
Abstract
The alveolar epithelium is composed of type I and type II cells. Flat squamous type I cells comprise 95 % of the alveoli surface area and play essential roles in mediating the gas exchange. Cuboidal type II cells occupy only 5 % of the area but have multiple functions including secreting surfactant, transporting ion and fluid, and modulating immunity. Type II cells also function as progenitor cells by self-renewal and differentiating into type I cells to maintain the alveoli cell homeostasis or to induce lung repair. This chapter focuses on the progenitor cell properties of alveolar type II cells in the adult lungs. In vitro and in vivo studies have shown that type II cells can behave as alveolar stem cells and play important roles in the repair of various types of lung injury. Some subgroups of type II cells may play more active roles in the repair by displaying higher potential for proliferation and transition from type II to type I cells. The initiation of the repairing program by type II cells depends on the changing microenvironment in response to injuries. Several signaling pathways and molecules involved in the type II cell repair programs are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Lung alveoli are lined by a continuous epithelial layer consisting of type I and type II cells. Type I cells constitute approximately 8 % of the total parenchymal lung cells but form more than 90 % of the alveolar surface area (Weibel 2009; Schneeberger 1997). Type I cells have a thin, flat, squamous shape with multiple branches spread over a large area; this shape facilitates their close contact with the basement membrane and capillary endothelial cells, allowing for efficient gas exchange (Schneeberger 1997; Weibel 2009). Type II cells usually reside at the corners of alveoli and are sometime referred to as “corner cells” (Mason and Shannon 1997; Weibel 2009) (Fig. 2.1a–b). Type II cells constitute approximately 15 % of all lung cells, but because of their cuboidal shape, they occupy only approximately 5 % of the alveolar surface area (Mason and Shannon 1997; Weibel 2009).

A model showing type II cell progenitor functions following alveoli injury. (a) Schematic representation of alveoli. (b) Section of mouse lung alveoli region stained by type II cell marker Sp-C (red) and type I cell marker T1α (green). Scale bar = 50 μm. (c) Upon alveolar epithelial injury, some type II cells are activated, undergo proliferation and transition into type I cells to restore alveolar barrier integrity
Alveolar type II cells have multiple functions. One of the most important functions of these cells is to synthesize and secrete surfactant (Mason and Shannon 1997; Mason 2006). Surfactants are synthesized in type II cell-specific organelles called “lamellar bodies.” Each type II cell contains approximately 150 lamellar bodies. This structure has a mean diameter of 1 μm, contains multiple phospholipid bilayers, and gives type II cells a unique morphology (Mason and Shannon 1997; Mason 2006; Mason and Crystal 1998). In this organelle, the phospholipids are packed with the surfactant proteins Sp-A, Sp-B, Sp-C, and Sp-D, which constitute surfactant (Andreeva et al. 2007; Mason and Shannon 1997). When secreted into the alveolar space, surfactant maintains alveolar surface tension and prevents lung collapse (Mason and Shannon 1997; Mason and Crystal 1998). While Sp-A, Sp-B, and Sp-D are also produced in some bronchiolar cells (Mason and Shannon 1997; Walker et al. 1986; Fehrenbach 2001), Sp-C is produced only in type II cells and is considered a type II cell marker (Kalina et al. 1992; Mason and Shannon 1997; Fehrenbach 2001).
Other than producing surfactant, type II cells also play roles in the transepithelial transport of ions and fluids (Mason and Shannon 1997; Mason 2006), modulation of lung inflammatory responses (Mason 2006), and maintenance of epithelial homeostasis and integrity by acting as progenitor cells through proliferation and differentiating into alveolar type I cells (Mason and Shannon 1997; Mason 2006; Stripp 2008; Rock and Hogan 2011). The function of type II cells as alveoli epithelial progenitor cells during injury repair is discussed in this chapter.
Type II cell hyper-proliferation is an important feature in the resolution and repair phase following lung injury (Matthay et al. 2012; Ware and Matthay 2000; Shimabukuro et al. 2003). Evidence indicates that type II cells function as progenitor cells for the re-epithelialization of injured alveoli by converting into type I cells (Evans et al. 1973, 1975) (Fig. 2.1c). In the 1970s, Evans et al. performed classical experiments using NO2 to damage the alveolar surface and showed that 3H-TdR was first incorporated into proliferating type II cells and later labeled type I cells (Evans et al. 1973, 1975). In addition, it has been shown that isolated type II cells can differentiate into type I cells in culture (Dobbs 1990). However, knowledge of the detailed cellular and molecular mechanisms of the progenitor cell properties of type II cells has only recently started to emerge.
2 Type II Cells Exhibit Progenitor Cell Phenotypes In Vitro
No reported cell line exhibits all the major properties of type II cells. However, techniques have been developed to isolate type II cells from human, rat, and mouse lungs (Kikkawa and Yoneda 1974; Dobbs 1990; Corti and Brody 1996). Basically, lung samples are separated into single cell suspensions using enzymes, such as dispase, and the type II cells are enriched using differential sedimentation. The type II cells can be further purified using antibodies to surface antigens of different cell types (Dobbs 1990; Corti and Brody 1996) (Fig. 2.2a).

Type II cells are able to differentiate into type I cells in culture. (a) Freshly isolated mouse type II cells stained with modified Papanicolaou method (Dobbs 1990). Dark blue dots are the lamellar bodies (Dobbs 1990). (b) After culturing on plastic surface, some type II cells underwent transition into type I-like cell and expressed type I cell marker T1α. This cell also expressed GFP in cytoplasm. (c) When co-cultured with lung fibroblasts in 3D matrigel, some type II cells formed alveoli-like structures that contain Sp-C expressing type II cells and T1α expressing type I cells. Scale bar = 10 μm
Isolated type II cells can be cultured on mixture of collagen, laminin, or fibronectin substrata on which they maintain their type II cell phenotypes (such as forming lamellar bodies and secreting surfactant) for approximately 1 week; however, the cells have a tendency to differentiate into flat type I-like cells (Dobbs 1990; Rice et al. 2002; Gobran and Rooney 2004; Rannels et al. 1987; Paine and Simon 1996). If isolated type II cells are cultured on plastic in the presence of 5–10 % fetal bovine serum and without exogenous extracellular matrix, they undergo a morphological change, lose their lamellar bodies, and differentiate into flat type I-like cells within 2–7 days (Dobbs 1990). Some of these type II cell-derived flat cells express type I cell markers such as aquaporin 5 and T1α (Williams 2003) (Fig. 2.2b). Thus, in vitro studies have suggested that type II cells may be precursors of type I cells.
Recently, a new 3D culturing technique of culturing type II cells has been developed (Barkauskas et al. 2013; McQualter et al. 2010; Chen et al. 2012a). When isolated type II cells are cultured in Matrigel (a mixture of extracellular matrix components secreted by Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells) (Kleinman and Martin 2005), they form cysts consisting of polarized monolayers of type II cells, and the cells secrete surfactant into the lumen (Dodelet and Pasquale 2000). However, when type II cells are co-cultured with lung mesenchymal cells in Matrigel (Barkauskas et al. 2013; McQualter et al. 2010; Chen et al. 2012a), they form 3D alveoli-like structures with flat type I-like cells lining the lumen and cuboidal type II-like cells facing the matrix (Fig. 2.2c). By mixing cells from different lineage-labeled lines, it appears that each of the alveolar-like spheres can be derived from a single cell (Barkauskas et al. 2013). These results show that at least some subgroups of type II cells have stem cell characteristics that include the ability to expand clonally and differentiate into multiple lineages.
From the above studies, the behavior of type II cells varies depending on the culture conditions. Developing new culture techniques will lead to novel discoveries of the progenitor cell aspects of type II cells. Some semi-in vivo culture methods, such as growing cells in subcutaneous Matrigel plugs or in a renal capsule (Chapman et al. 2011; Lee et al. 2014), may provide additional insight into type II cell progenitor functions.
3 Type II Cells Act as Progenitor Cells During Lung Repair in Animal Models
To study the progenitor cell behavior of type II cells during alveolar repair in vivo, it is important to have proper animal models to mimic various alveolar injuries; these studies also provide a bridge between patients and laboratory research. Because of their size and the availability of genetic approaches, mouse and rat models are widely used. Typically, three types of agents are used to target the alveolar epithelium in experimental animals: (1) the inhalation of gasses, such as toxic NO2 (Evans et al. 1973, 1975); because high concentrations of O2 can cause alveolar damage in rodents, hyperoxia is a frequently used alveolar injury model (Pogach et al. 2007); (2) the administration of chemicals or antibiotics, such as acid (Matute-Bello et al. 2008) or bleomycin (Flozak et al. 2010); and (3) the introduction of pathogens such as the intratracheal injection of Pseudomonas aeruginosa bacteria (Sadikot et al. 2005; Liu et al. 2011) or the H1N1 influenza virus (Kumar et al. 2011) (see Table 2.1).
The mechanisms that cause alveolar damage vary in different injury models. NO2 causes cell death by nitrating oxidants and the subsequent inflammatory responses (Persinger et al. 2001). In the hyperoxia model, high oxygen concentrations (usually 80–100 %) release reactive oxygen species or free radicals derived from O2, causing necrosis or apoptosis of alveolar epithelia cells (Pagano and Barazzone-Argiroffo 2003). Hyperoxia also leads to the activation of NFκB signaling and inflammatory responses that further enhance alveolar damage (Matute-Bello et al. 2008). One limitation of the hyperoxia model is that, in humans with normal lungs, exposure to 100 % oxygen does not cause clinical or pathological lung injury (Matute-Bello et al. 2008). The acid aspiration model is usually accomplished by the instillation of HCl into the trachea. The acid causes alveolar cell death, increased alveolar barrier permeability and an acute inflammatory response (Matute-Bello et al. 2008). Bleomycin is an antineoplastic antibiotic (Matute-Bello et al. 2008; Wansleeben et al. 2013) that, when administered into the lung, forms a complex with oxygen and metals, such as Fe, leading to the production of oxygen radicals that cause alveolar cell death (Matute-Bello et al. 2008; Wansleeben et al. 2013). At the same time, the bleomycin-treated lung undergoes an inflammatory response that increases damage (Wansleeben et al. 2013; Matute-Bello et al. 2008). The intratracheal instillation of bacteria causes pneumonia. Some bacteria such as Pseudomonas aeruginosa produce toxins that penetrate the cell membrane, causing cell death (Sadikot et al. 2005). The H1N1 influenza virus causes cell death and an inflammatory response (Wansleeben et al. 2013; Hendrickson and Matthay 2013; Kumar et al. 2011) (Table 2.1).
Excessive inflammation is a factor common to most of these models and plays an important role in causing cellular damage (Matute-Bello et al. 2011). The only exception listed in Table 2.1 is a model of diphtheria toxin-induced lung injury (Barkauskas et al. 2013). Using a knock-in mouse in which the expression of diphtheria toxin in type II cells is induced following tamoxifen injection, there does not appear to be a clear inflammatory response (Barkauskas et al. 2013) (Table 2.1).
Some types of injuries are reversible; for example, Pseudomonas aeruginosa-induced lung injury is usually repaired in approximately 7–10 days (Liu et al. 2011). By contrast, some injuries lead to chronic disease; for example, bleomycin injection can induce acute lung injury in the early phase; later, the lung frequently develops fibrosis (Flozak et al. 2010; Chapman et al. 2011; Zhao et al. 2002). Recent findings indicate that the lung alveoli may utilize different repair mechanisms in response to different types of damage. For example, a group of putative progenitor cells called BASC (for bronchioalveolar stem cells) appear to migrate and proliferate in response to bleomycin-induced injury (Barkauskas et al. 2013), but they do not respond to hyperoxia (Rawlins et al. 2009). This result is not surprising because the pathological mechanism as well as the extent of injury varies in different models. Most agents cause damage in multiple cell types in the alveoli, some injuries may be localized to certain foci, and others affect a more extended region. One might hypothesize that certain local facultative progenitor cells may be sufficient to repair a local mild injury, while putative stem cells reside in certain niches in the alveoli or airway may be mobilized in response to a more severe injury (Vaughan and Chapman 2013). Therefore, it is important to compare several injury models to study the mechanisms of alveolar repair.
Type II cells have been shown to be involved in repair in different injury models (Table 2.1). In response to NO2, type II cells proliferate, as revealed by 3H labeling (Evans et al. 1973, 1975). In addition, in pulse labeling experiments, the 3H label appeared first in type II cells and later in type I cells, suggesting that type II cells give rise to type I cells (Evans et al. 1973, 1975). Following hyperoxia or acid aspiration, type II cells engage in hyper-proliferation (Lee et al. 2006; Pogach et al. 2007; Desai et al. 2014). Following bleomycin-induced injury, type II cells proliferate, and lineage tracing experiments using this injury model showed that type II cells also differentiate into type I cells (Barkauskas et al. 2013; Rock et al. 2011). In a Pseudomonas aeruginosa-induced injury model (Liu et al. 2011), type II cells undergo proliferation in the early phase and differentiate into type I cells in the later repair phase (Liu et al. 2011). Finally, in the unilateral pneumonectomy (PNX) model, type II cell proliferation also plays an important role in the compensatory growth of the remaining lung (Nolen-Walston et al. 2008). Therefore, the involvement of type II cells is a common repair mechanism following alveolar injury. However, the exact molecular mechanisms and the subgroups of type II cells that are involved in the repair of different injuries may differ.
4 Lineage Analysis of Type II Cells Involved in Alveoli Repair
Even though the historical experiments by Evans et al. discussed above (Evans et al. 1973, 1975) strongly suggested that type II alveolar cells behave as progenitor cells by proliferating and differentiating into type I cells following injury, it is only with the recent development of new genetic approaches that direct evidence has shown that type II cells give rise to type I cells during alveolar homeostasis and repair (Rock et al. 2011; Barkauskas et al. 2013; Desai et al. 2014).
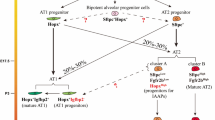
By adopting the yeast Cre DNA recombination enzyme and the cis-element loxP site (Soriano 1999), new genetic lineage tracing techniques have been developed in mouse models by coupling three elements together: an inducible Cre recombinase-expressing system; a type II cell-specific promoter; and a reporter allele tagged with a fluorescent protein (Kretzschmar and Watt 2012). Figure 2.3 shows an example of this system. With the administration of certain chemicals, in this case, tamoxifen, into mice, Cre recombinase is expressed in Sp-C-expressing type II cells, and Cre activates the expression of a fluorescent protein, such as GFP, which will permanently label the type II cells in adult mice; the fate of these labeled cells can be traced any time thereafter (Rock et al. 2011; Desai et al. 2014). Therefore, even if these type II cells later differentiate into another cell type, the cells can still be visualized by the fluorescent signals.

A typical lineage tracing study shows progenitor function of type II cells. (a) A double transgenic (or knock-in) mouse harbors (1) an inducible Cre-expressing system, in this case a Cre-ER in which the expression of Cre recombinase is dependent on tamoxifen (TAM). The Cre expression is also under the control of a type II cell-specific promoter Sp-C; (2) a reporter tagged with a fluorescent protein (in this case GFP). But GFP is normally not expressed because a stop sequence flanked by two loxP sites reside upstream of it. With the administration of TAM into this mouse, Cre is expressed in Sp-C-expressing type II cells, and Cre deletes the stop sequence to activate the GFP expression in type II cells through the constitutive ROSA promoter, and then GFP will permanently label the type II cells as well as their progenies. (b) Schematic representation of a typical result of the lineage tracing studies. Mice were injected with TAM before injury, so that the type II cells were permanently labeled with GFP while type I cells were unlabeled. Following alveoli injury, some type II cells underwent transition into type I cells; therefore, some GFP-labeled type I cells appeared (arrow). In addition, some type II cells without GFP labeling appeared in the lung (arrowhead), suggesting that some Sp-C− cells gave rise to type II cells
Using this lineage tracing mouse line and low doses of tamoxifen, Barkauskas et al. showed that only a small number of type II cells were labeled with the fluorescent proteins. When traced after 12–30 weeks, it was found that these labeled cells formed small clusters, indicating that type II cells undergo a slow self-renewal (Barkauskas et al. 2013). The same studies also showed that the labeled type II cells slowly differentiated into type I cells during steady-state tissue maintenance. Immediately after the mice were injected with tamoxifen, only type II cells were labeled by fluorescent proteins. However, after a 24-week chase, some flat cells expressing type I cell markers were also labeled with the same fluorescent signals, indicating that the lineage-labeled type II cells had converted into type I cells. These results are consistent with another lineage tracing study using a different mouse line (Desai et al. 2014) and support the conclusions from earlier cell tracing studies using 3H-labeled thymidine showing that the steady state turnover time of type II cells is on the order of 4–6 weeks (Spencer and Shorter 1962; Fehrenbach 2001).
From these studies, type II cells have been shown to be relatively quiescent in the absence of injury. However, in response to alveolar injury, type II cells are activated and enter the repairing program (Mason and Shannon 1997; Barkauskas et al. 2013; Desai et al. 2014). Using the above lineage tracing mouse lines, it is shown that after injury, the conversion of type II cells into type I cells was greatly accelerated (Barkauskas et al. 2013).
Furthermore, in another study that traced the type II cells following Pseudomonas aeruginosa-induced injury, it is found that some lineage-labeled type II cells lost the type II cell marker Sp-C at 5–7 days post-injury, and these cells did not yet express or only expressed faint level of type I cell markers. These may represent the intermediate cells undergoing type II to I transition (Liu et al. 2011).
Another question related to the progenitor function of type II cells in repair is how injured type II cells are replenished. Because type II cells undergo hyper-proliferation following injury (Mason and Shannon 1997), one might hypothesize that type II cells are able to repair themselves solely by self-renewal. However, recent lineage tracing studies have shown that type II cells may not be the only source for self-replenishment (Barkauskas et al. 2013; Chapman et al. 2011). It has been shown using lineage tracing mouse lines (Fig. 2.2), that, following bleomycin-induced injury, the percentage of lineage-labeled type II cells versus the total number of type II cells decreased in the injured areas. These results indicate that some non-lineage-labeled cells, i.e., cells that did not express Sp-C before the injury, gave rise to type II cells during repair (Barkauskas et al. 2013; Chapman et al. 2011).
5 Subgroups of Type II Cells Display Progenitor Features
Type II cells have long been considered a homogeneous population. However, recent studies have indicated that type II cells are heterogeneous and consist of subpopulations. For example, studies using transgenic mice that express GFP under the control of the Sp-C promoter have revealed that certain type II cells express higher levels of GFP than others (Lee et al. 2013; Roper et al. 2003). It would, therefore, be informative to separate type II cells into subpopulations because some of these subgroups may be particularly important in the repair of injured alveoli.
It has been found that approximately 5–10 % of type II cells express low levels of CC10. CC10, also known as CCSP or Scgbb1a1, is a secretory protein expressed at high levels in Club cells located in the airways. A number of Sp-C+CC10+ type II cells are located in the alveolar region; the function of these cells is unknown. A rare population of SP-C+CC10+ cells located at terminal bronchioles is known as BASC. BASCs show certain stem cell properties in vitro including forming colonies when co-cultured with mouse embryonic fibroblasts (MEFs) (Kim et al. 2005a, b) as well as multi-lineage differentiation when cultured in Matrigel (Kim et al. 2005a, b; Lee et al. 2014).
Some subpopulations of type II cells appear following injury. Studies by Reddy et al. showed that type II cells isolated from hyperoxia-treated rats can be separated into two populations after a brief culture; some cells expressed lower levels of E-cadherin than others (Driscoll et al. 2000; Reddy et al. 2004). Furthermore, the E-cadherinhigh population appeared to be relatively quiescent, while the E-cadherinlow population was more proliferative and expressed higher levels of telomerase activity (Driscoll et al. 2000). Therefore, certain type II cells appear to show more progenitor cell properties than their counterparts.
A subgroup of type II cells expressing the progenitor cell marker Sca-1 appear in the early repair phase following Pseudomonas aeruginosa-induced injury (Liu et al. 2011). These cells are mostly derived from Sp-C-expressing type II cells and can account for more than 40 % of the total type II cells following injury (Liu et al. 2011). This Sca-1+ subpopulation of type II cells exhibits a higher proliferation rate and also has a higher potential to convert into type I cells compared with the Sca-1− counterpart (Liu et al. 2011, 2014). These cells express higher levels of the transcription factor FoxM1 than the Sca-1− type II cells, and FoxM1 is required for the alveolar repair process (Liu et al. 2011). Thus, the Sca-1+ subgroup of type II cells appear to be responsible for the repair.
The results from the above studies indicate that certain subpopulations of type II cells manifesting progenitor cell features appear at the repair phase following injury. However, it is unclear whether these putative regenerative type II cells appearing post-injury are derived from the expansion of existing stem cell pools located in an undefined niche or whether they are derived from quiescent, terminally differentiated type II cells. It would also be interesting to identify which signals or factors induce the formation of progenitor-like type II cell subgroups following injury.
Another point is that, in most studies, Sp-C is considered a type II cell-specific marker. In other words, type II cells are defined as Sp-C+ cells. However, it has been observed that some type II cells express lower levels of Sp-C than others (Chapman et al. 2011; Liu et al. 2011; Lee et al. 2013). In addition, it has been reported that a small number of epithelial cells located in alveoli do not show type I cell phenotypes and do not express detectable levels of Sp-C (Chapman et al. 2011). It remains to be determined whether these cells can be regarded as a subtype of type II cells or type II-like cells. Among the Sp-C− non-type I alveolar epithelial cells is a group of cells expressing the α6β4 integrin (Chapman et al. 2011); α6β4 integrin double-positive cells have progenitor cell features and may participate in repair following bleomycin-induced injury (Chapman et al. 2011).
6 Cellular and Molecular Mechanisms Regulating Type II Cell Progenitor Properties
The cellular and molecular mechanisms underlying how type II cells engage in the repair of a damaged alveolar barrier is unclear. It is known that type II cells can convert into type I cells; however, the detailed cellular processes involved in this transition are unknown. One possibility is that certain relatively undifferentiated subgroups of type II cells located at unknown locations differentiate into type I cells. Another possibility is that differentiated, mature type II cells undergo trans-differentiation and give rise to type I cells. It is also possible that mature type II cells first undergo dedifferentiation into intermediate precursors and then differentiate into type I cells. Electron microscopy studies suggest that there are intermediate cell types appearing in NO2-injured lung that show morphological characters of both type II and type I cells (Evans et al. 1975). It has also been reported that alveolar cells co-expressing type II and type I cell surface markers appear following Staphylococcus aureus-induced lung injury (Clegg et al. 2005). The existence of intermediate cells supports the transition of type II to type I cells.
A possible mechanism to initiate type II cell activation into the repair process may be signals related to the injury. Recent studies have suggested that the inflammatory milieu that forms following most types of alveolar injury can create alveolar regenerative signals (Pociask et al. 2013; Buckley et al. 2011). In injuries induced by hyperoxia, the generation of oxidants may also signal the initiation of the repair process (Pogach et al. 2007). Type II cells, in response to unknown signals related to specific injuries, can start proliferation and a type I cell transition, and they may also migrate to the injured region and engage in repair.
Little is known regarding the molecules that regulate the type II cell activation that leads to alveolar repair. One would expect that the transcriptional programs involved in embryonic lung development, such as those under the control of the FGF and Wnt pathways (Cardoso 2001; Morrisey and Hogan 2010; Warburton et al. 2008), may be activated post-injury and may contribute to repair. However, even though FGF and Wnt signaling appear to be involved in alveolar repair (Tanjore et al. 2013; Flozak et al. 2010; Ghosh et al. 2013), several transcription factors (Id2, Erm, Gata6, and Elf5) that are involved in alveolar development have not been shown to have elevated expression in type II cells following Pseudomonas aeruginosa-induced injury (Liu et al. 2011; Liu and Hogan 2002). Therefore, even if there may be some correlations, the repair process does not completely recapitulate the developmental process in embryogenesis.
Several growth factors appear to be able to regulate certain aspects of the progenitor properties of type II cells (Table 2.2). IGF expression is enhanced following hyperoxia-induced injury, and exogenous IGF promotes the type II to type I transition in vitro (Narasaraju et al. 2006). FGF7 (KGF) induces cultured type II cells to proliferate, while preventing them from type I cell differentiation (Qiao et al. 2008; Portnoy et al. 2004; Borok et al. 1998). EGF and HGF can also stimulate cultured type II cells to proliferate (Mason et al. 1994; Portnoy et al. 2004; Desai et al. 2014). The intratracheal injection of HGF or KGF stimulates type II cell proliferation in the lung (Panos et al. 1996; Yano et al. 2000; Fehrenbach et al. 1999, 2003; Ware and Matthay 2002). By contrast, TGFβ expression in the bleomycin-injured lung suggests that it plays a negative regulatory role in the proliferation of type II cells during early repair phase (Khalil et al. 1994). In culture, TGFβ inhibits type II cell proliferation (Khalil et al. 1994) but promotes the conversion of cultured type II cells into type I cells (Zhao et al. 2013), while BMP4, another member of the TGF superfamily, antagonizes this differentiation (Zhao et al. 2013).
The Wnt/β-Catenin signaling pathway plays an important role in the differentiation of lung epithelial cells during development (Li et al. 2005; Shu et al. 2005), and recent studies have shown that Wnt/β-Catenin signaling activity is elevated following bleomycin-induced injury (Tanjore et al. 2013; Flozak et al. 2010). Inhibition of this signaling in cultured type II cells prevented their conversion into type I cells (Tanjore et al. 2013; Flozak et al. 2010; Ghosh et al. 2013). In addition, the microRNA miR375 appears to play a role in the type II cell proliferation/differentiation process through the regulation of the Wnt/β-Catenin signaling pathway (Wang et al. 2013) (Table 2.2).
Thus, FGF, TGF, and Wnt are signaling molecules that are possibly released in response to alveolar damage that may initiate the type II cell-dependent repair process. However, many of these results were generated by expression analysis and in vitro studies; it remains to be determined whether all these factors function in the in vivo context of alveolar repair.
It is also important to understand the transcriptional mechanisms that reprogram type II cells to acquire the progenitor property and initiate the repair process. Recently, it has been found that the forkhead transcription factor FoxM1 plays an essential role in the type II cell-mediated repair of alveolar injury induced by Pseudomonas aeruginosa infection (Liu et al. 2011). FoxM1 expression is elevated in the Sca-1+ subpopulation of type II cells that appear at the early repair phase following alveolar injury (Liu et al. 2011), and genetic studies using mouse models that disrupt FoxM1 in type II cells showed that FoxM1 is required in these cells for their proliferation and transition into type I cells that lead to repair (Liu et al. 2011) (Table 2.2).
Finally, epigenetic modifications may also play important roles in the progenitor cell function of type II cells (Marconett et al. 2013).
7 Regulation of Type II Cell Progenitor Properties by the Microenvironment
As discussed above, in vitro culture experiments showed that the progenitor cell features of type II cells differ in different extracellular matrices, and they show distinct behaviors when co-cultured with other cell types (Rice et al. 2002; Dobbs 1990; Demaio et al. 2009; Shannon et al. 1987; Sannes 1991; Leiner et al. 2006; Olsen et al. 2005). These results indicate that the progenitor cell properties of type II cells are tightly controlled by the cell–matrix and cell–cell interactions related to their particular environments before or after injury. In response to most acute lung injuries, many changes occur, including increases in microvascular permeability, edema and the proliferation of fibroblasts, deposit of collagen matrix and leukocyte recruitment (Ware and Matthay 2000; Matthay et al. 2012). All these factors contribute to an altered microenvironment, and it is likely that the initiation of type II cell activation is triggered by a combination of these factors.
Type II cells are normally in close contact with lung stromal cells in the basal layer. The interaction of type II cells with stromal cells, including fibroblasts, following injury has been studied extensively (Fehrenbach 2001; Thannickal et al. 2004; Mason and Crystal 1998). Fibroblast-derived soluble factors can promote the proliferation of type II cells (Fehrenbach 2001). Recently, a population of PDGFα-expressing stromal cells that include fibroblasts and lipofibroblasts have been identified that normally reside in close proximity with type II cells, and they might form a niche to maintain type II cell stemness (Barkauskas et al. 2013; Chen et al. 2012b). When co-cultured with type II cells, these cells promote type II cell self-renewal as well as differentiation toward type I cell (Barkauskas et al. 2013). Thus, fibroblasts and lipofibroblasts located adjacent to type II cells could play a role in regulating type II cell progenitor behavior through direct cell–cell contact as well as by secreting soluble factors such as growth factors or by remodeling the extracellular matrix (El Ghalbzouri and Ponec 2004).
One important feature of the lung is that alveoli are highly vascularized to facilitate gas exchange, and capillary endothelial cells reside in close proximity to alveolar epithelial cells (Komarova and Malik 2010; Bhattacharya 2005). Recent studies have revealed that alveolar capillary endothelial cells play a role in promoting alveolar regeneration following unilateral pneumonectomy (Ding et al. 2011). Controlling MMP14-dependent release of EGFR ligands appears to be one of the mechanisms that endothelial cells use to instruct type II cells to initiate the repair process (Ding et al. 2011).
Inflammatory cells, such as macrophages and neutrophils, are recruited to the alveolar space, and these cells release various inflammatory factors (Matthay et al. 2012; Ware and Matthay 2000). Among these factors are TNF, IL-1, IL-6, IL-8, and IL-10 (Fehrenbach 2001; Matthay et al. 2012). Many of these factors stimulate the proliferation, migration, and changing of cell fate of various target cells through NFκB signaling (dos Santos et al. 2012; Chen and Greene 2004; Zhang et al. 2009), and it is highly likely that these inflammatory mediators also play a role in directing type II cells to acquire progenitor phenotypes. In fact, it has been shown that IL-1β was able to stimulate migration and proliferation of cultured type II cells (Yang et al. 1999; Geiser et al. 2000). It is also possible that inflammatory cells send regeneration signals to type II cells through cell–cell contact. It has been reported that neutrophil transmigration through cultured lung alveolar type II cells activates Wnt/β-Catenin signaling in type II cells, and this is a possible mechanism for repair initiation (Zemans et al. 2011).
8 Potential Therapy of Lung Disease Utilizing the Progenitor Cell Properties of Type II Cells
Studies of the progenitor properties of type II cells could lead to the discovery of novel therapeutic approaches for the treatment of chronic and acute lung diseases. Discovering the distinct molecular elements of the transition from a quiescent to a regenerative type II cell could result in the identification of new pharmaceutical targets to accelerate lung repair. If we can dissect type II cell-mediated alveolar repair into a sequential process, it may be possible to intervene pharmacologically at each step to promote recovery and prevent or reverse chronic lung disease related to injuries.
Furthermore, because type II cells can be obtained through in vitro differentiation from iPS cells or lung biopsies, a potential therapeutic approach would be transplanting type II cells into injured lungs (Huang et al. 2014; Longmire et al. 2012). In fact, there is one report showing that by transplanting normal type II cells into bleomycin-injured rat lungs, the fibrosis formation induced by bleomycin is delayed or even reversed (Serrano-Mollar et al. 2007). The identification of subgroups of type II cells that function in regeneration will substantially improve such cell transplantation therapies by using distinct populations of type II cells.
Finally, recent lineage tracing studies showed that some lung adenocarcinoma cells are derived from type II cells (Desai et al. 2014; Xu et al. 2012). Therefore, understanding the regulation of type II cell progenitor properties may shed light on the mechanisms of lung cancer initiation.
Abbreviations
- BASC:
-
Bronchioalveolar stem cells
- BMP4:
-
Bone morphogenic protein 4
- CC10:
-
Club Cell 10 kDa protein
- EGFR:
-
Epidermal growth factor receptor
- Elf5:
-
E74-like factor 5
- Erm:
-
Ets-related molecule
- FGF:
-
Fibroblast growth factor
- FoxM1:
-
Forkhead box protein M1
- GATA6:
-
GATA binding protein 6
- GFP:
-
Green fluorescent protein
- HGF:
-
Hepatocyte growth factor
- Id2:
-
Inhibitor of DNA binding 2
- IGF:
-
Insulin-like growth factor
- IL-6/8/10:
-
Interleukin 6/8/10
- iPS:
-
Induced pluripotent stem cells
- KGF:
-
Keratinocyte growth factor
- MEF:
-
Mouse embryonic fibroblast
- MMP14:
-
Matrix metalloproteinase 14
- PDGF:
-
Platelet-derived growth factor
- SPA/B/C/D:
-
Surfactant protein A/B/C/D
- TGFβ:
-
Transforming growth factor beta
- Wnt:
-
Wingless protein
References
Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA (2007) Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 293(2):L259–L271
Barkauskas CE, Michael J, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM (2013) Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123(7):3025–3036
Bhattacharya J (2005) Alveolocapillary cross-talk: Giles F. Filley lecture. Chest 128(6 Suppl):553S–555S
Borok Z, Lubman RL, Danto SI, Zhang XL, Zabski SM, King LS, Lee DM, Agre P, Crandall ED (1998) Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin 5. Am J Respir Cell Mol Biol 18(4):554–561. doi:10.1165/rcmb.18.4.2838
Buckley S, Shi W, Carraro G, Sedrakyan S, Da Sacco S, Driscoll BA, Perin L, De Filippo RE, Warburton D (2011) The milieu of damaged alveolar epithelial type 2 cells stimulates alveolar wound repair by endogenous and exogenous progenitors. Am J Respir Cell Mol Biol 45(6):1212–1221
Cardoso W (2001) Molecular regulation of lung development. Annu Rev Physiol 63:471–494
Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH (2011) Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest 121(7):2855–2862
Chen LF, Greene WC (2004) Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol 5(5):392–401
Chen H, Matsumoto K, Brockway BL, Rackley CR, Liang J, Lee JH, Jiang D, Noble PW, Randell SH, Kim CF, Stripp BR (2012a) Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells 30(9):1948–1960. doi:10.1002/stem.1150
Chen L, Acciani T, Le Cras T, Lutzko C, Perl AK (2012b) Dynamic regulation of platelet-derived growth factor receptor alpha expression in alveolar fibroblasts during realveolarization. Am J Respir Cell Mol Biol 47(4):517–527. doi:10.1165/rcmb.2012-0030OC
Clegg GR, Tyrrell C, McKechnie SR, Beers MF, Harrison D, McElroy MC (2005) Coexpression of RTI40 with alveolar epithelial type II cell proteins in lungs following injury: identification of alveolar intermediate cell types. Am J Physiol Lung Cell Mol Physiol 289(3):L382–L390
Corti M, Brody AR, Harrison JH (1996) Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14(4):309–315
Demaio L, Tseng W, Balverde Z, Alvarez JR, Kim KJ, Kelley DG, Senior RM, Crandall ED, Borok Z (2009) Characterization of mouse alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 296(6):L1051–L1058. doi:10.1152/ajplung.00021.2009
Desai TJ, Brownfield DG, Krasnow MA (2014) Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507(7491):190–194. doi:10.1038/nature12930
Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY, Rafii S (2011) Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147(3):539–553. doi:10.1016/j.cell.2011.10.003
Dobbs LG (1990) Isolation and culture of alveolar type II cells. Am J Physiol 258(4 Pt 1):L134–L147
Dodelet VC, Pasquale EB (2000) Eph receptors and ephrin ligands: embryogenesis to tumorigenesis. Oncogene 19(49):5614–5619
dos Santos G, Kutuzov MA, Ridge KM (2012) The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol 303(8):L627–L633
Driscoll B, Buckley S, Bui KC, Anderson KD, Warburton D (2000) Telomerase in alveolar epithelial development and repair. Am J Physiol Lung Cell Mol Physiol 279(6):L1191–L1198
El Ghalbzouri A, Ponec M (2004) Diffusible factors released by fibroblasts support epidermal morphogenesis and deposition of basement membrane components. Wound Repair Regen 12(3):359–367
Evans MJ, Cabral LJ, Stephens RJ, Freeman G (1973) Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol 70(2):175–198
Evans MJ, Cabral LJ, Stephens RJ, Freeman G (1975) Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp Mol Pathol 22(1):142–150
Fehrenbach H (2001) Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2(1):33–46
Fehrenbach H, Kasper M, Tschernig T, Pan T, Schuh D, Shannon JM, Muller M, Mason RJ (1999) Keratinocyte growth factor-induced hyperplasia of rat alveolar type II cells in vivo is resolved by differentiation into type I cells and by apoptosis. Eur Respir J 14(3):534–544
Fehrenbach A, Bube C, Hohlfeld JM, Stevens P, Tschernig T, Hoymann HG, Krug N, Fehrenbach H (2003) Surfactant homeostasis is maintained in vivo during keratinocyte growth factor-induced rat lung type II cell hyperplasia. Am J Respir Crit Care Med 167(9):1264–1270
Flozak AS, Lam AP, Russell S, Jain M, Peled ON, Sheppard KA, Beri R, Mutlu GM, Budinger GS, Gottardi CJ (2010) {beta}-catenin/TCF signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem 285(5):3157–3167
Geiser T, Jarreau PH, Atabai K, Matthay MA (2000) Interleukin-1beta augments in vitro alveolar epithelial repair. Am J Physiol Lung Cell Mol Physiol 279(6):L1184–L1190
Ghosh MC, Gorantla V, Makena PS, Luellen C, Sinclair SE, Schwingshackl A, Waters CM (2013) Insulin-like growth factor-I stimulates differentiation of ATII cells to ATI-like cells through activation of Wnt5a. Am J Physiol Lung Cell Mol Physiol 305(3):L222–L228. doi:10.1152/ajplung.00014.2013
Gobran LI, Rooney SA (2004) Pulmonary surfactant secretion in briefly cultured mouse type II cells. Am J Physiol Lung Cell Mol Physiol 286(2):L331–L336
Hendrickson CM, Matthay MA (2013) Viral pathogens and acute lung injury: investigations inspired by the SARS epidemic and the 2009 H1N1 influenza pandemic. Semin Respir Crit Care Med 34(4):475–486. doi:10.1055/s-0033-1351122
Huang SX, Islam MN, O’Neill J, Hu Z, Yang YG, Chen YW, Mumau M, Green MD, Vunjak-Novakovic G, Bhattacharya J, Snoeck HW (2014) Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol 32(1):84–91. doi:10.1038/nbt.2754
Kalina M, Mason RJ, Shannon JM (1992) Surfactant protein C is expressed in alveolar type II cells but not in Clara cells of rat lung. Am J Respir Cell Mol Biol 6(6):594–600
Khalil N, O’Connor RN, Flanders KC, Shing W, Whitman CI (1994) Regulation of type II alveolar epithelial cell proliferation by TGF-beta during bleomycin-induced lung injury in rats. Am J Physiol 267(5 Pt 1):L498–L507
Kikkawa Y, Yoneda K (1974) The type II epithelial cell of the lung. I. Method of isolation. Lab Invest 30(1):76–84
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel SM, Crowley D, Bronson RT, Jacks T (2005a) Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121(6):823–835
Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV (2005b) The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem 280(23):22278–22286
Kleinman HK, Martin GR (2005) Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol 15(5):378–386
Komarova Y, Malik AB (2010) Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72:463–493. doi:10.1146/annurev-physiol-021909-135833
Kretzschmar K, Watt FM (2012) Lineage tracing. Cell 148(1–2):33–45. doi:10.1016/j.cell.2012.01.002
Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, de Wang Y, Lim B, Chow VT, Crum CP, Xian W, McKeon F (2011) Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147(3):525–538
Lee JC, Reddy R, Barsky L, Weinberg K, Driscoll B (2006) Contribution of proliferation and DNA damage repair to alveolar epithelial type 2 cell recovery from hyperoxia. Am J Physiol Lung Cell Mol Physiol 290(4):L685–L694
Lee JH, Kim J, Gludish D, Roach RR, Saunders AH, Barrios J, Woo AJ, Chen H, Conner DA, Fujiwara Y, Stripp BR, Kim CF (2013) Surfactant protein-C chromatin-bound green fluorescence protein reporter mice reveal heterogeneity of surfactant protein C-expressing lung cells. Am J Respir Cell Mol Biol 48(3):288–298. doi:10.1165/rcmb.2011-0403OC
Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng YH, Ryeom S, Kim CF (2014) Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 156(3):440–455. doi:10.1016/j.cell.2013.12.039
Leiner KA, Newman D, Li CM, Walsh E, Khosla J, Sannes PL (2006) Heparin and fibroblast growth factors affect surfactant protein gene expression in type II cells. Am J Respir Cell Mol Biol 35(5):611–618. doi:10.1165/rcmb.2006-0159OC
Li C, Hu L, Xiao J, Chen H, Li JT, Bellusci S, Delanghe S, Minoo P (2005) Wnt5a regulates Shh and Fgf10 signaling during lung development. Dev Biol 287(1):86–97
Liu Y, Hogan BL (2002) Differential gene expression in the distal tip endoderm of the embryonic mouse lung. Gene Expr Patterns 2:229–233
Liu Y, Suresh Kumar V, Zhang W, Rehman J, Malik AB (2014) Activation of type II cells into regenerative Sca-1+ cells during alveolar repair. Am J Respir Cell Mol Biol. [Epub ahead of print]
Liu Y, Sadikot RT, Adami GR, Kalinichenko VV, Pendyala S, Natarajan V, Zhao YY, Malik AB (2011) FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med 208(7):1473–1484
Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, Kwok LW, Mou H, Rajagopal J, Shen SS, Dowton AA, Serra M, Weiss DJ, Green MD, Snoeck HW, Ramirez MI, Kotton DN (2012) Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 10(4):398–411. doi:10.1016/j.stem.2012.01.019
Marconett CN, Zhou B, Rieger ME, Selamat SA, Dubourd M, Fang X, Lynch SK, Stueve TR, Siegmund KD, Berman BP, Borok Z, Laird-Offringa IA (2013) Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet 9(6):e1003513. doi:10.1371/journal.pgen.1003513
Mason RJ (2006) Biology of alveolar type II cells. Respirology 11:S12–S15
Mason RJ, Crystal RG (1998) Pulmonary cell biology. Am J Respir Crit Care Med 157(4 Pt 2):S72–S81
Mason RJ, Shannon JM (1997) Alveolar type II cells. In: Crystal RG, West JB, Weibel ER, Barnes PJ (eds) The lung: scientific foundations, vol 1, 3, 2nd edn. Lippincott Williams & Wilkins, Philadelphia, pp 543–556
Mason RJ, Leslie CC, McCormick-Shannon K, Deterding RR, Nakamura T, Rubin JS, Shannon JM (1994) Hepatocyte growth factor is a growth factor for rat alveolar type II cells. Am J Respir Cell Mol Biol 11(5):561–567
Matthay MA, Ware LB, Zimmerman GA (2012) The acute respiratory distress syndrome. J Clin Invest 122(8):2731–2740
Matute-Bello G, Frevert CW, Martin TR (2008) Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295(3):L379–L399. doi:10.1152/ajplung.00010.2008
Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM (2011) An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44(5):725–738. doi:10.1165/rcmb.2009-0210ST
McQualter JL, Yuen K, Williams B, Bertoncello I (2010) Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A 107(4):1414–1419. doi:10.1073/pnas.0909207107
Morrisey EE, Hogan BL (2010) Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 18(1):8–23
Narasaraju TA, Chen H, Weng T, Bhaskaran M, Jin N, Chen J, Chen Z, Chinoy MR, Liu L (2006) Expression profile of IGF system during lung injury and recovery in rats exposed to hyperoxia: a possible role of IGF-1 in alveolar epithelial cell proliferation and differentiation. J Cell Biochem 97(5):984–998
Nolen-Walston RD, Kim CF, Mazan MR, Ingenito EP, Gruntman AM, Tsai L, Boston R, Woolfenden AE, Jacks T, Hoffman AM (2008) Cellular kinetics and modeling of bronchioalveolar stem cell response during lung regeneration. Am J Physiol Lung Cell Mol Physiol 294(6):L1158–L1165. doi:10.1152/ajplung.00298.2007
Olsen CO, Isakson BE, Seedorf GJ, Lubman RL, Boitano S (2005) Extracellular matrix-driven alveolar epithelial cell differentiation in vitro. Exp Lung Res 31(5):461–482
Pagano A, Barazzone-Argiroffo C (2003) Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad Sci 1010:405–416
Paine R, Simon RH (1996) Expanding the frontiers of lung biology through the creative use of alveolar epithelial cells in culture. Am J Physiol 270(4 Pt 1):L484–L486
Panos RJ, Patel R, Bak PM (1996) Intratracheal administration of hepatocyte growth factor/scatter factor stimulates rat alveolar type II cell proliferation in vivo. Am J Respir Cell Mol Biol 15(5):574–581
Persinger RL, Blay WM, Heintz NH, Hemenway DR, Janssen-Heininger YM (2001) Nitrogen dioxide induces death in lung epithelial cells in a density-dependent manner. Am J Respir Cell Mol Biol 24(5):583–590
Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF (2013) IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182(4):1286–1296
Pogach MS, Cao Y, Millien G, Ramirez MI, Williams MC (2007) Key developmental regulators change during hyperoxia-induced injury and recovery in adult mouse lung. J Cell Biochem 100(6):1415–1429
Portnoy J, Curran-Everett D, Mason RJ (2004) Keratinocyte growth factor stimulates alveolar type II cell proliferation through the extracellular signal-regulated kinase and phosphatidylinositol 3-OH kinase pathways. Am J Respir Cell Mol Biol 30(6):901–907
Qiao R, Yan W, Clavijo C, Mehrian-Shai R, Zhong Q, Kim KJ, Ann D, Crandall ED, Borok Z (2008) Effects of KGF on alveolar epithelial cell transdifferentiation are mediated by JNK signaling. Am J Respir Cell Mol Biol 38(2):239–246
Rannels SR, Yarnell JA, Fisher CS, Fabisiak JP, Rannels DE (1987) Role of laminin in maintenance of type II pneumocyte morphology and function. Am J Physiol 253(6 Pt 1):C835–C845
Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL (2009) The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4(6):525–534
Reddy R, Buckle YS, Doerken M, Barsky L, Weinberg K, Anderson KD, Warburton D, Driscoll B (2004) Isolation of a putative progenitor subpopulation of alveolar epithelial type 2 cells. Am J Physiol Lung Cell Mol Physiol 286(4):L658–L667
Rice WR, Conkright JJ, Na CL, Ikegami M, Shannon JM, Weaver TE (2002) Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol 283(2):L256–L264
Rock JR, Hogan BL (2011) Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu Rev Cell Dev Biol 27:493–512
Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL (2011) Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A 108(52):E1475–E1483
Roper JM, Staversky RJ, Finkelstein JN, Keng PC, O’Reilly MA (2003) Identification and isolation of mouse type II cells on the basis of intrinsic expression of enhanced green fluorescent protein. Am J Physiol Lung Cell Mol Physiol 285(3):L691–L700
Sadikot RT, Blackwell TS, Christman JW, Prince AS (2005) Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171(11):1209–1223
Sannes PL (1991) Structural and functional relationships between type II pneumocytes and components of extracellular matrices. Exp Lung Res 17(4):639–659
Schneeberger EE (1997) Alveolar type I cells. In: Crystal RG, West JB, Weibel ER, Barnes PJ (eds) The lung: scientific foundations. Lippincott Williams & Wilkins, Philadelphia, pp 535–542
Serrano-Mollar A, Nacher M, Gay-Jordi G, Closa D, Xaubet A, Bulbena O (2007) Intratracheal transplantation of alveolar type II cells reverses bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 176(12):1261–1268
Shannon JM, Mason RJ, Jennings SD (1987) Functional differentiation of alveolar type II epithelial cells in vitro: effects of cell shape, cell-matrix interactions and cell-cell interactions. Biochim Biophys Acta 931(2):143–156
Shimabukuro DW, Sawa T, Gropper MA (2003) Injury and repair in lung and airways. Crit Care Med S524–531
Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, Piccolo S, Birchmeier W, Whitsett JA, Millar SE, Morrisey EE (2005) Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Dev Biol 283(1):226–239
Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21(1):70–71
Spencer H, Shorter RG (1962) Cell turnover in pulmonary tissues. Nature 194:880
Stripp BR (2008) Hierarchical organization of lung progenitor cells: is there an adult lung tissue stem cell? Proc Am Thorac Soc 5(6):695–698
Tanjore H, Degryse AL, Crossno PF, Xu XC, McConaha ME, Jones BR, Polosukhin VV, Bryant AJ, Cheng DS, Newcomb DC, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE (2013) beta-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am J Respir Crit Care Med 187(6):630–639
Thannickal VJ, Toews GB, White ES, Lynch JP, Martinez FJ (2004) Mechanisms of pulmonary fibrosis. Annu Rev Med 55:395–417
Vaughan AE, Chapman HA (2013) Regenerative activity of the lung after epithelial injury. Biochim Biophys Acta 1832(7):922–930. doi:10.1016/j.bbadis.2012.11.020
Walker SR, Williams MC, Benson B (1986) Immunocytochemical localization of the major surfactant apoproteins in type II cells, Clara cells, and alveolar macrophages of rat lung. J Histochem Cytochem 34(9):1137–1148
Wang Y, Huang C, Reddy Chintagari N, Bhaskaran M, Weng T, Guo Y, Xiao X, Liu L (2013) miR-375 regulates rat alveolar epithelial cell trans-differentiation by inhibiting Wnt/beta-catenin pathway. Nucleic Acids Res 41(6):3833–3844. doi:10.1093/nar/gks1460
Wansleeben C, Barkauskas CE, Rock JR, Hogan BL (2013) Stem cells of the adult lung: their development and role in homeostasis, regeneration, and disease. Wiley Interdiscip Rev Dev Biol 2(1):131–148
Warburton D, Perin L, Defilippo R, Bellusci S, Shi W, Driscoll B (2008) Stem/progenitor cells in lung development, injury repair, and regeneration. Proc Am Thorac Soc 5(6):703–706
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342(18):1334–1349
Ware LB, Matthay MA (2002) Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol 282(5):L924–L940
Weibel ER (2009) What makes a good lung? Swiss Med Wkly 139(27–28):375–386
Williams MC (2003) Alveolar type I cells: molecular phenotype and development. Annu Rev Physiol 65:669–695
Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, Hogan BL, Onaitis MW (2012) Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci U S A 109(13):4910–4915. doi:10.1073/pnas.1112499109
Yang GH, Osanai K, Takahashi K (1999) Effects of interleukin-1 beta on DNA synthesis in rat alveolar type II cells in primary culture. Respirology 4(2):139–145
Yano T, Mason RJ, Pan T, Deterding RR, Nielsen LD, Shannon JM (2000) KGF regulates pulmonary epithelial proliferation and surfactant protein gene expression in adult rat lung. Am J Physiol Lung Cell Mol Physiol 279(6):L1146–L1158
Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, Yang IV, De Langhe S, Reynolds SD, Mason RJ, Kahn M, Henson PM, Colgan SP, Downey GP (2011) Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci U S A 108(38):15990–15995. doi:10.1073/pnas.1110144108
Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, Morrisey EE, Overbeek PA, Scheidereit C, Millar SE, Schmidt-Ullrich R (2009) Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell 17(1):49–61
Zhao J, Shi W, Wang YL, Chen H, Bringas PJ, Datto MB, Frederick JP, Wang XF, Warburton D (2002) Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282(3):L585–L593
Zhao L, Yee M, O’Reilly MA (2013) Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-beta and BMP signaling. Am J Physiol Lung Cell Mol Physiol 305(6):L409–L418. doi:10.1152/ajplung.00032.2013
Acknowledgements
I am grateful to the grant support from National Institutes of Health (HL105947-01 to YL), and I thank Dr. Varsha Suresh Kumar for providing images.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Liu, Y. (2015). Type II Cells as Progenitors in Alveolar Repair. In: Firth, A., Yuan, JJ. (eds) Lung Stem Cells in the Epithelium and Vasculature. Stem Cell Biology and Regenerative Medicine. Springer, Cham. https://doi.org/10.1007/978-3-319-16232-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-16232-4_2
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16231-7
Online ISBN: 978-3-319-16232-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)