye partition coefficient (630 ± 46 for PC liposomes), and V L , V W are the volumes of the lipid and aqueous phases, respectively. Subsequent estimate of the surface density of lipid-bound acceptors and linear density of HR- and TR-associated acceptors (the number of ThT molecules per protein monomer) enabled us to assess the number of various ThT species (N L , N HR and N TR ) in the box.
Our initial assumption was that the fibrils retained their helical or twisted ribbon morphology when interacting with the lipid vesicles. The donors were confined to a plane parallel to the membrane surface and located at a distance r FD from the fibril axis. The lengths of the helical (l HR ) and twisted ribbon (l TR ) fibrils in the box relative to the box edge l b were defined by parameters \( {s}_{HR}={l}_{HR}/{l}_b \) and \( {s}_{TR}={l}_{TR}/{l}_b \). The coordinates of ThT molecules distributed over the fiber structure were generated as previously described (Girych et al. 2014). Our goal was to find out if any sets of theoretical parameters {r FD , s HR , s TR } were consistent with the experiment. The uncertainty in the mutual orientation of Laurdan emission and ThT absorption transition moments was minimized by setting the lower and upper limits for orientation factor (κ 2min and κ 2max ) using the information on the fluorophore rotational mobility derived from the fluorescence anisotropy measurements (Dale et al. 1979). Although the values of r FD , s HR and s TR were varied in the widest physically plausible range, no good agreement between the experimental FRET data and the simulation results could be achieved. This result suggested that 1-83/G26R/W@8 fibrils undergo lipid-induced morphological changes probably involving the unwinding of the helical and twisted ribbon fibers into more planar ribbons.
Since ThT is thought to reside along the surface side-chain grooves running parallel to the long fibril axis (Krebs et al. 2005; Biancalana et al. 2008; Teoh et al. 2011), in our simulations the fibril-bound acceptors were arranged along the lines parallel to bilayer surface and separated from the donor plane by certain distances, d HR and d TR . Two alternative scenarios were considered in regard to the orientation factor: (i) κ 2 is fixed and varies between κ 2min and κ 2max , or (ii) κ 2 depends on the donor-acceptor distance suggesting that the orientational behavior of the fibril-associated ThT resembles that of the membrane fluorophores whose transition moments are symmetrically distributed within the cones (Domanov and Gorbenko 2002). Only the latter scenario provided successful theoretical description of the experimental FRET profiles.
Obviously, when a certain fibril fragment attaches to the membrane surface, superficial grooves undergo structural changes due to the rearrangement of the hydrogen-bonded network at the lipid surface. Accordingly, ThT binding sites acquire the properties intermediate between those found on the fibrillar structure and in the lipid phase, and the orientational properties of ThT as an energy acceptor for Laurdan become similar to those of membrane-bound dyes. For this reason, only simulation-based FRET analysis with distance-dependent orientation factor could adequately fit the observed FRET data.
Solid line in Fig. 6.9 represents the simulation results providing the best agreement between experiment and theory, which was achieved by using the following parameter set: l HR ~29 nm, l TR ~6 nm, d HR ~1 nm, d TR ~1 nm. Notably, after converting into planar ribbons, helical and twisted ribbon fibers became structurally indistinguishable, yet the stoichiometries of ThT binding to high-affinity sites in HR and low-affinity sites in TR remained distinct, resulting in different linear densities of the bound dye in HR and TR. Given that Laurdan molecules are located in the planar interface between polar and non-polar parts of lipid bilayer, our estimates for donor-acceptor separation suggest that linear array of the fibril-bound ThT species is located at the water-lipid interface, with ThT-binding grooves facing the aqueous phase. In this orientation, the amyloid core-forming C-terminal segment Q41–S58 must reside within the fibril-membrane contact area.
To verify this idea, we employed the online server HeliQuest to obtain the mean hydrophobicity (<H>), hydrophobic moment (μH), net charge (z) and, eventually, lipid discrimination factor (D) characterizing lipid binding affinity of a given polypeptide fragment (Gautier et al. 2008; Keller 2011). HeliQuest analysis of 1-83/G26R/W@8 sequence showed that this peptide contains four most probable membrane-binding regions, namely R10–R27, K23–K40, L44–R61, and S52–Q69, with the strongest lipid-binding potential in L44–R61. This corroborates the model assumption that the β-sheet in residues 41–58 faces the membrane surface. Since the distance between the β-sheets in our proposed β-strand-loop-β-strand structure is ~1 nm, our estimates for d HR and d TR are consistent with bilayer penetration by the β-strands to the depth ~1 nm, i.e. to the level of the initial acyl chain carbons.
Furthermore, the values of l HR and l TR allowed us to characterize fibril-lipid binding in terms of the number of lipid molecules per protein monomer. Considering the surface area per lipid headgroup as 0.65 nm2, the box with edge length 29 nm contains 1,294 lipid molecules, while the total length of the untwisted fibrils is ~35 nm, corresponding to ~75 protein monomers. Hence, the number of lipid molecules per protein monomer in a planar ribbon configuration of 1-83/G26R/W@8 fibrils that are associated with PC liposomes is circa 17. Similar estimates for PC/Chol liposomes produced the parameters l HR ~17 nm, l TR = 0, d HR ~2 nm, with the number of lipid molecules per protein monomer circa 34. These results suggest that the presence of 30 mol% cholesterol in the PC bilayer impairs fibril penetration into the polar membrane region and reduces the fibril-lipid contact area.
Notably, helical and twisted ribbon fibers are rather rigid structures, as judged from their persistence length of about 2 μM determined directly from AFM images as described in (Adamcik et al. 2011). Nevertheless, adsorption of lipid vesicles along the fibril length (Fig. 6.10a, b) apparently produces local untwisting of the helical and twisted ribbons, thereby increasing their flexibility and extending the contact area with the membrane surface. In summary, the results reported in this section suggest that lipid membranes can not only trigger amyloid formation, but also modulate the structural morphology of fibrillar assemblies.

Transmission electron micrographs illustrating adsorption of 1-83/G26R/W@8 fibril on PC vesicle (a) and schematic illustration of untwisting of the 1-83/G26R/W@8 fibrils on a lipid bilayer template drawn approximately to scale (b). Scale bar in panel (a) is 50 nm
6.5 Concluding Remarks
In the past decades, a complex problem of protein-lipid interactions acquired a new intriguing facet concerning the role of cell membranes in initiating the growth of amyloid fibrils and the ensuing cytotoxic action of pre-fibrillar and fibrillar protein aggregates. It became increasingly clear that physicochemical, structural and morphological characteristics of amyloid assemblies are important in determining their membrane-mediated toxicity. On one hand, these characteristics can be fine-tuned by the membrane environment. On the other hand, membrane responses to pathogenic aggregated species are dictated by collective properties of the lipid bilayer, such as the surface charge and curvature (addressed by Uversky, Chap. 2 in this volume), dielectric permeability, viscosity and lateral pressure profiles, elasticity, etc., as well as by the exact chemical nature of individual membrane constituents and their conformational features. In the present chapter we focused mainly on the emerging understanding of the molecular processes involved in the interactions between lipid membranes and mature amyloid fibrils. Our fluorescence studies performed with two model amyloidogenic proteins, lysozyme and 1-83/G26R/W@8 apoA-I peptide, support the following hypotheses. First, superficial fibril binding to lipid bilayers is followed by the changes in lipid packing density and level of hydration in the interfacial bilayer region, with little or no perturbation of the hydrophobic core. Second, fibrillar assemblies can displace membrane-associated proteins. Third, morphological characteristics of protein fibrils can be modulated by the lipid bilayer. Finally, cholesterol can reduce lipid-associating and membrane-modifying abilities of amyloid aggregates. Although extrapolation of these model studies to the in vivo effects of mature amyloid fibrils on the membranes is not straightforward, our ideas provide the basis for a deeper understanding of the membrane-mediated cytotoxicity mechanisms of amyloid assemblies.
Abbreviations
- 1-83/G26R/W@8:
-
N-terminal 1-83 fragment of apoA-I with G26R mutation
- AFM:
-
Atomic force microscopy
- apoA-I:
-
Apolipoprotein A-I
- AV-PC:
-
Anthrylvinyl-labeled PC
- Aβ:
-
Amyloid-β peptide
- Chol:
-
Cholesterol
- CL:
-
Cardiolipin
- cyt c :
-
Cytochrome c
- FRET:
-
Förster resonance energy transfer
- GP:
-
Generalized fluorescence polarization of Laurdan
- HR:
-
Helical ribbon
- PC:
-
Phosphatidylcholine
- PG:
-
Phosphatidylglycerol
- PS:
-
Phosphatidylserine
- ThT:
-
Thioflavin T
- TR:
-
Twisted ribbon
References
Adachi E, Nakajima H, Mizuguchi C, Dhanasekaran P, Kawashima H, Nagao K, Akaji K, Lund-Katz S, Phillips MC, Saito H (2013) Dual role of an N-terminal amyloidogenic mutation in apolipoprotein A-I: destabilization of helix bundle and enhancement of fibril formation. J Biol Chem 288:2848–2856
Adamcik J, Mezzenga R (2012a) Study of amyloid fibrils via atomic force microscopy. Curr Opin Colloid Interface Sci 17:369–376
Adamcik J, Mezzenga R (2012b) Protein fibrils from polymer physics perspective. Macromolecules 45:1137–1150
Adamcik J, Castelletto V, Bolisetty S, Hamley IW, Mezzenga R (2011) Direct observation of time-resolved polymorphic states in the self-assembly of end-capped heptapeptides. Angew Chem 50(24):5495–5498
Aisenbrey C, Borowik T, Byström R, Bokvist M, Lindström F, Misiak H, Sani M, Gröbner G (2008) How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur Biophys J 37:247–255
Al Kayal T, Nappini S, Russo E, Berti D, Bucciantini M, Stefani M, Baglioni P (2012) Lysozyme interaction with negatively charged lipid bilayers: protein aggregation and membrane fusion. Soft Matter 8:4524–4534
Anderluh G, Gutierrez-Aguirre I, Rabzelj S, Ceru S, Kopitar-Jerala N, Macek P, Turk V, Zerovnik E (2005) Interaction of human stefin B in the prefibrillar oligomeric form with membranes. Correlation with cellular toxicity. FEBS J 272:3042–3051
Arispe N, Rojas E, Pollard H (1993) Alzheimer’s disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminium. Proc Natl Acad Sci U S A 90:567–571
Ascenzi P, Polticelli F, Marino M, Santucci R, Coletta M (2011) Cardiolipin drives cytochrome c proapoptotic and antiapoptotic actions. Life 63:160–165
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE (2003) A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci 23:2665–2674
Biancalana M, Koide S (2010) Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta 1804:1405–1412
Biancalana M, Makabe K, Koide A, Koide S (2008) Aromatic cross-strand ladders control the structure and stability of beta-rich peptide self-assembly mimics. J Mol Biol 383:205–213
Biancalana M, Makabe K, Koide A, Koide S (2009) Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J Mol Biol 385:1052–1063
Bucciantini M, Cecchi C (2010) Biological membranes as protein aggregation matrices and targets of amyloid toxicity. Methods Mol Biol 648:231–243
Bucciantini M, Forzan M, Russo E, Martino C, Pieri L, Formigli L, Quercioli F, Soria S, Pavone F, Savistchenko J, Meliki R, Stefani M (2012) Toxic effects of amyloid fibrils on cell membranes: the importance of ganglioside GM1. FASEB J 26:818–831
Bucciantini M, Rigacci S, Stefani M (2014) Amyloid aggregation: role of biological membranes and the aggregate-membrane system. J Phys Chem Lett 5:17–527
Butterfield SM, Lashuel HA (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed 49:5628–5654
Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 23:655–664
Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, Chiti F (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 6:140–147
Canale C, Torrassa S, Rispoli P, Relini A, Rolandi R, Bucciantini A, Stefani M, Gliozzi A (2006) Natively folded Hypf-N and its early amyloid aggregates interact with phospholipid monolayers and destabilize supported lipid bilayers. Biophys J 91:1–14
Carulla N, Caddy GL, Hall DR, Zurdo J, Gairi M, Feliz M, Giralt E, Robinson CV, Dobson CM (2005) Molecular recycling within amyloid fibrils. Nature 436:54–558
Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298
Cecchi C, Baglioni S, Fiorillo C, Pensalfini A, Liguri G, Nosi D, Rigacci S, Bucciantini M, Stefani M (2005) Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates. J Cell Sci 118:3459–3470
Conchillo-Sole O, de Groot NS, Avilés F, Vendrell J, Daura X, Ventura S (2007) AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8:65
Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TPJ, Dobson CM, Klenerman D (2012) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149(5):1048–1059
Dale R, Eisinger J, Blumberg W (1979) The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys J 26:161–194
Dante S, Haus T, Brandt A, Dencher NA (2008) Membrane fusogenic activity of the Alzheimer’s peptide Abeta(1-42) demonstrated by small-angle neutron scattering. J Mol Biol 376:393–404
Das M, Mei X, Jayaraman S, Atkinson D, Gursky O (2014) Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing – a common mechanism of apolipoprotein A-I misfolding in familial amyloidosis and atherosclerosis. FEBS J 281:2525–2542
de Planque RR, Raussen V, Contera SA, Rijkers DTS, Liskamp RMJ, Ruysschaert JM, Ryan JF, Separovic F, Watts A (2007) β-sheet structured β-amyloid (1-40) perturbs phosphatidylcholine model membranes. J Mol Biol 368:982–987
Delano WL (2005) The case for open-source software in drug discovery. Drug Discov Today 10:213–217
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280:17294–17300
Dima RI, Thirumalai D (2002) Exploring protein aggregation and self-propagation using lattice models: phase diagram and kinetics. Protein Sci 11:1036–1049
Domanov YA, Gorbenko GP (2002) Analysis of resonance energy transfer in model membranes: role of orientational effects. Biophys Chem 99:143–154
Eckert GP, Cairns NJ, Maras A, Gattaz WF, Müllera WE (2000) Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the Hippocampus: specific changes in Alzheimer’s disease. Dement Geriatr Cogn Disord 11:181–186
Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Höppener JW (2008) Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci U S A 105:6033–6038
Fang F, Szleifer I (2003) Competitive adsorption in model charged protein mixtures: equilibrium isotherms and kinetics behavior. J Chem Phys 119:1053–1065
Forloni G (1996) Neurotoxicity of β-amyloid and prion peptides. Curr Opin Neurol 9:492–500
Frare E, Polverino de Laureto P, Zurdo J, Dobson C, Fontana A (2004) A highly amyloidogenic region of hen lysozyme. J Mol Biol 340:1153–1165
Gautier R, Douguet D, Antonny B, Drin G (2008) HeliQuest: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24:2101–2102
Gharibyan AL, Zamotin V, Yanamandra K, Moskaleva OS, Margulis BA, Kostanyan IA, Morozova-Roche LA (2007) Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J Mol Biol 365:1337–1349
Giannakis E, Pacifico J, Smith DP, Hung LW, Masters CL, Cappai R, Wade JD, Barnham KJ (2008) Dimeric structures of α-synuclein bind preferentially to lipid membranes. Biochim Biophys Acta 1778:1112–1119
Girych M, Gorbenko G, Trusova V, Adachi E, Mizuguchi C, Nagao K, Kawashima H, Akaji K, Phillips M, Saito H (2014) Interaction of thioflavin T with amyloid fibrils of apolipoprotein A-I N-terminal fragment: resonance energy transfer study. J Struct Biol 185:116–124
Gorbenko GP, Kinnunen PKJ (2006) The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem Phys Lipids 141:72–82
Gorbenko G, Trusova V (2011) Effect of oligomeric lysozyme on structural state of model membranes. Biophys Chem 154:73–81
Gorbenko GP, Ioffe VM, Kinnunen PKJ (2007) Binding of lysozyme to phospholipid bilayers: evidence for protein aggregation upon membrane association. Biophys J 93:140–153
Gorbenko G, Trusova V, Sood R, Molotkovsky J, Kinnunen PKJ (2012) The effect of lysozyme amyloid fibrils on cytochrome c–lipid interactions. Chem Phys Lipids 165:769–776
Gray HB, Winkler JR (2010) Electron flow through metalloproteins. Biochim Biophys Acta 1797:1563–1572
Gursky O, Mei X, Atkinson D (2012) The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry 51:10–18
Hawe A, Sutter M, Jiskoot W (2008) Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res 25:1487–1499
Hill SE, Miti T, Richmond T, Muschol M (2011) Spatial extent of charge repulsion regulates assembly pathways for lysozyme amyloid fibrils. PLoS One 6:e18171
Hirano A, Uda K, Maeda Y, Akasaka T, Shiraki K (2010) One-dimensional protein-based nanoparticles induce lipid bilayer disruption: carbon nanotube conjugates and amyloid fibrils. Langmuir 26:17256–17259
Hirano A, Yoshikawa H, Matsushita S, Yamada Y, Shiraki K (2012) Adsorption and disruption of lipid bilayers by nanoscale protein aggregates. Langmuir 28:3887–3895
Huang B, He J, Ren J, Yan XY, Zeng CM (2009) Cellular membrane disruption by amyloid fibrils involved intermolecular disulfide cross-linking. Biochemistry 48:5794–5800
Ioffe VM, Gorbenko GP (2005) Lysozyme effect on structural state of model membranes as revealed by pyrene excimerization studies. Biophys Chem 114:199–204
Joy T, Wang J, Hahn A, Hegele RA (2003) ApoA-I related amyloidosis: a case report and literature review. Clin Biochem 36:641–645
Karpovich DS, Blanchard GJ (1995) Relating the polarity-dependent fluorescence response to vibronic coupling. Achieving a fundamental understanding of the py polarity scale. J Phys Chem 99:3951–3958
Keller R (2011) New user-friendly approach to obtain an Eisenberg plot and its use as a practical tool in protein sequence analysis. Int J Mol Sci 21:5577–5591
Kelly JW (2002) Towards an understanding of amyloidogenesis. Nat Struct Biol 9:323–325
Kim DH, Frangos JA (2008) Effects of amyloid β-peptides on the lysis tension of lipid bilayer vesicles containing oxysterols. Biophys J 95:620–628
Kinnunen PKJ (2009) Amyloid formation on lipid membrane surfaces. Open Biol J 2:163–175
Knowles TPJ, Buehler MJ (2011) Nanomechanics of functional and pathological amyloid materials. Nature 6:469–479
Krebs MR, Bromley EH, Donald AM (2005) The binding of thioflavin T to amyloid fibrils: localization and implications. J Struct Biol 149:30–37
Kremer JJ, Pallitto MM, Sklansky DJ, Murphy RM (2000) Correlation of beta-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry 39:10309–10318
Kremer JJ, Sklansky DJ, Murphy RM (2001) Profile of changes in lipid bilayer structure caused by β-amyloid peptide. Biochemistry 40:8563–8571
Lagerstedt JO, Cavigiolio G, Roberts LM, Hong HS, Jin LW, Fitzgerald PG, Oda MN, Voss JC (2007) Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 46:9693–9699
Lashuel HA, Lansbury PT (2006) Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q Rev Biophys 39:167–201
Lee AG (2003) Lipid–protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta 1612:1–40
Lee AG (2004) How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta 1666:62–87
Lee CC, Sun Y, Huang H (2012) How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys J 102:1059–1068
Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L (2004) A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J Mol Biol 342:345–353
Lopes DHJ, Meister A, Gohlke A, Hauser A, Blume A, Winter R (2007) Mechanism of IAPP fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy (IRRAS). Biophys J 93:3132–3141
Loura LM, Canto AM do., Martins J (2013) Sensing hydration and behavior of pyrene in POPC and POPC/cholesterol bilayers: a molecular dynamics study. Biochim Biophys Acta 1828:1094–1101
Lúcio AD, Vequi-Suplicy CC, Fernandez RM, Lamy MT (2010) Laurdan spectrum decomposition as a tool for the analysis of surface bilayer structure and polarity: a study with DMPG, peptides and cholesterol. J Fluoresc 20:473–482
Ma X, Sha Y, Lin K, Nie S (2002) The effect of fibrillar Aβ1-40 on membrane fluidity and permeability. Protein Pept Lett 9:173–178
Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC (2005) Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A 102:315–320
Malisauskas M, Ostman J, Darinskas A, Zamotin V, Liutkevicius E, Lundgren E, Morozova-Roche LA (2005) Does the cytotoxic effect of transient amyloid oligomers from common equine lysozyme in vitro imply innate amyloid toxicity? J Biol Chem 280:6269–6275
Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D’Hooge R, De Strooper B, Schymkowitz J, Rousseau F (2008) Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J 27:224–233
Matsuzaki K (2011) Formation of toxic amyloid fibrils by amyloid beta-protein on ganglioside clusters. Int J Alzheimers Dis 956104:1–7
Meratan AA, Ghasemi A, Nemat-Gorgani M (2011) Membrane integrity and amyloid cytotoxicity: a model study involving mitochondria and lysozyme fibrillation products. J Mol Biol 409:826–838
Milanesi L, Sheynis T, Xue WF, Orlova EV, Hellewell AL, Jelinek R, Hewitt EW, Radford SE, Saibil HR (2012) Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc Natl Acad Sci U S A 109:20455–20460
Molotkovsky J, Manevich E, Gerasimova E, Molotkovskaya I, Polessky V, Bergelson L (1982) Differential study of phosphatidylcholine and sphingomyelin in human high-density lipoproteins with lipid-specific fluorescent probes. Eur J Biochem 122:573–579
Nakajima A (1971) Solvent effect on the vibrational structure of the fluorescence and absorption spectra of pyrene. Bull Chem Soc Jpn 44:3272–3277
Nelson R, Sawaya MR, Balbirnie M, Madsen A, Riekel C, Grothe R, Eisenberg D (2005) Structure of the cross-β spine of amyloid-like fibrils. Nature 435:773–778
Nichols WC, Gregg RE, Brewer HB, Benson MD (1990) A mutation in apolipoprotein A-I in the Iowa type of familial amyloidotic polyneuropathy. Genomics 8:318–323
Nicolay J, Gatz S, Liebig G, Gulbins E, Lang F (2007) Amyloid induced suicidal erythrocyte death. Cell Physiol Biochem 19:175–184
Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV (2006) Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem 281:13828–13836
Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, Bellotti V (2006) Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 13:191–205
Palsdottir H, Hunte C (2004) Lipids in membrane protein structures. Biochim Biophys Acta 1666:2–18
Parasassi T, Gratton E (1995) Membrane lipid domains and dynamics as detected by Laurdan fluorescence. J Fluoresc 8:365–373
Parasassi T, De Stasio G, Ravagnan G, Rusch RM, Gratton E (1991) Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys J 60:179–189
Parasassi T, Krasnowska EK, Bagatolli L, Gratton E (1998) Laurdan and Prodan as polarity-sensitive fluorescent membrane probes. J Fluoresc 8:365–373
Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Souter AK, Totty N, Nguyen O, Blake CC, Terry CJ, Feest TG, Zalin AM, Hsuan JJ (1993) Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 362:553–557
Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 307:262–265
Petkova AT, Yau WM, Tycko R (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45:498–512
Phillips MC (2013) New insights into the determination of HDL structure by apolipoproteins. J Lipid Res 54:2034–2048
Qiu L, Buie C, Reay A, Vaughn MW, Cheng KH (2011) Molecular dynamics simulations reveal the protective role of cholesterol in β-amyloid protein-induced membrane disruptions in neuronal membrane mimics. J Phys Chem B 115:9795–9812
Quist A, Doudewski I, Lin H, Azimova R, Ng D, Frangine B, Kagan B, Ghiso J, Lal R (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A 102:10427–10432
Relini A, Torrassa S, Rolandi R, Gliozzi A, Rosano C, Canale C, Bolognesi M, Plakoutsi G, Bucciantini M, Chiti F, Stefani M (2004) Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J Mol Biol 338:943–957
Relini A, Cavalleri O, Rolandi R, Gliozzi A (2009) The two-fold aspect of the interplay of amyloidogenic proteins with lipid membranes. Chem Phys Lipids 158:1–9
Sanchez SA, Tricerri MA, Gratton E (2012) Laurdan generalized polarization fluctuations measures membrane packing micro-heterogeneity in vivo. Proc Natl Acad Sci U S A 109:7314–7319
Sawaya MR, Sambashivan S, Nelson R, Ivanova M, Sievers S, Apostol M, Thompson M, Balbirnie M, Wiltzius J, McFarlane H, Madsen A, Riekel C, Eisenberg D (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447:453–457
Sciacca MF, Brender JR, Lee DK, Ramamoorthy A (2012) Phosphatidylethanolamine enhances amyloid-fiber dependent membrane fragmentation. Biochemistry 51:7676–7684
Serpell LC (2000) Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502:16–30
Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME (2006) Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci U S A 103:15806–15811
Smith DP, Tew DJ, Hill AF, Bottomley SP, Masters CL, Barnham KJ, Cappai R (2008) Formation of a high affinity lipid-binding intermediate during the early aggregation phase of α-synuclein. Biochemistry 47:1425–1434
Smith P, Brender J, Ramamoorthy A (2009) The induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides. The case of islet amyloid polypeptide. J Am Chem Soc 131:4470–4478
Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ (2001) Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci 21:7576–7586
Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijf B, Hoppener JWM, Killian JA (2004) Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett 577:117–120
Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T (2004) Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-β peptide. FASEB J 838:836–838
Squier T (2001) Oxidative stress and protein aggregation during biological aging. Exp Gerontol 36:1539–1550
Stefani M (2004) Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim Biophys Acta 1739:5–25
Stefani M (2007) Generic cell dysfunction in neurodegenerative disorders: role of surfaces in early protein misfolding, aggregation, and aggregate cytotoxicity. Neuroscientist 13:519–531
Stefani M (2008) Protein folding and misfolding on surfaces. Int J Mol Sci 9:2515–2542
Stefani M (2010) Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J 277:4602–4613
Stefani M, Dobson C (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 81:678–699
Stsiapura VI, Maskevich AA, Kuznetsova IM (2007) Computational study of thioflavin T torsional relaxation in the excited state. J Phys Chem A 111:4829–4835
Sulatskaya AI, Maskevich AA, Kuznetsova IM, Uversky VN, Turoverov KK (2010) Fluorescence quantum yield of Thioflavin T in rigid isotropic solution and incorporated into the amyloid fibrils. PLoS One 5:e15385
Tabner B, Turnbull S, El-Agnaf O, Allsop D (2002) Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic Biol Med 32:1076–1083
Tartaglia GG, Vendruscolo M (2008) The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev 37:1395–1401
Teoh CL, Pham CL, Todorova N, Hung A, Lincoln CN, Lees E, Lam YH, Binger KJ, Thomson NH, Radford SE, Smith TA, Müller SA, Engel A, Griffin MD, Yarovsky I, Gooley PR, Howlett GJ (2011) A structural model for apolipoprotein C-II amyloid fibrils: experimental characterization and molecular dynamics simulations. J Mol Biol 4:1246–1266
Tilley SJ, Saibil HR (2006) The mechanism of pore formation by bacterial toxins. Curr Opin Struct Biol 16(2):230–236
Tokunaga Y, Sakakibara Y, Kamada Y, Watanabe K, Sugimoto Y (2013) Analysis of core region from egg white lysozyme forming amyloid fibrils. Int J Biol Sci 9:219–227
Valincius G, Heinrich F, Budvytyte R, Vanderah DJ, McGillivray DJ, Sokolov Y, Hall JE, Losche M (2008) Soluble amyloid β-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: implications for cell toxicity. Biophys J 95:4845–4861
van Rooijen BD, Claessens MMA, Subramaniam V (2009) Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim Biophys Acta 1788:1271–1278
Verdier Y, Zarandi M, Penke B (2004) Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer’s disease. J Pept Sci 10:229–248
Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, Mantyh PW (1998) Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18:2161–2173
Williams TL, Serpell LC (2011) Membrane and surface interactions of Alzheimer’s Aβ peptide: insights into the mechanism of cytotoxicity. FEBS J 278:3905–3917
Williams AD, Shivaprasad S, Wetzel R (2006) Alanine scanning mutagenesis of Ab (1–40) amyloid fibril stability. J Mol Biol 357:1283–1294
Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 108(10):4194–4199
Wu C, Wang Z, Lei H, Duan Y, Bowers MT, Shea JE (2008) The binding of thioflavin T and its neutral analog BTA-1 to protofibrils of the Alzheimer’s disease Abeta(16-22) peptide probed by molecular dynamics simulations. J Mol Biol 384:718–729
Wu C, Biancalana M, Koide S, Shea JE (2009) Binding modes of thioflavin-T to the single-layer beta-sheet of the peptide self-assembly mimics. J Mol Biol 394:627–633
Wu C, Bowers MT, Shea JE (2011) On the origin of the stronger binding of PIB over thioflavin T to protofibrils of the Alzheimer amyloid-β peptide: a molecular dynamics study. Biophys J 100:1316–1324
Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE (2009) Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem 284:34272–34282
Yoshiike Y, Akagi T, Takashima A (2007) Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry 46:9805–9812
Zbilut JP, Colosimo A, Conti F, Colafranceschi M, Manetti C, Valerio M, Webber CL Jr, Giuliani A (2003) Protein aggregation/folding: the role of deterministic singularities of sequence hydrophobicity as determined by nonlinear signal analysis of acylphosphatase and Aβ (1–40). Biophys J 85:3544–3557
Acknowledgements
The authors thank Prof. Kenichi Akaji and Dr. Hiroyuki Kawashima (Kyoto Pharmaceutical University) for their help with AFM, and Dr. Rohit Sood (Aalto University) for his aid with transmission electron microscopy. This work was supported by the grant from Fundamental Research State Fund of Ukraine (project number F54.4/015 to G. G.) and Grant-in-Aid for Scientific Research 25293006 (to H. S.) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gorbenko, G., Trusova, V., Girych, M., Adachi, E., Mizuguchi, C., Saito, H. (2015). Interactions of Lipid Membranes with Fibrillar Protein Aggregates. In: Gursky, O. (eds) Lipids in Protein Misfolding. Advances in Experimental Medicine and Biology, vol 855. Springer, Cham. https://doi.org/10.1007/978-3-319-17344-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-17344-3_6
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17343-6
Online ISBN: 978-3-319-17344-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)