
Abstract
The relatively high levels of aneuploidy identified in IVF embryos recently have raised the opportunity to improve pregnancy rates and liveborn outcomes with IVF by identifying and transferring only one chromosomally balanced embryo. With the increased use of CGH, it has been observed that any of the chromosomes can be involved in aneuploidy and even de novo segmental aneuploidies may contribute to implantation failure and miscarriage in a typical IVF cycle. The previous limitations in FISH complexity had meant that the full benefits of aneuploidy screening were not initially realised. The impact of reliable whole chromosome amplification methods and the availability of quality controlled commercial microarrays has seen strong uptake of total chromosome screening by many clinics around the world with substantial changes in implantation rates for the selected embryos reported in most cases. In spite of these benefits, the cost of the aCGH process appears to have limited the availability for a significant number of patients. The price of arrays has reduced in the last few years, but this remains a substantial cost burden in many situations. An alternative approach that can still meet the criteria of total chromosome screening but is potentially more cost-effective could bring the benefits of total chromosome screening to an even wider patient group. Partial genome sequencing (PGS) or low-pass sequencing is one alternative that can be used to rapidly karyotype whole-genome-amplified samples from human blastocysts. We compared these with array-based comparative genomic hybridization (aCGH) karyotypes. Readily available open-source software was customised and then used to map the individual Next Generation Sequencing (NGS) reads to chromosomes providing considerable flexibility for resolving both total chromosomes as well as segmental variations. Qualitatively, there was complete diagnostic agreement between the two approaches. Quantitatively, the sequencing approach also offered simple implementation of objective measures of levels of mosaicism within the multi-cell trophectoderm biopsy piece aiding final decision-making regarding suitability for transfer of the nominated (single) embryo. Depending on individual laboratory capabilities, either technique can have shortened protocols that enable next day transfer of one euploid embryo. Standard protocols for aCGH or PGS can fit timing wise with cleavage stage biopsy. Embryos biopsied on day 5 of cycle will only be transferrable the next day, while slower developing embryos biopsied on day 6 may need to be frozen and transferred at a later time. While reports of day 3 blastomere biopsy show improved pregnancy rates after screening, the final outcomes do not appear to be as good as blastocyst stage biopsy, thus not realising the full potential of screening. Applied correctly, simplified, cost-effective routine aneuploidy analysis via aCGH or NGS promises high live birth rates per single transferred embryo.
Author Contributions:
All authors contributed to the preparation of the manuscript. PB and DL performed the laboratory components and WR developed the SeqVar algorithms from publicly available software.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Assisted reproduction treatment employing in vitro fertilisation (IVF) often results in a surplus of embryos potentially suitable for transfer. The transfer of several embryos at once can enhance immediate pregnancy rates, but it also increases the chance of multiple pregnancy, with its risks of serious complications during pregnancy and the perinatal period [1–3]. This led to a trend of electively transferring a single embryo at a time [3–6]. It has been revealed recently that a significant percentage of early embryos, however, harbour substantial chromosomal anomalies which may be incompatible with implantation (or with establishment of normal gestation), so, in the absence of effective screening, elective single embryo transfer can appear to reduce the immediate, first-transfer IVF pregnancy rate [1–3]. Traditionally, a hierarchy of best embryos (or best remaining embryo for transfer after cryostorage) has been inferred from a combination of developmental and morphological features based on cell number, cleavage rate, blastomere fragmentation fraction, presence of intracellular vacuoles, and, most recently, the ability to form blastocysts suitable for transfer on day 5 or 6. With the exception of cleavage rate and blastulation, these factors are subjective [7, 8] and are ultimately inadequate for choosing the embryo with the best potential for initiating a normal, singleton gestation; a morphologically normal embryo can fail to implant, or might initiate a pregnancy only to miscarry later, because of chromosomal aneuploidy.
As many as 65 % of clinical miscarriages in the first trimester have a major abnormality of chromosomal copy number identifiable with classical low-resolution karyotyping on products of conception (POC) [9–11]. While aneuploidy may involve any (or several) of the 24 chromosomes, some typically larger chromosomes appear to be so crucial that their aneuploidies are lethal during pre-implantational development; they are almost never observed among tested clinical POC. Recent reports show that more than 50 % of oocytes from women in their mid-30s can be aneuploid [12]. Given similar aneuploidy rates identified in embryos, even among relatively young IVF patients [13], identifying such embryos and excluding them from transfer logically offers the possibility to increase implantation rates substantially using those that test normal and should also decrease the risk of miscarriage among any pregnancies that follow. While such preimplantation screening of embryos with partial characterization of chromosomes using fluorescent in situ hybridization (FISH) has been performed for nearly 20 years, no objective demonstrable improvement in IVF outcomes was reported, irrespective of whether biopsies were performed on day 3, where embryos are essentially 8 identical cells, or on morphologically normal day 5 and day 6 blastocysts, when there are typically more than 100 cells and differentiation has occurred of outer trophectoderm (TE, the future placenta) and the inner cell mass (ICM, or embryo proper). If any incomplete analysis of potential chromosomes involved in aneuploidy at either stage or, in the case of blastocysts, any mosaic observation that then rules out the transfer of that embryo, then such preimplantation genetic screening potentially disadvantages live birth rates per embryo transfer event compared with standard IVF practices [14].
The debate on what constitutes effective screening for aneuploidy has been protracted, but it is clear that FISH, while very convenient, falls far short. The particular blastomere or cells removed and tested from an embryo may be euploid and considered normal. In contrast, if the blastomere were aneuploid, this could be representing a true meiotic non-disjunction or may be a mosaic in the embryo arising from anaphase lag, chromosome gain, or mitotic non-disjunction followed by trisomy and monosomy mixtures among clonally surviving daughter cells. Such mosaic aneuploidy has been attributed to loose cell cycle controls during rapid cell mitosis in the early embryo [15–17] and is paralleled by confined placental mosaicism observations in otherwise healthy pregnancy outcomes. Depending on the ‘dosage’ and level of survival disadvantage of the mitotically derived aneuploid cell line for the embryo, partial or complete resolution can take place naturally [18]; given the chance, this will lead to a normal gestational outcome in at least some cases. Accordingly, the clinical significance of such occurrences at the embryonic stage is unknown. But secondly, and perhaps most importantly, many instances of meiotically founded aneuploidy are missed through the limited number of chromosomes able to be examined with FISH [19].
A method of total chromosome screening employing comparative genomic hybridization (CGH) of metaphases at single-cell level following DNA amplification by degenerate oligonucleotide-primed PCR was developed and reported by Wells and colleagues as long ago as 1999 and applied to human preimplantation blastomeres from three embryos of normal appearance a year later [20, 21]. The following year the technique was also employed clinically and led to a normal infant [22].
The lengthy hybridization time required for classical metaphase CGH meant that biopsied embryos by necessity needed to be frozen and cryostored, with transfer delayed to a subsequent cycle, a process which was considered at the time to be suboptimal for biopsied embryos mainly due to the impact of traditional freezing methods on embryo viability [23]. Furthermore, when comparing CGH to FISH, the test preparation and laboratory personnel skill base needed for testing the multiple embryos available in PGD cycles was more complex, time-consuming, and expensive than the use of FISH which was more readily applied to multiple biopsy specimens simultaneously using suitably trained staff available in most laboratories. CGH for preimplantation embryo karyotyping languished clinically. In 2008, however, Fragouli, Wells, and colleagues [24] (still using classical metaphase CGH techniques) gave the first indications that the potential of total chromosome screening of day 5 blastocysts [25] in combination with vitrification (a refined method of freezing embryos [26]) could realise the improvement sought—but had proven elusive—using FISH [20, 21].
In summary, the key developments that made karyotyping preimplantation human embryos a routine clinical prospect with improved outcomes have comprised (1) the demonstrated safety and reliability of trophectoderm biopsy at the stage of blastocyst [4, 6, 27–29]; (2) the advent and application of an efficient vitrification process for the storage of biopsied embryos [26]; (3) the improved reliability of whole-genome amplification (WGA); and (4) the reduction in cost and improved utility of comprehensive molecular cytogenetic methods that employ array CGH or single nucleotide polymorphisms [23–25, 30–32] and more recently next generation or second generation sequencing [33].
This study was aimed at comparing two methods of molecular karyotyping (microarray analysis vs. partial genome sequencing) in assigning the chromosomal status of embryos that were otherwise defined as clinically useable by traditional embryologic criteria.
Methods
Embryo Culture and Biopsy
All embryo analyses were carried out under a National Health and Medical Research Council (NHMRC) licence for human embryo research (Licence 309702B) and under a protocol approved by Genea’s formally constituted and NHMRC-registered Human Research Ethics Committee. Seven couples donated 25 clinically useable frozen embryos that had become excess to their reproductive needs. Embryos had been stored for up to 9 years in liquid nitrogen.
Patients had been stimulated for multiple egg retrieval using standard protocols [29]. Embryos were cultured to blastocysts in MINC incubators (Cook Australia Pty Ltd) under 89 % nitrogen/5 % oxygen/6 % CO2; excess blastocysts were cryopreserved using standard slow-freezing protocols. Stage-specific culture medium (Sydney IVF Media Suite version 2, Cook IVF, Eight Mile Plains, Queensland) was used for each step. After thawing, embryos were allowed to re-expand in blastocyst medium. Embryos were removed from the zona pellucida, biopsied according to standard protocols [27] and, where possible, the ICM was identified and kept as a discrete sample for analysis (there was no attempt at removing any attached trophectoderm cells as visually they were considered numerically much less than the ICM cells).
Whole-Genome Amplification
In total, 176 tissue samples were isolated from the 25 embryos, each consisting of about 8–10 cells. All samples were placed into individual PCR tubes and subjected to whole-genome amplification (WGA) using PicoPLEX (Rubicon Genomics, Inc. Ann Arbor, MI). After purification of amplified products (QIAquick PCR purification kit, Qiagen), the WGA product was quantified (NanoDrop 1000, Thermo Scientific); 168 amplifications were considered to have amplified effectively and yielded the manufacturers’ suggested final DNA amount (3–6 μg per amplified sample). Two samples from each embryo were selected for both array and NGS analyses.
Array CGH Analysis
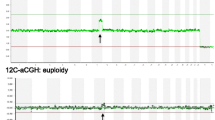
Two samples from each embryo were compared by array CGH—ICM (where available) and one trophectoderm product. An aliquot of purified labelled WGA was hybridised to Agilent 8x60k oligonucleotide arrays using standard protocols. The WGA product (600 ng) was labelled using the Agilent ULS labelling system (Genomic DNA ULS Labelling Kit, Agilent Technologies) and 300–400 ng used for each subarray. Control DNA was similarly whole genome amplified, purified, labelled with the alternative ULS fluorophore reagent, combined in equal amount with WGA product, and hybridised for 16–20 h at 65 °C. After washing (Oligo aCGH Wash Buffer 1, Agilent Technologies) at room temperature for 5–10 min and then washed at 37 °C (Oligo aCGH Buffer 2, Agilent Technologies) for 1 min, slides were scanned at 3 μm (Agilent G2565CA Microarray Scanner, Agilent Technologies). The resultant TIFF image was extracted (Feature Extraction 10.7.3.1, Agilent Technologies) and analyses performed using Agilent Genomic Workbench (Version 7.0.4.0, Agilent Technologies) (Moving average: triangular algorithm, 20 Mb window; ADM-2 aberration algorithm; fuzzy zero; Normalisation: GC correction 10 Kb). The plotted microarray outputs for each of the embryo biopsy samples were read visually and independently by at least two trained observers. These reads were used to assign the ploidy status for each piece with the embryo status considered to be the result of the ICM when it was available. aCGH moving average plots for example embryos are presented in Fig. 12.1a–d with individual biopsy pieces from each single embryo overlayed.


(a–d) Upper panel in each section shows chromosome profiles determined by partial genome sequencing. The lower panel in each section shows overlaid array CGH profile
Partial Genome Sequencing: Low Depth Sequencing
A second aliquot of WGA product from 50 of the initial samples (two samples from each embryo) was used for sequencing using the Ion Torrent Personal Genome Machine (PGM) system (Life Technologies, Melbourne, Australia). WGA from each biopsy piece produced a range of long amplicon products that were then fragmented (Ion Xpress Plus Fragment Library Kit, Life Technologies) to yield blunt-ended DNA fragments of c. 250 base pairs. Fragments were then ‘library prepared’ (Ion Plus Fragment Library Kit, Life Technologies) and indexed using Ion Torrent barcodes (Ion Xpress Barcodes 1-48, Life Technologies). Template preparation was carried out using the OneTouch System (Ion OneTouch 200 Template Kit, Life Technologies) and sequenced using the 200 base read kit (Ion Xpress 200 Sequencing Kit, Life Technologies).
We analysed the initial data using the standard software supplied with the Ion Torrent Suite 3.2 PGM sequencer. The cumulative sequence reads for individual chromosomes were plotted (Fig. 12.2). Each autosome chromosome cumulative score was obtained from the Ion Torrent Suite output and characterised as a simple fraction of the total autosome read from that biopsy piece. Mean reads and standard deviations (SDs) for each chromosome from each run were calculated (with the previous array-identified abnormal chromosomes being excluded from individual chromosome normal range calculations) and a Z-score table was generated. The Ion Torrent sequencing data and the reads obtained are presented in Table 12.1. These preliminary analyses on their own were found to be adequate for simple chromosome aneuploidy assessment for clinical diagnostic purposes but were insufficient for some segmental losses (and presumably gains). Therefore, we devised and applied further algorithms.

Preliminary chromosome coverage output (Ion Torrent Suite 3.2) for the inner cell mass of embryo G20. Partial sequencing reads are arranged in conventional order of chromosome number (1–22), followed by the sex chromosomes (X and Y) and mitochondrial chromosome (chrM). There is monosomy of chromosome 13
SeqVar Algorithm
Our SeqVar algorithm set was adapted from an open-sourced algorithm [34] that calculates the Poisson probability of difference between two samples of a number of mapped reads in small windows that tile each chromosome. SeqVar thus detects significant over- and under-representation of mapped reads of the sample under test compared to the control sample and adjusts for global variation between the test and control samples across all chromosomes using Poisson distribution. The software output includes visual plots of segmental gains and losses respectively above and below a threshold value on any chromosome and marks the chromosome when the difference is significant (Fig. 12.3).

SeqVar output indicating a segmental deletion at 5p in two samples from embryo B3. (a) At a total genome coverage of c. 245,000 reads, (b) at c. 400,000 reads. PGS methodology readily permits increasing the resolution in additional examinations of remaining extant sample to clarify areas of ambiguity or concern
Detection and Display of Segmental Variations
The procedure for detecting segmental variations involves two steps:
-
1.
Aligning the reads to the February 2009 human genome reference sequence GRCh37/hg19. A higher number of reads mapped to the genome increases the statistical power of variation calls and enables the detection of smaller deletions. The 3-stage Ion Torrent Mapping Alignment Program was used to align the sequence and to filter putative sequencing errors. This was able to align an average of 91–193 % of sequencing reads that passed the Torrent Quality Control step.
-
2.
Detecting segmental variations from the mapped reads. Our approach employs a sliding window (of varying size depending on the total number of mapped reads) across the entire genome using the Poisson distribution for subsequent calculation. In each window, the number of reads counted that mapped to the input sample and a normal reference sample were used to calculate the probability that any difference between the input and reference samples is statistically significant. Because the Y-chromosome (chrY) is small, the number of reads that map to it can vary materially between samples. Within each sample, however, the ratio between the number of reads that map to chrY and the total number of mapped reads is reasonably consistent: for male samples, median ratio Y/Total = 5.7 × 10−3, ±SD = 4 × 10−4; for female samples, median = 1.6 × 10−3, ±SD = 3 × 10−4. To further reduce false-positive chrY calls, the SeqVar detection algorithm checks this ratio before calling a copy number variation on chromosomes X and Y.
In the absence of an accepted nomenclature for sequence-derived karyotyping, comparable results for aCGH and PGS are given here principally in the familiar banded-chromosome nomenclature of classical cytogenetics. Chromosome-mapping outputs obtained from sampling across the genome are based on relative imbalances in DNA copy number for both CGH and PGS, however, and produce visibly comparable chromosome-based displays. The current international nomenclature for reporting virtual karyotypes from arrays can therefore be provided in addition to the banded-chromosome-based terminology.
Results
Among the 25 available embryos from the seven patients (average age = 34.4, range = 29–40 year), we observed an effective euploidy rate of 15/25 (60 %), a prevalence comparable to that reported for blastocysts from similar age cohorts by other authors [31, 32, 34, 35]. Control normal and abnormal karyotypes obtained by array CGH are illustrated in Fig. 12.1a–d. The median number of embryos per patient was 3 (range = 2–6). Of the 25 embryos, 11 were uniformly normal. Seven embryos were uniformly aneuploid and three embryos that displayed mosaicism across TE and ICM were also considered abnormal.
Four embryos revealed one or more mosaic aneuploidies confined to TE, a phenomenon that (a) generally indicates isolated mitotic aneuploidy generally arising from anaphase lag during rapid cleavage [15, 36]; (b) is usually overlooked clinically when, at the earlier, 8-cell-or-earlier stage, only one cell is sampled for preimplantation testing; or (c) through divisional disadvantage is ordinarily followed by cell-line dilution and extinction [37], or lingering low-level placental mosaicism of uncommon clinical importance [38]. Alternatively, discrete cell analysis of a multicellular TE biopsy (as occurs with FISH), by revealing occasional aneuploid cells, can be over-responded to if it is elected not to transfer the blastocyst on this basis [14].
Initial sequence data were obtained using software supplied with the Ion Torrent Suite 3.2 (Fig. 12.2). Individual partial genome sequence results for each sample were de-convoluted for each chromosome and scored. Total sequence reads per chromosome for each embryo were converted to a fraction of the total sequence reads for the autosomes from that embryo and a Z-score table was constructed (Table 12.1). Most analyses were performed with c. 40,000–c. 250,000 such hits per sample. The fractional reads per chromosome were then used to calculate a fraction mean and the standard deviation of the fraction mean. A score greater than 3 standard deviations (SDs) above or below the population mean for the particular chromosome was considered a necessary and sufficient deviation to indicate highly probable aneuploidy (trisomy or monosomy, respectively). In cases of doubt (Fig. 12.3), the already amplified DNA was tested again at resolutions up to c. 800,000 hits. All aneuploidies identified on array CGH were confirmed by NGS, with typical individual array-based aneuploid ascertained chromosome fraction scores appearing 4–8 SDs away from the fraction mean.
The log2 ratio between the human genome reference sequence and the ‘test’ sequence in the SeqVar methodology led to identification of every loss and gain detected by aCGH. On aCGH, two blastocysts (8 %) showed an intrachromosomal segmental aneuploidy with a uniform loss of a substantial part of one chromosome: one case of loss of 5p and one case of loss of 6q14-tel; a similar segmental aneuploidy prevalence among blastocysts has been reported by others [32]. Mean hit analysis using SeqVar readily detected the significant proportional deviation for the 6q deletion analysis, but the 5p loss was equivocal at c. 245,000 reads; testing at increased resolution rendered this segmental aneuploidy obvious. As is the case with aCGH, NGS output plots were visually inspected for anomalies, paying particular attention to focal or segmental within-chromosome losses or gains that reach log2 ratios outside the range of −1.0 to +0.58 or −1.0. The Agilent CGH array employs a software-based centralization algorithm that balances overall gains and losses and renders the sample’s most common ploidy the new zero point—a step acknowledged to lead to erroneous calls for highly aberrant genomes (Agilent Genomic Workbench 7.0 handbook: CGH Interactive Analysis, p. 476). This step is not required with the Ion Torrent/SeqVar direct sequencing strategy, where limits are precisely predefined numerically prior to analysis.
Discussion
From a simple biological perspective, the unsuitability for transfer of any embryo that has significant chromosomal imbalance(s) is unquestioned. What has caught the attention of clinics throughout the world, however, is the relatively high level of aneuploidy amongst otherwise clinically suitable embryos as well the diverse nature of the chromosomes involved. The use of whole chromosome analysis methods is having a significant impact on the ability to identify and transfer genetically suitable embryos with subsequent implantation rates compared to their standard IVF patients nearly doubling in some clinics. The application and benefits of CGH are now receiving worldwide acknowledgement. The use of commercial microarrays has simplified the approach to total ploidy analysis and has permitted many laboratories to offer this service. However, the cost of array CGH can be prohibitive and potentially excludes an even wider uptake, at least in some countries around the world. New technologies such as NGS are now offering a different approach to the same solution of total chromosome analysis. Currently, we show the process timing for arrays and NGS is similar (see Fig. 12.4).

Workflow schedule for partial genome sequencing and for aCGH sequencing and alignment timing is for low hit analysis. As resolution need is increased, then sequencing and alignment times increase to approximately 8 h
Employing massive parallel sequencing with an average of 8–12 million reads per sample of embryo trophectoderm, Yin and coworkers showed that next generation sequencing technologies can reveal aneuploidies and unbalanced chromosomal rearrangements; their methodology, however, required 10–17 days of lab time [33]—a time frame that while suitable for IVF/cryocycle transfer is not appropriate for fresh transfer. We report the similar use of NGS technology but using a reduced-representation (‘partial’) approach (see Simpson et al. [39] for a methodological review) to comprehensively study morphologically normal human IVF embryos with a sample of trophoblast and to disclose chromosomal aneuploidies utilising economically low numbers of reads across the genome. The methods employ commercially available NGS chips and equipment available to most IVF laboratories experienced with molecular genetic testing for preimplantation genetic diagnosis. Using different modes of analysis, NGS is also able to identify segmental chromosome losses as well as quantify, in an objective way, the relative abundance of individual chromosomes and so disclose mosaicism to various levels.
We show that complete karyotypes via NGS for biopsied blastocysts can also be available overnight, as is the case with microarrays based on CGH, the present standard [25], while potentially providing some useful advantages.
First, by electively increasing the number of hits per genome or chromosome, we can flexibly increase intrachromosomal resolution. For clinically infertile couples undergoing IVF, as few as 40,000 reads per whole genome enable reliable counting of whole chromosomes to avoid transferring grossly aneuploid embryos. A clinical need for higher levels of within-chromosomal resolution can become apparent during low-resolution screening sequencing (Fig. 12.3) or can be planned in advance for preimplantation genetic diagnosis in families with a known intrachromosomal segmental CNV or small segment reciprocal translocations. We show that 400,000 or more reads detect relatively small segmental losses within chromosomes and also may enable greater discernment of blastocyst mosaicism.
Second, as equipment manufacturers produce improvements in NGS chip capacity, the number of sequencing tests performed per fixed price lab NGS run is increasing with little change in cost of materials. The Ion 316 chip we used delivered about 2.5 million mappable reads, enabling simple but full karyotypic analysis of up to 50 indexed embryos in one sequencing run. Process improvements in commercially available sequencing kits that decrease the time needed for testing to a single day can be expected in due course, enabling potential for same-day results and the transfer of the embryo or embryos starting with the fresh treatment cycle in which eggs have been retrieved and fertilised. Routine CGH with IVF thus offers the promise of clinical scale karyotyping of all embryos before transfer or cryostorage, at an increasingly economical cost.
Finally, it could be that in some circumstances NGS with the Ion Torrent/SeqVar algorithms is able to resolve genomic complexities beyond the resolution of standard aCGH and reduce the necessity for array customisation in such cases or when there are highly aberrant genomes such as the mosaic states seen in blastocysts [37]. We are presently investigating this possibility further by applying array aCGH and NGS in parallel to a series of aneuploidy-exclusion trophectoderm biopsy cases in our clinical service.
These developments bring blastocyst-based clinical IVF to the point where whole-genome karyotyping can be used to potentially screen all embryos before transfer and thus to substantially decrease chromosomally abnormal conceptions from compromising reproductive objectives. Early experience revealed that whole-genome preimplantation screening for aneuploidy had the capacity to increase pregnancy rates to over 50 % per embryo transferred [23]. By reducing or eliminating chromosomally abnormal embryos [9, 10], the routine use of CGH can be expected also to reduce miscarriage risk by approximately half. These predicted outcomes represent significant improvements over standard IVF practice and even over natural conception [40, 41]. Moreover, by ensuring high rates of implantation, the transfer of chromosomally normal embryos one at a time ought to lead IVF practitioners to limit multiple embryo transfers and thus to reduce IVF-associated perinatal mortality and morbidity from multiple pregnancy [3, 5, 28].
Which approach to take—array or NGS? There are different laboratory technical and equipment requirements for the arrays compared to the sequencing approach, and these differences may be one of the deciding factors on which technology a clinic can or should employ. It is conceivable that array implementation (aCGH) is the best approach for some small to medium clinics with variable loads and insufficient resources to support specialised scientists for NGS, whereas partial genome sequencing (NGS) may be more suited to a bigger clinic or even a service centre with greater resources in staffing. The final decisions may need to be based on what is most appropriate for the individual clinic. Either array-based or NGS-based embryo molecular karyotyping has the opportunity to improve transfer outcomes for most clinics. With regard to transfer of a tested embryo, which is best—fresh or frozen? Recent reports seem to suggest that a cycle involving embryo storage and subsequent transfer in a non-stimulated situation possibly offers the best outcomes with highest implantation rates and healthiest pregnancies, as the impact of the stimulation protocol on endometrial receptivity may play a larger part on final cycle outcomes than was attributed previously [42, 43]. This would mean that immediate requirements for a speedy analysis protocol may be of lesser importance as would any consideration of protocol changes for biopsy on day 5 compared to day 6. In addition, biopsy followed by vitrification would permit even larger numbers of laboratories to outsource total aneuploid screening through service suppliers and avoid incurring the added burden of expensive capital equipment acquisition and maintenance or supporting further specialised staff.
Note: Life Technologies has now released a software package called ‘Ion Reporter’ that performs similar functions to the bioinformatics reported herein. This means even more laboratories can now readily access analysis platforms for aneuploidy and segmental chromosome assessment by sequencing.
References
Catt J, Wood T, Henman M, Jansen R. Single embryo transfer in IVF to prevent multiple pregnancies. Twin Res. 2003;6:536–9.
Land JA, Evers JLH. Risks and complications in assisted reproduction techniques: report of an ESHRE consensus meeting. Hum Reprod. 2003;18:455–7.
Henman M, Catt JW, Wood T, Bowman MC, de Boer KA, Jansen RPS. Elective transfer of single fresh blastocysts and later transfer of cryostored blastocysts reduces the twin pregnancy rate and can increase the IVF live birth rate in younger women. Fertil Steril. 2005;84:1620–7.
de Boer KA, Catt JW, Jansen RPS, Leigh D, McArthur S. Moving to blastocyst biopsy for preimplantation genetic diagnosis and single embryo transfer at Sydney IVF. Fertil Steril. 2004;82:295–8.
Van Steirteghem A. A European perspective on single embryo transfer. In: Gerris J, Adamson GD, De Sutter P, Racowsky C, editors. Single embryo transfer. New York, NY: Cambridge University Press; 2009. p. 283–8.
Jansen RPS, McArthur SJ. Ovarian stimulation, blastocyst culture and preimplantation genetic screening for elective single embryo transfer. In: Gerris J, Adamson GD, De Sutter P, Racowsky C, editors. Single embryo transfer. New York, NY: Cambridge University Press; 2009. p. 93–108.
Montag M, Liebenthron J, Köster M. Which morphological scoring system is relevant in human embryo development? Placenta. 2011;32:s252–6.
Machtinger R, Racowsky C. Morphological systems of human embryo assessment and clinical evidence. Reprod Biomed Online. 2013;26:210–21.
Boué A, Boué J, Gropp A. Cytogenetics of pregnancy wastage. Adv Hum Genet. 1985;14:1–57.
Eiben B, Bartels I, Bähr-Porsch S, Borgmann S, Gatz G, Gellert G, et al. Cytogenetic analysis of 750 spontaneous abortions with the direct-preparation method of chorionic villi and its implications for studying genetic causes of pregnancy wastage. Am J Hum Genet. 1990;47:656–63.
Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Med Genet. 2005;7:251–63.
Delhanty JDA. Mechanisms of aneuploidy induction in human oogenesis and early embryogenesis. Cytogenet Genome Res. 2005;111:237–44.
Mantzouratou A, Delhanty JDA. Aneuploidy in the human cleavage stage embryo. Cytogenet Genome Res. 2011;133:141–8.
Jansen RPS, Bowman MC, de Boer KA, Leigh DA, Lieberman DB, McArthur SJ. What next for preimplantation screening (PGS)? Experience with blastocyst biopsy and testing for aneuploidy. Hum Reprod. 2008;23:1476–8.
Coonen E, Derhaag JG, Dumoulin JCM, van Wissen LCP, Bras M, Janssen M, Evers JLH, Geraedts JPM. Anaphase lagging mainly explains chromosomal mosaicism in human preimplantation embryos. Hum Reprod. 2004;19:316–24.
Daphnis DD, Delhanty JDA, Jerkovic S, Geyer J, Craft I, Harper JC. Detailed FISH analysis of day 5 human embryos reveals the mechanisms leading to mosaic aneuploidy. Hum Reprod. 2005;20:129–37.
Harrison RH, Kuo H-C, Scriven PN, Handyside AH, Mackie Ogilvie M. Lack of cell cycle checkpoints in human cleavage stage embryos revealed by a clonal pattern of chromosomal mosaicism analysed by sequential multicolour FISH. Zygote. 2000;8:217–24.
Avo Santos M, Teklenburg G, Macklon NS, Van Opstal D, Schuring-Blom GH, Krijtenburg PJ, de Vreeden-Elbertse J, Fauser BC, Baart EB. The fate of the mosaic embryo: chromosomal constitution and development of Day 4, 5 and 8 human embryos. Hum Reprod. 2010;25:1916–26.
Baart EB, van den Berg I, Martini E, Eussen HJ, Fauser BCJM, Van Opstal D. FISH analysis of 15 chromosomes in human day 4 and 5 preimplantation embryos: the added value of extended aneuploidy detection. Prenat Diagn. 2007;27:55–63.
Wells D, Sherlock JK, Handyside AH, Delhanty JD. Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res. 1999;27:1214–8.
Wells D, Delhanty JDA. Comprehensive chromosomal analysis of human preimplantation embryos using whole genome amplification and single cell comparative genomic hybridization. Mol Hum Reprod. 2000;6:1055–62.
Wilton L, Williamson R, McBain J, Edgar D, Voullaire L. Birth of a healthy infant after preimplantation confirmation of euploidy by comparative genomic hybridization. N Engl J Med. 2001;345:1537–41.
Wells D, Alfarawati S, Fragouli E. Use of comprehensive chromosomal screening for embryo assessment: microarray and CGH. Mol Hum Reprod. 2008;14:703–10.
Fragouli E, Lenzi M, Ross R, Katz-Jaffe M, Schoolcraft WB, Wells D. Comprehensive molecular cytogenetic analysis of the human blastocyst stage. Hum Reprod. 2008;23:2596–608.
Schoolcraft WB, Fragouli E, Stevens J, Munné S, Katz-Jaffe MG, Wells D. Clinical application of comprehensive chromosomal screening at the blastocyst stage. Fertil Steril. 2010;94:1700–6.
Kuwayama M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: the Cryotop method. Theriogenology. 2007;67:73–80.
McArthur SJ, Leigh D, Marshall JT, de Boer KA, Jansen RPS. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil Steril. 2005;84:1628–36.
McArthur SJ, Leigh D, Marshall JT, Gee AJ, De Boer KA, Jansen RPS. Blastocyst trophectoderm biopsy and preimplantation genetic diagnosis for familial monogenic disorders and chromosomal translocations. Prenat Diagn. 2008;28:434–42.
Jansen RPS. Benefits and challenges brought by improved results from in vitro fertilization. Intern Med J. 2005;35:108–17.
Fragouli E, Alfarawati S, Daphnis DD, Goodall NN, Mania A, Griffiths T, Gordon A, Wells D. Cytogenetic analysis of human blastocysts with the use of FISH, CGH and aCGH: scientific data and technical evaluation. Hum Reprod. 2011;26:480–90.
Johnson DS, Cinnioglu C, Ross R, Filby A, Gemelos G, Hill M, Ryan A, Smotrich D, Rabinowitz M, Murray MJ. Comprehensive analysis of karyotypic mosaicism between trophectoderm and inner cell mass. Mol Hum Reprod. 2009;16:944–9.
Bisgnano A, Wells D, Harton G, Munné S. PGD and aneuploidy screening for 24 chromosomes: advantages and disadvantages of competing platforms. Reprod Biomed Online. 2011;23:677–85.
Yin XY, Tan K, Vajta G, Jiang H, Tan YQ, Zhang CL, et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol Reprod. 2013;88:1–6.
Clouston HJ, Herbert M, Fenwick J, Murdoch AP, Wolstoneholm J. Cytogenetic analysis of human blastocysts. Prenat Diagn. 2002;22:1143–52.
Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80–8.
Vanneste E, Voet T, Melotte C, Debrock S, Sermon K, Staessen C, Liebaers I, Fryns JP, D’Hooghe T, Vermeesch JR. What next for preimplantation genetic screening? High mitotic chromosome instability rate provides the biological basis for the low success rate. Hum Reprod. 2009;24(11):2679–82.
Barbash-Hazan S, Frumkin T, Malcov M, Yaron Y, Cohen T, Azem F, Amit A, Ben-Yosef D. Preimplantation aneuploid embryos undergo self-correction in correlation with their developmental potential. Fertil Steril. 2009;92:890–6.
Kalousek DK, Vekemans M. Confined placental mosaicism. J Med Genet. 1996;33(7):529–33.
Simpson JL, Rechitsky S, Kuliev A. Next-generation sequencing for preimplantation genetic diagnosis. Fertil Steril. 2013;99:1203–4.
Jansen RPS. Elusive fertility: fecundability in perspective. Fertil Steril. 1995;64:252–4.
Leridon H. Levels of natural fertility. In: Human fertility, the basic components. Chicago, IL: University of Chicago Press; 1977. p. 104–20.
Roy TK, Bradley CK, Bowman MC, McArthur SJ. Single-embryo transfer of vitrified-warmed blastocysts yields equivalent live-birth rates and improved neonatal outcomes compared with fresh transfers, Chapter 14 Microarrays. Fertil Steril. 2014;101:1294–301.
Barnhart KT. Introduction: are we ready to eliminate the transfer of fresh embryos in in vitro fertilization? Fertil Steril. 2014;102(1):1–2.
International Standing Committee on Human Cytogenetic Nomenclature. In: Shaffer LG, McGowan-Jordon J, Schmid M, editors. ISCN 2013: An international system for human cytogenetic nomenclature. Basel: Karger; 2013. p. 121–8.
Acknowledgements
We thank Omar Chami, Ph.D., at Genea for his contribution towards the administration associated with the 25 blastocysts contributed to the project under NHMRC Licence 309702B and for thawing the embryos for further culture and preparation leading to the analyses.
Competing Interests DL, SMcA, and RPSJ are shareholders in Genea Limited (formerly Sydney IVF, Ltd), an unlisted public company.
Conflict of Interest The authors declare no conflict.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Barahona, P., Leigh, D., Ritchie, W., McArthur, S.J., Jansen, R.P.S. (2015). Array CGH and Partial Genome Sequencing for Rapidly Karyotyping IVF Blastocysts Before Single Transfer. In: Sills, E. (eds) Screening the Single Euploid Embryo. Springer, Cham. https://doi.org/10.1007/978-3-319-16892-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-16892-0_12
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16891-3
Online ISBN: 978-3-319-16892-0
eBook Packages: MedicineMedicine (R0)