
Abstract
Public Health practices focus on the implementation of programmes for health improvement and disease prevention (Khoury et al., Am J Prev Med 40(4):486–493, 2011). Public health initiatives in disease were initially targeted to prevent infectious diseases. Partly due to the availability of vaccines and anti-microbial therapy and partly due to better standard of living, the world is free of diseases such as small pox, almost free of polio and the prevalence of infections such as malaria and HIV is steadily on the decline. This has meant that the human race is living longer with the result that non-communicable diseases have become a global public health priority. Preventing non-communicable diseases is a more logical approach than treating them, even more so when modifiable, common lifestyle risk factors share a role in the onset and progression of the disease. Preventive genetics plays a crucial role in the identification of subjects at risk at a very early age, which would thus give public health officials the necessary time to take appropriate action.
Genetic tests can be classified into carrier, diagnostic and predictive testing. In carrier testing, the tests are directed towards the identification of carriers of autosomal recessive or X-linked genetic disorders to prevent disease. Preventive genetics can be defined as using genetics for the prevention of a future disease that has a genetic component either in the individual tested or in future offspring. Diagnostic testing is the process that identifies the current disease status of the subject and includes, among others, prenatal and newborn screening. The implementation of screening programmes allow the detection of genetic disorders at an early stage, so as to prevent these conditions or their serious consequences. Predictive testing determines whether a subject with a positive history but no symptoms of the disease, is at risk of developing the disorder at a future date. In this chapter, we will discuss the application of genetic screening tests to monogenic disorders and complex disorders with monogenic subsets, in view of the current practices. The multifactorial aetiology of complex disorders involves multiple gene effects and gene-lifestyle interactions that cannot be singled out to give a strong predictive value. However, a subset of the complex disorders are caused by highly penetrant genetic mutations. Hence, in this chapter we shall also address predisposing syndromes with high predictive value. In addition, the need of biobanks will be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Public health genomics
- Preventive genetics
- Predictive genetics
- Screening programmes
- Monogenic disorders
- Monogenic syndromes
- Biobanks
1 Screening Programmes
According to the UK National Health Services, the term screening signifies a public health service aimed at identifying individuals that, though apparently healthy, are at risk of or are already affected by a particular disease or its complications. This identification could be through a particular medical or biochemical test or a questionnaire. Once identified, the affected individuals could then be offered further information, complementary tests or treatment in a bid to reduce their risk of developing the disease or its complication. The use of genetic testing to improve healthcare, requires implementation of programmes as part of public health practice [1]. In their 2004 paper on the history of medical screening, Morabia and Zhang [2] identified the US army’s 1917 screening programme, aimed to exclude individuals with clear psychological disorders from joining the army, as the first reported instance of a “screening programme.” This programme consisted of the administration of psychological tests to officers, drafted and enlisted soldiers. Since this early example of a screening programme, other initiatives mostly directed towards the general public and aimed at the prevention or early treatment of important health conditions, have spread across the world .
There are three main types of organised screening programmes, namely population screening, newborn screening (NBS) and cascade screening. Population-based screening involves testing the majority of the population, which may either be defined as the whole population of a country or a specific population at risk (such as Ashkenazi Jews for Tay-Sachs disease, or women over a specific age for breast cancer screening). A highly targeted population for screening of genetic disorders is that of women at the prenatal or pre-conception stage, due to their high accessibility and ease of retraceability. Whereas pre-conception screening allows a wider choice of reproductive options than prenatal screening (i.e. opting to have no children, using a sperm donor, or pre-implantation genetic diagnosis (PGD) and selective implantation of embryos created through in-vitro fertilisation (IVF) in the former, against elective termination of pregnancy in the latter), antenatal groups are easier to target. Meanwhile, NBS involves screening of all newborns in order to detect (generally and preferably) early-onset diseases before the occurrence of overt symptoms. Several genetic metabolic disorders, diagnosed through biochemical tests rather than genetic tests, constitute the core group of disorders for which established newborn screening programmes exist worldwide. These include phenylketonuria (PKU), for which the first NBS programme was set up in the early 1960s, maple syrup urine disease (MSUD) and congenital hypothyroidism (CH). Other specific disorders that are tested for within certain regions or ethnic groups include the haemoglobinopathies (by isoelectric focusing [IEF] and high-performance liquid chromatography [HPLC]) and Cystic Fibrosis (CF; by immunoreactive trypsinogen [IRT]). Cascade screening starts with an index case showing symptoms (the proband) and testing family members for mutations predisposing them to the same disease. Unlike the other forms of screening, cascade screening specifically targets individuals considered at high risk of acquiring the disease due to family history and is restricted to genetic (i.e. DNA) tests .
In the late 1960s, technological advances in medicine enabled the spread of screening in various fields of medicine but, at the same time, brought forward topics of controversy as well as ethical implications . Under this scenario, the World Health Organisation commissioned a report on screening from James M. G. Wilson and Gunner Jungner. In their 1968 report, Wilson and Jungner [3] recounted their pre-occupation that, while the “central idea of early disease detection and treatment is essentially simple”, achieving its success of “bringing to treatment those with previously undetected disease”, as opposed to “avoiding harm to those persons not in need of treatment”, is not as easy as it might appear. In an attempt to simplify the process of screening, Wilson and Jungner proposed a set of criteria that has been adopted as the gold standard in the establishment of all screening programmes. According to these criteria, screening programmes should be considered for conditions fitting in within the following:
-
1.
The condition should be an important health problem.
-
2.
There should be a treatment for the condition.
-
3.
Facilities for diagnosis and treatment should be available.
-
4.
There should be a latent stage of the disease.
-
5.
There should be a test or examination for the condition.
-
6.
The test should be acceptable to the population.
-
7.
The natural history of the disease should be adequately understood.
-
8.
There should be an agreed policy on whom to treat.
-
9.
The total cost of finding a case should be economically balanced in relation to medical expenditure as a whole.
-
10.
Case-finding should be a continuous process, not just a "once and for all" project.
However, these criteria were targeted towards screening for diseases of significant burden in general and did not take into account certain aspects pertaining to genetic diseases, such as the serious debilitating nature of certain rare genetic conditions and the inheritable nature of these disorders. The accelerated rate of discovery of new disease-causing genes has opened up a whole new dimension of diagnosis by using genetic testing to detect diseases before the first clinical signs and/or symptoms appear, even before the disease starts its pathological course. Conversely, these advancements have also caused public health to lag behind in the introduction or expansion of genetic screening programmes, mostly because the decision-making process requires extensive risk-assessments and the implementation of pilot studies, as well as control and standardisation of such programmes [4]. This has resulted in different countries applying different criteria, most of which are based on the original ones by Wilson and Jungner but which take into account the mentioned additional factors, leading to a lack of standardisation or consensus. In the following sections, disorders for which screening programmes are either already in place or may be considered in the near future will be described, with the application of screening criteria to the decision-making process. In addition, the use of next-generation sequencing for newborn screening will be discussed as a future strategy in public health genomics .
2 Monogenic Disorders
Monogenic disorders are mainly rare disorders, caused by single-gene modifications that are present in all the cells of the body and following Mendelian modes of inheritance, i.e. dominant, recessive or X-linked [5]. Though the phenotype in monogenic disorders is almost dependent on a single genetic component, it is also influenced by the individual’s genome (modifier genes), as well as environmental and lifestyle factors. Rare, or orphan, disorders are most commonly defined as disorders affecting 5/10,000 persons or less, but collectively they contribute to a significant degree of morbidity and mortality. Furthermore, eighty percent of rare disorders have genetic origins [6], and the advent of next-generation sequencing has brought about the accelerated discovery of new causative mutations [7]. There are also several monogenic disorders which are relatively common, either world-wide or in specific populations. Being easier to identify, either by biochemical or genetic tests, these disorders were the first genetic conditions for which population screening programmes were established .
2.1 Inborn Errors of Metabolism
An important subset of monogenic disorders consists of the inborn errors of metabolism. Neonatal screening for inborn errors of metabolism has been in place since the early 1960s, following the development of a fast and cheap blood test for phenylketonuria (PKU) [8]. After the success of the first newborn screening programme, maple syrup urine disease (MSUD) screening by the Guthrie (“heel prick”) method was soon added. Up to the introduction of tandem mass spectrometry (MS/MS) technology in newborn screening, individual diseases where tested on a one test, one disorder system that is both expensive and relatively inefficient. The use of MS/MS has expanded the number of inborn errors of metabolism disorders that can be tested from a single dried blood spot, in one single analytical run. To date, MS/MS can identify 45 different disorders, including amino acid disorders, fatty acid oxidation disorders, and organic acidemias, from a single blood spot taken in the neonatal period [9]. Almost all of the national newborn screening programmes in the world include PKU, with medium-chain acyl-coA dehydrogenase deficiency (MCADD) found in a majority of programmes. Other disorders that are actively being added to the list are homocystinuria (HCU), maple syrup urine disease (MSUD) and glutaric aciduria type 1 (GA1).
2.1.1 Phenylketonuria (PKU)
PKU is an autosomal recessive disorder with a reported prevalence as high as 1 in 4500 in Ireland [10] to as low as less than 1 in 100,000 in Finland [11]. In its typical form, it is a result of a mutant phenylalanine hydroxylase (PAH) enzyme, which catalyses the conversion of phenylalanine to tyrosine. Reduced activity of PAH results in an accumulation of phenylalanine and its metabolite phenylpyruvate. This accumulation of phenylalanine leads to intellectual disability, seizures and other symptoms. The major problem is an inability of the brain to utilise other large neutral amino acids (LNAA), such as tryptophan and tyrosine, since the large quantities of phenylalanine block the blood-brain barrier’s large neutral amino acid transporter (LNAAT). Thus, the brain is starved from amino acids that are essential for proper synthesis of neurotransmitters. Management of PKU consists in low phenylalanine diet and possibly the oral administration of tetrahydrobiopterin that is a cofactor in the oxidation process of phenylalanine. To be successful in achieving normal brain development, the diet needs to be initiated as early as possible and continued throughout life.
2.1.2 Medium-Chain Acyl-coA Dehydrogenase Deficiency (MCADD)
Medium-chain acyl-coA dehydrogenase deficiency (MCADD) is a disorder of fatty acid oxidation, in particular the conversion of medium chain fatty acids (MCFA) into acetyl-CoA. It is most prevalent in individuals of Northern European Caucasian descent, with reported worldwide prevalence of between 1:10000 and 1: 27000 [12]. The inability to convert MCFA into Acetyl-CoA results in a deficiency of fatty acid breakdown during periods of metabolic stress, such as a period of fasting or illness. The presenting symptoms include hypoglycaemic attacks, vomiting and encephalopathy that can lead to coma and sudden death. As studies have shown that over 25 % of undiagnosed children die during their first attack, with 16 % of the survivors having severe neurological deficiencies [13], early diagnosis and start of treatment is essential. The latter consists of a diet which is high in carbohydrates and low in fatty acids, in particular during periods of risk, and avoidance of fasting [14].
2.1.3 Homocystinuria (HCU)
Homocystinuria (HCU) is another rare, autosomal recessive condition, where the body is unable to convert homocysteine (derived from methionine) into cystathionine due to the reduced activity of the enzyme cystathionine beta-synthase. Thus, in a similar way to phenylketonuria, homocysteine and to an extent methionine, accumulate in the body while there is a deficiency of cystathionine. The reported worldwide prevalence is of 1 in 344,000, with certain countries such as Ireland (1: 65000) reporting comparatively very high rates [15]. The symptoms of HCU include developmental delay of the brain, behavioural changes such as mood and personality disturbances, dislocation of the lens of the eye, disproportionate length of limbs compared to the body, osteoporosis and higher risk of vascular thrombosis. Treatment consists of vitamin B6 (a cofactor of cystathionine beta-synthase) and a low methionine diet. In addition, the administration of oral betaine helps to convert homocysteine back into methionine and thus reduce the levels of homocysteine.
2.1.4 Maple Syrup Urine Disease (MSUD)
Maple syrup urine disease (MSUD) gets its name from the presence of a distinctive sweet smelling urine reminiscent of maple syrup. The condition can be the result of mutations in any one of the four genes that code for the 4 subunits (E1α, E1β, E2, and E3) of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC). The lack of BCKDC leads to a gradual increase of the branched-chain amino acids isoleucine, leucine and valine, resulting in a build-up of toxic ketoacids in the blood and consequently in the urine. Like most of the other inborn errors of metabolism, the condition is inherited in an autosomal recessive way. As the disorder can be due to the lack or reduced activity of any one of the 4 subunits, its clinical picture and prognosis is variable, but brain damage with mental problems as well as physical problems, such as lethargy, hypotonia, seizures, pancreatitis and ketoacidosis, are common events. The management of MSUD requires a life-long diet where the level of these essential branched chain amino acids is kept under strict control and at the barest minimum. If well managed from early years, those afflicted are usually able to live a normal life with little or no neurological damage. The prevalence of this disorder is reported to be approximately 1 in 200,000 [9], with increased incidence in persons of Amish, Mennonite, and Ashkenazi Jewish descent [16–18].
2.1.5 Glutaric Aciduria Type 1 (GA1)
Glutaric aciduria type 1 (GA1) is another inherited error of metabolism involving the reduced capability of the breakdown of amino acids. Its worldwide prevalence is reported as 1:100,000 [19]. GA1 is caused by mutations within the GCDH gene that encodes for the enzyme glutaryl-CoA dehydrogenase. The enzyme is required for the metabolism of the amino acids hydroxylysine, lysine and tryptophan. This reduction of effective enzyme activity results in a build-up of intermediate metabolites 3-hydroxyglutaric acid and glutaconic acid. Both these metabolites are toxic to the basal ganglia, with initial symptoms such as macrocephaly occurring at birth in some affected individuals, though it is not unusual for symptoms to become apparent during adolescence or early adulthood. Without treatment, the condition invariably leads to encephalitis-like crisis that leaves severe sequelae such as developmental delay, neurologic deterioration, and cerebral palsy. Encephalitis is usually triggered by an intercurrent illness such as infection, fever or prolonged fasting. Treatment consists of dietary manipulation to ensure a low level of lysine and tryptophan, avoidance of prolonged fasting (less than 4–6 h) as well as treatment with supplements such as riboflavin and L-carnitine.
The collective importance of the health burden constituted by inborn errors of metabolism, together with the cost-effective and acceptable patient testing and management strategies, make them ideal candidates for newborn testing programmes; in fact, several countries already have such programmes set-up, with a significant number offering extended screening through MS/MS (Table 3).
2.2 Cystic Fibrosis
Cystic fibrosis (CF) is the most common lethal autosomal recessive condition among Caucasians, with a prevalence of around 1/2500 live births [20] and a high mutation carrier rate at 1/25 [21]. It is caused by mutations in the CFTR gene encoding the cystic fibrosis transmembrane conductance regulator protein. The latter functions mainly as a chloride ion channel and is present in the epithelial cell membranes of several tissues having secretory functions, such as the lungs, sweat glands, gastrointestinal tract and pancreas [22]. Mutations in CFTR result in the loss or impaired function of the channel, leading to the pathological hallmarks of the disease. The main manifestations are in the lung and pancreas: 85–90 % of affected children develop pancreatic insufficiency by the first year of life, while pulmonary insufficiency causes more than 80 % of CF-related mortality. In the lungs, increased mucous viscosity inhibits its normal clearance by cilia and coughing; the excess mucous forms plaques with hypoxic spaces which can be colonised by opportunistic bacteria such as Pseudomonas aeruginosa. Chronic infections of the airways with inflammation and infiltration by polymorphonuclear cells are followed by bronchiectasis (irreversible dilation of the airways), hypoxaemia (low blood oxygen concentration) and hypercarbia (abnormally high levels of circulating carbon dioxide). Pancreatic insufficiency occurs due to intrapancreatic duct obstruction by viscous secretions, with autolysis and fat replacement; insulin insufficiency or carbohydrate intolerance are in fact commonly present in CF patients [23]. Most affected males are also infertile [24].
Fortunately, life expectancy for CF patients has increased to an average of 37 years through new management strategies, although no definitive cure is yet available, and new models predict newborns with CF today to have a life expectancy of around 50 years [25]. Since it is very important to diagnose the disease and provide treatment at an early stage, CF is one of the most important conditions included in newborn screening programmes (Table 3). The main testing strategy involves the measurement of immunoreactive trypsinogen (IRT) from heel-prick blood samples, followed by the sweat test and genetic mutation analyses [24]. More than 95 % of affected newborns have no recorded family history of CF [21], which provides an argument in favour of prenatal or (preferably) pre-conception screening for mutations to predict the risks of having an affected child. However, carrier screening for CFTR mutations has its disadvantages. Firstly, around 1000 mutations in the CFTR gene have been reported since its association with the disease; although in most populations, up to 90 % of affected individuals should have one of a few mutations included in a pan-ethnic gene panel, there may still be mutations which are not covered by the panel and this residual risk must be clearly stated both while obtaining informed consent and while reporting the results, to avoid instilling a false sense of security in mutation-negative patients [26, 27]. This might be overcome in the near future by high-throughput screening of the whole CFTR gene, but the latter in turn will identify sequence variations of undetermined significance and thus more research into each detectable variant will be required [21]. Secondly, not all mutations have been associated with classical CF; other CFTR-related conditions such as male infertility may constitute incidental findings and require careful genetic counselling [26]. Only the functional effects of a few mutations have been established to date, and associations between genotype and phenotype are weak, due to variability in the environmental and genetic background as well as phenotype heterogeneity in patients having the same mutations [23].
The recent discovery of mutation-specific orphan drugs for CF, most notably VX-770 for patients carrying the G551D mutation [28] and PTC124 for patients expressing premature stop codons [29], further highlights the importance of genetic testing for CF for early detection and treatment in target patients. Although these mutations constitute only a small fraction of CF patients (ca. 5 and 10 %, respectively) and, subsequently, treatment costs are still very high, research for new drugs is ongoing and will undoubtedly make population genetic screening more feasible in the near future.
2.3 The Haemoglobinopathies
Heamoglobinopathies comprise a heterogeneous group of autosomal recessive inherited disorders resulting in the production of abnormal or reduced synthesis of normal globin chains that constitute the building blocks of haemoglobin. Those conditions that completely or partially abolish the production of globin chains are known as thalassaemia, while the conditions that result in abnormal globin chain production constitute chain variants. The haemoglobin molecule is composed of two pairs of globin chains with each globin attached to a haem moiety. In adult haemoglobin, the two pairs of globin chains are known as alpha (α) and beta (β). The alpha chains are coded for by a pair of identical alpha genes (HBA1 and HBA2) on chromosome 16, thus an individual has four alleles. In contrast, the beta globin chains are encoded by a single gene (HBB) on chromosome 11 with an individual having two alleles. The worldwide distribution of haemoglobinopathies follows the worldwide distribution of endemic malaria [30]. As a result of human migratory patterns, haemoglobinopathies have become a major health issues in developed countries where the disease was not endemic. Since early identification and treatment of haemoglobinopathies improves morbidity and mortality rates, newborn, antenatal and/or cascade screening for these conditions has been established in various countries in Europe (Table 3) and around the world.
2.3.1 Haemoglobin Variants
In general, haemoglobin variants are due to mutations within the β or α globin genes, though foetal haemoglobin variants as a result of mutations in one of the two γ globin genes (HBG1 and HBG2) have also been reported. Individuals that carry only one mutated gene are known as carriers. Though considered as an autosomal recessive condition, it is more accurate to define a trait as autosomal codominant, as the globin chain variants are expressed and are present in the heamoglobin of ‘carriers’. In contrast, carriers are usually considered healthy as the trait does not normally result in clinical consequences. Haemoglobin variant homozygotes or compound heterozygotes that result in significant functional alterations of the resultant haemoglobin would usually present with symptoms. The clinically relevant variants are easily identified through simple and relatively cheap electrophoretic techniques supplemented by HPLC methods to verify and quantify the variants. The worldwide clinically important variants include sickle cell haemoglobin, also known as HbS (β6 (A3) glutamic acid→valine), haemoglobin C (β6 (A3) glutamic acid→lysine) and haemoglobin E (β26 (B8) glutamic acid→lysine).
HbS is the most widespread haemoglobin variant, occurring mostly in persons of African origin but also found in persons of Mediterranean ethnicity and in the Indian subcontinent. The single point mutation substitutes a valine residue for glutamic acid in codon 6. This substitution induces the polymerisation of the deoxygenated haemoglobin variant through the formation of hydrophobic bonds between the inserted valine residues of adjacent haemoglobin molecules. This polymerisation results in deformation of the red blood cells that take the sickle cell shape form. Such deformed blood cells obstruct the microcirculation of sickle cell patients. Thus, any physiological stress that reduces oxygenation or increases oxygen requirements, results in the rapid polymerisation of HbS and the precipitation of sickle cell crises, with risks of strokes at a very young age. Treatment involves daily prophylactic antibiotics, transfusions and hydroxyurea treatment to reduce sickle cell crises.
In haemoglobin C, the glutamic acid to lysine change is in the same position to that of HbS but instead of polymerisation, the haemoglobin precipitates within the red cell and damages the membrane. Damaged red blood cells increase the viscosity of the blood, are haemolysed and sequestered both in the bone marrow and spleen. HbC disease is not as severe as HbS and might not be diagnosed before adulthood. Though mild, patients can still experience joint pains as well as gall stone problems. HbC is most common in persons of African ancestry.
Haemoglobin E is the most common beta globin variant in the Far East, where carrier frequency can reach 25 %, mainly in Thai and Chinese. The HbE mutation activates a cryptic splice site, leading to a slightly reduced rate of synthesis in addition to some instability. Thus in the homozygous state, HbE is considered a very mild haemoglobinopathy with mild anaemia that does not usually require treatment. In contrast, the condition is very severe in compound βE/βThal heterozygotes.
2.3.2 α-Thalassaemia
As humans inherit two HBA genes from each parent and thus the normal genotype is αα/αα, the genetics of α-thalassaemia is somewhat complicated. Most of the alpha-thalassaemia abnormalities are the result of deletions of either one or two of the α-genes. Mutations that result in the inactivation of a single α-gene exist but are not common. The α-thalassaemias can be classified into two groups, α+ -thalassaemia and α0-thalassaemia (Table 1).
The homozygous α0-thalassaemia state is incompatible with life as no α-globin chains are produced from around the 6th week of foetal life, the α0-thalassaemic foetus is severely anaemic, oedematous and has all the features of severe intrauterine hypoxia. The child is usually stillborn late in pregnancy and the pregnancy is usually complicated with toxaemia and difficulty during delivery, in part due to an enlarged placenta. The compound heterozygous state (- -/- α) is a condition known as HbH disease, which is characterised by anaemia and an enlarged spleen and might require lifelong treatment with transfusions and iron chelation. The α + -thalassaemia and the heterozygous α0-thalassaemia do not require treatment and can be considered as healthy carriers. α-thalassaemia can be identified during newborn screening through the identification of an abnormal haemoglobin made up of tetramers of γ globin chains (γ4) called Hb Bart’s and in adults through the identification of HbH (β4).
2.3.3 β-Thalassaemia
Similar to α-thalassaemia, β-thalassaemia is prevalent in Mediterranean countries (with Cyprus having the highest prevalence), the Middle East, Central Asia, India, Southern China, and the Far East, as well as in countries along the north coast of Africa and in South America [31]. β-thalassaemia is mostly due to point mutations or small deletions, though rarely large deletions can also be the cause. Over 270 HBB gene mutations that give rise to a β-thalassaemia phenotype are listed in the database of human haemoglobin variants and thalassaemias ( http://globin.bx.psu.edu/hbvar/menu.html ). Each ethnic group has a small set of predominant mutations. β-Thalassaemia can be classified into three groups, β-thalassaemia trait or carriers (βA/βThal), β + -thalassaemia (homozygous state for mild or moderate mutations or compound heterozygous for a moderate and severe mutation) and β0-thalassaemia (homozygous state for severe mutations). Persons with the trait are considered healthy carriers. β+ - and β0-thalassaemia patients, require regular transfusions, depending on the severity of the conditions, together with regular iron chelation. Newborn screening for β-thalassaemia is complicated by the fact that the majority of haemoglobin in a normal newborn is HbF and thus is not affected by the presence of the thalassaemia. The early identification of β-thalassaemia compound heterozygotes and homozygotes requires the establishment of antenatal screening programmes so as to identify β-thalassaemia carrier expectant mothers. Antenatal diagnosis offers the possibility of counselling and allows targeting of the baby once it is born to avoid complications of the disease.
2.4 Hereditary Haemochromatosis
Hereditary haemochromatosis (HH) is a rather common monogenic disorder, affecting 1 in 200 to 1 in 500 of Caucasians [32] and characterised by increased absorption of dietary iron, with subsequent progressive deposition in organs including the liver, pancreas and heart [33]. It is most prevalent in Northern European populations but is found worldwide [34]. The associated gene, HFE, is related to the human leukocyte antigen (HLA)-A3 complex; two missense mutations, namely C282Y and H63D account for up to 95 % of probands. The HFE protein localises to the duodenal crypt cells, where dietary iron is absorbed, and negatively regulates absorption through association with cell-surface transferrin receptors. HFE mutations cause loss of HFE protein function, the C282Y mutation being more detrimental due to concurrent disruption of the association of HFE with β2-microglobulin, which is important for proper function [33].
HH initially presents with relatively non-specific symptoms, such as fatigue, joint and abdominal pain and palpitations, leading to frequent misdiagnosis. Later complications of iron deposition include cirrhosis, diabetes mellitus (DM) cardiomyopathy and primary hepatocellular carcinoma, which are also relatively common primary disorders. Symptoms generally appear between 40 and 60 years, with later onset in females attributed to iron loss through menstrual cycles, pregnancy and lactation [33, 35]. Management of HH is by periodic venesection or phlebotomy to remove blood and, consequently, excess iron; this treatment is effective, safe and inexpensive [33].
Although inheritance of HFE mutations is simple Mendelian, incomplete penetrance makes predictive testing difficult and provides an argument against population screening, which may inevitably have psychosocial consequences. On the other hand, it meets most of the WHO criteria which justify population screening (Table 2). Cascade screening for HFE is in fact implemented in many countries throughout Europe (Table 3).
2.5 Familial Hypercholesterolaemia
Familial hypercholesterolaemia (FH) is also rather common, with an estimated prevalence of 1/500 (0.2 %) in Caucasians. Some populations show yet higher frequencies attributed to founder effects [36]. FH is characterised by abnormally high plasma levels of low-density lipoproteins (LDL) and total cholesterol, with predisposition to early-onset coronary heart disease (CHD) due to the formation of atherosclerotic plaques. FH is mostly caused by mutations in the LDL receptor (LDLR; > 1000 mutations identified), apolipoprotein B (APOB; 9 mutations identified) or proprotein convertase stabilisin/kexin type 9 (PCSK9) genes, with autosomal dominant inheritance [37]. The latter implies that the inheritance of one mutation from just one parent will result in the disease phenotype; in fact, heterozygous patients constitute the vast majority of cases. Also, couples where one partner is affected (with a heterozygous genotype) have a 50 % chance of disease transmission to the offspring [38]. The high degree of risk among family members makes cascade screening for FH a valuable tool in the identification of affected individuals (Table 2); early detection of FH and subsequent treatment with statins significantly reduce morbidity and mortality from CHD associated with FH [38, 39].
Despite international efforts, an estimated 80 % of FH patients are still not being diagnosed [40]. An important pitfall is the phenotypic and genotypic heterogeneity of the disease. Until recently, diagnosis was mostly made by means of LDL measurements, with specific cut-off points according to age and family history (lower cut-offs are used for those having an affected first-degree relative than those having an affected second-degree relative, for example) [41]. However, LDL and total cholesterol levels are highly variable in FH patients, even after adjustment for gender, age and body mass index (BMI), and may also overlap with those of the general population, resulting in reduced sensitivity and specificity [42].
The inclusion of mutation analysis has been proved to increase the specificity and sensitivity of the diagnosis of FH [36]. In fact, European criteria for FH developed by the Simon Broome Register Group (UK) and the Dutch Lipid Clinic Network, include the presence of a functional FH mutation, even in the absence of other criteria, as diagnostic of “definite” FH [43, 44]. This is due to the dominant nature of the genotype and the high penetrance of mutations, which is close to 100 % [38]. Nonetheless, a wide genotypic variation is also observed: apart from populations with founder effects, which are characterised by a few mutations responsible for most cases, the situation is generally that of a large number of mutations giving rise to a highly heterogeneous population, such as in the UK, Italy and Germany [42]. Thus, each population must define the genotypic characteristics of its FH patients before a successful screening programme can be implemented, and even at this stage negative mutational analysis results do not necessarily exclude the presence of FH, since there might be mutations which are not included in the testing panel [38].
Table 2 describes how the Wilson and Jungner criteria apply to the monogenic disorders discussed above, for all of which some type of screening programme is currently implemented in most European countries (Table 3). It may be observed that the majority of the listed disorders do not satisfy the complete list of criteria, and that the most important criteria, which may have had the highest impact on the decision-making process, are the burden of the disease, the availability of a specific and acceptable treatment (or rather management, as no definitive treatment yet exists for any of the disorders), and the presence of an early stage during which the condition can be diagnosed (or predicted) and treatment initiated.
As can be observed in Table 3, all 26 European countries reviewed by Javaher et al. (2010) have taken up one form or another of newborn screening [45]. All states have NBS programmes for congenital hypothyroidism, and all except Malta for PKU.
It is evident that several factors played an important part in the decision-making process to set up the screening programmes , highlighting the importance of the previously described criteria. PKU can be defined as the classical type of disease ideal for NBS: although classified as a rare disease, it is of early onset (symptoms developing in the first few months of life), it is relatively easy to diagnose before symptoms develop and also easy to treat, and it has severe complications if left untreated. The same can be applied to congenital hypothyroidism, although it is estimated that only around 15 % of cases are genetic and 85 % are due to thyroid dysgenesis [46]; thus this disorder was not addressed in detail in this chapter. Newborn screening for other inborn errors of metabolism which are very rare has been made feasible through MS/MS, which is currently being applied in 11 European countries. It is important to note, however, that despite the ability of MS/MS to detect up to 45 disorders, most countries choose to report only a few of these disorders, mostly due to ethical and psychosocial reasons.
In the case of CF, NBS is generally by an established algorithm, involving a primary screening test for immunoreactive trypsinogen (IRT), followed by either repeated IRT testing or DNA testing. The second tier depends on whether repeat samples are taken from the newborns; where only one blood sample is available, a positive IRT is followed by DNA testing. The latter will defer in detection rate according to the number of mutations screened for, due to the large number of possible CFTR mutations causative of CF. Population-wide testing for carriers and couples at risk of having affected children are also dependent on mutation panels and thus include the inevitable degree of false-negative findings [47]. As has already been mentioned, psychosocial risks involved in genetic testing for CFTR mutations are also significant [11]. Cascade screening plays an important role since the mutations are narrowed down to those carried by the proband.
Currently, only cascade screening programmes are in place for HH and FH. This is due to the importance of family history in these disorders as well as the later onset, which makes screening targeted only at high-risk individuals more feasible. The screening process is generally initiated when the first case showing the symptoms and carrying a causative mutation, or proband, is found, and his/her relatives are followed-up to find whether they are carriers of the same mutation, in order to prevent the consequences of the disease.
The HH and FH cascade screening programmes provide valuable arguments in the implication of screening for monogenic subsets of common, multifactorial conditions. Being highly penetrant and autosomal dominant, LDLR mutations do not just confer a risk for FH but justify preventive measures, i.e. statin administration to prevent hypercholesterolaemia. Furthermore, FH in turn confers an increased risk of heart disease such as myocardial infarction. However, the low penetrance of HH mutations, which are very common especially in Northern Europe, makes screening for such mutations a risk status assessment, similar to that in multifactorial disorders. Thus, no population-wide screening programmes to detect HFE mutations are in place; rather, probands present with elevated iron levels in serum and their relatives are followed up to determine the risk of having inherited the same disorder. It is only at this level that genotyping of symptomless individuals take place [5].
An important observation to make is that there are significant discrepancies in the screening programmes present between European countries, and no consensus exists as yet as to which disorders should be screened for or, as in the case of MS/MS, reported [48].
Even more diverse are the European policies on population-based carrier screening programmes, targeting either pre-conception or prenatal individuals. In the case of such programmes, the epidemiology of the targeted disorders probably plays the most important part in their selection. A good model to illustrate this diversity are the population-wide carrier screening programmes for thalassaemia and other haemoglobinopathies established in several countries. Population-wide haemoglobinopathy screening is present in countries having higher prevalences, such as in the Mediterranean region, with ethnic-specific screening being preferred in countries such as Germany. Furthermore, in countries with high prevalence such as Cyprus, the population screening programme is run in parallel to a newborn screening programme, to ensure maximum coverage of affected individuals [45]. Increased public awareness and uptake of prenatal screening have largely contributed to the success of these programmes [49]. Other country specific differences involve pre-conception versus prenatal screening. While in Cyprus pre-conception screening of β-thalassaemia is carried out in all cases (followed by prenatal diagnosis in those cases that are at risk), in other countries such as the UK, screening is carried out prenatally.
3 The Way Forward—Public Health Genomics Perspectives on use of Next-Generation Sequencing for Newborn Screening
NBS started with the Guthrie test in the 1960s and rapidly expanded around the globe. It has been possible to test for a handful of disorders with this method. In the late 1990s, early 2000s, we have seen the introduction of MS/MS, which has been replacing the Guthrie test in many countries. With MS/MS, it is possible to expand the number of screened disorders extensively, without significantly increasing the overall costs.
In the transition from the Guthrie test to MS/MS, the major question was not the detectability of the disorders with this technology, but how to decide on which disorders to include in the NBS programmes. Although there has been a general consensus on screening criteria, such as the Wilson and Jungner criteria [3], various countries interpreted them differently or used modified criteria when selecting the disorders to be included in the NBS programmes. For example, Germany has been using three criteria [50], whereas the UK has been using 22 criteria, all of which must be fulfilled before a disorder is included in the NBS programme [51]. Additionally, various countries have different stakeholders and technology-push vs. market-pull dynamics for NBS, as well as different values, structures and processes in health technology assessment, all of which are rooted in differences in their health care systems [52]. These lead to different lists of disorders covered in NBS programmes, as seen in Table 3.
We are approaching another shift in NBS with the upcoming next-generation sequencing (NGS) technologies. Also termed as ‘massively parallel sequencing’ or ‘second generation sequencing’ [53], NGS has been reducing both the cost and time required to accomplish whole genome sequencing. With the rapidly decreasing cost of this technology, soon it will be possible to sequence the whole genome of an individual for less than 1000 US dollars. Once DNA sequencing technology is sufficiently robust and affordable, it will be possible for all babies to have their genomes sequenced at birth, replacing both newborn bloodspot screening and additional genetic tests required later in life [54]. This means that it will be possible to screen for a virtually unlimited number of disorders in NBS with almost no additional costs per disorder. However, it is not the technologic capacity to sequence entire genomes, but the analysis and interpretation of the generated data, that is the main bottleneck for the application of NGS for whole genome or exome sequencing [54].
The data generated from whole genome or exome sequencing in the newborn phase can be used not only to screen for monogenic disorders or monogenic subset of complex disorders, but also for pharmacogenetics (potential response to drugs), nutrigenetics (response to nutrients) and risk assessment programmes for major complex disorders, such as cardiovascular diseases or type 2 diabetes. Therefore, implementation of this technology will have an impact on the whole health care system, beyond the NBS programme.
There are several issues that need to be resolved when preparing for using NGS in the newborn phase. Some of these are presented very briefly below:
3.1 Human Resources
The knowledge, attitude and skills of health professionals to use genome-based knowledge are presently very limited. This shortcoming needs to be addressed effectively with professional education and training programmes. However, the effect of such training might be limited on health professionals that have already been practising in traditional ways for many years. The main target should be the under- and postgraduate education of health professionals.
Besides, to develop the tools for data analysis, bioinformaticians will be a crucial professional group required both in central levels and in local clinical services [54].
3.2 Informatics Capacity to Store and Process Data
Sequencing the whole genome or exome of large population groups will create massive amounts of data which need to be stored, retrieved and analysed. This requires data storage and processing infrastructures. Additionally, analytical tools to analyse and interpret the whole genome or exome must be developed.
3.3 Clinical Health Services
The systems must be in place to integrate the generated genome-based information in clinical health services. For example, in the first place, genome-based data can be used in the prescription of drugs which have significant pharmacogenetic interactions, such as warfarin . Nevertheless, the infrastructure to access such data and clinical work flows making use of the data must be in place, for NGS to be applicable to the healthcare setting.
3.4 Data
A recurring issue that arises when genome-based data are discussed is whether genetic data are different than other data concerning health. ‘Genetic exceptionalism’ claims that all genetic and genomic samples and data merit special protection, regardless of their medical sensitivity or predictive power. This claim has been rejected by various groups and reports, including the Public Health Genomics European Network [55] , the Ickworth Group (an international group consisting of experts from multiple disciplines which came together in Ickworth, UK) [56] and others [57]. The main underlying idea is that genetic data deserve no separate status; they must satisfy equally high standards of data protection and confidentiality as other types of health data.
In the future paradigm of health care, the line dividing health care provision from health research will likely get thinner. In particular, in the context of the ‘big data’ approach, datasets created from the regular health care data and various other data collected from individuals are envisioned to be used by data mining to provide insights to health and diseases. Whole genome or exome sequence data of large populations will provide a great opportunity for such research and to discover the genetic basis of various diseases.
For this future vision, having longitudinal (long-data) and large sets of data (big-data) is required, but not enough. For the development of new forms of prevention, diagnosis and treatment of complex diseases, abundant and intricate health data must be combined with innovative analysis strategies in a cross-disciplinary environment [57]. However, recent developments in the EU legislation on data protection impede use of health care data for research, due to the restrictions proposed on the use of health data, even for research purposes [58–60]. This is an important regulatory bottleneck that needs to be overcome.
Genetic data hold a very promising future. However, precautions are required to protect the owner of the data, i.e. the individual. Regulatory mechanisms must be in place to prevent any discrimination that may arise due to genetic characteristics of the individual. GINA—Genetic Information Non-Discrimination Act in the USA, which came into force in 2009, is one of the major examples of a legislation that protects individuals from being discriminated by employers or insurance companies based on their genetic data.
3.5 Ethical Issues
An important issue that needs to be considered for whole genome or exome sequencing in the newborn phase is that the cost of sequencing the whole genome will soon practically be not more expensive than sequencing targeted genes. Therefore, sequencing the whole genome (or exome) at once seems to be the most practical solution. However, this brings ethical discussions on issues such as protection of the future autonomy of the screened infant, the right not to know, and the issues around incidental findings .
Incidental findings are any findings which are outside the scope of the clinical enquiry [54]. They are “the results of a deliberate search for pathogenic or likely pathogenic alterations in genes that are not apparently relevant to a diagnostic indication for which the sequencing test was ordered” [61]. In the context of whole genome or exome sequencing, the issues that may arise from incidental findings span multiple clinical, genetic and social dimensions, such as racial ancestry, misattributed parentage, consanguinity, disease susceptibility, and reproductive risks [54]. Several strategies are discussed to manage the issue of incidental findings, such as classification of genes and disorders according to net benefits [54] and providing lists of conditions that must be reported when found [61].
4 Complex Disorders
Common complex diseases can be defined as a group of disorders with similar symptoms but having a variable aetiology. Though the genetic component within complex diseases is inherited in a similar way to the inheritance in monogenic or Mendelian disorders, the main difference is that, while in Mendelian disorders a single gene variant is compulsory to reach the critical threshold in developing the disease, with modifier genes and environmental factors modifying the phenotype, in complex disorders both the threshold to develop the disease as well as its severity, are modified by complex aetiological factors. In between these two ends of the genetic spectrum, one finds a group of disorders in which alterations in susceptibility genes would bring one close to the required threshold for the condition, but other genetic, environmental and lifestyle factors are required for the disease to establish itself. This group can be considered as that of ‘monogenic subtypes’ and includes such genes as BRCA1, BRCA2, MSH2 and MLH1.
Common complex disorders include cardiovascular disease (CVD), diabetes, cancer , dementia, auto-inflammatory and auto-immune diseases, amongst others. Preventive genetics in multifactorial disorders require an in-depth understanding of the genetic variability associated with the disorder, in order to define the complex disease into subtypes of variable aetiology. Genome-wide association studies (GWAS) have been used extensively to identify genetic variants associated with multifactorial diseases. Whereas current testing programmes include inherited monogenic diseases (Table 2), taken individually, the contributing genetic variants in multifactorial diseases still lack a high predictive value and hence genetic results are inconclusive and very difficult to interpret.
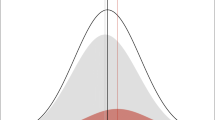
Of interest is the characterisation of monogenic subtypes within complex diseases that can be easily taken up into genetic testing programmes (Fig. 1). The term ‘monogenic subtype’ needs some clarification since the gene involved requires other contributing factors to cause the disease. The mutated gene in the monogenic subtypes within complex disorders, provides a cellular programme that is susceptible to the initiation of disease. The effect of the mutant will be exerted on tissues that normally express the gene. The resulting loss or gain of function will set a molecular threshold, requiring the contribution of additional factors, to initiate a specific disease. These susceptibility genes are masked by the presence of other variations that might occur during the disease state. Hence, low frequency susceptibility genes can only be identified through high penetrance in family studies.

Inherited susceptibility mutations in cancer. (Genes in red: mismatch repair genes; violet: phosphatases; green: apoptosis induction)
Major contributing genes in complex disorders have been identified due to their high penetrance, exemplified by BRCA1 and BRCA2 [62, 63] with increased risk of breast and ovarian cancers , hMSH2 with increased risk of hereditary nonpolyposis colorectal cancer (HNPCC) [64], and α-synuclein in Parkinson disease [65].
The genetic basis of colorectal cancer (CRC) is well defined, and is used as a model to describe the molecular events that promote the progression of disease in cooperation with the defects in inherited susceptibility genes. The risk to develop CRC has been associated with inherited mutations in the mismatch repair genes [66]. Similarly, DNA repair genes predispose for breast cancer, including BRCA1 and the Fanconi Anaemia (FA) genes BRCA2 (FANCD1), FANCJ (BRIP1), FANCN (PALB2) and FANCO (RAD51C) [67–69]. In addition, Fanconi genes are associated with other cancers, including acute myeloid leukaemia [70].
Inherited mutations in PTEN, p53, RB1, MEN1 and VHL give rise to predisposing syndromes, namely Cowden’s Disease [71], Li-Fraumeni syndrome [72], Retinoblastoma [73], Wermer syndrome [74], and von Hippel-Lindau syndrome [75], respectively. These inherited susceptibility genes have a low frequency with a high penetrance. Hence, these genes are candidates for a testing programme in specialised clinics, screening family members of patients, to identify risk and initiate preventive monitoring or discuss possible clinical solutions to reduce the risk significantly [76].
4.1 Familial Cancer Predisposing Syndromes
4.1.1 Familial Adenomatous Polyposis
Polyposis syndromes are exemplified by Familial adenomatous polyposis (FAP) characterised by APC gene mutations and a lifetime risk of CRC close to 100 %. APC is a tumour suppressor regulating the degradation of the transcription factor β-catenin, the effector molecule of the Wnt pathway. APC stabilises a protein complex that sequesters β-catenin in the cytoplasm, leading to proteosomal degradation. FAP is characterised by the presence of more than 100 colorectal adenomatous polyps prevalence [71]. Rectal bleeding indicates enlarged and numerous adenomas, a condition which is rare in children and adolescents. If untreated, the condition will develop into colorectal adenocarcinoma with an early age of onset. Surveillance, chemoprevention [77] and improved endoscopic treatment provide opportunities for better treatment of FAP [78] and decrease dependency on prophylactic cancer-preventive colorectal surgery [79].
4.1.2 Li-Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS; OMIM 151623) is an autosomal dominant cancer predisposition syndrome associated with p53 germline mutations [80]. Family studies show a high penetrance with early onset of sarcomas, breast cancer and other non-therapy-related neoplasms. The age of primary cancer onset ranges from 4 months to 49 years with a mean age of 25 years [81]. The variety of neoplasms and the early age of onset is attributed to mutant p53, a major gatekeeper of apoptosis in response to DNA damage. Of interest, mutations that result in truncation of the p53 protein are associated with higher cancer risk and earlier age of onset [82]. Defective p53 function results in the accumulation of mutations in proliferative tissues. The population frequency of germline p53 mutations in Europe and United States is around 1:5000 individuals [83]. Eligibility for genetic screening is determined by established clinical criteria that classify individuals with LFS. The classic LFS classification scheme requires a proband with sarcoma diagnosed before the age of 45, a first-degree relative with any cancer before 45 years of age and another first- or second-degree relative with any cancer diagnosed at under 45 years or with a sarcoma at any age [84]. Other classification schemes were designed based on further family studies and the Chompret criteria [85] enhances the predictive value of the classic LFS classification, resulting in a testing sensitivity of 95 % [81]. The Chompret criteria consider any proband with adrenocortical carcinoma at any age of onset eligible to p53 mutation analysis, irrespective of family history. In addition, the criteria include other proband neoplasms such as breast cancer and brain tumour diagnosed at an early age of 36 years. Presymptomatic testing for germline p53 mutations predicts the susceptibility to various neoplasms, imposing ethical issues due to lack of complete clinical surveillance, preventive measures and treatment recommendations. The high penetrance of germline p53 mutations in familial breast cancer patients predicts an average age of onset of 31 years [86], and hence provides eligibility for breast cancer screening in p53-mutant women who are in their mid-20s, followed by implementation of risk reduction strategies.
4.1.3 Cowden Syndrome
Cowden Syndrome (CS; MIM 158350) is a rare autosomal dominant cancer predisposition syndrome with a prevalence of 1 in 200,000 [87] and an age-related penetrance of around 80 % [88]. The susceptibility gene in CS is PTEN, predisposing individuals to breast, endometrial and thyroid cancer [89]. PTEN is a ubiquitously expressed phosphatase involved in the attenuation of the PI3K pathway, hence acting as a proliferation suppressor [90]. The clinical symptoms of CS include multi-organ hamartomatous polyps in the majority of the affected subjects [91]. In addition to Cowden Syndrome in adults, germline PTEN mutations result in Bannayan-Riley-Ruvalcaba syndrome (BRRS; MIM 153480) in children [92], collectively known as the PTEN hamartoma tumour syndrome (PHTS). The highest age-adjusted standardised incidence ratio of germline PTEN mutants occurs in thyroid cancer, followed by endometrial, kidney, breast and colorectal cancer and melanoma (Table 4). Of interest, promoter mutations in the PTEN gene were associated with breast cancer, while nonsense mutants were associated with colorectal cancers [93]. The age of onset of any of the core cancers within the family will establish the age for clinical observations, taken as 5 years less than the age of the youngest family member affected.
4.1.4 Lynch Syndrome
Lynch Syndrome (MIM 120435) is an autosomal dominant cancer predisposing syndrome caused by germline mutations in the mismatch repair (MMR) genes, including MLH1, MSH2, MSH6 and PMS2 [94]. Lynch Syndrome is also known as hereditary nonpolyposis colorectal cancer (HNPCC) syndrome and confers a high risk to early-onset colorectal and endometrial cancer [95]. In contrast to polyposis syndromes, HNPCC lacks characteristic diagnostic features and clinics depend on standardised criteria, such as the Amsterdam criteria, to establish diagnosis. Eligibility for carrier screening requires a family history characterised by immunohistochemistry-verified CRC cases in at least three relatives, including a first-degree relative, the presence of the disease in at least two successive generations, and one of the relatives diagnosed with CRC with an age of onset less than 50 years, with absence of polyposis syndrome [96]. In addition, the inheritance of syndrome-associated cancers within the family and testing for microsatellite instability are used to predict patients with HNPCC [96]. Lynch syndrome accounts for 2–5 % of the total CRC cases, as reflected in the high estimated lifetime risk of MLH1 and MSH2 germline mutation carriers (Table 4). Of interest, 10–15 % of CRCs have microsatellite instability, a useful marker of mismatch repair genes loss of function [97]. The majority of cases with no familial predisposition are caused by sporadic hypermethylation of the MLH1 gene promoter, supporting the high risk of inherited MLH1 mutants in the development of carcinoma. Lynch syndrome patients are also at risk of developing other cancers , including stomach, ovarian, renal, pancreatic, small intestinal, brain and skin tumours; it can thus be included in the subset of cancer-predisposing syndromes [98].
4.1.5 Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS; MIM 175200) is an autosomal dominant condition arising from germline mutations in the serine/threonine kinase gene (STK11/LKB1) [99]. Since PJS condition is rare, estimates of frequency within populations vary significantly. Patients present with mucocutaneous pigmentation and gastrointestinal polyposis [100] and have an increased risk of gastrointestinal cancers (as in most polyposis syndromes), breast ovarian, uterine, cervical, lung and testicular cancers [101, 102] (Table 4). Loss of heterozygosity (LOH) of the normal STK11 allele in 70 % of PJS patients, followed by the progression of hamartomas to adenocarcinomas, suggest that LKB1 is a tumour suppressor gene [103]. Interestingly, somatic mutations in LKB1 are common in lung cancer [104], supporting the involvement of inherited LKB1 mutants in the progression of disease.
4.2 Tools in Predictive (& Preventive) Genetics
Unlike in monogenic disorders , screening for complex diseases presents a greater challenge since the aim is that of risk assessment rather than presymptomatic diagnosis. Furthermore, the concept of personalised (genomic) medicine seems to conflict with the aim of public health, that is, to improve health from a population perspective [1]. However, predictive genetics reduces the burden imposed on the health care system by admission of fewer patients with advanced stages of disease; thus, its importance is being increasingly recognised. Family history and personal genomics (the assessment of individual genetic variations at multiple loci) are two important predictive genetics tools . These have a recognised value in the diagnosis of monogenic conditions (as observed in cascade screening programmes such as for HH); however, their value in the management of risk of complex diseases remains to be established [106]. Although assessment of family history provides information on the inheritance of a phenotype [107], the likelihood of an individual carrying a mutant susceptibility gene to develop cancer depends on other genetic variations (modifier genes), as well as on dietary, lifestyle and environmental factors influencing the age of onset and severity of the disease. The routine use of family history or personal genomics alone as a measure of risk of common complex conditions does not generate sufficient evidence in the primary care setting; however, when used together and in conjunction with evidence-based medicine (EBM- the application of population-derived data) , they comprise a very useful tool in the improvement of disease prevention [106]. Public health genomics shall play an essential role in designing more effective genetic screening programmes, by applying data derived from personal genomics to public health. This becomes especially important with the advent of cost-effective services for whole genome sequencing or microarrays detecting large panels of mutations, which may undoubtedly lead to over-diagnosis.
Cascade screening for genetic predisposition to cancer is a form of “systematic predictive testing”, where asymptomatic individuals at an increased risk due to their genotypic inheritance may benefit from surveillance programmes to ensure early detection of tumours.
Colorectal cancer is a major contributor to cancer morbidity and mortality. 15 % of CRCs are familial with 2–5 % caused by HNPCC and less than 1 % associated with polyposis syndromes (FAP and PJS). Following diagnosis of Lynch disease, family members benefit from clinical screening by colonoscopy [108]. Recommendations include offering a colonoscopy every 1 or 2 years starting at the age of 25 years or at 5 years before the youngest diagnosed member of the family, whichever is the earlier [109]. In the case of FAP families, individuals with an APC mutation should strongly consider a prophylactic colectomy before the age of 25 years. Referral of genetic testing for APC mutations will provide confirmation of familial adenomatous polyposis (FAP) or, in the case of family members not exhibiting adenomatous polyps, will evaluate relatives at risk and initiation of surveillance programmes. Surveillance should be offered for APC mutant members that defer surgery [110]. For patients diagnosed with PJS, risk reduction strategies include upper and lower endoscopy, breast examination, endoscopic ultrasound and CA19–9 tumour marker testing for pancreatic tumour surveillance and ultrasound, cervical cytology and CA125 testing tumour marker testing for ovarian cancer [111] .
Familial cases account for 10 % of breast cancer in Western countries. Susceptibility genes are inherited in an autosomal dominant pattern, but with limited penetrance. High risk families testing positive for the BRCA1 and BRCA2 mutant genes are associated with a four or more times higher incidence of the disease in close relatives. The use of surveillance programmes and/or risk reduction strategies in healthy BRCA1 and BRCA2 mutation carriers are instrumental to ensure a positive impact of genetic screening on the health care system. Risk-reducing mastectomy (RRM) supresses breast cancer development by 90 % [112]. Inherited mutations in p53 (Li-Fraumeni) and PTEN (Cowden syndrome) have a high incidence of breast cancer development, but the syndromes are very rare. Testing for mutations within these genes requires the use of further diagnostic criteria as detailed below.
Risk assessment tools for complex diseases take into consideration various contributing factors, including the incidence of disease in first-degree relatives; previous diagnostic test results; monitoring results (if any); and presence of genetic mutations/polymorphisms associated with the disease of interest. In the case of breast cancer, risk assessment includes age at first live birth, the use of hormone replacement therapy and other risk factors specifically associated with the disease. The breast cancer risk assessment tool (GAIL model) is used to measure contribution of variants to the calculated risk [113]. The selection of the genetic contribution to multifactorial diseases is not a simple task. Also, the interrogation of genetic variation through screening programmes or referral for testing depends on the minor allele frequency within a population and also on the penetrance within a family having a history of the phenotype. Less penetrant genes with higher prevalence are more significant from a public health point of view. For instance, the factor V Leiden mutation, with increased risk of thrombotic events [114], is integrated in the testing regime of health care genetic clinics.
Most of the common chronic disorders, such as asthma , cancer , cardiovascular diseases, obesity, diabetes, hypertension, psychiatric disorders, arthritis, Parkinson’s and Alzheimer’s, have a complex aetiology and pathophysiology. The current view is that these disorders are due to numerous small, additive genetic defects compounded with environmental and lifestyle causes. Considering that these conditions constitute a major health and economic burden and are the cause of substantial morbidity and mortality, a concerted action is required to elucidate the pathophysiology and thus open the road for successful treatments and preventive strategies. One major drawback in the determination of aetiological factors that singly confer a small increase in risk is the need of a large number of affected and unaffected individuals (‘cases’ and ‘controls’), so as to achieve statistically significant results. It is very difficult for single clinical and research centres to obtain such large numbers in a relatively short time and with reasonable budgets. For this reason, biobanks and their related databanks have become an important tool for the elucidation of the pathophysiology of these disorders.
5 Biobanks and Preventive Genetics
The Organisation for Economic Co-operation and Development (OECD) definition of a Biobank is “A collection of biological material and the associated data and information stored in an organised system, for a population or a large subset of a population.” This definition brings about the need to consider a number of terms, basically “biological material”, “associated data and information,” “stored in an organised system,” and “population or a large subset of a population.”
It is current practice, if not a legal requisite, for hospitals and laboratories to collect and store whole organs and tissues that have been excised for diagnostic or therapeutic aims. In addition to these archival banks, since the early 70s specialised collections have been initiated, targeting cells and their products including DNA, RNA and proteins. Collections of biological material, without any associated data, have very limited usefulness as biobanks. The collection of data, including medical histories, lifestyle, social and environmental information, increases the research value of these biobanks as the samples can now be separated into different case and control categories. The next important step in the establishing of a useful biobank is the establishment of associated databases that results in an easier and more efficient search and classification tool as compared to pen and paper processes. Finally, to be considered as a biobank, the samples have to be collected either from the whole population or from a subset of the population that has a particular disorder. In each case, the information collected can either be retrospective, transvers or prospective, depending on the final aim and use of the biobank.
5.1 Future Biobanks
The networking of biobanks from different countries or centres, in particular those where both the samples and data have been collected with standardised protocols, has the potential of becoming an ideal platform to collect the necessary data of thousands of individuals with the same medical condition. This has been the main impetus behind various international initiatives in forming large, virtual biobanks through the networking of individual databases corresponding to individual biobanks. Apart from data on demographic, environmental risk factors, health, lifestyle, nutrition and socioeconomic variables, these biobanks might also hold ‘omics’ (genomics, transcriptomics, proteomics and metabolomics) data. These data and related bioinformatics software offer advanced possibilities in understanding the disease pathophysiology, thus paving the way for therapeutic and prevention programmes for a number of chronic diseases.
6 Conclusion and Recommendations
Consensus is being sought as to which genetic diseases should be included in population screening programmes and new criteria are being defined to achieve this aim [115]. These emerging criteria include important psychosocial aspects of screening, which become especially important when the test is only an assessment of risk rather than a definitive prediction, as well as ethical procedures such as informed consent [5]. Probably, the most effective way of reaching a suitable conclusion is to organise the established and emerging criteria into a whole process of policy-making, involving a thorough assessment of public health requirements, evaluation of the involved tests and interventions, and the actual development of the policy and implementation of the screening programme (Fig. 2). It is only through rigorous planning and organisation that a screening programme may be truly efficacious and cost-effective. In addition, the decision to integrate genetic testing for the identity of carriers at pre-conception, prenatal or cascade screening is important for autosomal recessive and low penetrance disorders. Furthermore, adverse results in both types of conditions lead to decision-making which involves many psychological and ethical issues [49, 116, 117]. This is especially because, in screening for complex or untreatable conditions, the benefits conferred do not necessarily involve treatment/management, but other life-plans such as reproductive or lifestyle choices.

A global effort should be made to standardise the design of such screening programmes, since integration of preventive and predictive genetics into the diverse health care systems would always remain under the responsibility of national/regional health authorities. Hence, guidelines should be set-up and implemented by leading regulatory bodies, such as the European Medicines Agency and the US FDA, under the recommendation of global experts in the field. Finally, standardisation of screening programmes may be also achieved by setting up regional centres of expertise, for example across the European Union. Such centres would provide standardisation and cost-effectiveness by carrying out tests for rare genetic conditions for all the participating countries.
References
Khoury MJ, Bowen MS, Burke W, Coates RJ, Dowling NF, Evans JP, Reyes M, St Pierre J (2011) Current priorities for public health practice in addressing the role of human genomics in improving population health. Am J Prev Med 40(4):486–493. doi:10.1016/j.amepre.2010.12.009
Morabia A, Zhang FF (2004) History of medical screening: from concepts to action. Postgrad Med J 80(946):463–469. doi:10.1136/pgmj.2003.018226
Wilson JM, Jungner YG (1968) Principles and practice of mass screening for disease. Bol Oficina Sanit Panam 65 (4):281–393
Andermann A, Blancquaert I, Beauchamp S, Déry V (2008) Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ 86(4):241–320
Becker F, van El CG, Ibarreta D, Zika E, Hogarth S, Borry P, Cambon-Thomsen A, Cassiman JJ, Evers-Kiebooms G, Hodgson S, Janssens AC, Kaariainen H, Krawczak M, Kristoffersson U, Lubinski J, Patch C, Penchaszadeh VB, Read A, Rogowski W, Sequeiros J, Tranebjaerg L, van Langen IM, Wallace H, Zimmern R, Schmidtke J, Cornel MC (2011) Genetic testing and common disorders in a public health framework: how to assess relevance and possibilities. Eur J Hum Genet 19(Suppl 1):S6–S44. doi:10.1038/ejhg.2010.249
IRDiRC (2012) International rare diseases research. http://www.irdirc.org. Accessed 22 Apr 2014
Gilissen C, Hoischen A, Brunner HG, Veltman JA (2011) Unlocking Mendelian disease using exome sequencing. Genome Biol 12(9). doi:10.1186/gb-2011-12-9-228
Guthrie R, Susi A (1963) A simple phenylalanine method for detecting Phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Ozben T (2013) Expanded newborn screening and confirmatory follow-up testing for inborn errors of metabolism detected by tandem mass spectrometry. Clin Chem Lab Med 51(1):157–176. doi:10.1515/cclm-2012-0472
O’Neill CA, Eisensmith RC, Croke DT, Naughten ER, Cahalane SF, Woo SL (1994) Molecular analysis of PKU in Ireland. Acta Paediatr Suppl 407:43–44
Guldberg P, Henriksen KF, Sipila I, Guttler F, de la Chapelle A (1995) Phenylketonuria in a low incidence population: molecular characterisation of mutations in Finland. J Med Genet 32(12):976–978
Oerton J, Khalid JM, Besley G, Dalton RN, Downing M, Green A, Henderson M, Krywawych S, Leonard J, Andresen BS, Dezateux C (2011) Newborn screening for medium chain acyl-CoA dehydrogenase deficiency in England: prevalence, predictive value and test validity based on 1.5 million screened babies. J Med Screen 18(4):173–181. doi:10.1258/jms.2011.011086
Wilcken B, Leung KC, Hammond J, Kamath R, Leonard JV (1993) Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 341(8842):407–408. doi:10.1016/0140-6736(93)92993-4
Leonard JV, Dezateux C (2009) Newborn screening for medium chain acyl CoA dehydrogenase deficiency. Arch Dis Child 94(3):235–238. doi:10.1136/adc.2007.134957
Yap S, Naughten E (1998) Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis 21(7):738–747
Carleton SM, Peck DS, Grasela J, Dietiker KL, Phillips CL (2010) DNA carrier testing and newborn screening for maple syrup urine disease in Old Order Mennonite communities. Genet Test Mol Biomarkers 14(2):205–208. doi:10.1089/gtmb.2009.0107
Edelmann L, Wasserstein MP, Kornreich R, Sansaricq C, Snyderman SE, Diaz GA (2001) Maple syrup urine disease: identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population. Am J Hum Genet 69(4):863–868. doi:10.1086/323677
Puffenberger EG (2003) Genetic heritage of the Old Order Mennonites of southeastern Pennsylvania. Am J Med Genet C Semin Med Genet 121C(1):18–31. doi:10.1002/ajmg.c.20003
Kolker S, Christensen E, Leonard JV, Greenberg CR, Burlina AB, Burlina AP, Dixon M, Duran M, Goodman SI, Koeller DM, Muller E, Naughten ER, Neumaier-Probst E, Okun JG, Kyllerman M, Surtees RA, Wilcken B, Hoffmann GF, Burgard P (2007) Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I). J Inherit Metab Dis 30(1):5–22. doi:10.1007/s10545-006-0451-4
Collins FS (1992) Cystic fibrosis: molecular biology and therapeutic implications. Science 256(5058):774–779. doi:10.1126/science.256.5058.774
Massie J, Delatycki MB (2013) Cystic fibrosis carrier screening. Paediatric Respir Rev 14(4):270–275. doi:10.1016/j.prrv.2012.12.002
Barrett PM, Alagely A, Topol EJ (2012) Cystic fibrosis in an era of genomically guided therapy. Human Mol Genet 21(R1):R66–R71. doi:10.1093/hmg/dds345
O’Sullivan BP, Freedman SD (2009) Cystic fibrosis. The Lancet 373(9678):1891–1904. doi:10.1016/s0140-6736(09)60327-5
Hurt K, Bilton D (2012) Cystic fibrosis. Medicine 40(5):273–276. doi:10.1016/j.mpmed.2012.02.006
Dodge JA, Lewis PA, Stanton M, Wilsher J (2007) Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 29(3):522–526. doi:10.1183/09031936.00099506
Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ, Subcommittee on Cystic Fibrosis screening AoGSCAACoMG (2001) Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med 3(2):149–154. doi:10.109700125817-200103000-00010
Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW (2004) Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 6(5):387–391. doi:10.1097/01.gim.0000139506.11694.7c
Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordonez CL, Campbell PW, Ashlock MA, Ramsey BW (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363(21):1991–2003. doi:10.1056/NEJMoa0909825
Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL (2010) Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 182(10):1262–1272. doi:10.1164/rccm.201001-0137OC
Roberts DJ, Williams TN (2003) Haemoglobinopathies and resistance to malaria. Redox Rep 8(5):304–310. doi:10.1179/135100003225002998
Galanello R, Origa R (2010) Beta-thalassemia. Orphanet J Rare Dis 5:11. doi:10.1186/1750-1172-5-11
Limdi JK, Crampton JR (2004) Hereditary haemochromatosis. QJM 97(6):315–324. doi:10.1093/qjmed/hch065
Hanson EH, Imperatore G, Burke W (2001) HFE gene and hereditary hemochromatosis: A HuGE review. Am J Epidemiol 154(3):193–206. doi:10.1093/aje/154.3.193
Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J, Robson KJH (2000) Geography of HFE C282Y and H63D Mutations. Genetic Test 4(2):183–198. doi:10.1089/10906570050114902
Niederau C, Fischer R, Pürschel A, Stremmel W, Häussinger D, Strohmeyer G (1996) Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 110(4):1107–1119
Austin MA, Hutter CM, Zimmern RL, Humphries SE (2004) Familial hypercholesterolemia and coronary heart disease: A HuGE association review. Am J Epidemiol 160(5):421–429. doi:10.1093/aje/kwh237
Varret M, Abifadel M, Rabès JP, Boileau C (2008) Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet 73(1):1–13. doi:10.1111/j.1399-0004.2007.00915.x
Ned RM, Sijbrands EJG (2011) Cascade screening for familial hypercholesterolemia (FH). PLoS Curr 3:RRN1238. doi:10.1371/currents.RRN1238
Neil A, Cooper J, Betteridge J, Capps N, McDowell I, Durrington P, Seed M, Humphries SE (2008) Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J 29(21):2625–2633. doi:10.1093/eurheartj/ehn422
Williams RR, Hamilton-Craig I, Kostner GM, Hegele RA, Hayden MR, Pimstone SN, Faergeman O, Schuster H, Steinhagen-Thiessen E, Beisiegel U, Keller C, Czeizel AE, Leitersdore E, Kastelein JC, Defesche JJP, Ose L, Leren TP, Seftel HC, Raal FJ, Marais AD, Eriksson M, Keller U, Miserez AR, Jeck T, Betterridge DJ, Humphries SE, Day INM, Kwiterovich PO, Lees RS, Stein E, Illingworth R, Kane J, Boulyjenkov V (1996) MED-PED: An integrated genetic strategy for preventing early deaths. In: Dr KBP, Dr VB, Dr YC (eds) Genetic approaches to noncommunicable diseases. Springer, Berlin, pp 35–45
Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN (1993) Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 72(2):171–176
Bertolini S, Cantafora A, Averna M, Cortese C, Motti C, Martini S, Pes G, Postiglione A, Stefanutti C, Blotta I, Pisciotta L, Rolleri M, Langheim S, Ghisellini M, Rabbone I, Calandra S (2000) Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol 20(9):e41–e52. doi:10.1161/01.ATV.20.9.e41
Scientific Steering Committee on behalf of the Simon Broome Register Group (1991) Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 303(6807):893–896
World Health O (1999) Familial hypercholesterolaemia: report of a second WHO consultation. WHO, Geneva
Javaher P, Nyoungui E, Kaariainen H, Kristoffersson U, Nippert I, Sequeiros J, Schmidtke J (2010) Genetic screening in Europe. Public Health Gen 13(7–8):524–537. doi:10.1159/000294998
Rastogi MV, LaFranchi SH (2010) Congenital hypothyroidism. Orphanet J Rare Dis 5:17. doi:10.1186/1750–1172-5-17
Comeau AM, Accurso FJ, White TB, Campbell PW 3rd, Hoffman G, Parad RB, Wilfond BS, Rosenfeld M, Sontag MK, Massie J, Farrell PM, O’Sullivan BP (2007) Guidelines for implementation of cystic fibrosis newborn screening programs: Cystic Fibrosis Foundation workshop report. Pediatrics 119(2):e495–518. doi:10.1542/peds.2006-1993
Loeber JG, Burgard P, Cornel MC, Rigter T, Weinreich SS, Rupp K, Hoffmann GF, Vittozzi L (2012) Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis 35(4):603–611. doi:10.1007/s10545-012-9483-0
Godard B, ten Kate L, Evers-Kiebooms G, Aymé S (2003) Population genetic screening programmes: principles, techniques, practices, and policies. Eur J Hum Genet 11(S2):S49–S87. doi:10.1038/sj.ejhg.5201113
Gemeinsamer Bundesausschuss (G-BA) Mandate of the Federal Joint Committee. (2014) http://www.english.g-ba.de/special-topics/prevention/mandate/. Accessed 05 June 2014
UK National Screening Committee (2013) Programme appraisal criteria—Criteria for appraising the viability, effectiveness and appropriateness of a screening programme. http://www.screening.nhs.uk/criteria. Accessed 05 Jun 2014
Sanderman LK (2011) Newborn screening in Europe: lessons learned from tandem mass spectrometry in Germany and the UK. (Master of Science) Maastricht University, Maastricht
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724
Wright C, Burton H, Hall A, Moorthie S, Pokorska-Bocci A, Sagoo G, Sanderson S, Skinner R (2011) Next steps in the sequence. The implications of whole genome sequencing for health in the UK. PHG Foundation, Cambridge
Schröder P, Zimmern R (2007) PHGEN Working Group 2– The status of genetic information and genetic testing. Consequences for Public Health Genomics and the Public Health Genomics European Network (PHGEN). http://ec.europa.eu/health/screening_genetics/genomics/index_en.htm, Accessed 05 Jun 2014
Burke W, Burton H, Hall AE, Karmali M, Khoury MJ, Knoppers B, Meslin EM, Stanley F, Wright CF, Zimmern RL (2010) Extending the reach of public health genomics: what should be the agenda for public health in an era of genome-based and “personalized” medicine? Genet Med 12(12):785–791
EAPM (2013) Innovation and patient access to personalised medicine—Report from Irish Presidency Conference March 20th/21st 2013. http://euapm.eu/wp-content/uploads/2012/07/EAPM-REPORT-on-Innovation-and-Patient-Access-to-Personalised-Medicine.pdf. Accessed 05 Jun 2014
Wellcome Trust (2013) Impact of the draft European Data Protection Regulation and proposed amendments from the rapporteur of the LIBE committee on scientific research. http://www.wellcome.ac.uk/stellent/groups/corporatesite/@policy_communications/documents/web_document/wtvm054713.pdf. Accessed 16 Nov 2013
Ploem MC, Essink-Bot ML, Stronks K (2013) Proposed EU data protection regulation is a threat to medical research. BMJ 346:f3534. doi:10.1136/bmj.f3534
Brice P (2013) EU legal amendments threaten genomic medicine and research. PHG Foundation. http://www.phgfoundation.org/news/ 14842 /. Accessed 16 Nov 2013
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG (2013) ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 15(7):565–574. doi:10.1038/gim.2013.73
Easton DF, Ford D, Bishop DT (1995) Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet 56(1):265–271
Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M, the Breast Cancer Linkage Consortium (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 62(3):676–689. doi:10.1086/301749
Lynch HT, Smyrk T, Lynch JF (1998) Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): historical journey from pedigree anecdote to molecular genetic confirmation. Oncology 55(2):103–108.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10(8):789–799. doi:10.1038/nm1087
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR (2007) PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 39(2):165–167. doi:10.1038/ng1959
Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38(11):1239–1241. doi:10.1038/ng1902
Somyajit K, Subramanya S, Nagaraju G (2010) RAD51C: a novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 31(12):2031–2038. doi:10.1093/carcin/bgq210
Lensch MW, Tischkowitz M, Christianson TA, Reifsteck CA, Speckhart SA, Jakobs PM, O'Dwyer ME, Olson SB, Le Beau MM, Hodgson SV, Mathew CG, Larson RA, Bagby GC, Jr. (2003) Acquired FANCA dysfunction and cytogenetic instability in adult acute myelogenous leukemia. Blood 102(1):7–16. doi:10.1182/blood-2002–09-2781
Garber JE, Offit K (2005) Hereditary cancer predisposition syndromes. J Clin Oncol 23(2):276–292. doi:10.1200/JCO.2005.10.042
McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, Thomas DM, Mitchell G (2014) Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. doi:10.1038/nrclinonc.2014.41
Francke U (1976) Retinoblastoma and chromosome 13. Cytogenet Cell Genet 16(1–5):131–134
Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ (1997) Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276(5311):404–407
Gnarra JR, Duan DR, Weng Y, Humphrey JS, Chen DY, Lee S, Pause A, Dudley CF, Latif F, Kuzmin I, Schmidt L, Duh FM, Stackhouse T, Chen F, Kishida T, Wei MH, Lerman MI, Zbar B, Klausner RD, Linehan WM (1996) Molecular cloning of the von Hippel-Lindau tumor suppressor gene and its role in renal carcinoma. Biochim Biophys Acta 1242(3):201–210
Hirshfield KM, Rebbeck TR, Levine AJ (2010) Germline mutations and polymorphisms in the origins of cancers in women. J Oncol 2010:1–11. doi:10.1155/2010/297671
Samoha S, Arber N (2005) Cyclooxygenase-2 inhibition prevents colorectal cancer: from the bench to the bed side. Oncology 69(Suppl 1):33–37. doi:10.1159/000086630
Lynch PM (2007) Prevention of colorectal cancer in high-risk populations: the increasing role for endoscopy and chemoprevention in FAP and HNPCC. Digestion 76(1):68–76. doi:10.1159/000108395
Rozen P, Samuel Z, Rabau M, Goldman G, Shomrat R, Legum C, Orr-Urtreger A (2001) Familial adenomatous polyposis at the Tel Aviv Medical Center: demographic and clinical features. Fam Cancer 1(2):75–82
Malkin D, Li FP, Strong LC, Fraumeni JF, Jr., Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, Friend SH (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250(4985):1233–1238
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, Han JH, Lowstuter K, Longmate J, Sommer SS, Weitzel JN (2009) Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27(8):1250–1256. doi:10.1200/jco.2008.16.6959
Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA (2003) Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 63(20):6643–6650
Lalloo F, Varley J, Ellis D, Moran A, O’Dair L, Pharoah P, Evans DG (2003) Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 361(9363):1101–1102. doi:10.1016/S0140-6736(03)12856-5
Li FP, Fraumeni JF, Jr., Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, Miller RW (1988) A cancer family syndrome in twenty-four kindreds. Cancer Res 48(18):5358–5362
Chompret A, Abel A, Stoppa-Lyonnet D, Brugieres L, Pages S, Feunteun J, Bonaiti-Pellie C (2001) Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 38(1):43–47
Hwang SJ, Lozano G, Amos CI, Strong LC (2003) Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am J Hum Genet 72(4):975–983. doi:10.1086/374567
Farooq A, Walker LJ, Bowling J, Audisio RA (2010) Cowden syndrome. Cancer Treat Rev 36(8):577–583. doi:10.1016/j.ctrv.2010.04.002
Hobert JA, Eng C (2009) PTEN hamartoma tumor syndrome: an overview. Genet Med 11(10):687–694. doi:10.1097/GIM.0b013e3181ac9aea
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16(1):64–67. doi:10.1038/ng0597-64
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308):1943–1947
Scheper MA, Nikitakis NG, Sarlani E, Sauk JJ, Meiller TF (2006) Cowden syndrome: report of a case with immunohistochemical analysis and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 101(5):625–631. doi:10.1016/j.tripleo.2005.06.026
Gorlin RJ, Cohen MM, Jr., Condon LM, Burke BA (1992) Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet 44(3):307–314. doi:10.1002/ajmg.1320440309
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C (2012) Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18(2):400–407. doi:10.1158/1078-0432.CCR-11-2283
Vasen HF, Boland CR (2005) Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA 293(16):2028–2030. doi:10.1001/jama.293.16.2028
Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Nogues C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frebourg T, Sobol H, Lasset C, Bonaiti-Pellie C (2011) Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305(22):2304–2310. doi:10.1001/jama.2011.743
Vasen HF, Watson P, Mecklin JP, Lynch HT (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116(6):1453–1456
Boland CR, Goel A (2010) Microsatellite instability in colorectal cancer. Gastroenterology 138 (6):2073–2087 e2073. doi:10.1053/j.gastro.2009.12.064
Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ (1999) Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 81(2):214–218. doi:10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L
Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M (1998) Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18(1):38–43. doi:10.1038/ng0198-38
Tomlinson IP, Houlston RS (1997) Peutz-Jeghers syndrome. J Med Genet 34(12):1007–1011
Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA (2000) Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 119(6):1447–1453. doi:10.1053/gast.2000.20228
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61(5):759–767. doi:10.1016/0092-8674(90)90186-I
Gruber SB, Entius MM, Petersen GM, Laken SJ, Longo PA, Boyer R, Levin AM, Mujumdar UJ, Trent JM, Kinzler KW, Vogelstein B, Hamilton SR, Polymeropoulos MH, Offerhaus GJ, Giardiello FM (1998) Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res 58(23):5267–5270
Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D (2002) Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res 62(13):3659–3662
Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, Trimbath JD, Giardiello FM, Gruber SB, Offerhaus GJ, de Rooij FW, Wilson JH, Hansmann A, Moslein G, Royer-Pokora B, Vogel T, Phillips RK, Spigelman AD, Houlston RS (2006) Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 12(10):3209–3215. doi:10.1158/1078-0432.CCR-06-0083
Khoury MJ, Feero WG, Valdez R (2010) Family history and personal genomics as tools for improving health in an era of evidence-based medicine. Am J Prev Med 39(2):184–188. doi:10.1016/j.amepre.2010.03.019
Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ (2010) Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 31:69–87. doi:10.1146/annurev.publhealth.012809.103621
Jarvinen HJ, Mecklin JP, Sistonen P (1995) Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 108(5):1405–1411. doi:10.1016/0016-5085(95)90688-6
Burke W, Daly M, Garber J, Botkin J, Kahn MJ, Lynch P, McTiernan A, Offit K, Perlman J, Petersen G, Thomson E, Varricchio C (1997) Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2. Cancer Genetics Studies Consortium. JAMA 277(12):997–1003
De Cosse JJ, Bulow S, Neale K, Jarvinen H, Alm T, Hultcrantz R, Moesgaard F, Costello C (1992) Rectal cancer risk in patients treated for familial adenomatous polyposis. The Leeds Castle Polyposis Group. Br J Surg 79(12):1372–1375
Dunlop MG (2002) Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 51(Suppl 5):V21–27
van Sprundel TC, Schmidt MK, Rookus MA, Brohet R, van Asperen CJ, Rutgers EJ, Van't Veer LJ, Tollenaar RA (2005) Risk reduction of contralateral breast cancer and survival after contralateral prophylactic mastectomy in BRCA1 or BRCA2 mutation carriers. Br J Cancer 93(3):287–292. doi:10.1038/sj.bjc.6602703
Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, Thun MJ, Cox DG, Hankinson SE, Kraft P, Rosner B, Berg CD, Brinton LA, Lissowska J, Sherman ME, Chlebowski R, Kooperberg C, Jackson RD, Buckman DW, Hui P, Pfeiffer R, Jacobs KB, Thomas GD, Hoover RN, Gail MH, Chanock SJ, Hunter DJ (2010) Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362(11):986–993. doi:10.1056/NEJMoa0907727
Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH (1995) High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 85(6):1504–1508
Andermann A, Blancquaert I, Beauchamp S, Costea I (2011) Guiding policy decisions for genetic screening: developing a systematic and transparent approach. Public Health Gen 14(1):9–16. doi:10.1159/000272898
Nussbaum RL, McInnes RR, Willard HF (2007) Thompson & Thompson Genetics in Medicine. Elsevier Health Sciences, Makati City
Health Council of the Netherlands (2008) Screening: between hope and hype. http://www.emgo.nl/files/Bonn%202010%20genetic%20screening%20criteria%20in%20the%20age%20of%20genomics.ppt. Accessed 20 Sep 2014
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Grech, G., Scerri, C., Scerri, J., Cesuroglu, T. (2015). Preventive and Predictive Genetics: A perspective. In: Grech, G., Grossman, I. (eds) Preventive and Predictive Genetics: Towards Personalised Medicine. Advances in Predictive, Preventive and Personalised Medicine, vol 9. Springer, Cham. https://doi.org/10.1007/978-3-319-15344-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-15344-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-15343-8
Online ISBN: 978-3-319-15344-5
eBook Packages: MedicineMedicine (R0)