
Abstract
Post-transplant lymphoproliferative disorders (PTLD) are a serious complication after solid organ or allogeneic hematopoietic stem cell transplantation and include a range of diseases from benign proliferations to malignant lymphomas. Risk factors for developing PTLD include Epstein-Barr virus (EBV) infection, recipient age, transplanted organ, type of immunosuppression, and genetics. Uncontrolled proliferation of EBV-infected B cells is implicated in EBV-positive PTLD, whereas the pathogenesis of EBV-negative PTLD may be similar to non-Hodgkin’s lymphoma in the general population. The World Health Organization (WHO) classifies PTLD into four categories: early lesions, polymorphic PTLD, monomorphic PTLD, and classical Hodgkin’s lymphoma (cHL). Treatment is aimed at cure of PTLD, while maintaining transplanted organ function. However, there are no established guidelines for the treatment of PTLD. Immune suppression reduction (ISR) is the first line of treatment in most cases, with more recent data suggesting early use of rituximab. In more aggressive forms of PTLD, upfront chemotherapy may offer a better and more durable response. Sequential therapy using rituximab followed by chemotherapy has demonstrated promising results and may establish a standard of care. Novel therapies including anti-viral agents, adoptive immunotherapy, and monoclonal antibodies targeting cytokines require further study in the prevention and treatment of PTLD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Post-transplant lymphoproliferative disorder
- PTLD
- Lymphoma
- Non-Hodgkin’s lymphoma
- Epstein-Barr virus (EBV)
- Rituximab
- Chemotherapy
- Anti-viral therapy
- Adoptive immunotherapy
1 Introduction
Post-transplant lymphoproliferative disorders (PTLD) include a wide range of diseases from benign hyperplasia to malignant lymphomas that occur after solid organ transplantation (SOT) and allogeneic hematopoietic cell transplantation (allo-HCT) in the setting of immunosuppression. PTLD are the most common post-SOT malignancy in children. In adults, PTLD are the second most common post-SOT malignancy, after non-melanoma skin cancer [1, 2]. PTLD were first described in the late 1960s in patients following renal transplantation [3, 4]. More than 40 years later, PTLD remain a serious and, at times, fatal complication of transplantation. In SOT, it is the most common cause of cancer-related mortality [5–7].
2 Epidemiology
Though there are many commonalities between PTLD in SOT and allo-HCT, distinct differences exist in the epidemiology and pathophysiology of the disease. In both SOT and allo-HCT, many cases are associated with Epstein-Barr virus (EBV) [8].
For adult SOT recipients, PTLD are seen in up to 10–15 % of all recipients and the highest incidence is after small bowel transplantation (20 %), followed by lung (10 %), heart (6 %), liver (2.8 %), and renal (2.3 %) transplantation [8–11]. The incidence of PTLD is significantly higher in children compared to adults, owing to a high rate of primary EBV infection after transplantation [8]. While early studies found the highest incidence of PTLD to be in the first year post-transplant, more recent data suggest a median onset of PTLD after SOT to be 30–40 months [12, 13].
For allo-HCT patients, the incidence of PTLD is significantly lower. In several large retrospective studies, the incidence of PTLD in patients following allo-HCT has been 0.5–2.5 %, with peak incidence between 2 to 6 months post-transplant [9, 14–16].
3 PTLD Risk Factors
3.1 Solid Organ Transplantation
Various risk factors for post-SOT PTLD have been identified, including recipient age, transplanted organ, characteristics of immunosuppressive therapy, and EBV status [8, 9, 17]. First recognized in 1985, EBV infection plays an integral role in PTLD [18]. The risk of PTLD after SOT is highest in those who develop a primary EBV infection after transplantation, specifically when an EBV-seronegative recipient receives an allograft from an EBV-seropositive donor [19]. Over 95 % of the world’s population has been exposed to EBV by adulthood [8]; as such, primary EBV infections are more worrisome in the pediatric population [20]. EBV-seronegative recipients have a 10–76 times greater incidence of PTLD [17, 19, 21–25].
The type of SOT and the type of immunosuppression used contribute to the risk of PTLD. Early PTLD are likely due to the combined intensity of immunosuppression, while late PTLD are related to the duration of immunosuppression [9]. Specific immunosuppressive agents have also been associated with increasing risks of PTLD, such as cyclosporine [26, 27], tacrolimus [8], OKT3 (a T cell depleting anti-CD3 monoclonal antibody used to prevent and treat acute rejections) [6, 9], and anti-thymocyte globulin (ATG) [6].
Other risk factors implicated in PTLD include hepatitis C [28], cytomegalovirus (CMV) [9, 29], or HHV-8 [30] and age younger than 10 or older than 60 years [6]. The combination of multiple risk factors (CMV mismatch, OKT3 exposure, and pre-transplant EBV-seronegative recipient) can increase the risk of PTLD up to 500-fold compared to patients with no risk factors [17].
3.2 Allogeneic Hematopoietic Cell Transplantation
The most important risk factor in the development of PTLD after allo-HCT is T-cell depletion of the donor marrow or peripheral blood stem cell product [15]. Other factors include the degree of HLA mismatching, EBV serology, degree and severity of graft versus host disease (GVHD), and age older than 50 at time of transplant [14, 15, 31, 32].
As in SOT, EBV-seronegative recipients who are grafted from EBV-seropositive donors are at significantly higher risk for PTLD. Although rare, PTLD are still possible in patients with EBV-seronegative donors [14]. CMV seropositive status for donors or recipients has also been associated with increased risk for PTLD after allo-HCT [14]. The degree of HLA mismatch can increase the relative risk (RR) of developing PTLD by up to 8.9 times the general population [31, 32]. Patients who undergo myeloablative conditioning regimens and receive T-cell depleting antibodies are at higher risk for EBV-associated PTLD [33]. Agents that selectively target T cells and/or NK cells are associated with a higher risk of PTLD than those that deplete both T and B cells, such as alemtuzumab [15, 34–36]. If multiple risk factors are combined, patients at a particularly high risk for PTLD can be identified.
3.3 Genetics
Host and donor genetic variation in human leukocyte antigen (HLA) loci and genes for several cytokines have been implicated in PTLD. In one study, donor and recipient HLA-A26 and B38 haplotypes were independent risk factors for developing PTLD, while donor HLA-A1, B8, and DR3 haplotypes were protective against PTLD [37]. A separate study found HLA-B donor–recipient mismatching alone to be associated with PTLD in renal transplant patients [38].
Cytokine polymorphisms have been also implicated as risk factors for PTLD. These include the genes encoding interleukin (IL)-10, IL-6, interferon gamma (IFN-γ), transforming growth factor-beta (TGF-β), tumor necrosis factor-alpha (TNF-α) promoter, and TNF-α receptor [9, 39–43].
4 Pathogenesis
In Europe and USA, the majority (approximately 85 %) of PTLD cases arise from B cells and of these, more than 80 % are associated with EBV infection [8]. Over 90 % of the world population is exposed to EBV by adulthood [44]. Though usually acquired in infancy, EBV can cause infectious mononucleosis (IM) in up to 50 % of adolescents [10].
The virus gains entry into hosts via salivary exchange and infects B cells by binding to CD21. It then replicates by lysis and proliferation of infected B cells. EBV-infected latent B cells then begin to express multiple latent membrane proteins (LMP) and EB nuclear antigens (EBNA). Recognizing the viral antigens, the host mounts a primary CD8+ cytotoxic T lymphocyte (CTL) response affecting both lytic and latent cells. This response leads to a decrease in EBV-infected B cells; however, EBV establishes itself in memory B cells for the duration of the host’s life. LMP1 upregulates anti-apoptotic genes and moves the infected cells into the latent phase [8]. Though infected memory B cells express a restricted range of viral antigens, these limited antigens produce a secondary CTL response, which in turn creates a balance of proliferation and destruction of infected B cells which persists throughout life [45]. Thereafter, EBV can be present in up to 1 in 106 circulating B cells [10].
In addition to EBV infection, other contributing factors are likely necessary, such as allo-antigens and cytokines such as interleukin (IL)-10 and IFN-α, for the development of PTLD [45].
Most of the studied pathogenesis in PTLD is linked to EBV. However, there is limited literature on the pathogenesis of non-EBV-related PTLD, which may be similar to non-Hodgkin’s lymphoma in the general population [8]. In fact, some authors have suggested that late occurring PTLD should be considered a distinct entity from early PTLD [46]. EBV-negative PTLD typically present as a late complication of transplantation, with a median time of 50–60 months, and has more aggressive features [47, 48]. Given the increased survival of patients following SOT and allo-HCT, the incidence of EBV-negative PTLD may be on the rise [47, 49].
5 Clinical Presentation
Patients with PTLD may have an array of clinical signs and symptoms, depending on the organ system and degree of organ involvement. As previously discussed, PTLD may present at any time after transplantation. Some patients can present with clinical emergencies such as intestinal perforations or fulminant PTLD with disseminated disease mimicking septic shock [8, 50]. Lymphadenopathy alone is less commonly seen as a presenting sign, when compared to the non-transplant population. Commonly, extra-nodal organ involvement is seen [51]. Symptoms are often due to dysfunction of the organ involved, but patients can also develop constitutional (or “B”) symptoms [11]. Extra-nodal sites may include the central nervous system (CNS), skin, gastrointestinal tract, lungs, renal, skin, and bone marrow [12, 47, 52, 53]. Among these, gastrointestinal involvement is most commonly reported (22–25 %) [52, 54]. Allografts themselves are less frequently involved, with exception of lung transplants [8].
6 Pathology, Diagnosis, and Staging
Current diagnosis and classification of PTLD is based on the 2008 World Health Organization (WHO) system [55]. Although it is sometimes difficult to clearly distinguish between these lesions, the WHO divides PTLD into 4 main histologic categories as detailed below (see also Table 1).
-
1.
Early lesions (Fig. 1a). These are typically seen within a year of transplantation. In this type, lymphoid tissues maintain normal architecture by definition and present with one of two distinct histological patterns—plasmacytic hyperplasia or IM-like form. In the former, scattered EBV-positive immunoblasts are seen in the background of sheets of polytypic, mature appearing plasma cells. The latter histology resembles IM, demonstrating paracortical expansion by variable numbers of immunoblasts and unremarkable lymphocytes. The immunoblasts include EBV-infected B cells [11].
Fig. 1 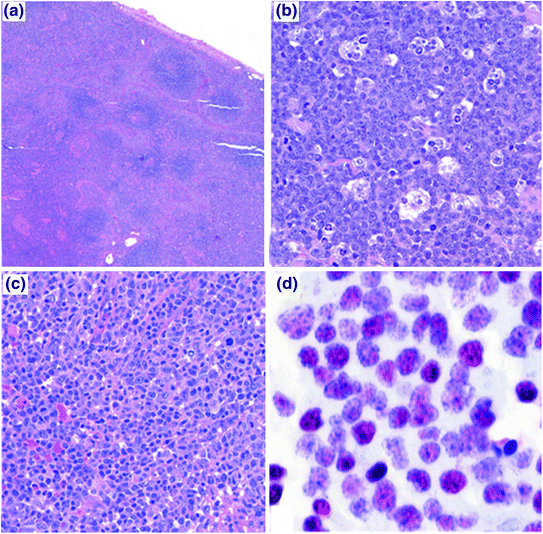
The histopathology of PTLD. a Infectious mononucleosis-like “early” PTLD lesion. b Monomorphic PTLD, Burkitt lymphoma type. c Polymorphic PTLD. d Positive EBV in situ hybridization stain in a PTLD
-
2.
Polymorphic PTLD (Fig. 1c). In this type, the lymphoid tissue architecture is effaced or a destructive extra-nodal mass is observed. As the name implies, the lymphoid cells are polymorphic and include small- to medium-sized lymphocytes with variable nuclear atypia, immunoblasts, and mature plasma cells. Necrosis may be observed [11]. These can be monoclonal or polyclonal, as establishing clonality is dependent on the analysis technique and clonal burden. Importantly, these do not meet diagnostic criteria for specific WHO-defined B-cell or T- /NK-cell lymphoma categories [56].
-
3.
Monomorphic PTLD (Fig. 1b). This is the most common type of PTLD. These are monoclonal proliferations that can be separated into specific B-cell and T-cell lymphomas, using the same WHO criteria/classification as in non-transplant patients. B-cell PTLD are more common and include diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL) (Fig. 1b), plasma cell myeloma, and plasmacytoma-like PTLD. DLBCL accounts for the majority of monomorphic PTLD cases [11].
T-cell PTLD are rare and include peripheral T-cell lymphoma, hepatosplenic T-cell lymphoma, and anaplastic large cell lymphoma (ALCL). Up to 90 % of T-cell PTLD are EBV negative [57], while the majority of NK cell PTLD are EBV positive [58]. Compared to B-cell PTLD, T-cell PTLD usually occur later and carry a poorer prognosis. T-cell PTLD cases have been associated with human T-lymphotrophic virus type 1 (HTLV-1) and may be a factor in the rising incidence of T-cell lymphomas in Japan [59].
-
4.
Classical Hodgkin’s Lymphoma (cHL)—This is the rarest form of PTLD and usually presents late after transplantation. It is histopathologically identical to cHL, showing Reed–Sternberg cells in a variable inflammatory cell milieu.

An excisional tissue biopsy is the preferred specimen for PTLD diagnosis. If an excisional biopsy is not feasible, for example, in case of suspected extra-nodal involvement, core needle biopsy and needle aspiration may be diagnostic [28]. Tissue should be examined for histology, immunophenotyping, EBER-ISH (EBV-encoded small nuclear RNA—in situ hybridization), and cytogenetic studies for classification.
Basic laboratory tests should include complete blood count with differential, comprehensive metabolic panel, lactate dehydrogenase, and uric acid. Patients should also be checked for EBV viral load, HIV, and hepatitis serologies. There is insufficient data at this time to recommend following serial EBV viral loads to assess response to therapy. Modern imaging, usually computed tomography (CT) scanning with or without positron emission tomography (PET) scanning, is an essential tool in diagnosis and staging and should include neck, chest, abdomen, and pelvis. If cytopenias are present, bone marrow biopsy may be warranted [11].
Similar to Hodgkin’s and non-Hodgkin’s lymphoma in the non-transplant setting, the Ann Arbor classification is typically used for staging.
7 Prognosis
Due to the large variability of disorders encompassed in PTLD, no reliable prognostic scoring system exists. In a French study of 500 patients with PTLD post-renal transplant, the authors constructed a 5-point prognostic score based on the following: age older than 55 years, serum creatinine greater than 1.5 g/dl, elevated LDH, disseminated PTLD, and monomorphic histology [60]. Patients were risk stratified into low risk (0 risk factors), moderate risk (1 risk factor), high risk (2–3 risk factors), or very high risk (4–5 risk factors). Five-year OS was 92, 83, 59, and 25 %, respectively. Another study of 80 PTLD patients after SOT noted 3 prognostic factors: CNS involvement, bone marrow involvement, and hypoalbuminemia [61]. Three-year survival was 93 % with 0 risk factors, 68 % with 1 risk factor, and 11 % with 2–3 risk factors. CNS involvement has been associated with poor prognosis in more than one study, though this may be improving with the use of rituximab and high-dose methotrexate [11, 61].
8 Treatment
There are no uniformly applicable guidelines for the treatment of PTLD, due to the wide spectrum of disease and scarcity of prospective phase II and III studies. The goals of treatment in PTLD are twofold: first, to eliminate the PTLD and second, to preserve the transplanted graft [56]. The majority of evidence available for the treatment of PTLD has been seen in SOT (particularly CD20-positive B-cell PTLD), with limited data available regarding PTLD after allo-HCT or T-cell PTLD.
In general, the initial therapeutic intervention for PTLD consists of immune suppression reduction (ISR) [62, 63]. Unfortunately, only about half of patients respond to ISR. In addition, it can take several weeks before a response is evident after ISR [13, 64]. Many other treatment options have been studied and are used either following, or in conjunction with ISR (see Table 2 and Fig. 2). These include the use of rituximab (a monoclonal antibody directed against CD20), chemotherapy regimens, local therapy with radiation or surgery, EBV-specific CTL infusions, and more recent novel therapies.

Treatment algorithm for PTLD
8.1 Immune Suppression Reduction (ISR)
In most cases of PTLD, ISR is the first step in treatment. ISR should partially restore the ability of CTLs to eliminate EBV-infected lymphocytes [8]. There are, however, several potential drawbacks associated with ISR—the most significant being graft rejection. In addition, ISR as monotherapy has a relatively slow time to response (on average>2–4 weeks) [65] and lower efficacy compared to ISR combined with rituximab and/or chemotherapy. Reported overall response rates (ORR) to ISR vary from 0 to 74 % [64, 66–68], with durable responses of less than 30 % of patients in several studies [13, 64, 66, 67].
In older studies, patients with early lesions and polyclonal PTLD appeared to respond better to ISR, while those with monoclonal tumors [46], in particular with bcl-6 expression, were less likely to respond [69, 70]. However, in a more recent study, monomorphic versus polymorphic histology did not predict for response to ISR [66].
In theory, EBV-positive PTLD should have a higher likelihood to respond to ISR; however, recent evidence suggests this may not necessarily be the case. In a recent study of SOT patients, EBV positivity was not a predictor of response—with only non-bulky disease (<7 cm) and age <50 at diagnosis being predictive [66]. Some factors such as high LDH and multi-organ dysfunction or involvement have inconsistently been associated with poor response to ISR [65, 66]. Multiple factors can be combined to help predict response to ISR. For example, using LDH elevation, hepatitis C infection, bone marrow or liver involvement, and B symptoms, 3-year overall survival (OS) was 100 % in patients with none of these factors, 79 % with one, and 8 % with two or more factors [66].
The specific dose modifications for ISR in an individual patient are based on multiple factors, such as the transplanted organ, extent of PTLD, perceived risk of rejection, symptoms, physical exam, and laboratory data. Decisions regarding ISR should be pursued in close collaboration with the transplant team. Anti-proliferative agents such as azathioprine and mycophenolate should be dose-reduced or discontinued, if possible [11]. Guidelines suggest dose reduction of 25 % in limited disease. With extensive disease in non-critically ill patients, cyclosporine/tacrolimus is typically reduced to 50 %, with discontinuation of azathioprine/mycophenolate and continuation of prednisone at 7.5–10 mg daily [71]. In critically ill patients, it may be necessary to discontinue all immune suppressing agents except prednisone 7.5–10 mg daily. Patients need to be followed closely to assess disease response and to monitor for graft rejection. Though there are risks with ISR, the majority of evidence supports the use of ISR as the first step in management of PTLD in most cases. In patients with aggressive disease, low predicted response to ISR alone or a contraindication to ISR may necessitate consideration of alternative or augmented therapies.
8.2 Rituximab
Rituximab, a chimeric monoclonal antibody targeting the B-cell surface protein CD20, has improved outcomes in many B-cell lymphomas. Data showing rituximab to be superior to other treatment approaches started to emerge around 2005 [72]. Prior to the rituximab era, the 3-year OS for PTLD ranged from 30 to 50 % but has now improved to >60 % [62, 72–75].
In the first trial of rituximab monotherapy use after ISR failure in PTLD, an ORR of 44 % was seen, with a median OS of 15 months [74]. Since then, several other studies have shown ORR ranging from 44 to 68 % and median OS up to 42 months (Table 2).
More recently, in a large, multicenter retrospective analysis, Evens et al. reviewed 80 SOT PTLD patients to establish the impact of the introduction of rituximab on outcomes [61]. All patients in the study had ISR, with 74 % of patients receiving rituximab with or without chemotherapy. With regard to first-line therapy, patients had a 73 % OS when rituximab was a part of the regimen compared to 33 % without rituximab. The 3-year progression-free survival (PFS) was 70 % with rituximab and 21 % without.
8.3 Chemotherapy
Prior to rituximab, chemotherapy was the primary treatment in patients who failed ISR. Several regimens have been described for use in PTLD. Anthracycline-based chemotherapy with rituximab, such as R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) is the most widely used regimen [71, 76–78]. It is preferred in patients for whom a rapid clinical response is required, such as those with multi-organ involvement or rapidly declining clinical status. Although the risk of toxicity and treatment-related mortality (TRM) can be as high as 25–50 % [79], the ORR is higher than rituximab monotherapy, ranging between 65 and 100 % [71]. Given the increased risk of TRM, chemotherapy should be reserved for those who fail to achieve appropriate response with ISR or ISR and rituximab, who have highly aggressive histology, or who require a rapid clinical response such as patients presenting with severe organ dysfunction or rapidly declining clinical status [71]. In some scenarios, it may be prudent to avoid certain chemotherapy drugs if possible, such as anthracyclines in heart transplant patients and high-dose methotrexate in renal transplant patients.
8.4 Sequential Therapy
Many experts recommend a sequential or staged approach to the treatment of PTLD, in which patients generally receive ISR or ISR + rituximab as a starting point. An assessment of response is made 4–8 weeks later and, if the response is insufficient, additional therapy is then administered [63, 71, 80, 81]. This strategy was recently tested in a large prospective study (the “PTLD-1” trial) and may provide the foundation for a standard approach in the future [81]. In this study, 70 patients who failed ISR were then treated with 4 weekly doses of rituximab followed by 4 cycles of CHOP. Following rituximab, there was a 60 % ORR (with 20 % CR). After completion of rituximab × 4 and CHOP × 4, the ORR increased to 90 % (with 68 % CR) [81]. In this study, the planned therapy was rituximab × 4 followed by CHOP × 4, so it is unclear whether the patients who achieved CR with rituximab alone might have maintained their responses without chemotherapy.
In practice, a commonly utilized approach is to initially implement ISR or ISR + rituximab and then, for those who have not achieved CR, to subsequently apply chemotherapy. The “sequential therapy” approach of the PTLD-1 trial was modified to test this “risk-adapted” approach, after an interim analysis demonstrated that response to rituximab correlated with OS [82]. In the “risk-adapted” approach, patients achieving CR with 4 weeks of rituximab received 4 additional doses of rituximab, whereas those who did not achieve CR were then treated with 4 cycles of R-CHOP. In a preliminary analysis (n = 90), an ORR of 93 % and CR rate of 78 % were seen. The CR rate to rituximab alone was 27 %, with an additional 51 % going on to achieve CR after CHOP. Only 13 % of patients achieving CR with rituximab alone went on to relapse. Treatment-related mortality was low at 8 %. Comparing to previous data from the “sequential therapy” part of the PTLD-1 trial, it appears that (1) omitting chemotherapy from those achieving CR to rituximab is safe, and (2) for those who have progressive disease with rituximab, R-CHOP is more effective than CHOP. Those who failed to achieve CR after rituximab × 4 ± R-CHOP × 4 (22 %) did not benefit from additional R-CHOP and had nearly 80 % risk of progressive lymphoma in the following 1–2 years. Therefore, with this approach, most patients were able to achieve CR, while the relatively high treatment-related toxicity seen with CHOP was avoided except for patients who truly required chemotherapy [82]. Given these promising results, further study may help establish this “response-adapted” approach as a standard of care in CD20-positive PTLD [63].
8.5 Localized Therapy
Localized therapy using either radiation therapy (RT) or surgery can be utilized in carefully selected PTLD patients. When combined with ISR, this may be curative in patients with PTLD localized to a single site (Ann Arbor Stage I disease) [63]. RT may also have a significant role in the treatment of CNS or limited-stage PTLD, with some studies demonstrating complete responses [83, 84]. Incorporation of RT may also allow for an abbreviated course of chemotherapy for those with limited-stage disease or avoidance of chemotherapy for those who are unlikely to tolerate chemotherapy.
8.6 Primary CNS PTLD
Primary CNS PTLD is unique in both its presentation and treatment compared to other forms of PTLD. CNS involvement occurs in up to 15–22 % of PTLD, most of which are primary CNS PTLD, and has consistently been associated with poor outcomes across multiple studies [6, 12, 61, 75, 84–87].
In a recent multicenter international retrospective analysis of 84 cases of primary CNS PTLD over a 14-year period, it was found that although 83 % of cases occurred late (>1 year post-transplant), over 90 % were EBV positive [87]. The large majority (79 %) were associated with renal transplantation, presented with a median time to onset of 54 months, and 79 % had diffuse large B-cell lymphoma (DLBCL) histology. Median PFS and OS for the cohort were 8 months and 17 months, respectively.
There are no clear guidelines for the treatment of primary CNS PTLD. Options include the use of ISR, rituximab, high-dose methotrexate (HD-MTX), high-dose cytarabine, and whole brain radiation [63]. The role of rituximab is poorly defined in this setting, given its poor penetration across the blood brain barrier [71]. Response to first-line therapy is the most important prognostic factor [85, 87]. In a review of 289 patients with primary CNS PTLD after SOT, it was reported that 32 of the 39 cases of CR occurred in patients who received RT [84]. Overall, however, the outcomes were poor, with <20 % of patients achieving long-term remission. In general, treatment with ISR alone is rarely effective for primary CNS PTLD. For now, treatment should be approached in a manner akin to the primary CNS lymphoma, including whole brain RT, HD-MTX, and high-dose cytarabine, bearing in mind that HD-MTX is likely to have increased toxicity in renal transplant patients [71, 87, 88].
8.7 Burkitt PTLD
Burkitt PTLD accounts for less than 5 % of PTLD cases [63]. It is a highly aggressive malignancy with frequent extra-nodal manifestations, high proliferative index, high latent EBV infection, and rapid tumor growth. Translocations involving c-myc and the heavy or light chain immunoglobulin loci underlie the aggressive biology. Burkitt Lymphoma, in the non-transplant population, has been successfully treated with short-duration intensive combination chemotherapy, along with aggressive CNS prophylaxis [89].
Unfortunately, there are no consensus guidelines or treatment protocols for the treatment of Burkitt PTLD. Due to the aggressive nature of the disease, ISR alone or ISR + rituximab are not recommended. Instead, most clinicians proceed directly to a combined approach involving ISR, rituximab, and chemotherapy. In a small study, CR was achieved in 5/5 patients prospectively treated with ISR and R-CHOP [90]. The role of intrathecal chemoprophylaxis is unclear in Burkitt PTLD. However, extrapolating from the non-transplant setting, we recommend CNS prophylaxis in cases of Burkitt PTLD as well. Highly aggressive regimens typically used for BL in the non-transplant setting should be used with extreme caution in PTLD patients due to the increased risk for toxicity [91].
8.8 Hodgkin’s and Hodgkin-like PTLD
Hodgkin PTLD is the rarest form of PTLD; as a result, treatment data are limited to a few case reports and series [92, 93]. The approach to treatment for Hodgkin PTLD is similar to that employed in Hodgkin’s lymphoma (HL). This typically involves chemotherapy or combined modality treatment using chemotherapy and RT [94]. The most commonly used chemotherapy regimen for HL in the USA is ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). In one study of 7 patients with Hodgkin PTLD treated with standard HL therapy along with ISR, 4 patients achieved CR [93]. In another series, 4 out of 4 patients treated with standard regimens achieved CR [92].
8.9 Anti-viral Therapy
Anti-viral agents, such as acyclovir and ganciclovir, have been studied as either prophylaxis or treatment for PTLD. These agents require activation by EBV thymidine kinase (EBV-TK), an enzyme expressed in infected replicating cells, and are therefore ineffective against latent EBV-infected B cells [95, 96]. Arginine butyrate can be used to induce EBV-TK in infected latent B cells, with subsequent administration of ganciclovir to eliminate these cells. Results utilizing arginine butyrate and ganciclovir are encouraging [95], but further trials are required to prove clear clinical benefit. Prophylactic use of these agents has had mixed results [71]. Outside of clinical trials, anti-viral agents are not currently recommended for routine prophylaxis or treatment of PTLD.
8.10 Adoptive Immunotherapy
Adoptive T-cell therapy with EBV-specific cytotoxic T lymphocytes (CTL) has also been used with success in treating PTLD. Since the pathogenesis of PTLD can be from EBV infection or reactivation due to an impaired CTL response, EBV-specific CTLs are thought to possibly restore this response [97]. However, one study reported favorable response even in EBV-negative PTLD [98].
In SOT patients, PTLD originate from recipient origin (i.e. EBV reactivation), and therefore, EBV-specific CTL from the recipient are required to effectively target infected B cells [8]. In patients unresponsive to ISR, autologous EBV CTL, in combination with rituximab and/or chemotherapy, have shown some therapeutic success [99, 100]. In contrast to SOT, post-allo-HCT PTLD are typically of donor origin and effective immunotherapy against PTLD would require donor CTL for tumor targeting. In one recent study, 49 allo-HCT patients with biopsy-proven EBV-positive PTLD were treated with EBV CTL infusions (many with ISR or prior rituximab) and resulted in a sustained CR rate of 68 %. In a phase II study, 33 patients with both SOT and allo-HCT PTLD who failed conventional therapy were infused with HLA-matched donor EBV-specific CTL, resulting in an ORR of 64 % [97]. Due to a lack of consensus on the use of EBV CTL in PTLD treatment, this approach is currently recommended for use only in the context of clinical trials [71].
8.11 Novel Therapies
Multiple newer therapies are being investigated for the treatment and prevention of PTLD. These include monoclonal antibodies against cytokines and mammalian target of rapamycin (mTOR) inhibitors (such as sirolimus). IL-6 is elevated in most PTLD patients, and monoclonal antibodies targeting IL-6 are under investigation [101]. Other targets with promise include IL-10 and interferon alpha (INF-α) [102–106].
Activation of the mTOR signaling pathway has been implicated in all PTLD subtypes [8], and sirolimus has shown anti-proliferative properties in PTLD [107]. However, confounding this, use of mTOR inhibitors post-transplant has been associated with an increased risk of PTLD when compared to non-mTOR inhibitors [108]. Further study is required to understand the clinical relevance of this pathway and its inhibitors in PTLD.
9 Prevention
Many studies have looked at approaches to identify patients at risk for PTLD so that measures can be taken to prevent its development. These include monitoring for primary EBV infection and monitoring for reactivation of EBV with PCR post-transplant. Though a rising EBV viral load is concerning, it does not consistently predict the development of PTLD [109]. Monitoring EBV viral loads and pre-emptive modulation of immunosuppressive regimens with or without rituximab therapy can decrease the incidence of PTLD and related mortality [110–112]. In a recent prospective study, 299 heart transplant patients were monitored for EBV reactivation (viral loads >105) or primary infection in which case immunosuppression was tapered and response assessed in 1 month. Patients that continued to have high viral loads received one dose of rituximab. In this cohort, only one patient developed PTLD, responsive to ISR, and one patient died due to respiratory complications from PTLD. Mean follow-up time for the entire group of patients was 2.11 years. None of the patients had evidence of transplant rejection by biopsy [110]. Anti-viral therapies, aimed to decrease lytic replication of EBV-infected B cells, have also been investigated to reduce viral loads. Patients receiving acyclovir and ganciclovir have been shown to have decreased risk of PTLD when compared to those without anti-viral therapy in small studies [113–115]. However, this has not been proven in larger studies. Though promising, this approach is only applicable to EBV-positive PTLD, and further study is needed before routine implementation can be recommended.
10 Conclusions
More than 40 years since recognition, PTLD remain a serious complication for patients undergoing allo-HCT and SOT. The understanding of the pathophysiology of PTLD continues to expand. With the identification of multiple risk factors, timely intervention, and more effective treatment, the morbidity and mortality from PTLD have decreased. However, there are no clearly established consensus guidelines for the treatment of PTLD, due in part to the scarcity of large phase 3 prospective trials and heterogeneity of the disease. For patients with minimal disease and good prognostic markers, PTLD may resolve with ISR or ISR+ rituximab. The results of the PTLD-1 trial using sequential therapy are very promising and may establish a standard of care for PTLD treatment.
Continued research on new and novel therapies for PTLD, ideally in the form of multicenter prospective trials, is needed to further improve outcomes for PTLD .
References
Feng S, Buell JF, Chari RS, DiMaio JM, Hanto DW (2003) Tumors and transplantation: the 2003 third annual ASTS state-of-the-art winter symposium. Am J Transpl 3(12):1481–1487
Boubenider S, Hiesse C, Goupy C, Kriaa F, Marchand S, Charpentier B (1997) Incidence and consequences of post-transplantation lymphoproliferative disorders. J Nephrol 10(3):136–145
Doak PB, Montgomerie JZ, North JD, Smith F (1968) Reticulum cell sarcoma after renal homotransplantation and azathioprine and prednisone therapy. Br Med J 4(5633):746–748
Penn I, Hammond W, Brettschneider L, Starzl TE (1969) Malignant lymphomas in transplantation patients. Transpl Proc 1(1):106–112
Leblond V, Sutton L, Dorent R, Davi F, Bitker MO, Gabarre J et al (1995) Lymphoproliferative disorders after organ transplantation: a report of 24 cases observed in a single center. J Clin Oncol 13(4):961–968
Opelz G, Dohler B (2004) Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transpl 4(2):222–230
Newell KA, Alonso EM, Whitington PF, Bruce DS, Millis JM, Piper JB et al (1996) Posttransplant lymphoproliferative disease in pediatric liver transplantation. Interplay between primary epstein-barr virus infection and immunosuppression. Transplantation 62(3):370–375
Taylor AL, Marcus R, Bradley JA (2005) Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit Rev Oncol Hematol 56(1):155–167
Cockfield SM (2001) Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transpl Infect Dis 3(2):70–78
Burns D, Crawford DH (2004) Epstein–Barr virus-specific cytotoxic T-lymphocytes for adoptive immunotherapy of post-transplant lymphoproliferative disease. Blood Rev 18(3):193–209
Parker A, Bowles K, Bradley JA, Emery V, Featherstone C, Gupte G et al (2010) Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant recipients—BCSH and BTS guidelines. Br J Haematol 149(5):675–692
Leblond V, Dhedin N, Mamzer Bruneel MF, Choquet S, Hermine O, Porcher R et al (2001) Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol 19(3):772–778
Ghobrial IM, Habermann TM, Maurer MJ, Geyer SM, Ristow KM, Larson TS et al (2005) Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol 23(30):7574–7582
Sundin M, Le Blanc K, Ringden O, Barkholt L, Omazic B, Lergin C et al (2006) The role of HLA mismatch, splenectomy and recipient epstein-barr virus seronegativity as risk factors in post-transplant lymphoproliferative disorder following allogeneic hematopoietic stem cell transplantation. Haematologica 91(8):1059–1067
Landgren O, Gilbert ES, Rizzo JD, Socie G, Banks PM, Sobocinski KA et al (2009) Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 113(20):4992–5001
Juvonen E, Aalto SM, Tarkkanen J, Volin L, Mattila PS, Knuutila S et al (2003) High incidence of PTLD after non-T-cell-depleted allogeneic haematopoietic stem cell transplantation as a consequence of intensive immunosuppressive treatment. Bone Marrow Transpl 32(1):97–102
Walker RC, Marshall WF, Strickler JG, Wiesner RH, Velosa JA, Habermann TM et al (1995) Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis 20(5):1346–1353
Ho M, Miller G, Atchison RW, Breinig MK, Dummer JS, Andiman W et al (1985) Epstein-barr virus infections and DNA hybridization studies in posttransplantation lymphoma and lymphoproliferative lesions: the role of primary infection. J Infect Dis 152(5):876–886
Walker RC, Paya CV, Marshall WF, Strickler JG, Wiesner RH, Velosa JA et al (1995) Pretransplantation seronegative epstein-barr virus status is the primary risk factor for posttransplantation lymphoproliferative disorder in adult heart, lung, and other solid organ transplantations. J Heart Lung Transpl 14(2):214–221
Dharnidharka VR, Sullivan EK, Stablein DM, Tejani AH, Harmon WE (2001) North American pediatric renal transplant cooperative study (NAPRTCS). Risk factors for posttransplant lymphoproliferative disorder (PTLD) in pediatric kidney transplantation: a report of the North American pediatric renal transplant cooperative study (NAPRTCS). Transplantation 71(8):1065–1068
Younes BS, McDiarmid SV, Martin MG, Vargas JH, Goss JA, Busuttil RW et al (2000) The effect of immunosuppression on posttransplant lymphoproliferative disease in pediatric liver transplant patients. Transplantation 70(1):94–99
Cockfield SM, Preiksaitis JK, Jewell LD, Parfrey NA (1993) Post-transplant lymphoproliferative disorder in renal allograft recipients. Clinical experience and risk factor analysis in a single center. Transplantation 56(1):88–96
Swinnen LJ, Costanzo-Nordin MR, Fisher SG, O’Sullivan EJ, Johnson MR, Heroux AL et al (1990) Increased incidence of lymphoproliferative disorder after immunosuppression with the monoclonal antibody OKT3 in cardiac-transplant recipients. N Engl J Med 323(25):1723–1728
Ellis D, Jaffe R, Green M, Janosky JJ, Lombardozzi-Lane S, Shapiro R et al (1999) Epstein-barr virus-related disorders in children undergoing renal transplantation with tacrolimus-based immunosuppression. Transplantation 68(7):997–1003
Swerdlow AJ, Higgins CD, Hunt BJ, Thomas JA, Burke MM, Crawford DH et al (2000) Risk of lymphoid neoplasia after cardiothoracic transplantation. A cohort study of the relation to epstein-barr virus. Transplantation 69(5):897–904
Gao S, Chaparro SV, Perlroth M, Montoya JG, Miller JL, DiMiceli S et al (2003) Post-transplantation lymphoproliferative disease in heart and heart–lung transplant recipients: 30-year experience at Stanford university. J Heart Lung Transpl 22(5):505–514
Libertiny G, Watson CJE, Gray DWR, Welsh KI, Morris PJ (2001) Rising incidence of post-transplant lymphoproliferative disease in kidney transplant recipients. Br J Surg 88(10):1330–1334
Buda A, Caforio A, Calabrese F, Fagiuoli S, Pevere S, Livi U et al (2000) Lymphoproliferative disorders in heart transplant recipients: role of hepatitis C virus (HCV) and epstein-barr virus (EBV) infection. Transpl Int 13(1):S402–S405
Manez R, Breinig MC, Linden P, Wilson J, Torre-Cisneros J, Kusne S et al (1997) Posttransplant lymphoproliferative disease in primary epstein-barr virus infection after liver transplantation: the role of cytomegalovirus disease. J Infect Dis 176(6):1462–1467
Kapelushnik J, Ariad S, Benharroch D, Landau D, Moser A, Delsol G et al (2001) Post renal transplantation human herpesvirus 8-associated lymphoproliferative disorder and kaposi’s sarcoma. Br J Haematol 113(2):425–428
Bhatia S, Ramsay NK, Steinbuch M, Dusenbery KE, Shapiro RS, Weisdorf DJ et al (1996) Malignant neoplasms following bone marrow transplantation. Blood 87(9):3633–3639
Witherspoon RP, Fisher LD, Schoch G, Martin P, Sullivan KM, Sanders J et al (1989) Secondary cancers after bone marrow transplantation for leukemia or aplastic anemia. N Engl J Med 321(12):784–789
Gerritsen EJ, Stam ED, Hermans J, van den Berg H, Haraldsson A, van Tol MJ et al (1996) Risk factors for developing EBV-related B cell lymphoproliferative disorders (BLPD) after non-HLA-identical BMT in children. Bone Marrow Transpl 18(2):377–382
Hale G, Waldmann H (1998) Risks of developing epstein-barr virus-related lymphoproliferative disorders after T-cell-depleted marrow transplants. CAMPATH users. Blood 91(8):3079–3083
Weinstock DM, Ambrossi GG, Brennan C, Kiehn TE, Jakubowski A (2006) Preemptive diagnosis and treatment of epstein-barr virus-associated post transplant lymphoproliferative disorder after hematopoietic stem cell transplant: an approach in development. Bone Marrow Transpl 37(6):539–546
Fox CP, Burns D, Parker AN, Peggs KS, Harvey CM, Natarajan S et al (2013) EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transpl 49:280–286
Reshef R, Luskin MR, Kamoun M, Vardhanabhuti S, Tomaszewski JE, Stadtmauer EA et al (2011) Association of HLA polymorphisms with post-transplant lymphoproliferative disorder in solid-organ transplant recipients. Am J Transpl 11(4):817–825
Bakker NA, van Imhoff GW, Verschuuren EA, van Son WJ, van der Heide JJ, Lems SP et al (2005) HLA antigens and post renal transplant lymphoproliferative disease: HLA-B matching is critical. Transplantation 80(5):595–599
Van Buskirk AM, Malik V, Xia D, Pelletier RP (2001) A gene polymorphism associated with posttransplant lymphoproliferative disorder. Transpl Proc 33(1-2):1834
Lee TC, Savoldo B, Barshes NR, Rooney CM, Heslop HE, Gee AP et al (2006) Use of cytokine polymorphisms and epstein-barr virus viral load to predict development of post-transplant lymphoproliferative disorder in paediatric liver transplant recipients. Clin Transpl 20(3):389–393
Babel N, Vergopoulos A, Trappe RU, Oertel S, Hammer MH, Karaivanov S et al (2007) Evidence for genetic susceptibility towards development of posttransplant lymphoproliferative disorder in solid organ recipients. Transplantation 84(3):387–391
Thomas RV, McAulay K, Higgins C, Wilkie G, Crawford DH (2005) Interferon gamma (IFN-gamma) polymorphism in posttransplantation lymphoproliferative disease. Blood. 106(4):1502, 3; author reply 1503
McAulay KA, Haque T, Crawford DH (2009) Tumour necrosis factor gene polymorphism: a predictive factor for the development of post-transplant lymphoproliferative disease. Br J Cancer 101(6):1019–1027
Nourse JP, Jones K, Gandhi MK (2011) Epstein-barr virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transpl 11(5):888–895
Lim WH, Russ GR, Coates PT (2006) Review of epstein-barr virus and post-transplant lymphoproliferative disorder post-solid organ transplantation. Nephrology (Carlton) 11(4):355–366
Dotti G, Fiocchi R, Motta T, Gamba A, Gotti E, Gridelli B et al (2000) Epstein-barr virus-negative lymphoproliferate disorders in long-term survivors after heart, kidney, and liver transplant. Transplantation 69(5):827–833
Leblond V, Davi F, Charlotte F, Dorent R, Bitker MO, Sutton L et al (1998) Posttransplant lymphoproliferative disorders not associated with epstein-barr virus: a distinct entity? J Clin Oncol 16(6):2052–2059
Nelson BP, Nalesnik MA, Bahler DW, Locker J, Fung JJ, Swerdlow SH (2000) Epstein-barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity? Am J Surg Pathol 24(3):375–385
Duvoux C, Pageaux GP, Vanlemmens C, Roudot-Thoraval F, Vincens-Rolland AL, Hezode C et al (2002) Risk factors for lymphoproliferative disorders after liver transplantation in adults: an analysis of 480 patients. Transplantation 74(8):1103–1109
Orjuela M, Gross TG, Cheung YK, Alobeid B, Morris E, Cairo MS (2003) A pilot study of chemoimmunotherapy (cyclophosphamide, prednisone, and rituximab) in patients with post-transplant lymphoproliferative disorder following solid organ transplantation. Clin Cancer Res 9(10 Pt 2):3945S–3952S
Bates WD, Gray DW, Dada MA, Chetty R, Gatter KC, Davies DR et al (2003) Lymphoproliferative disorders in oxford renal transplant recipients. J Clin Pathol 56(6):439–446
Caillard S, Lelong C, Pessione F, Moulin B, French PTLD (2006) Working group. Post-transplant lymphoproliferative disorders occurring after renal transplantation in adults: report of 230 cases from the French registry. Am J Transpl 6(11):2735–2742
Oton AB, Wang H, Leleu X, Melhem MF, George D, Lacasce A et al (2008) Clinical and pathological prognostic markers for survival in adult patients with post-transplant lymphoproliferative disorders in solid transplant. Leuk Lymphoma 49(9):1738–1744
Subklewe M, Marquis R, Choquet S, Leblond V, Garnier JL, Hetzer R et al (2006) Association of human leukocyte antigen haplotypes with posttransplant lymphoproliferative disease after solid organ transplantation. Transplantation 82(8):1093–1100
Swerdlow S, Campo E, Harris NL, Jaffe E, Pileri S, Stein H (2008) WHO classification of tumours of haematopoietic and lymphoid tissues (international agency for research on cancer, lyon, France)
Jagadeesh D, Woda BA, Draper J, Evens AM (2012) Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 13(1):122–136
Dockrell DH, Strickler JG, Paya CV (1998) Epstein-barr virus-induced T cell lymphoma in solid organ transplant recipients. Clin Infect Dis 26(1):180–182
Kwong YL, Lam CC, Chan TM (2000) Post-transplantation lymphoproliferative disease of natural killer cell lineage: a clinicopathological and molecular analysis. Br J Haematol 110(1):197–202
Rajakariar R, Bhattacharyya M, Norton A, Sheaff M, Cavenagh J, Raftery MJ et al (2004) Post transplant T-cell lymphoma: a case series of four patients from a single unit and review of the literature. Am J Transpl 4(9):1534–1538
Caillard S, Porcher R, Provot F, Dantal J, Choquet S, Durrbach A et al (2013) Post-transplantation lymphoproliferative disorder after kidney transplantation: report of a nationwide French registry and the development of a new prognostic score. J Clin Oncol 31(10):1302–1309
Evens AM, David KA, Helenowski I, Nelson B, Kaufman D, Kircher SM et al (2010) Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol 28(6):1038–1046
Al-Mansour Z, Nelson BP, Evens AM (2013) Post-transplant lymphoproliferative disease (PTLD): risk factors, diagnosis, and current treatment strategies. Curr Hematol Malig Rep 8(3):173–183
Zimmermann H, Trappe RU (2013) EBV and posttransplantation lymphoproliferative disease: what to do? Hematol Am Soc Hematol Educ Program 2013:95–102
Knight JS, Tsodikov A, Cibrik DM, Ross CW, Kaminski MS, Blayney DW (2009) Lymphoma after solid organ transplantation: risk, response to therapy, and survival at a transplantation center. J Clin Oncol 27(20):3354–3362
Tsai DE, Hardy CL, Tomaszewski JE, Kotloff RM, Oltoff KM, Somer BG et al (2001) Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: analysis of prognostic variables and long-term follow-up of 42 adult patients. Transplantation 71(8):1076–1088
Reshef R, Vardhanabhuti S, Luskin MR, Heitjan DF, Hadjiliadis D, Goral S et al (2011) Reduction of immunosuppression as initial therapy for posttransplantation lymphoproliferative disorder (bigstar). Am J Transpl 11(2):336–347
Swinnen LJ, Mullen GM, Carr TJ, Costanzo MR, Fisher RI (1995) Aggressive treatment for postcardiac transplant lymphoproliferation. Blood 86(9):3333–3340
Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K (2005) Posttransplant lymphoproliferative disorders after renal transplantation in the united states in era of modern immunosuppression. Transplantation 80(9):1233–1243
Cesarman E, Chadburn A, Liu YF, Migliazza A, Dalla-Favera R, Knowles DM (1998) BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood 92(7):2294–2302
Penn I (1998) The role of immunosuppression in lymphoma formation. Springer Semin Immunopathol 20(3–4):343–355
Parker A, Bowles K, Bradley JA, Emery V, Featherstone C, Gupte G et al (2010) Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients—BCSH and BTS guidelines. Br J Haematol 149(5):693–705
Ghobrial I, Habermann T, Ristow K, Ansell S, Macon W, Geyer S et al (2005) Prognostic factors in patients with post-transplant lymphoproliferative disorders (PTLD) in the rituximab era. Leuk Lymphoma 46(2):191–196
Jain AB, Marcos A, Pokharna R, Shapiro R, Fontes PA, Marsh W et al (2005) Rituximab (chimeric anti-CD20 antibody) for posttransplant lymphoproliferative disorder after solid organ transplantation in adults: long-term experience from a single center. Transplantation 80(12):1692–1698
Choquet S, Leblond V, Herbrecht R, Socie G, Stoppa AM, Vandenberghe P et al (2006) Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood 107(8):3053–3057
Maecker B, Jack T, Zimmermann M, Abdul-Khaliq H, Burdelski M, Fuchs A et al (2007) CNS or bone marrow involvement as risk factors for poor survival in post-transplantation lymphoproliferative disorders in children after solid organ transplantation. J Clin Oncol 25(31):4902–4908
Fohrer C, Caillard S, Koumarianou A, Ellero B, Woehl-Jaegle ML, Meyer C et al (2006) Long-term survival in post-transplant lymphoproliferative disorders with a dose-adjusted ACVBP regimen. Br J Haematol 134(6):602–612
Mamzer-Bruneel MF, Lome C, Morelon E, Levy V, Bourquelot P, Jacobs F et al (2000) Durable remission after aggressive chemotherapy for very late post-kidney transplant lymphoproliferation: a report of 16 cases observed in a single center. J Clin Oncol 18(21):3622–3632
Giraldi E, Provenzi M, Fiocchi R, Colledan M, Cornelli P, Torre G et al (2011) Fludarabine, cyclophosphamide, doxorubicin (FCD), and rituximab: a remission induction therapy for aggressive pediatric post-transplant lymphoproliferative disease (PTLD). Pediatr Blood Cancer 57(2):324–328
Elstrom RL, Andreadis C, Aqui NA, Ahya VN, Bloom RD, Brozena SC et al (2006) Treatment of PTLD with rituximab or chemotherapy. Am J Transpl 6(3):569–576
Shah NM, Fenske TS (2013) Post-transplant lymphoproliferative disorders. Hematology 8(Part 2):1
Trappe R, Oertel S, Leblond V, Mollee P, Sender M, Reinke P et al (2012) Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol 13(2):196–206
Trappe RU, Dierickx D, Reinke P, Neuhaus R, Morschhauser F, Zaucha JM et al (2012) Interim analysis of the largest prospective trial to date in adult CD20-positive post-transplant lymphoproliferative disorder (PTLD): introducing risk-stratified sequential treatment (RSST). J Clin Oncol 30 (suppl; abstract 8030)
Dotti G, Fiocchi R, Motta T, Mammana C, Gotti E, Riva S et al (2002) Lymphomas occurring late after solid-organ transplantation: influence of treatment on the clinical outcome. Transplantation 74(8):1095–1102
Penn I, Porat G (1995) Central nervous system lymphomas in organ allograft recipients. Transplantation 59(2):240–244
Buell JF, Gross TG, Hanaway MJ, Trofe J, Roy-Chaudhury P, First MR et al (2005) Posttransplant lymphoproliferative disorder: significance of central nervous system involvement. Transpl Proc 37(2):954–955
Cavaliere R, Petroni G, Lopes MB, Schiff D (2010) International primary central nervous system lymphoma collaborative group. Primary central nervous system post-transplantation lymphoproliferative disorder: an international primary central nervous system lymphoma collaborative group report. Cancer 116(4):863–870
Evens AM, Choquet S, Kroll-Desrosiers AR, Jagadeesh D, Smith SM, Morschhauser F et al (2013) Primary CNS posttransplant lymphoproliferative disease (PTLD): an international report of 84 cases in the modern era. Am J Transpl 13(6):1512–1522
Taj MM, Messahel B, Mycroft J, Pritchard-Jones K, Baker A, Height S et al (2008) Efficacy and tolerability of high-dose methotrexate in central nervous system positive or relapsed lymphoproliferative disease following liver transplant in children. Br J Haematol 140(2):191–196
Blum KA, Lozanski G, Byrd JC (2004) Adult burkitt leukemia and lymphoma. Blood 104(10):3009–3020
Zimmermann H, Reinke P, Neuhaus R, Lehmkuhl H, Oertel S, Atta J et al (2012) Burkitt post-transplantation lymphoma in adult solid organ transplant recipients: sequential immunochemotherapy with rituximab (R) followed by cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or R-CHOP is safe and effective in an analysis of 8 patients. Cancer 118(19):4715–4724
Xicoy B, Ribera J, Esteve J, Brunet S, Sanz M, Fernández-Abellán P et al (2003) Post-transplant burkitt’s leukemia or lymphoma. Study of five cases treated with specific intensive therapy (PETHEMA ALL-3/97 trial). Leuk Lymphoma 44(9):1541–1543
Bierman PJ, Vose JM, Langnas AN, Rifkin RM, Hauke RJ, Smir BN et al (1996) Hodgkin’s disease following solid organ transplantation. Ann Oncol 7(3):265–270
Garnier JL, Lebranchu Y, Dantal J, Bedrossian J, Cahen R, Assouline D et al (1996) Hodgkin’s disease after transplantation. Transplantation 61(1):71–76
Diehl V, Stein H, Hummel M, Zollinger R, Connors JM (2003) Hodgkin’s lymphoma: biology and treatment strategies for primary, refractory, and relapsed disease. Hematology Am Soc Hematol Educ Program 2003(1):225–247
Perrine SP, Hermine O, Small T, Suarez F, O’Reilly R, Boulad F et al (2007) A phase 1/2 trial of arginine butyrate and ganciclovir in patients with epstein-barr virus-associated lymphoid malignancies. Blood 109(6):2571–2578
Mentzer SJ, Perrine SP, Faller DV (2001) Epstein–barr virus post-transplant lymphoproliferative disease and virus-specific therapy: pharmacological re-activation of viral target genes with arginine butyrate. Transpl Infect Dis 3(3):177–185
Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P et al (2007) Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood 110(4):1123–1131
Nalesnik MA, Rao AS, Furukawa H, Pham S, Zeevi A, Fung JJ et al (1997) Autologous lymphokine-activated killer cell therapy of epstein-barr virus-positive and -negative lymphoproliferative disorders arising in organ transplant recipients. Transplantation 63(9):1200–1205
Comoli P, Labirio M, Basso S, Baldanti F, Grossi P, Furione M et al (2002) Infusion of autologous epstein-barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood 99(7):2592–2598
Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA et al (2010) Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 115(5):925–935
Haddad E, Paczesny S, Leblond V, Seigneurin JM, Stern M, Achkar A et al (2001) Treatment of B-lymphoproliferative disorder with a monoclonal anti-interleukin-6 antibody in 12 patients: a multicenter phase 1–2 clinical trial. Blood 97(6):1590–1597
Davis CL, Wood BL, Sabath DE, Joseph JS, Stehman-Breen C, Broudy VC (1998) Interferon-alpha treatment of posttransplant lymphoproliferative disorder in recipients of solid organ transplants. Transplantation 66(12):1770–1779
Komrokji RS, Oliva JL, Zand M, Felgar R, Abboud CN (2005) Mini-BEAM and autologous hematopoietic stem-cell transplant for treatment of post-transplant lymphoproliferative disorders. Am J Hematol 79(3):211–215
Williams KM, Higman MA, Chen AR, Schwartz CL, Wharam M, Colombani P et al (2008) Successful treatment of a child with late-onset T-cell post-transplant lymphoproliferative disorder/lymphoma. Pediatr Blood Cancer 50(3):667–670
Bobey NA, Stewart DA, Woodman RC (2002) Successful treatment of posttransplant lymphoproliferative disorder in a renal transplant patient by autologous peripheral blood stem cell transplantation. Leuk Lymphoma 43(12):2421–2423
Mahe B, Moreau P, Le Tortorec S, Hourmant M, Harousseau JL, Milpied N (1994) Autologous bone marrow transplantation for cyclosporin-related lymphoma in a renal transplant patient. Bone Marrow Transpl 14(4):645–646
Majewski M, Korecka M, Kossev P, Li S, Goldman J, Moore J et al (2000) The immunosuppressive macrolide RAD inhibits growth of human epstein-barr virus-transformed B lymphocytes in vitro and in vivo: a potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc Natl Acad Sci USA 97(8):4285–4290
Kirk AD, Cherikh WS, Ring M, Burke G, Kaufman D, Knechtle SJ et al (2007) Dissociation of depletional induction and posttransplant lymphoproliferative disease in kidney recipients treated with alemtuzumab. Am J Transpl 7(11):2619–2625
Green M, Soltys K, Rowe DT, Webber SA, Mazareigos G (2009) Chronic high epstein-barr viral load carriage in pediatric liver transplant recipients. Pediatr Transpl 13(3):319–323
Choquet S, Varnous S, Deback C, Golmard JL, Leblond V (2014) Adapted treatment of epstein-barr virus infection to prevent posttransplant lymphoproliferative disorder after heart transplantation. Am J Transpl 14(4):857–866
Lee TC, Savoldo B, Rooney CM, Heslop HE, Gee AP, Caldwell Y et al (2005) Quantitative EBV viral loads and immunosuppression alterations can decrease PTLD incidence in pediatric liver transplant recipients. Am J Transpl 5(9):2222–2228
Jang JY, Kim KM, Lee YJ, Lee SG, Chi HS (2008) Quantitative epstein-barr virus viral load monitoring in pediatric liver transplantation. Transpl Proc 40(8):2546–2548
Darenkov IA, Marcarelli MA, Basadonna GP, Friedman AL, Lorber KM, Howe JG et al (1997) Reduced incidence of epstein-barr virus-associated posttransplant lymphoproliferative disorder using preemptive antiviral therapy. Transplantation 64(6):848–852
Davis CL, Harrison KL, McVicar JP, Forg PJ, Bronner MP, Marsh CL (1995) Antiviral prophylaxis and the epstein barr virus-related post-transplant lymphoproliferative disorder. Clin Transpl 9(1):53–59
Keay S, Oldach D, Wiland A, Klassen D, Schweitzer E, Abruzzo LV et al (1998) Posttransplantation lymphoproliferative disorder associated with OKT3 and decreased antiviral prophylaxis in pancreas transplant recipients. Clin Infect Dis 26(3):596–600
Oertel SH, Verschuuren E, Reinke P, Zeidler K, Papp-Vary M, Babel N et al (2005) Effect of anti-CD 20 antibody rituximab in patients with post-transplant lymphoproliferative disorder (PTLD). Am J Transpl 5(12):2901–2906
Blaes AH, Peterson BA, Bartlett N, Dunn DL, Morrison VA (2005) Rituximab therapy is effective for posttransplant lymphoproliferative disorders after solid organ transplantation: results of a phase II trial. Cancer 104(8):1661–1667
Gonzalez-Barca E, Domingo-Domenech E, Capote FJ, Gomez-Codina J, Salar A, Bailen A et al (2007) Prospective phase II trial of extended treatment with rituximab in patients with B-cell post-transplant lymphoproliferative disease. Haematologica 92(11):1489–1494
Swinnen LJ, LeBlanc M, Grogan TM, Gordon LI, Stiff PJ, Miller AM et al (2008) Prospective study of sequential reduction in immunosuppression, interferon alpha-2B, and chemotherapy for posttransplantation lymphoproliferative disorder. Transplantation 86(2):215–222
Gross TG, Orjuela MA, Perkins SL, Park JR, Lynch JC, Cairo MS et al (2012) Low-dose chemotherapy and rituximab for posttransplant lymphoproliferative disease (PTLD): a children’s oncology group report. Am J Transpl 12(11):3069–3075
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Singavi, A.K., Harrington, A.M., Fenske, T.S. (2015). Post-transplant Lymphoproliferative Disorders. In: Evens, A., Blum, K. (eds) Non-Hodgkin Lymphoma. Cancer Treatment and Research, vol 165. Springer, Cham. https://doi.org/10.1007/978-3-319-13150-4_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-13150-4_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13149-8
Online ISBN: 978-3-319-13150-4
eBook Packages: MedicineMedicine (R0)