
Abstract
Peripheral T-cell lymphomas (PTCLs) are an uncommon group of lymphoproliferative disorders accounting for approximately 10–15 % of all non-Hodgkin lymphomas (NHL) in Western countries. Although PTCLs are associated with poor prognosis, outcomes vary with disease subtype. The standard of care has been anthracycline-based induction combination chemotherapy, however, with the exception of low-risk ALK-positive anaplastic large cell lymphoma, relapse rates are high. Therefore, consolidation with autologous stem cell transplantation is usually recommended for patients deemed candidates, and with aggressive subtypes. In recent years, a number of novel agents including pralatrexate, histone deacetylase inhibitors, immunotoxins, proteasome inhibitors, aurora kinase inhibitors and the CD30 antibody-drug conjugate brentuximab vedotin, have shown promise in the treatment of PTCLs. Studies are underway to explore the activity of these newer agents used in the frontline setting.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
T-cell lymphomas may be broadly divided into those arising from precursor T cells and those arising from mature T cells, which are referred to as peripheral T-cell lymphomas or PTCLs [1]. PTCLs are a heterogenous group of lymphoproliferative disorders characterized by clonal expansion of a population of post-thymic T cells and NK cells [2], and in general, this group is marked by a poorer prognosis than its B-cell counterparts. The PTCLs are less common than B-cell non-Hodgkin’s lymphomas (NHL), accounting for approximately 10–15 % of lymphoid malignancies in North America [1]. Consequently, large randomized clinical trials have been difficult, and treatment is often adapted from studies in B-cell neoplasms. However, these lymphomas demonstrate distinctive biology and molecular characteristics and, with the exception of the more indolent cutaneous T-cell lymphomas, are usually aggressive [1]. Prognosis remains poor due to poor response rates to frontline therapy, leading to a dismal overall and failure-free survival [3]. More recently, however, the landscape has begun to change, with a number of important new therapies developed in the last few years and a better understanding of the biology of the various subtypes of PTCL.
2 Definition and Classification
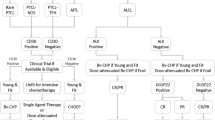
The classification of mature T-cell lymphomas has undergone a number of revisions. With the development of the Revised European-American Lymphoma (REAL) classification, T-cell lymphomas were recognized as a distinct entity for the first time [4]. In 2008, the fourth edition of the World Health Organization (WHO) Classification of Tumors of Hematopoietic and Lymphoid Tissues subdivided PTCLs into four categories: nodal, extra-nodal, disseminated/leukemic, and cutaneous [5]. As noted earlier, the last subtype is distinguished, with few exceptions, by a more indolent behavior. Within the nodal subgroup, the most prevalent disease is PTCL not otherwise specified (NOS), accounting for approximately 26 % of cases, followed by angioimmunoblastic lymphoma (AITL), which constitutes 18 % of cases. The anaplastic large cell lymphomas (ALCL) are subdivided by the presence of a mutation in the anaplastic lymphoma kinase (ALK); ALK-positive (ALK+) lymphomas account for 6.6 % of cases, while ALK− cases represent 5.5 % [3].
The extra-nodal subgroup of PTCLs comprises different entities usually named after the tissue in which they manifest, e.g., hepatosplenic T-cell lymphoma. The leukemic subtypes include adult T-cell leukemia/lymphoma (ATLL, HTLV-1 associated), chronic large granular lymphocytic leukemia (LGL), prolymphocytic T-cell leukemia, and aggressive NK cell leukemia (Table 1).
Epidemiologically, the PTCLs are geographically more common in Asia than in North America, which is due to both a true increase in PTCLs, including the HTLV-driven adult T-cell leukemia/lymphoma, and a relative decrease in the B-cell NHLs [6]. The International T-cell and natural killer/T-cell lymphoma study [7] provided further insight into the epidemiology of this group of diseases. While PTCL-NOS predominates in the United States and Europe (accounting for 34 % of cases), ATLL and NK/T-cell lymphomas are the most common types in Asia (25 and 22 % of cases, respectively). Other geographic trends include a higher prevalence of ALK+ALCL in the United States (16 % of cases) and high rates of AITL in Europe (29 % of cases).
This review will focus on the most common nodal entities, as the management of less common PTCLs can be quite different.
Prognosis of PTCLs differs by subtype. Based on data from the international T-cell lymphoma project (IPTCL) and the British Columbia Cancer Agency (BCCA), with most patients receiving an upfront anthracycline-containing chemotherapy regimen, the 5-year overall survival (OS) of PTCL-NOS was 32–35 % and the 5-year failure-free survival (FFS) was 20–29 %. AITL has a similar 5-year OS, but FFS was worse at 13–18 %. ALCL fared better, particularly the ALK+ subtype with a 5-year OS of 58–70 % and FFS of 28–60 % [8].
3 Clinical Presentation and Diagnosis
Among patients with PTCL, 38 % present with nodal disease alone, 49 % with both nodal and extra-nodal disease, and 13 % with extra-nodal disease only (commonly skin and gastrointestinal tract). The bone marrow is involved in one-fifth of patients. Approximately half of patients present with stage IV disease, and one-third have B-symptoms. Eosinophilia and pruritus is often seen, and hemophagocytic syndrome may be present [7, 9, 10].
Although pathologists can accurately distinguish B cell from T-cell lymphomas, elucidation of subtypes has been difficult due to molecular heterogeneity. T-cell antigens (CD2, CD3, CD4, CD5, CD7, and CD52) are variably expressed, and clonal T-cell receptor (TCR) gene rearrangements are characteristic but not always seen due to relative genomic stability of the TCR genes [11]. Gene expression profiling (GEP) has helped in the differential diagnosis of some subtypes; for example, AITL has been shown to have follicular T-lymphocyte derivation, and PTCL-NOS seems to be derived from activated rather than resting T cells [12, 13].
4 Management of Peripheral T-Cell Lymphomas
4.1 Frontline Therapy of Aggressive PTCL
Despite advances in the understanding of the biology of PTCLs, the standard first-line or induction chemotherapy regimens for these disorders have been derived from the treatment of B-cell neoplasms. Cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), mainstay backbone chemotherapy for aggressive B-cell neoplasms, is the regimen typically used.
In a single-institution study with 208 PTCL patients who received frontline CHOP, complete remission (CR) was obtained in 57 %, 5-year OS was 28.5 % (22.3–36.3), and 5-year event-free survival (EFS) was 18.4 % (13.4–25.3) [14]. In general, outcomes with CHOP are inferior in PTCL compared to aggressive B-cell lymphomas [15], with the exception of ALK + ALCL. In a study by Akagi et al. [16], CR rate was 39 % in PTCL versus 67 % in DLBCL (P < 0.008), and 3-year OS was 26 % versus 50 %, respectively (P = 0.005). However, patients with ALK + ALCL patients have acceptable overall survival rates (58–70 %) with a frontline anthracycline-based regimen like CHOP, compared to ALK-ALCL (49 %), AITL or, PTCL-NOS (32–35 %) [17]. An attempt to improve these outcomes was made by adding etoposide to CHOP, with encouraging results. In an important analysis of a series of studies by the German high-grade non-Hodgkin’s Lymphoma study group, a total of 343 patients were analyzed with 289 belonging to one of the four major subtypes of PTCL [18], (ALCL ALK+ = 78, ALCL ALK− = 113, PTCL-NOS = 70, and AITL = 28). These patients were given either conventional CHOP or CHOP plus etoposide (CHOEP). Three-year event-free survival (EFS) and OS were 75.8 and 89.8 % (ALK + ALCL), 45.7 and 62.1 % (ALK-ALCL), 50.0 and 67.5 % (AITL), and 41.1 and 53.9 % (PTCL-NOS), respectively, between the two groups. Of note, the ALK + ALCL patients did particularly well with CHOEP, with a significant improvement in both EFS and OS (P <0.001 for both endpoints). Patients with AITL did better than those with ALK- ALCL, but the difference was not statistically significant. Across both treatment groups, patients with international prognostic index (IPI) >1 did worse, and those with ALK + ALCL fared better. In younger patients, with normal LDH, CHOEP significantly improved 3-year EFS (75.4 % vs. 51.0 %); however, OS was not significantly affected (P = 0.176). When ALK + ALCL was excluded, the difference in EFS between CHOP and CHOEP was not significant in younger patients but showed a trend toward improvement. Older patients (>60 years of age) did worse with the addition of etoposide. More intensive regimens have been used in an attempt to improve the response rates and survival over CHOP/CHOEP, without much success. In a single-institution study from the MD Anderson Cancer Center, 135 patients with PTCL other than mycosis fungoides were treated with either CHOP (37 %) or more intensive regimens, such as hyperfractionated cyclophosphamide, vincristine, doxorubicin and dexamethasone (HyperCVAD) or hyperCVAD/ESHAP (etoposide, methylprednisolone, cytarabine, and cisplatin) [19]. The 3-year OS was 62 % for CHOP versus 56 % for more intensive regimens. After excluding the ALK+ ALCL patients who traditionally do better, the 3-year OS was 43 % for CHOP versus 49 % for more intensive therapies. Another prospective trial comparing CHOP/ESHAP to CHOP alone with a plan for autologous transplant in first CR did not show any advantage for the more intensive regimen [20]. Etoposide, ifosfamide, cisplatin alternating with doxorubicin, bleomycin, vinblastine, and dacarbazine (VIP-reinforced ABVD; VIP-rABVD) were compared to the CHOP/21 regimen in 88 patients with PTCL by The Groupe Ouest Est d’Etude des Leucemies et Autres Maladies du Sang (GOELAMS). Two-year EFS in the two groups was nearly identical (41 % vs. 45 %) with a median overall survival of 42 months for each arm [21]. Finally, gemcitabine-based regimens have been evaluated for PTCL and have shown good response rates but no improvement in OS when compared to CHOP/CHOEP; they also appear to be associated with more toxicity [22–24].
CD52 is expressed in up to 42 % of patients with PTCL, and attempts have been made to exploit this possible therapeutic target by adding alemtuzumab, an anti-CD52 monoclonal antibody, to CHOP (CHOP-C) [25]. In a phase I/II trial, 24 patients received 8 cycles of CHOP with alemtuzumab. The CR rate was 71 %, and at a median follow-up of 16 months, median duration of response (DOR) was 11 months [26]. Serious infectious complications were observed, including febrile neutropenia, CMV and JC virus reactivation, pulmonary aspergillosis, and staphylococcal sepsis. In a larger phase II trial (41 patients), patients received CHOP or CHOEP followed by alemtuzumab consolidation (29 patients) if a clinical response was attained following CHO(E)P. CR rate was 58.5 %; EFS and OS at 3 years in the whole intent-to-treat population were 32.3 and 62.5 %, respectively, and 42.4 and 75.1 % in the patients who received alemtuzumab [27]. Two randomized phase III trials are underway (ACT-1 and ACT-2) comparing CHOP to CHOP-C.
Bortezomib, a proteasome inhibitor, has activity in relapsed PTCL. Early, small phases I and II studies with PTCL found an ORR of 62–67 % [28]. A large multicenter phase II trial with 46 patients evaluated bortezomib plus CHOP in the frontline setting; although ORR was 76 % (CR = 65 %), 3-year OS was 47 %, and PFS was 35 %, thus similar to the results achieved with CHOP alone [29].
Other studies are underway, attempting to improve response rates combining other novel agents to CHOP, or using maintenance strategies.
5 The Role of Transplantation in Frontline Therapy
5.1 Autologous Stem Cell Transplant
High-dose chemotherapy with autologous stem cell rescue (HDT/ASCR) has been investigated as consolidative therapy in PTCL given the poor outcomes with available frontline chemotherapy, particularly in non-ALK + ALCL subtypes. However, there are no prospective randomized trials comparing chemotherapy alone with chemotherapy followed by HDT/ASCR. A meta-analysis of 21 phase I/II trials showed a trend toward survival advantage for HDT/ASCR in comparison with historical controls (HR 0.81, 95 % CI 0.31–2.13). The presence of CR at transplant and good IPI scores significantly affected survival [30]. Various other retrospective and non-randomized prospective studies report PFS and OS in the range of 35–58 % and 44–65 % with HDT/ASCR following induction chemotherapy. Again, CR at transplant, favorable-risk IPI scores, and ALK + ALCL subtype were good prognostic factors [20, 31–38].
In a phase II study by the Nordic Lymphoma Group [39], 160 patients with biopsy proven PTCL (treatment-naïve) underwent induction with 6 cycles of bi-weekly CHOEP (etoposide omitted for age >60 years) followed by HDT/ASCR in responders. Most patients presented with advanced stage disease, and 72 % had IPI scores of 2 or more. One hundred and fifteen patients underwent ASCT. The 5-year overall and progression-free survival (PFS) were 51 and 44 %, respectively. Patients with ALK-ALCL fared the best, but not much better than historical controls treated with induction chemotherapy alone.
Reimer and colleagues conducted another large prospective study evaluating ASCT in first remission after induction chemotherapy with CHOP. Eighty-three patients were enrolled with 55 going on to consolidation with transplant. The 3-year OS rate was 48 % but was more favorable (71 %) in patients who underwent ASCT [37].
Randomized trials are still needed to establish HDT/ASCR as a standard frontline consolidative approach. It remains an option for patients with significant responses to induction chemotherapy.
5.2 Allogeneic Transplant
In principle, allogeneic transplant for PTCL would utilize a graft-versus-lymphoma effect (as well as infusing a lymphoma-free graft) and might perform better than HDT/ASCR [40]. However, there are no randomized, phase III trials evaluating allogeneic transplant in the frontline setting, and most of the retrospective analyses have been conducted in the relapsed/refractory setting.
A study by Kanakry et al. reviewed the outcomes of 44 consecutive, related-donor allogeneic transplants for PTCL, 24 with reduced-intensity conditioning (RIC) and 20 with myeloablative conditioning (MAC), and included 18 reduced-intensity/haploidentical transplants. The estimated 2-year PFS was 40 % and OS was 43 %, with a tendency toward superior outcomes for those undergoing transplant in first remission (P = 0.08). Of note, approximately 50 % of the patients presented with poor-risk or chemorefractory disease, and 25 % received transplant with active disease. Relapse in RIC and MAC regimens were comparable, with less treatment-related mortality (TRM) with RIC [41]. A recent phase II study evaluated frontline autologous and allogeneic transplants in different age groups with CHOP-alemtuzumab conditioning (n = 86) and found that auto/allotransplants improved DFS in younger patients only [42].
In summary, allogeneic transplant is usually recommended in the relapsed setting but can be an option in first remission in select poor-risk patients.
6 Treatment of Relapsed/Refractory Disease
Traditionally, refractory or relapsed PTCL has been treated with second-line chemotherapy regimens often followed by autologous or allogeneic transplant. However, the recent development of therapies specific to T-cell lymphomas is rapidly changing this paradigm. We will briefly discuss the role of transplant and then discuss chemotherapy and other novel therapies in this setting.
There are no large randomized trials evaluating transplant in relapsed and refractory disease, and our practices are derived mainly from retrospective data. A retrospective study by Le Gouill et al. reported on 77 patients with PTCLs (84 % ALCL, AITL, or PTCL-NOS) treated with allotransplant. PFS and OS were 53 and 57 %, respectively, with a 43 months follow-up following allogeneic transplant [40]. Many of these patients had received at least two prior lines of therapy including autologous transplant (25 %). Of note, AITL patients fared the best (OS rate 80 %) followed by PTCL-NOS and ALCL (63 and 55 %, respectively). TRM was 21 % at 100 days and 34 % at 5 years. A study by Kyriakou et al. [43] found similar PFS/OS for AITL patients, with chemotherapy-sensitive disease faring better. Similar outcomes have been seen in other retrospective analyses; however, early TRM remains a significant problem. It has been difficult to compare autologous and allogeneic transplant in the relapsed/refractory setting, but in general, results appear to be better with allogeneic transplant. However, a recent retrospective study by Smith et al. [44] suggested a possible benefit in favor of autologous transplant. Of 241 patients with PTCL, 115 underwent autologous and 126 allogeneic transplant. Patients receiving autologous versus allogeneic transplant had significantly greater adjusted 3-year OS (59 % vs. 47 %, P = 0.046) and lower NRM or non-relapse mortality (P < 0.001). In the absence of mature data from trials of novel agents, transplant following chemotherapy remains the recommended option for patients considered good candidates.
7 Non-transplant Therapies for Relapsed/Refractory PTCLS
Transplant may not always be an option for PTCL patients with relapsed/refractory disease due to presence of co-morbidities or unavailability of donors, and even when it is possible, it does not have robust outcomes. There is a clear need for the development of therapies directed specifically toward T-cell neoplasms, and the field has advanced significantly in the last few years. Once again, there is a paucity of large randomized phase III studies, so we will review mainly data from phase II studies, as well as ongoing trials.
7.1 Chemotherapy
Among the various chemotherapeutic agents that have been investigated in the relapsed setting, the nucleoside analog gemcitabine has shown significant activity. In a small study, 13 patients with PTCL-NOS and mycosis fungoides (MF) received single agent gemcitabine, and 9 of 13 patients responded [45]. The long-term outcome data for gemcitabine monotherapy in relapsed PTCL (PTCL-NOS and MF) were published in 2010 by the same group [46]. Five out of 20 patients with PTCL-NOS had continuous CR at the time of final follow-up, with a median duration of CR of 34 months (range 15–60 months).
In a small trial, 24 elderly patients received treatment with gemcitabine in combination with oxaliplatin and dexamethasone; the ORR was 25 % and the median OS was 14 months [47].
The other chemotherapeutic agent that has shown promise in the treatment of PTCLs is bendamustine. Bendamustine is a “dual-structure” drug consisting of an alkylating portion, derived from nitrogen mustard, and a purine analog portion. An open-label phase II trial of bendamustine was conducted in patients with relapsed PTCL (primarily AITL and PTCL-NOS). Patients had received a median of one previous line of therapy, thus not heavily pretreated [48]. In the intention to treat (ITT) analysis, the ORR was 50 % (CR 28 %), and the PFS and OS were 3.6 and 6.2 months, respectively. However, only 25 % of patients completed the planned six cycles of bendamustine. Common reasons for early discontinuation included toxicities (cytopenia and infections) and disease progression. Future studies will likely incorporate bendamustine into multidrug regimens.
Forodesine is a potent inhibitor of the enzyme purine nucleoside phosphorylase, which leads to intracellular accumulation of deoxyguanosine triphosphate (dGTP), causing reduced proliferation of lymphocytes and apoptosis [49]. In a phase I–II study, patients received oral forodesine. A maximum tolerated dose (MTD) was not reached, and the optimal biologic dose was determined as being 80 mg/m2 daily. Thirty-six patients received this dose and the ORR was 39 %. Long-term data are awaited.
8 Antibody-Directed Therapy
8.1 Brentuximab Vedotin
Systemic ALCL is characterized by strong CD30 positivity, and the search for an effective monoclonal antibody has been an active area of research in the field of PTCL. Brentuximab vedotin (BV) is an antibody–drug conjugate, comprising an anti-CD30 monoclonal antibody conjugated to the potent antimicrotubule agent monomethyl auristatin E (MMAE). The monoclonal antibody binds to CD30-positive neoplastic T cells and is internalized; MMAE is then cleaved from the molecule exerting its action through inhibition of microtubule formation. Hence, a potent chemotherapeutic agent can be delivered in a targeted manner [50]. A phase II multicenter study by Pro et al. evaluated the activity of BV in 58 patients with systemic ALCL who had relapsed after at least one prior line of therapy. The ORR was 86 % (CR = 57 %, PR = 29 %) [51] with a median overall response duration of 12.6 months, and CR duration of 13.2 months. Grade 3–4 adverse events included neutropenia, thrombocytopenia, and peripheral sensory neuropathy. On the basis of this study, BV was approved by the FDA for use in relapsed systemic ALCL.
A recent phase II multicenter study evaluated the efficacy and safety of brentuximab vedotin in relapsed/refractory PTCL other than ALCL. Of 34 evaluable patients (PTCL-NOS n = 22, AITL n = 13), ORR was 41 % with a median PFS of 6.7 months. Interestingly, there was no correlation between CD30 expression and response [52, 53]. A retrospective analysis of brentuximab vedotin in CD30-positive relapsed lymphomas in older patients showed a 100 % RR in systemic ALCL, with tolerable toxicities [54]. A strategy of combining standard frontline therapy (CHP) with BV was shown to be safe and effective in a phase I trial and is now being tested in an international randomized phase III trial comparing CHOP to CHP-BV as upfront therapy for CD30-positive PTCL. The results are eagerly awaited at present [55].
8.2 Denileukin Diftitox
Denileukin Diftitox (DD) is a unique genetically engineered chimeric protein, which combines IL-2 with certain domains of the diphtheria toxin (membrane translocating and cytotoxic domains). A phase II study in PTCL (excluding cutaneous) demonstrated an ORR of 48 % and median duration of response was 6 months. In CD25-positive (i.e., IL-2-receptor-positive) PTCL, a greater ORR (61.5 %) was observed [56]. As a follow-up to this, DD was evaluated as frontline therapy for PTCL in combination with CHOP in a phase II trial. In the ITT analysis, the overall response rate was 65 % and median progression-free survival was 12 months [57].
8.3 Folate Analogs: Pralatrexate
Pralatrexate is a novel folate analog with improved membrane transport and polyglutamylation within tumor cells [58]. Early phase I/II studies of pralatrexate in patients with relapsed B- or T-cell lymphomas established a significant activity in PTCL, with an ORR of 54 % [59]. In an important prospective phase II trial, 115 PTCL patients (109 evaluable) received pralatrexate 30 mg/m2 weekly for 6 out of 7 weeks. Overall response rate was 29 % [60], and median PFS and OS were 3.5 months and 14.5 months, respectively. The most common grade 3/4 adverse events were thrombocytopenia (32 %), mucositis (22 %), neutropenia (22 %), and anemia (18 %).
Pralatrexate was the first FDA-approved agent for the treatment of patients with relapsed/refractory PTCL.
8.4 Histone Deacetylase Inhibitors
The histone deacetylase (HDAC) inhibition is an important therapeutic strategy based on the principle that increased histone acetylation leads to enhanced tumor-suppressor gene transcription, cell cycle regulation, apoptosis induction, DNA repair, protein acetylation, and induction of autophagy [61]. Romidepsin, a potent selective HDAC-I inhibitor, was evaluated in a single-arm, phase II, international prospective trial which enrolled 131 patients. Patients with relapsed/refractory disease received romidepsin at a dose of 14 mg/m2 [2]. The ORR was 25 %, including 15 % complete response/unconfirmed complete response (CR/CRu). The median duration of response was 17 months, and among patients who achieved CR/CRu, 89 % had not progressed at 13.4 months. Toxicities (cytopenias and infections) were tolerable. Consequently, this drug was FDA approved for relapsed/refractory PTCL [62]. Long-term follow-up data for this study was recently reported [63], and 10 of the 19 patients who achieved CR/CRu had durable responses. Median progression-free survival was 29 months, with significantly longer survival in patients achieved CR/CRu for ≥12 months.
The success of romidepsin led to the exploration of other HDAC inhibitors. Belinostat or PXD 101, a hydroxyamate HDAC I and II inhibitor, was evaluated in a phase I and then phase II multicenter clinical trials. In 19 patients with relapsed/refractory T-cell lymphoma, the ORR was 32 % and median duration of response 268+ days [64]. A larger phase II trial was recently reported, and enrolled 129 patients with relapsed/refractory PTCL following at least one prior therapy [65]. ORR was 26–28 %, with median duration of response 8.3 months. Additionally, belinostat was very well tolerated, suggesting that it can be used in combination with other agents. Other HDAC inhibitors now under investigation include panobinostat and vorinostat.
8.5 Aurora Kinase Inhibitors
Aurora A is a mitotic kinase implicated in oncogenesis and has been found to be upregulated in PTCL, most strongly in ALK+ ALCL, followed by ALK-ALCL and PTCL-NOS [66]. The Aurora A kinase inhibitor alisertib was shown to have preclinical activity against PTCL cell lines [67]. A phase II trial evaluated its efficacy against a variety of B- and T-cell lymphomas. In this trial, patients received alisertib at a dose of 50 mg twice daily for 7 days on 21-day cycles, until PD or unacceptable toxicities. ORR in 48 enrolled patients was 27 and 50 % in the subset of patients with PTCL (n = 8). On the basis of these results, a phase III study of alisertib versus investigator’s choice of treatment is underway for patients with relapsed PTCL [68, 69].
9 Summary
In summary, PTCL represents a unique group of neoplasms characterized by marked molecular heterogeneity and poor response to conventional chemotherapy regimens. The addition of etoposide to CHOP (CHOEP) appears to be an effective up-front therapy in select patients, and alternative regimens to CHOP are currently being explored. Various approaches to consolidation have been studied, including stem cell transplant, chemotherapy, and novel immunotherapies. However, consolidation with autologous transplant is still considered the standard approach with the exception of low-risk ALK+ALCL, even in the absence of randomized phase III trials. Allogeneic transplant is usually reserved for relapsed disease and has variable outcomes. The novel agents romidepsin, pralatrexate, and brentuximab vedotin (for systemic ALCL), are currently FDA approved in the relapsed or refractory setting. The future of treating PTCL will likely involve incorporation of these and other novel agents in frontline regimens, and a number of studies are already exploring newer combinations.
References
Armitage JO (2013) The aggressive peripheral T-cell lymphomas: 2013. Am J Hematol 88:910–918
Savage KJ (2007) Peripheral T-cell lymphomas. Blood Rev 21:201–216
Foss FM et al (2011) Peripheral T-cell lymphoma. Blood 117:6756–6767
Harris NL et al (1994) A revised European-American classification of lymphoid neoplasms: a proposal from the International lymphoma study group. Blood 84:1361–1392
Campo E et al (2011) The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117:5019–5032
Anderson JR, Armitage JO, Weisenburger DD (1998) Epidemiology of the non-Hodgkin’s lymphomas: distributions of the major subtypes differ by geographic locations. non-Hodgkin’s lymphoma classification project. Ann Oncol Off J Eur Soc Med Oncol ESMO 9:717–720
Vose J, Armitage J, Weisenburger D (2008) International T-cell lymphoma project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol Off J Am Soc Clin Oncol 26:4124–4130
Moskowitz AJ, Lunning M, Horwitz SM (2014) How I treat the peripheral T cell lymphomas. Blood 123: 2636. doi:10.1182/blood-2013-12-516245
Weisenburger DD et al (2011) Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the International peripheral T-cell Lymphoma project. Blood 117:3402–3408
Falini B et al (1990) Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood 75:434–444
Weiss LM, Picker LJ, Grogan TM, Warnke RA, Sklar J (1988) Absence of clonal beta and gamma T-cell receptor gene rearrangements in a subset of peripheral T-cell lymphomas. Am J Pathol 130:436–442
Iqbal J et al (2010) Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 115:1026–1036
Agostinelli C et al (2008) Peripheral T cell lymphoma, not otherwise specified: the stuff of genes, dreams and therapies. J Clin Pathol 61:1160–1167
Broussais-Guillaumot F et al (2013) Peripheral T-cell lymphomas: analysis of histology, staging and response to treatment of 208 cases at a single institution. Leuk Lymphoma 54:2392–2398
Coiffier B et al (2002) CHOP chemotherapy plus Rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346:235–242
Akagi T et al (2011) Comparison of long-term clinical outcomes of CHOP chemotherapy between Japanese patients with nodal peripheral T-cell lymphomas and those with diffuse large B-cell lymphoma in the study group of the Tohoku Hematology Forum. J Clin Exp Hematop (JCEH) 51:29–35
Savage KJ et al (2008) ALK-anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the international peripheral T-cell lymphoma project. Blood 111:5496–5504
Schmitz N et al (2010) Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 116:3418–3425
Escalón MP et al (2005) Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: the M. D. Anderson Cancer Center experience. Cancer 103:2091–2098
Mercadal S et al (2008) Intensive chemotherapy (high-dose CHOP/ESHAP regimen) followed by autologous stem-cell transplantation in previously untreated patients with peripheral T-cell lymphoma. Ann Oncol Off J Eur Soc Med Oncol ESMO 19:958–963
Simon A et al (2010) Upfront VIP-reinforced-ABVD (VIP-rABVD) is not superior to CHOP/21 in newly diagnosed peripheral T cell lymphoma. Results of the randomized phase III trial GOELAMS-LTP95. Br J Haematol 151:159–166
Arkenau H-T et al (2007) Gemcitabine, cisplatin and methylprednisolone for the treatment of patients with peripheral T-cell lymphoma: the Royal Marsden Hospital experience. Haematologica 92:271–272
Kim JG et al (2006) CHOP plus etoposide and gemcitabine (CHOP-EG) as front-line chemotherapy for patients with peripheral T cell lymphomas. Cancer Chemother Pharmacol 58:35–39
Mahadevan D et al (2013) Phase 2 trial of combined cisplatin, etoposide, gemcitabine, and methylprednisolone (PEGS) in peripheral T-cell non-Hodgkin lymphoma: Southwest Oncology Group Study S0350. Cancer 119:371–379
Piccaluga PP, Agostinelli C, Righi S, Zinzani PL, Pileri SA (2007) Expression of CD52 in peripheral T-cell lymphoma. Haematologica 92:566–567
Gallamini A et al (2007) Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 110:2316–2323
Binder C et al (2013) CHO(E)P-14 followed by alemtuzumab consolidation in untreated peripheral T cell lymphomas: final analysis of a prospective phase II trial. Ann Hematol 92:1521–1528
Zinzani PL et al (2007) Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol 25:4293–4297
Kim SJ et al (2012) Bortezomib in combination with CHOP as first-line treatment for patients with stage III/IV peripheral T-cell lymphomas: a multicentre, single-arm, phase 2 trial. Eur J Cancer Oxf Engl 1990(48):3223–3231
Yin J, Wei J, Xu JH, Xiao Y, Zhang YC (2014) Autologous stem cell transplantation as the first-line treatment for peripheral T cell lymphoma: results of a comprehensive meta-analysis. Acta Haematol 131:114–125
Ahn J-S et al (2013) Autologous stem cell transplantation with busulfan, cyclophosphamide, and etoposide as an intensifying frontline treatment in patients with peripheral T cell lymphomas: a multicenter retrospective trial. Ann Hematol 92:789–797
Schetelig J et al (2003) Long-term disease-free survival in patients with angioimmunoblastic T-cell lymphoma after high-dose chemotherapy and autologous stem cell transplantation. Haematologica 88:1272–1278
Jantunen E et al (2004) Autologous stem cell transplantation in adult patients with peripheral T-cell lymphoma: a nation-wide survey. Bone Marrow Transplant 33:405–410
Feyler S et al (2007) The role of high-dose therapy and stem cell rescue in the management of T-cell malignant lymphomas: a BSBMT and ABMTRR study. Bone Marrow Transplant 40:443–450
Kyriakou C et al (2008) High-dose therapy and autologous stem-cell transplantation in angioimmunoblastic lymphoma: complete remission at transplantation is the major determinant of outcome-lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol Off J Am Soc Clin Oncol 26:218–224
Yang D-H et al (2009) Prognostic factors and clinical outcomes of high-dose chemotherapy followed by autologous stem cell transplantation in patients with peripheral T cell lymphoma, unspecified: complete remission at transplantation and the prognostic index of peripheral T cell lymphoma are the major factors predictive of outcome. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 15:118–125
Reimer P et al (2009) Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol Off J Am Soc Clin Oncol 27:106–113
Corradini P et al (2006) Long-term follow-up of patients with peripheral T-cell lymphomas treated up-front with high-dose chemotherapy followed by autologous stem cell transplantation. Leukemia 20:1533–1538
D’ Amore F et al (2012) Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol Off J Am Soc Clin Oncol 30:3093–3099
Le Gouill S et al (2008) Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol Off J Am Soc Clin Oncol 26:2264–2271
Kanakry JA et al (2013) Outcomes of related donor HLA-identical or HLA-haploidentical allogeneic blood or marrow transplantation for peripheral T cell lymphoma. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 19:602–606
Corradini P et al (2014) Intensified chemo-immunotherapy with or without stem cell transplantation in newly diagnosed patients with peripheral T-cell lymphoma. Leukemia. doi:10.1038/leu.2014.79
Kyriakou C et al (2009) Allogeneic stem cell transplantation is able to induce long-term remissions in angioimmunoblastic T-cell lymphoma: a retrospective study from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol Off J Am Soc Clin Oncol 27:3951–3958
Smith SM et al (2013) Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol Off J Am Soc Clin Oncol 31:3100–3109
Zinzani PL et al (1998) Therapy with gemcitabine in pretreated peripheral T-cell lymphoma patients. Ann Oncol Off J Eur Soc Med Oncol ESMO 9:1351–1353
Zinzani PL et al (2010) Gemcitabine as single agent in pretreated T-cell lymphoma patients: evaluation of the long-term outcome. Ann Oncol Off J Eur Soc Med Oncol (ESMO) 21:860–863
Yao Y et al (2013) Gemcitabine, oxaliplatin and dexamethasone as salvage treatment for elderly patients with refractory and relapsed peripheral T-cell lymphoma. Leuk Lymphoma 54:1194–1200
Damaj G et al (2013) Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol Off J Am Soc Clin Oncol 31:104–110
Forero-Torres A, Foss F, Olsen E, Pinter-Brown L, Kim Y (2009) Long-term treatment of CTCL with the oral PNP inhibitor, forodesine. J Clin Oncol 27:15s
Fanale MA et al (2012) A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res 18:248–255
Pro B et al (2012) Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol Off J Am Soc Clin Oncol 30:2190–2196
Oki Y (2013) Safety and efficacy of brentuximab vedotin for treatment of relapsed or refractory mature T-/NK-cell lymphomas. http://onlinelibrary.wiley.com/doi/10.1002/hon.2055/full
Horwitz SM et al (2014) Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 123:3095–3100
Gopal AK et al (2014) Brentuximab vedotin in patients aged 60 years or older with relapsed or refractory CD30-positive lymphomas: a retrospective evaluation of safety and efficacy. Leuk Lymphoma. doi:10.3109/10428194.2013.876496
A Phase III Study of Brentuximab Vedotin plus CHP versus CHOP Therapy for Patients with Previously Untreated CD30-Positive Peripheral T-Cell Lymphoma. Meml Sloan Kettering Cancer Cent http://www.mskcc.org/cancer-care/trial/13-055
Dang NH et al (2007) Phase II trial of denileukin diftitox for relapsed/refractory T-cell non-Hodgkin lymphoma. Br J Haematol 136:439–447
Foss FM et al (2013) A multicenter phase II trial to determine the safety and efficacy of combination therapy with denileukin diftitox and cyclophosphamide, doxorubicin, vincristine and prednisone in untreated peripheral T-cell lymphoma: the CONCEPT study. Leuk Lymphoma 54:1373–1379
Foss FM (2011) Evaluation of the pharmacokinetics, preclinical and clinical efficacy of pralatrexate for the treatment of T-cell lymphoma. Expert Opin. Drug Metab. Toxicol. 7:1141–1152
O’Connor OA et al (2009) Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol Off J Am Soc Clin Oncol 27:4357–4364
O’Connor OA et al (2011) Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol Off J Am Soc Clin Oncol 29:1182–1189
Harrison SJ et al (2012) A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax(®)). Epigenomics 4:571–589
Coiffier B et al (2012) Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol Off J Am Soc Clin Oncol 30:631–636
Coiffier B et al (2014) Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol J Hematol Oncol 7:11
Zain JM, O’Connor O, Zinzani PL, Norman A, Brown de NP (2010) Multicenter, open-label trial of PXD 101 in patients with relapsed/refractory peripheral T-cell lymphoma. J Clin Oncol 28
O’Connor OA et al (2013) Belinostat, a novel pan-histone deacetylase inhibitor (HDACi), in relapsed or refractory peripheral T-cell lymphoma (R/R PTCL): results from the BELIEF trial. J Clin Oncol 31:8507
Kanagal-Shamanna R et al (2013) Differential expression of aurora-A kinase in T-cell lymphomas. Mod. Pathol. Off. J. US. Can. Acad. Pathol. Inc 26:640–647
Qi W et al (2013) Alisertib (MLN8237) an investigational agent suppresses Aurora A and B activity, inhibits proliferation, promotes endo-reduplication and induces apoptosis in T-NHL cell lines supporting its importance in PTCL treatment. Leuk Res 37:434–439
Friedberg JW et al (2014) Phase II study of alisertib, a selective aurora a kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol Off J Am Soc Clin Oncol 32:44–50
National Cancer Institute (2000) In Clin Internet (National Library of Medicine). http://clinicaltrials.gov/ct2/show/NCT01466881
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gooptu, M., Rhoades, R., Pro, B. (2015). Current Management of Peripheral T-Cell Lymphomas. In: Evens, A., Blum, K. (eds) Non-Hodgkin Lymphoma. Cancer Treatment and Research, vol 165. Springer, Cham. https://doi.org/10.1007/978-3-319-13150-4_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-13150-4_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13149-8
Online ISBN: 978-3-319-13150-4
eBook Packages: MedicineMedicine (R0)